

Abstract
Bacteriorhodopsin (BR) and sensory rhodopsin II (SRII), homologous photoactive proteins in haloarchaea, have different molecular functions. BR is a light-driven proton pump, whereas SRII is a phototaxis receptor that transmits a light-induced conformational change to its transducer HtrII. Despite these distinctly different functions, a single residue substitution, Ala215 to Thr215 in the BR retinal-binding pocket, enables its photochemical reactions to transmit signals to HtrII and mediate phototaxis. We pursued a crystal structure of the signaling BR mutant (BR_A215T) to determine the structural changes caused by the A215T mutation and to assess what new photochemistry is likely to be introduced into the BR photoactive site. We crystallized BR_A215T from bicelles and solved its structure to 3.0 Å resolution to enable an atomic-level comparison. The analysis was complemented by molecular dynamics simulation of BR mutated in silico. Three main conclusions regarding the roles of photoactive site residues in signaling emerge from the comparison of BR_A215T, BR, and SRII structures: (i) the Thr215 residue in signaling BR is positioned nearly identically with respect to the retinal chromophore as in SRII, consistent with its role in producing a steric conflict with the retinal C14 group during photoisomerization, proposed earlier to be essential for SRII signaling from vibrational spectroscopy and motility measurements; (ii) Tyr174–Thr204 hydrogen bonding, critical in SRII signaling and mimicked in signaling BR, is likely auxiliary, for example, to maintain Thr204 in the proper position for the steric trigger to occur; and (iii) the primary role of Arg72 in SRII is spectral tuning and not signaling.
Keywords: microbial rhodopsins, sensory rhodopsin II, bacteriorhodopsin, light sensor, phototaxis
Introduction
The molecular functions of bacteriorhodopsin (BR) and sensory rhodopsin II (SRII), two homologous photochemically reactive proteins in haloarchaeal membranes, are distinctly different. BR is a light-driven pump that uses the energy of a photon to translocate an ion vectorially across the membrane, whereas SRII is a phototaxis receptor that transmits a light-induced conformational change to the membrane-bound transducer protein HtrII, which in turn modulates a cytoplasmic kinase that controls the motility of the cell.1–4 X-ray crystal structures of BR5 [Protein Data Bank (PDB) ID: 1C3W] and SRII6 (PDB ID: 1JGJ) show similar overall structures. However, the detailed structures differ greatly since there is only 26% residue identity. There is greater residue conservation in the retinal-binding pockets of the proteins, but even residues that are conserved are, in some cases, in different positions and exhibit different interactions (Fig. 1). Despite these functional and structural differences and as a remarkable testament to the ease by which nature can evolve a new function in a protein, a single residue substitution, Thr for Ala215 in the BR retinal-binding pocket, enables the photochemical reactions of this BR mutant A215T (“signaling BR”) to transmit signals to an HtrII molecule to mediate phototaxis by the cell.7 Two additional mutations on the lipid facing surface of BR, corresponding to residues in SRII that form hydrogen bonds with HtrII in the SRII–HtrII interface, greatly enhance the signaling efficiency by BR_A215T, presumably by aligning the mutant BR and HtrII for more efficient signal transfer.7
Fig. 1.

Comparison of crystal structures of native BR (PDB ID: 1C3W), A215T mutant BR, and SRII (PDB ID: 1JGJ). (a) Relative positions of Tyr185/Tyr174, Asp212/Asp201, Tyr57/Tyr51, and Thr215/Thr204 (Ala215 in native BR) in the three proteins. The hydroxyl group of Thr215/Thr204 is shown as a dark-yellow sphere and as a dark-blue sphere (respectively). Asp212 of BR_A215T is rotated ~60° around the χ2 angle relative to both native BR and SRII. (b) Relationship between Arg82/Arg72 and retinal in all three proteins. The position of Arg82 in the native and mutant BR structures is almost identical. (c) Relative positions of retinal and Thr215/Thr204 in BR_A215T (yellow) and SRII (blue). Distance between threonine and retinal represented by an arrow. (d) Distance between hydroxyl oxygens of Tyr185/Tyr174 and Thr215/Thr204 in BR_A215T (red) and SRII (blue) during the last 10 ns of the molecular dynamics simulations. A broken line at 3.2 Å represents the hydrogen-bond cutoff. While the Tyr174–Thr204 hydrogen bond remains strong throughout the duration of the SRII simulation, the bond is weak in BR_A215T and fluctuates between 3 and 4 Å.
We pursued a crystal structure of signaling BR to compare with the structures of native BR and SRII in order to determine the structural changes induced by the A215T mutation and to assess what new photochemistry is likely to be introduced into the BR photoactive site by the mutation. Additionally, a BR_A215T structure would identify features of SRII not found in BR that are also not in BR_A215T, indicating that they are not essential for signaling. Toward these ends, we succeeded in crystallizing signaling BR and solved its structure to 3.0 Å resolution.
Crystallization and structure of signaling BR
After unsuccessful attempts to crystallize BR_A215T using the cubic lipid phase method,5,8 we were able to grow type I membrane protein crystals using the bicelle method developed by Faham and Bowie,9 which has been employed in a number of membrane protein structure determinations.10–12 Bicelle-grown crystals of BR_A215T diffract to 3.0 Å resolution and belong to space group C2 (Table 1). This is the same space group, albeit with different unit cell dimensions, that was reported for wild-type BR crystals obtained by vapor diffusion,13 for which a 2.9-Å structure was published subsequently (PDB code: 1BRR).14 However, only two of the unit cell dimensions are similar, whereas the shortest axis has a length of 80.2 Å for 1BRR type II crystals and a length of 61.5 Å for BR_A215T type I crystals. In both cases, the asymmetric unit contains three molecules that form a C3 trimer. The structure of BR_A215T was solved using molecular replacement with the native BR structure5 (PDB ID: 1C3W) as the search model. Retinal was not included in the search to avoid model bias, but all three independent molecules showed strong electron density extending from Lys216 in Fo − Fc maps. Because of the limited resolution, non-crystallographic symmetry (NCS) restraints were placed on the backbone of the three monomers during all steps of refinement. The A215T mutation was apparent in the Fo − Fc difference map. Other features observed in the electron density were lipid molecules and residues 157–161, which were not part of the search model. The missing residues were built in using the D85S/F219L BR double mutant15 (PDB ID: 1JV6) as a model. We confirmed 40 ordered waters by superimposing the mutant structure on the native structure and by looking for unassigned density that overlap water molecules. Additional water molecules were placed in regions of unassigned density after all protein residues and lipid molecules had been accounted for.
Table 1.
Data collection and refinement statistics
| Data collection | |
| Beamline | 9.1, SSRL, Menlo Park, CA |
| Wavelength (Å) | 0.979 |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 61.452, 106.139, 124.139 |
| α, β, γ (°) | 90.0, 95.08, 90.0 |
| No. of molecules in the asymmetric unit | 3 |
| Resolution range (Å) | 50.0–3.0 |
| Mosaicity range (°) | 0.34–1.15 |
| Total observations | 53,674 |
| Unique reflections | 15,545 |
| Redundancy | 3.5 (2.9) |
| Completeness (%) | 97.5 (92.1) |
| Rmerge (%) | 16.2 (49.1) |
| Average I/σ | 9.43 (2.34) |
| Data processing program | HKL2000 |
| Refinement | |
| Refinement programs | CNS, Refmac5 |
| Resolution range (Å) | 50.0–3.0 |
| R-factor (%) | 23.5 |
| Rfree (%) | 28.8 |
| Solvent content (%) | 54.8 |
| No. of atoms, average B (Å2) | |
| Protein | 5241, 47.2 |
| Water | 46, 24.1 |
| Retinal | 60, 34.5 |
| Lipid | 325, 55.6 |
| RMSD from ideal bond lengths (Å) | 0.010 |
| RMSD from ideal bond angles (°) | 1.55 |
| Ramachandran plot (preferred/allowed/outliers) (%) | 94.0/6.0/0.0 |
The overall structure of BR_A215T does not vary significantly from the native structure (backbone RMSD: 0.6 Å). When individual helices are compared between native and mutant BR, the backbone RMSD of each of the seven helix pairs is below 0.5 Å. However, there is a significant change near the active site of all three monomers in the BR_A215T structure. Both BR and SRII have an aspartic acid residue (residue 212 and 201, respectively) that is coordinated by two tyrosine residues (185/174 and 57/51) via hydrogen bonding between the carboxyl groups of the aspartic acids and the hydroxyl groups of the tyrosines. While BR_A215T retains the hydrogen bond between Asp212:OD2 and Tyr57:OH (Fig. 2a and b), the Asp212:OD1-to-Tyr185:OH bond is broken, and the distance between the two atoms lies outside of the typical hydrogen-bond cutoff distance of 3.2 Å. The carboxylate of Asp212 is rotated about 60° around the χ2 angle relative to both native BR and SRII (Fig. 1a). To confirm the validity of this conformation, we removed the NCS restraints on the BR_A215T residues in question, rotated the side chain of Asp212 to the most common rotamer (as found in BR and SRII), and performed 10 cycles each of TLS (translation/libration/screw-motion) and restrained refinement. For all three independent molecules, the carboxylate of Asp212 moved back to the new conformation, resulting in the loss of hydrogen bonding with Tyr185. 2Fo − Fc density maps also support that the Asp212 side chain is rotated.
Fig. 2.
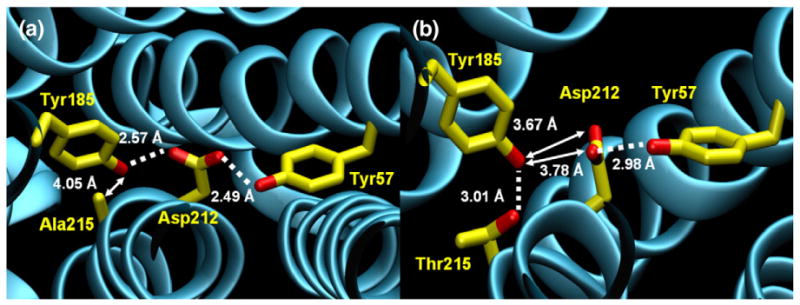
Region of mutation in native and mutant BR crystal structures. Broken lines denote hydrogen bonds, and arrows denote distances. (a) Crystal structure of native BR, with emphasis on Ala215, Tyr185, Asp212, and Tyr57. Asp212 is coordinated by the hydroxyls of two tyrosine residues (57 and 185) via hydrogen bonds. The nonpolar Ala215:CB remains 4 Å from Tyr185. (b) Crystal structure of A215T mutant BR (monomer A), with an emphasis on Thr215, Tyr185, Asp212, and Tyr57. Asp212 continues to accept a hydrogen bond from Tyr57 but is now rotated too distant from Tyr185 for a hydrogen bond. The mutated residue, Thr215, is closer to Tyr185 than the original alanine, forming a hydrogen bond. The mutated residue, Thr215, is closer to Tyr185 than the original alanine, forming a hydrogen bond. Here, we define a hydrogen bond geometrically as having a donor–acceptor distance of <3.2 Å and a donor–hydrogen–acceptor angle of >150°. Range of distances for all three monomers of A215T: Y185(OH) to T215(OG1), 2.90–3.08 Å; Y185(OH) to D212 (OD1), 3.45–3.67 Å; Y185(OH) to D212(OD2), 3.47–3.78 Å; and Y57(OH) to D212(OD2), 2.94–3.27 Å.
Tyr174 in SRII (the homolog of Tyr185 in BR) forms a key hydrogen bond with Thr204 (Ala215 in BR).16 Disruption of this interaction in SRII causes a loss of a steric hindrance17 upon isomerization of retinal, which is a prerequisite for signal transfer. A similar hindrance is introduced by the A215T mutation in BR,18 suggesting that the steric hindrance is a sufficient addition to the structural changes characteristic of microbial rhodopsin photo-cycles to confer a phototaxis signaling function. In the BR_A215T structure, the distance between Tyr185:OH and Thr215:OG1 is 3.01 Å (Fig. 2b). Hydrogen bonding is favored between Tyr185 and Thr215 in the ground state, coupled with a decrease in interaction between Tyr185 and Asp212.
A key factor that causes the difference in absorption maximum between BR and SRII is the positioning of arginine 82/72 with respect to the retinal.19 The guanidinium group of Arg72 in SRII is flipped 180° relative to the guanidinium group of Arg82 in native BR. We observed no change in the position of Arg82 in BR_A215T (Fig. 1b). The small spectral shift between native BR and BR_A215T can be explained by the introduction of the threonine residue near the retinal molecule. The threonine residues in BR_A215T and SRII are in nearly identical positions with respect to the retinal (Fig. 1c), and it appears that the threonine is causing a shift in absorbance toward shorter wavelengths from that of native BR. However, the absence of a change in the arginine position results in the spectral shift not being nearly as large as the spectral difference between BR and SRII.
Molecular dynamics
Using native BR (PDB ID: 1C3W)5 as a starting structure, we mutated Ala215 to a threonine in silico for molecular dynamics simulations. We run 100-ns simulations of three systems—native BR, BR_A215T, and SRII—in parallel, each in a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) bilayer in excess water. Interestingly, the molecular dynamics simulation predicted the very same disruption of the hydrogen bond between Asp212 and Tyr185 in BR_A215T, yet this hydrogen bond remained intact in the control simulations (Fig. 3a and b). The Tyr185:OH-to-Asp212:OD1 distance shows very little fluctuation during a 10-ns sampling of native BR trajectory, yet the same distance measurement in the trajectory of BR_A215T remains above the hydrogen-bond-distance cutoff of 3.2 Å (Fig. 3c and d).
Fig. 3.
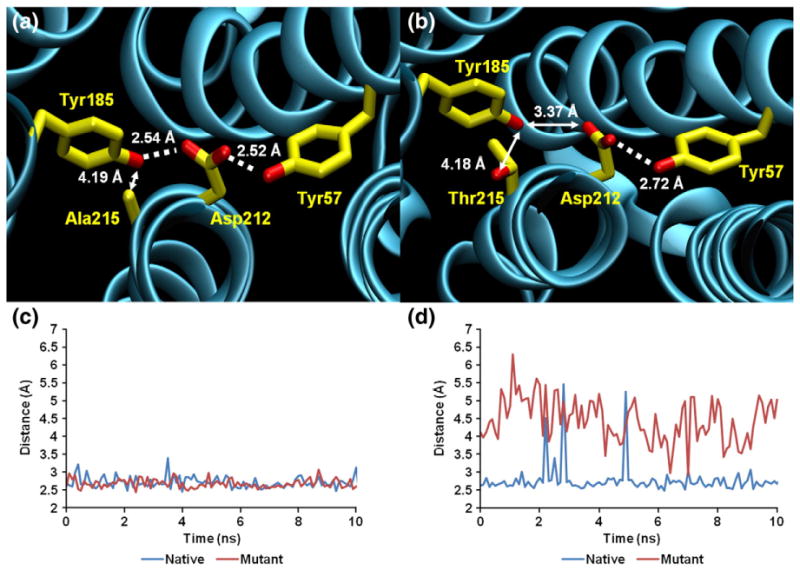
Region of mutation in native and mutant BR molecular dynamics simulations. Broken lines denote hydrogen bonds, and arrows denote distances. (a) Snapshot of native BR molecular dynamics simulation taken after ~80 ns. Similar to the crystal structure, Asp212 is coordinated by Tyr185 and Tyr57 through hydrogen bonds. The nonpolar Ala215:CB is >4 Å from Tyr185. (b) Snapshot of A215T mutant BR molecular dynamics simulation at ~80 ns. Asp212 continues to accept a hydrogen bond from Tyr57 but is too distant from Tyr185 to accept a second hydrogen bond. Tyr185 is still >4 Å from the mutated residue, Thr215. (c) Distance between Asp212:OD2 and Tyr57:OH in both native and mutant simulations over the last 10 ns of trajectory. (d) Distance between Asp212:O12 and Tyr185:OH in both native and mutant simulations over the last 10 ns of trajectory. Hydrogen bonds are defined as in Fig. 2.
The distance between Tyr185 and Thr215 decreased over time and reached a baseline around 4.0 Å. However, we saw that the distance fluctuates between 3 and 4 Å once equilibrium was established (Fig. 1d), suggesting that a weak hydrogen bond forms intermittently. The simulation also provided us with an in-depth look at the arrangement of water molecules near the active site as a function of time, something that was not attainable through crystallography alone. The mutant BR showed an arrangement of waters that is different from those of BR and SRII. Substitution of the nonpolar Ala215 with threonine changed the electrostatic environment in the region of the retinal pocket. In the A215T simulation, the equilibrated system includes a network of several hydrogen-bonded waters that extend from the retinal pocket to the backbone oxygen of Thr46 near the cytosolic region of the protein (Fig. 4). This organization of water molecules was not seen in either the native BR or the SRII simulations. In the BR_A215T system, the complex that is formed between Tyr185, Thr215, and Asp212 is mediated by water molecules; the connections between Tyr185 and Asp212 and between Thr215 and Tyr185 are mediated in the simulation via a single water molecule (Fig. 5a and b). It is interesting that the additional waters are only present in A215T. It is possible that the additional water in the retinal pocket optimizes the geometry between Thr215–Tyr185–Asp212 in the A215T structure. We also note that the Tyr185–Asp212 interaction is similar in both the native BR and the SRII structures yet is altered in the A215T structure. Our interpretation is that the precise position and environment of this pair is not essential for signaling, although we cannot exclude that the exact path in which the signaling activity is propagated to the HtrII surface differs between A215T_BR and SRII.
Fig. 4.
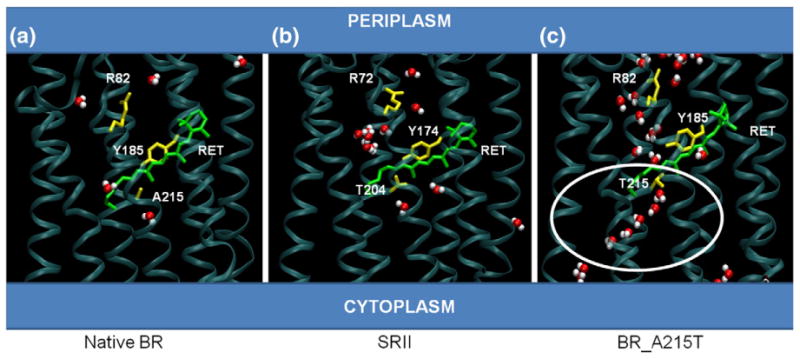
Water coordination near photoactive sites of BR, SRII, and BR_A215T. (a) Snapshot of native BR molecular dynamics simulation taken at ~80 ns. Water molecules (red and white) are localized between retinal and Arg82. (b) Snapshot of native SRII simulation taken at ~80 ns. Water molecules (red and white) are localized near retinal and Arg72, although at least two molecules are seen toward the cytoplasmic region of the protein. (c) Snapshot of BR_A215T mutant simulation taken at ~80 ns. A chain of water molecules (red and white) is visible from the cytoplasmic region above the retinal, and this extends into a region between retinal and Arg82, toward the periplasmic region. This arrangement is not observed in native BR or SRII.
Fig. 5.
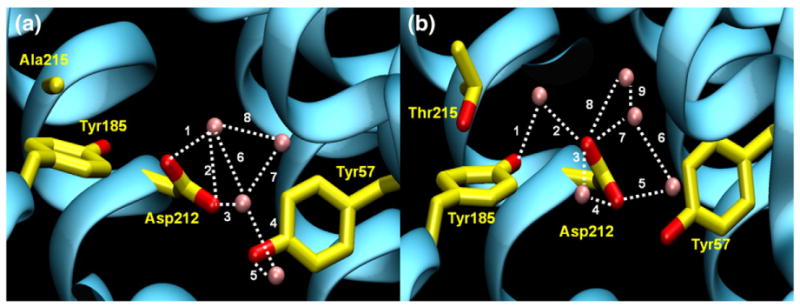
Water molecules near the mutation site in both native and mutant BR molecular dynamics simulations. Possible hydrogen bonding involving water molecules indicated with broken lines. (a) Snapshot of native BR molecular dynamics simulation at ~80 ns. Four water molecules (shown as pink spheres) are within 5 Å of Tyr185, Asp212, and Tyr57. Distances of bonds are (1) 2.70 Å, (2) 3.16 Å, (3) 2.61 Å, (4) 2.80 Å, (5) 2.96 Å, (6) 2.87 Å, (7) 3.14 Å, and (8) 2.89 Å. (b) Snapshot of A215T mutant BR molecular dynamics simulation at ~80 ns. Five water molecules (shown as pink spheres) are within 5 Å of Tyr185, Asp212, Tyr57, and Thr215. Distances of bonds are (1) 2.68 Å, (2) 2.56 Å, (3) 2.84 Å, (4) 2.55 Å, (5) 2.82 Å, (6) 2.80 Å, (7) 2.92 Å, (8) 2.84 Å, and (9) 3.02 Å. Hydrogen bonds are defined as in Fig. 2.
The photoactive site of signaling BR: Implications for the phototaxis signaling mechanism
Mutational disruption of the hydrogen-bonded Thr204–Tyr174 pair in the SRII photoactive site by substituting Phe for Tyr174 or Ala for Thr204 results in loss of phototaxis signaling,16 and the Thr–Tyr pair has been shown by cryogenic FTIR (Fourier transform infrared spectroscopy) to be responsible for a steric conflict with retinal in the SRII–HtrII complex during photoisomerization.17 The A215T mutation creates a similar steric conflict in BR18 and converts the BR molecule into a functional SRII-like photosensor.7 Based on these observations, the Thr–Tyr hydrogen-bonded pair was proposed to provide a key component of a steric trigger activation mechanism in both SRII and signaling BR. The A215T structure shows that Thr215 is located in a nearly identical position with respect to the retinal as Thr204 in SRII, and its hydroxyl group is within 3.01 Å of that of Tyr185, similar to the 3.20 Å in SRII. The BR_A215T structure and simulation data are consistent with Thr215 forming a hydrogen bond with Tyr185, mimicking the Thr204–Tyr174 pair in SRII. However, the structure shows large differences in the interactions and environment of Tyr185 in BR_A215T compared to Tyr174 in SRII. Mutation of Tyr174 to Phe in SRII causes loss of signaling and loss of the steric conflict during retinal photoisomerization.16 The different local environments of Tyr185 in BR_A215T and Tyr174 in SRII argue that Tyr174 itself is essential because of an auxiliary rather than direct role in signaling, for example, to maintain Thr204 in the proper position for the steric trigger to occur.
There are two major structural differences between BR and SRII photoactive sites.5,6 First, a dramatic difference is that Arg82 is flipped ~180° so that its positive charge points away from the retinylidene Schiff base in SRII and toward the Schiff base in BR. Second, the hydrogen-bonded Thr–Tyr pair exists in SRII and creates a steric conflict with the retinal during photoisomerization, whereas BR lacks the hydrogen-bonded pair since it lacks the Thr residue. Based on these facts, the BR_A215T structure gives us two important insights into the signaling mechanism.
The position of SRII Arg72 (Arg82 in BR) is at most only of minor importance for signaling since it is not flipped in BR_A215T. This conclusion was not evident before the BR_A215T structure since the different position of the Arg in SRII compared to BR produces several major differences in hydrogen bonding. As concluded from molecular orbital calculations,19 the repositioning of Arg82 explains well the shift in absorption maximum of SRII (490 nm) compared to that of BR (568 nm), and the fact that A215T without the flipped Arg is not so strongly blue shifted (550 nm) further supports that explanation. Therefore, the new structure provides compelling evidence that the primary role of Arg72 in SRII is spectral tuning and not signaling.
A major question is whether the Tyr or the Thr residue or both further propagates the signal to the interface. Both are hydrogen bonded to residues in or near the interface. The BR_A215T structure argues strongly that it is Thr204 that is crucial for the signal propagation. The Tyr (a) is greatly altered in its hydrogen bonding in A215T compared to BR and SRII and (b) has lost its connection to Asp212, a key residue between the Tyr and the interface. If the Tyr were a crucial residue in the post-isomerization signaling reactions, one would expect it to be unable, being so greatly altered from the SRII case, and the steric trigger to still function in signaling. In contrast, the introduced Thr in BR_A215T assumes a position with respect to the retinal and the surrounding residues essentially identical with that of Thr204 in SRII. Therefore, the result favors that Thr204 in SRII propagates the signal out of the retinal pocket to the interface. This result from the structure is in line with vibrational spectra from FTIR of C14D-labeled retinal, indicating that the steric constraint during isomerization is between Thr204 and the C14H group of retinal.20 In this view, the lack of signaling function (and the lack of steric hindance) in the SRII mutant Y174F would be explained in terms of the hydrogen bond between Thr and Tyr being necessary to maintain Thr204 in the proper position for the steric trigger to occur in SRII.
Methods
Crystallization
Halobacterium salinarum cells transformed with a BR_A215T expression plasmid were cultured in CM medium plus mevinolin as described previously.16 BR_A215T purple membrane (λmax, 554 nm) was isolated essentially as described for native BR.21 The concentrated suspension of cells in 4 M NaCl was dialyzed overnight against 0.1 M NaCl. The dialysate was washed twice with H2O, layered over a sucrose step gradient (30%, 50%, and 60%), and centrifuged 18 h at 100,000g. The purple band was collected, and sucrose was removed by centrifugation and washing twice with H2O and finally resuspended at 10 mg/ml. The suspension was mixed with 5 mg of polar lipid extract22 from H. salinarum strain S9 in 1% octylglucoside and incubated at 4 °C with gentle agitation for 1 h. The protein–lipid mixture was combined in a 4:1 ratio with a 40% 1,2-dimyristoyl-sn-glycero3-phosphocholine/3-(chloramidopropyl)-dimethylammonio-2-hydroxyl-1-propane-sulfonate (2.8:1) bicellar solution following the procedure of Faham and Bowie.9 Crystals were grown in hanging drops at 28 °C using the vapor-diffusion method. Drops contained 6 μl of protein/bicelle mix plus 2.5 μl of well solution [3.0 M NaH2PO4 (pH 3.6)].
The polar lipid extract was assayed by negative mode electrospray mass spectrometry and HPLC to assess lipid content (analysis conducted by Avanti Polar Lipids, Inc., Alabaster, AL). Ninety-nine percent of the extract consisted of phosphatidylglycerol, phosphatidylglycerol sulfate, archaeal glycocardiolipin, glycolipid sulfate, and phosphatidylglycerophosphate methyl ester, similar to those obtained from cells.23
X-ray diffraction
Crystals of BR_A215T diffracted to 3.0 Å resolution at the Stanford Synchrotron Radiation Laboratory, beamline 9-1. Data were collected at 100 K as 360 frames with 1° rotation each. Data were processed using HKL200024 (see Table 1 for crystallographic statistics), followed by molecular replacement using Phaser25 (CCP4 suite) with native BR5 (PDB ID: 1C3W) as the search model. Retinal was not included in the search to avoid bias, but all three independent molecules showed strong electron density extending from Lys216 in Fo − Fc maps contoured at 3 σ. Refinement and further model building were performed using CNS26 and Coot.27 Because the resolution was 3.0 Å, NCS restraints of backbone atoms were employed at all steps of refinement. The A215T mutation was apparent in the Fo − Fc difference map. Other features observed in the electron density were lipid molecules and residues 157–161, which were not part of the search model. These residues were built using the D85S/F219L BR double mutant15 (PDB ID: 1JV6) as a model. Lipid molecules were built manually using lipids from the native BR model5 (PDB ID: 1C3W). We identified 40 ordered waters and confirmed them by superimposing the mutant structure on the native structure and by looking for unassigned density overlapping water molecule from the native high-resolution BR model. Additional water molecules were placed into unassigned density after accounting for all protein residues and lipid molecules.
Molecular dynamics
Simulations of three systems—native BR, BR_A215T, and SRII—were run in parallel, each in a POPC bilayer (~230 lipid molecules) in excess water (~12,000 water molecules) with a single protein monomer and sufficient counterions to give the system a net zero charge. Each simulation was roughly 100 ns in length. We placed each monomer in a pre-equilibrated bilayer and relaxed the systems by running first for 100 ps with the protein backbone fixed, then 100 ps sequentially with backbone restraints of 50, 20, 10, 5, 2, and 1 kcal/mol/Å. All simulations were performed with the NAMD 2.6 software package.28 The CHARMM22 and CHARMM32 force fields29,30 were used for the protein and lipids, respectively, and the TIP3P force field31 was used for water. Parameters for the retinal were taken from quantum chemical calculations performed by Nina et al.32 and Tajkhorshid et al.33 The smooth particle Ewald method34,35 was used to calculate electrostatic interactions, and the short-range real-space interactions were cut off at 11 Å, employing a switching function. A reversible multiple-time-step algorithm36 was employed to integrate the equations of motion with time steps of 4 fs for electrostatic forces, 2 fs for short-range nonbonded forces, and 1 fs for bonded forces. All bond lengths involving hydrogen atoms were held fixed using the SHAKE37 and SETTLE38 algorithms. Molecular graphics and simulation analyses were performed using VMD 1.8.7.39 A Langevin dynamics scheme was used for temperature control, and a Nosé-Hoover-Langevin piston was used for pressure control.40,41
Acknowledgments
This work was supported by National Institutes of Health grants R37GM27750 (J.L.S.), R01AI07000 (H.L.), and P01GM86685 (D.J.T.); Department of Energy grant DE-FG02-07ER15867 (J.L.S.); National Science Foundation grant CHE-0750175 (D.J.T.); an endowed chair AU-0009 from the Robert A. Welch Foundation (J.L.S.); and a Chancellor’s Fellowship (H.L.). G.O. was supported by training grant T15LM07443 from the National Library of Medicine. Computer time was provided by the TeraGrid resources at the Texas Advanced Computing Center supported by the National Science Foundation and the University of California Shared Research Computing Services Pilot Project.
Abbreviations used
- BR
bacteriorhodopsin
- SRII
sensory rhodopsin II
- PDB
Protein Data Bank
- NCS
non-crystallographic symmetry
Footnotes
Accession code
Structure factors and refined coordinates have been deposited in the PDB under accession code 3T45.
References
- 1.Spudich JL, Yang CS, Jung KH, Spudich EN. Retinylidene proteins: structures and functions from archaea to humans. Annu Rev Cell Dev Biol. 2000;16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 2.Lanyi JK, Luecke H. Bacteriorhodopsin. Curr Opin Struct Biol. 2001;11:415–419. doi: 10.1016/s0959-440x(00)00226-8. [DOI] [PubMed] [Google Scholar]
- 3.Klare JP, Chizhov I, Engelhard M. Microbial rhodopsins: scaffolds for ion pumps, channels, and sensors. Results Probl Cell Differ. 2008;45:73–122. doi: 10.1007/400_2007_041. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki J, Spudich JL. Signal transfer in haloarchaeal sensory rhodopsin–transducer complexes. Photochem Photobiol. 2008;84:863–868. doi: 10.1111/j.1751-1097.2008.00314.x. [DOI] [PubMed] [Google Scholar]
- 5.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 Å resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 6.Luecke H, Schobert B, Lanyi JK, Spudich EN, Spudich JL. Crystal structure of sensory rhodopsin II at 2.4 angstroms: insights into color tuning and transducer interaction. Science. 2001;293:1499–1503. doi: 10.1126/science.1062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sudo Y, Spudich JL. Three strategically placed hydrogen-bonding residues convert a proton pump into a sensory receptor. Proc Natl Acad Sci USA. 2006;103:16129–16134. doi: 10.1073/pnas.0607467103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 11.Luecke H, Schobert B, Stagno J, Imasheva ES, Wang JM, Balashov SP, Lanyi JK. Crystallographic structure of xanthorhodopsin, the light-driven proton pump with a dual chromophore. Proc Natl Acad Sci USA. 2008;105:16561–16565. doi: 10.1073/pnas.0807162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, et al. The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schertler GF, Bartunik HD, Michel H, Oesterhelt D. Orthorhombic crystal form of bacteriorhodopsin nucleated on benzamidine diffracting to 3.6 Å resolution. J Mol Biol. 1993;234:156–164. doi: 10.1006/jmbi.1993.1570. [DOI] [PubMed] [Google Scholar]
- 14.Essen L, Siegert R, Lehmann WD, Oesterhelt D. Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin–lipid complex. Proc Natl Acad Sci USA. 1998;95:11673–11678. doi: 10.1073/pnas.95.20.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouhani S, Cartailler JP, Facciotti MT, Walian P, Needleman R, Lanyi JK, et al. Crystal structure of the D85S mutant of bacteriorhodopsin: model of an O-like photocycle intermediate. J Mol Biol. 2001;313:615–628. doi: 10.1006/jmbi.2001.5066. [DOI] [PubMed] [Google Scholar]
- 16.Sudo Y, Furutani Y, Kandori H, Spudich JL. Functional importance of the interhelical hydrogen bond between Thr204 and Tyr174 of sensory rhodopsin II and its alteration during the signaling process. J Biol Chem. 2006;281:34239–34245. doi: 10.1074/jbc.M605907200. [DOI] [PubMed] [Google Scholar]
- 17.Sudo Y, Furutani Y, Shimono K, Kamo N, Kandori H. Hydrogen bonding alteration of Thr-204 in the complex between pharaonis phoborhodopsin and its transducer protein. Biochemistry. 2003;42:14166–14172. doi: 10.1021/bi035678g. [DOI] [PubMed] [Google Scholar]
- 18.Sudo Y, Furutani Y, Spudich JL, Kandori H. Early photocycle structural changes in a bacteriorhodopsin mutant engineered to transmit photosensory signals. J Biol Chem. 2007;282:15550–15558. doi: 10.1074/jbc.M701271200. [DOI] [PubMed] [Google Scholar]
- 19.Ren L, Martin CH, Wise KJ, Gillespie NB, Luecke H, Lanyi JK, et al. Molecular mechanism of spectral tuning in sensory rhodopsin II. Biochemistry. 2001;40:13906–13914. doi: 10.1021/bi0116487. [DOI] [PubMed] [Google Scholar]
- 20.Ito M, Sudo Y, Furutani Y, Okitsu T, Wada A, Homma M, et al. Steric constraint in the primary photoproduct of sensory rhodopsin II is a prerequisite for light-signal transfer to HtrII. Biochemistry. 2008;47:6208–6215. doi: 10.1021/bi8003507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oesterhelt D, Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 22.Bergo V, Spudich EN, Scott KL, Spudich JL, Rothschild KJ. FTIR analysis of the SII540 intermediate of sensory rhodopsin II: Asp73 is the Schiff base proton acceptor. Biochemistry. 2000;39:2823–2830. doi: 10.1021/bi991676d. [DOI] [PubMed] [Google Scholar]
- 23.Corcelli A, Lattanzio VM, Mascolo G, Papadia P, Fanizzi F. Lipid–protein stoichiometries in a crystalline biological membrane: NMR quantitative analysis of the lipid extract of the purple membrane. J Lipid Res. 2002;43:132–140. [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips JC, Braun B, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feller SE, Brown CA, Nizza DT, Gawrisch K. Nuclear Overhauser enhancement spectroscopy cross-relaxation rates and ethanol distribution across membranes. Biophys J. 2002;82:1396–1404. doi: 10.1016/S0006-3495(02)75494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 32.Nina M, Smith JC, Roux B. Ab initio quantum chemical analysis of Schiff base-water interactions in bacteriorhodopsin. J Mol Struct. 1993;286:231–245. [Google Scholar]
- 33.Tajkhorshid E, Baudry J, Schulten K, Suhai S. Molecular dynamics study of the nature and origin of retinal’s twisted structure in bacteriorhodopsin. Biophys J. 2000;78:683–693. doi: 10.1016/S0006-3495(00)76626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 35.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 36.Grubmüller H, Heller H, Windemuth A, Schulten K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol Simul. 1991;6:121–142. [Google Scholar]
- 37.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 38.Miyamoto S, Kollman P. An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 39.Humphrey W, Dalke W, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 40.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 41.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]