

Abstract
Cataracts are a major cause of blindness. The most common forms of cataracts are age and UV related and develops mostly in the elderly, while congenital cataracts appear at birth or in early childhood. The Dahl salt-sensitive (SS/Jr) rat is an extensively used model of salt-sensitive hypertension that exhibits concomitant renal disease. In the mid 1980’s, cataracts appeared in a few animals in the Dahl S colony, presumably the result of a spontaneous mutation. The mutation was fixed and bred to establish the SS/Jr-Ctr substrain. The SS/Jr-Ctr substrain has been exclusively used by a single investigator to study the role of steroids and hypertension. Using a classical positional cloning approach, we localized the cataract gene with high-resolution to a less than 1 Mbp region on chromosome 9 using an F1 (SS/Jr-Ctr X SHR) X SHR backcross population. The 1 Mbp region contained only 13 genes, including 4 genes from the γ-crystallins (Cryg) gene family which are known to play a role in cataract formation. All of the γ-crystallins were sequenced and a novel point mutation in the start codon (ATG → GTG) of the Crygd gene was identified which led to the complete absence of CRYGD protein in the eyes of the SS/Jr-Ctr strain. In summary, the identification of the genetic cause in this novel cataract model may provide an opportunity to better understand the development of cataracts, particularly in the context of hypertension.
Keywords: Alternative initiation codon, Dahl salt-sensitive, spontaneous mutation
INTRODUCTION
Cataracts account for approximately 50 percent of the world’s blindness and affect more than 20 million Americans (Zambelli-Weiner, Crews et al.; Ryskulova, Turczyn et al. 2008). In general, cataract formation involves the aggregation of lens proteins, resulting in the clouding of the lens that disrupts the flow of light to the retina and leads to decreased clarity, dull color-contrast, and impaired vision (Michael and Bron 2011). There are several types of cataracts, including nuclear, cortical, and sub-capsular. Nuclear cataracts are the most common and typically take years to develop and primarily occur in the elderly. There are several medical conditions, such as diabetes and hypertension, trauma, or exposure to certain drugs that contribute to the development of cataracts (West 2007). In contrast, congenital cataracts, which can be nuclear, cortical, and sub-capsular, appear at birth or develop in early childhood (Churchill and Graw 2011). These can result from prenatal infection or be directly linked to specific genetic mutations. There have been numerous studies in humans and animal models that have identified genetic mutations in a number of genes, such as crystallins, gap junction proteins, membrane transport and channel proteins, and transcription factors that lead to cataracts (Graw 2009; Huang and He 2010).
The Dahl salt-sensitive (SS/Jr) rat is a widely studied model of salt-sensitive hypertension. The model has been extensively used to study the pathophysiology of hypertension (Zicha, Dobesova et al. 2012) as well as the genetic basis of hypertension (Garrett, Dene et al. 1998; Joe, Saad et al. 2009) and associated kidney disease (Garrett, Joe et al. 2006; Garrett, Gunning et al. 2007; Williams, Johnson et al. 2012). Originally, the Dahl S model was maintained as an outbred stock, but was later inbred by John Rapp by more than 20 generations of brother-sister mating (Rapp and Dene 1985). At some point, animals within the colony demonstrated the appearance of cataracts, presumably the result of a spontaneous mutation (Rapp 1986). Despite the development of cataracts in these animals, they appeared to be phenotypically similar with regard to the development of salt-sensitive hypertension compared to non-cataract Dahl S rats (Rapp 1986). Ultimately, the Dahl S animals that exhibited cataracts were provided exclusively to Elise Gomez-Sanchez and later denoted as SS/Jr-Ctr.
The SS/Jr-Ctr strain has been extensively used for studies investigating the role of steroids with hypertension (Gomez-Sanchez, Zhou et al. 1996; Gomez-Sanchez, Gomez-Sanchez et al. 2010; Oki, Gomez-Sanchez et al. 2012), but there has been a single report characterizing the cataracts exhibited in this strain (Margo, Gomez-Sanchez et al. 1987). Light and electron microscopy analysis of eyes from the SS/Jr-Ctr strain demonstrated lens capsular defects and marked degenerative changes of lens fibers when examined at advanced age. Lens fibers showed cell membrane ruptures and dissipation, liquefied lens proteins, and “dark” bodies inferring necrosis, followed by shrinking and calcification at 30–40 days after birth, and finally separation from the zonules and luxation (Margo, Gomez-Sanchez et al. 1987). However, the specific genetic defect that leads to the development of cataracts in the SS/Jr-Ctr has not been addressed.
The purpose of our study was to identify the gene and specific mutation(s) that are responsible for the development of cataracts in the eyes of the SS/Jr-Ctr strain and whether the genetic mutation could have some impact on the cardiovascular and renal complications exhibited by the strain. To achieve this goal, we performed a systematic genome wide linkage analysis for cataract status as well as related traits, sequencing of candidate genes, and ultimately demonstrated by western blot analysis that the gene mutation leads to an apparent knockout at the protein level.
MATERIAL AND METHODS
Animals, Study Design, and Segregating Population
All experiments were approved by the Institutional Animal Care and Use Committee. The Dahl salt-sensitive (SS/Jr) and the spontaneously hypertensive rat (SHR//NHsd) inbred strains are maintained at the University of Mississippi Medical Center. The SS/Jr-Ctr strain was obtained from E. Gomez-Sanchez at GV (Sonny) Montgomery VAMC, Jackson, MS.
A segregating population was derived from SS/Jr-Ctr and SHR to map the cataract mutation. SS/Jr-Ctr males (n=4) were breed to SHR females (n=7) to generate F1(SS/Jr-Ctr X SHR). F1 animals (n=57) were backcrossed to SHR (n=19), generating an F1(SS/Jr-Ctr X SHR) X SHR backcross population (n=146), denoted as BC. At day 21, the BC population was characterized for body weight (BW), eye weight (EW), presence or absence of cataract, eye to eye distance (EED), eye to nose difference (END), and histology.
Genotyping and Sequencing
DNA was obtained from spleen or tail biopsy and prepared using Wizard SV 96 Genomic DNA kit (Promega, San Luis Obispo, CA). The BC population was genotyped using two approaches: (1) whole genome single nucleotide polymorphism (SNP) genotyping (Kbioscience, UK). A total of 112 SNP markers were tested through-out the genome; and/or (2) microsatellite analysis (n=10) using standard PCR-ethidium bromide-gel electrophoresis or using a fluorescent-based approach on a Beckman Coulter CEQ8000 XL capillary sequencer (Beckman Coulter, Brea, CA) (Garrett, Joe et al. 2006; Garrett, Gunning et al. 2007).
Primers were designed from known sequences (www.ncbi.nlm.nih.gov or www.ensembl.org) to amply the coding regions of the γ-crystallin (Cryg) family b–e (Table 1). All primers were tagged with either M-13 Forward (5′ GTAAAACGACGGCCAGT 3′) or Reverse (5′ CAGGAAACAGCTATGAC 3′) for sequencing analysis. PCR was performed using SS/Jr, SS/Jr-Ctr, F1 (SS/Jr-Ctr X SHR), and SHR genomic DNA samples, purified using PureLink PCR Purification Kit (Invitrogen, Carlsbad, CA) and prepared for fluorescence-based DNA sequencing on CEQ8000 using DTCS Quick Start Kit (Beckman Coulter, Brea, CA). Sequencing reads were assessed for quality and aligned to the BN reference sequence using the DNASTAR’s Lasergene v7.2 software package. SNP were visually confirmed in trace files and identified variants were verified by direct sequencing from genomic DNA isolated from at least three rats per strain.
Table I.
Genomic DNA primers used to amplify γ-Crystallin gene family (Crygb-Crygf)
| Primer Name | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|
| Crygb Exon1–2 | TGTGTGATTTCCTGTGGAGG | TGCTCTAATTGAAACGACTTGG |
| Crygb Exon3 | GAGGTGTCGGGACAGGAAAC | GGAAGTTCCTTCCCATCTCTG |
| Crygc Exon1–2 | TCCGCTAACACAGCAACAAC | GACAAACAAACCCTATTCTCTGG |
| Crygc Exon3 | CGTTGTTTAGCTTGGGTGAG | TGTAGGGAGAGTTTACAGGTGG |
| Crygd Exon1–2 | CCTTTTGTGCTGTTCCTGC | GGCAACTGCAGTCAGGGTC |
| Crygd Exon3 | ATGGCTTGATGCAGCTGAG | CCAATAAACATGAGCAAACAGG |
| Cryge Exon1–2 | TTGTGCTGTTCCTGCCAAC | CGGCCACAGATGTTACTAGC |
| Cryge Exon3 | TGGGTTTCCAAGTTCAGGTC | CATCCTTTCTTTGCTGTGCC |
All Crygb-e genes consist of three exons. The first primer pair was used to amplify exon 1–2 (including intron 1) and the second primer pair was used to amplify exon 3.
Western-Blot Analysis
Whole eyeballs from SS/Jr, SS/Jr-Ctr, F1(SS/Jr-Ctr X SHR), and SHR rats were homogenized in RIPA lysis buffer (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), normalized, and 50–100ug of total protein were evaluated using standard polyacrylamide-gel electrophoresis on 4–10% Mini-PROTEAN TGX gels (Bio-Rad Laboratories, Hercules, CA) as previously described (Williams, Johnson et al. 2012). Samples were transferred to PDVF membranes using Trans-Blot Turbo System (Bio-Rad Laboratories, Hercules, CA). Blots were probed with monoclonal anti-CRYGD antibody (Sigma-Aldrich, St. Louis, MO) using 1:500 dilution and 1:000 goat anti-mouse (HRP) as secondary. Pierce ECL Substrate (Thermo Scientific) was prepared according to the manufacturers’ instructions and added to membranes. The membranes were imaged using a ChemiDoc XRS+ System (Bio-Rad). Equal loading of protein was confirmed by GAPDH (Millipore; MAB374) on same blot probed with CRYGD.
Histological Assessment
Whole eyeballs isolated from SS/Jr and SS/Jr-Ctr were fixed in buffered formalin and embedded in paraffin, cut into 5-μm sections and stained with hematoxylin and eosin (H&E). Unstained sections were stained with Von Kossa stain kit (American Mastertech Scientific, CA) to confirm calcium deposits observed under H&E. Histological images were captured using Nis-Elements image analysis software (Version 3.03, Nikon Instruments Inc., Melville, NY) on Nikon 55i microscope with DS-Fi1 5-Meg Color C digital camera (Nikon, Melville, NY).
Statistical and Genome-Wide Linkage Analysis
Linkage analysis was performed using Windows QTL Cartographer V2.5_010 (Li, Wang et al. 2006; Wang, Basten et al. 2012). The BC population was analyzed using interval mapping function for continuous traits (BW, EW, EED, and END) and category trait analysis for cataract status. The LOD threshold for significance was calculated by permutation testing (1:1000). Additional statistical comparisons of phenotypic data (e.g., eye weight) were evaluated by independent t-test (SPSS, Chicago, IL) and p< 0.05 was considered to be statistically significant. All data are presented as mean ± standard error (SE).
RESULTS
The presence of cataracts in the lens of SS/Jr-Ctr animals were immediately obvious from the time their eyes opened (~day 15–16). Eyes from SS/Jr-Ctr animals demonstrate an opaque white color in the center of the lens, while the lenses from wild-type SS/Jr animals appear transparent (Fig. 1). Eyes from SS/Jr-Ctr animals (or heterozygous animals) were significantly smaller than eyes from SS/Jr, regardless of sex (Fig. 2). The distance from eye to eye or eye to nose were also significantly reduced in the SS/Jr-Ctr compared SS/Jr. Histological assessment of eyes from SS/Jr-Ctr revealed the clear presence of a nuclear cataract, disruption of lens fibers and calcification, whereas SS/Jr eye sections demonstrated normal appearance (Fig. 3).
Figure 1. Gross appearance of nuclear cataract at 21 days of age.

(A) Normal appearance of eye in wild-type SS/Jr strain. (B) Appearance of cataract in eye of SS/Jr-Ctr strain which exhibits spontaneous cataracts. (C) Gross dissection of eye from wild-type and (D) cataract animals. The lens of the SS/Jr eye is clear and transparent, whereas the lens from the SS/Jr-Ctr eye is opaque and cloudy.
Figure 2. Measurement of eye weight in SS/Jr and SS/Jr-Ctr animals.
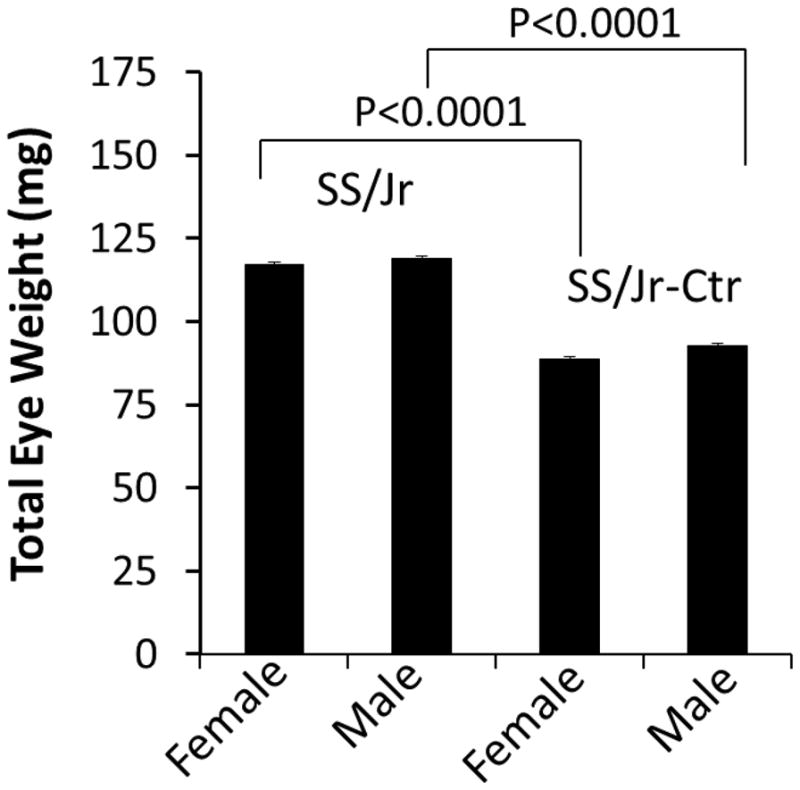
Total eye weight (left + right eye) is shown for each strain and sex. For either sex, eyes from SS/Jr-Ctr were significantly smaller than eyes from SS/Jr (n=6).
Figure 3. Representative whole-eye and high resolution images of lens from SS/Jr and SS/Jr-Ctr at 21 days of age.

(A–B) Longitudinal histology section through whole-eye (1X, H&E) which demonstrates the normal appearance of key anatomical structures in each strain, aside from obvious lens fiber changes observed in eye from SS/Jr-Ctr. (C–D) Higher magnification images (10X, H&E) showing normal lens appearance in eye from SS/Jr and disruption of lens fibers in eye from SS/Jr-Ctr. (E–F) Lens from SS/Jr-Ctr also demonstrates distinct calcification (40X, Von Kossa stain), whereas wild-type SS/Jr have normal appearance.
To map and identify the causative genetic variant(s) responsible for cataracts in the SS/Jr-Ctr strain, a segregating population was developed using the SS/Jr-Ctr and the spontaneously hypertensive rat (SHR). The SHR was selected as a contrasting strain because previous linkage analyses were performed to identify quantitative trait locus (QTL) for cardiovascular and renal traits in similar segregating populations (Garrett, Dene et al. 2003; Garrett, Joe et al. 2006). All F1(SS/Jr-Ctr X SHR) animals developed cataracts confirming the causative mutation was completely dominant. An F1(SS/Jr-Ctr X SHR) X SHR population was evaluated for several traits, including body weight (BW), eye weight (EW), eye to eye distance (EED), eye to nose difference (END) and presence or absence of cataracts. A total of 112 SNP markers were tested across the entire genome and genome-wide interval mapping was performed. A single, highly significant (LOD>28) locus was identified on chromosome 9 for cataract status (Fig. 4). QTL for EW (left, right, or total) were also identified at the same location as cataract status, but no QTL was observed for EED or END. A suggestive QTL for EW (LOD=2.6) was also observed on chromosome 15.
Figure 4. Genome-wide linkage analysis for cataract status in F1(SS/Jr-Ctr X SHR) X SHR population at 21 days of age.

(A) LOD plot of cataract status (category trait analysis-presence or absence). (B) LOD plot of continuous traits, including body weight, eye weight (left, right, and total), eye to eye or eye to nose distance. The chromosome location is shown on the x-axis and LOD score is shown on the y-axis. The significance threshold for significant quantitative trait locus (QTL) was determined by permutation testing and is denoted by the line around LOD~3.
To better define the region on chromosome 9 containing the cataract mutation, animals were classified by genotype at multiple markers (hapolytype analysis) (Fig. 5A). Five distinct haplotypes were observed in the region between 40.2 to 68.1 Mbp. Animals with haplotype 1 were homozygous SHR/SHR and exhibited no cataracts. Haplotype 2 carried the S allele between 40.2 and 45.3 Mbp and exhibited no cataracts, whereas animals with haplotype 3–5 all carried the S allele at 59.7 Mb and exhibited cataracts, suggesting that the location of the cataract was tightly linked to the marker at 59.7 Mb (range = 45.3 to 68.1 Mb). Three key recombinant animals were further genotyped using additional microsatellite markers to refine the location of cataract mutation (Fig. 5B). All three animals exhibited cataracts and shared the same genotype (S/SHR) only at microsatellite D9Rat85 (63.68 Mbp) and D9Mit2 (63.71 Mbp). Based on this data and the recombination interval for two of the animals (85867 and 85633), the cataract mutation could be localized to region from 62.9 to 63.8Mbp or ~900Kb (Fig. 5B).
Figure 5. Haplotype analysis and refinement of cataract mutation to rat chromosome 9.

(A) Location of the 95% confidence interval (CI) of the genomic interval linked to the cataract mutation (solid black bar) next to the physical map (left) and haplotype analysis of the F1(SS/Jr-Ctr X SHR) X SHR population (right). The haplotype analysis provides a refinement of the QTL based on genotype and absence of cataract (haplotype 1–2) or cataract status (haplotype 3–5). Haplotype 3–5 exhibit cataracts and share the SS/Jr-Ctr/SHR genotype at 09_0059708371 (denoted by the arrow). (B) Refinement of the cataract QTL using key recombinant animals from BC. All three animals exhibited cataracts and shared the SS/Jr-Ctr allele at only microsatellite markers D9Rat85 (63,689,889 Mb) to D9Mit2 (63,839,862 Mb). The genomic interval containing the cataract mutation is denoted by the boxed area.
Table 2 provides a summary of the 13 genes that reside in the refined genomic interval (~900Kb). Five genes (D4A9A7_RAT, Fam119a, F1M4U0_RAT, LOC100363697, and Pikfyve) were denoted as uncharacterized proteins and another 4 genes (Creb, Fzd5, Plekhm3, and Idh1) appear to have no biological role in the eye or lens. However, there were four genes in the interval that belong to the γ-crystallin family (Cryg, -b,-c,-d,-e) and mutations in these genes have been previously linked to the development of cataracts. The coding regions of all four Cryg genes were sequenced. No sequence differences were observed in Crygb, -c, or -e between the SS/Jr and SS/Jr-Ctr. However, a single-base mutation was observed in the start codon of exon 1 in the Crygd gene (Fig. 6). The start codon (ATG) in SS/Jr encodes a methionine, whereas in the SS/Jr-Ctr it is expected to encode a valine (GTG). Western blot analysis of eye homogenates demonstrated no CRYGD protein in SS/Jr-Ctr, but was clearly observed in wild-type SS/Jr animals (Fig. 6). Eyes from heterozygous animals [F1(SS/Jr-Ctr X SHR)] demonstrated a ~50% reduction in the amount of CRYGD protein compared to wild-type SS/Jr animals. Western blot analysis of homogenates from various organs demonstrated that the CRYGD protein appears specific to the eye.
Table II.
Genes located in refined cataract locus between 62.9and 63.8 Mb on chromosome 9
| Ensembl ID | Gene Name | Location (bp) | Description |
|---|---|---|---|
| ENSRNOG00000013412 | Creb1 | 63170785 | Cyclic AMP-responsive element-binding protein 1 |
| ENSRNOG00000031069 | D4A9A7_RAT* | 63237141 | Uncharacterized protein |
| ENSRNOG00000014212 | Fam119a | 63246137 | Uncharacterized /similar to methyltransferase like 21A (Mettl21A) |
| ENSRNOG00000023807 | F1M4U0_RAT | 63351853 | Uncharacterized /similar to cyclin Y-like 1 (Ccnyl1) |
| ENSRNOG00000014678 | Fzd5 | 63386990 | Frizzled-5 |
| ENSRNOG00000023760 | Plekhm3 | 63441846 | Peckstrin homology domain containing, family M, member 3 |
| ENSRNOG00000042709 | LOC100363697 | 63661748 | Uncharacterized /similar to aldo-keto reductase family 1 |
| ENSRNOG00000014790 | Cryge | 63698609 | Gamma-crystallin E |
| ENSRNOG00000032219 | Crygd | 63711051 | Gamma-crystallin D |
| ENSRNOG00000014950 | Crygc | 63720587 | Gamma-crystallin C |
| ENSRNOG00000032926 | Crygb | 63730906 | Gamma-crystallin B |
| ENSRNOG00000015020 | Idh1 | 63769401 | Isocitrate dehydrogenase |
| ENSRNOG00000015158 | Pikfyve | 63798657 | Similar to phosphatidylinositol 5-kinase, type III isoform 2 |
no orthologues (predicted only in rat)
Figure 6. Sequence and western blot analysis of whole eye and other organs from SS/Jr and SS/Jr-Ctr.
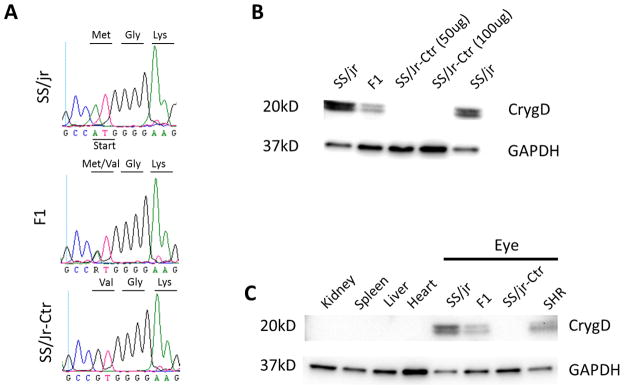
(A) DNA sequencing chromatographs shows a point mutation in the start codon (ATG → GTG) in SS/Jr-Ctr and F1 DNA compared to wild-type SS/Jr. The start codon (ATG) which encodes a methionine is predicated to be a valine in the SS/Jr-Ctr and F1. (B) Western blot analysis of eye. No CRYGD protein is evident in the SS/Jr-Ctr eye, but is observed in wild-type SS/Jr animals. In eyes from F1 animals, there is approximately 50% reduction in the amount of CRYGD protein observed. (C) Western blot analysis of homogenates from various organs and eye. No CRYGD protein is evident in tissues related to cardiovascular health and disease (heart or kidney).
DISCUSSION
Though the Dahl SS/Jr-Ctr strain has been around since the mid 1980’s an attempt had not been made to elucidate the underlying genetic cause of cataracts in the model. Using a classical positional cloning approach, the causative gene was localized with high-resolution to a less than 1 Mbp region on chromosome 9 containing only 13 genes, including 4 genes from the Cryg family. A novel point mutation in the start codon (ATG → GTG) of the Crygd gene was identified by sequencing. In eukaryotes, the start codon almost always codes for methionine (ATG) and alternative start codons (non-ATG) are rare (Hwang, Garza et al. 2005). The Crygd ATG start codon (methionine) is also conserved across all mammals (mouse, human, dog, etc.) and vertebrate species (data not shown). Thus, it was expected that the Crygd gene transcript would not be efficiently translated into protein in the SS/Jr-Ctr strain. Western blot analysis confirmed that the CRYGD protein was not expressed in the eye of SS/Jr-Ctr, but was clearly observed in the wild-type SS/Jr and SHR. Therefore, we conclude, the Met1Val substitution is almost certainly the cause of cataracts in the SS/Jr-Ctr, although definite evidence could only be established by additional experiments such as gene knockout.
The lens of the eye is composed of fiber cells that undergo differentiation involving changes in cell shape, expression of crystallin proteins, and degradation of cellular organelles that ultimately lead to the transparency of the lens (Michael and Bron 2011). Crystallins are the predominant proteins of the lens and are divided into two major families, α and β/γ (Andley 2007). The crystallin proteins contribute to the transparency and refractive properties of the lens by establishing a uniform concentration gradient in the lens. The α-crystallins, which are composed αA and αB, not only perform an important role in preserving lens transparency, but serve as molecular chaperones by preventing aberrant protein interactions (Andley 2007). The targeted disruption of αA-crystallin in mice as well as several spontaneous mutations in the gene result in cataracts (Chang, Hawes et al. 1996; Brady, Garland et al. 1997; Xia, Liu et al. 2006). There is no clear relationship between disruption of αB-crystallin and cataracts. In mice, Cryab−/− show general loss of body weight, muscle degeneration, abnormal skeletal development, but do not exhibit cataracts (Brady, Garland et al. 2001). In contrast, there are a number of known mutations in human αB-crystallin that result in dominant cataracts (Graw 2009), in addition to desmin-related myopathy or dilated cardiomyopathy (Vicart, Caron et al. 1998).
The β/γ-crystallins compose a large family of genes (>14 genes) that are organized as individual genes (Cryba1, Cryba2, Crygf, Crygs, CrygN), duplicates (Cryba4–Crybb1 and Crybb2–Crybb3) and one major cluster (Cryg, -b, -c, -d, -e) (Graw 2009). A key feature of the β/γ-crystallins is the Greek key motif which allows for the dense packing of proteins in the lens to minimize light scatter and provide transparency (Graw 2009). A second feature is the motif’s Ca2+ binding properties (Suman, Mishra et al. 2011). The role of Ca2+ in cataract development has been investigated by several groups (Duncan and Wormstone 1999; Sanderson, Marcantonio et al. 2000; Nagai, Ito et al. 2008). The relationship between Ca2+ and crystallin proteins could play a role in the calcification observed in the SS/Jr-Ctr lens.
Mutations in most β/γ-crystallins genes result in cataract formation in mice and humans (Graw 2009). Mice that exhibit mutations in Crygd (such as observed in SS/Jr-Ctr) demonstrate variability in onset, severity, and type of cataracts (Smith, Hawes et al. 2000; Graw, Löster et al. 2002; Wang, Cheng et al. 2007). This is likely related to the location of the mutation and its functional consequence (i.e. protein domain). In general, the loss of solubility of mutated crystallin proteins has been frequently suggested as the cause for cataract formation as most Crygd mouse mutants exhibit only single amino acid changes (Andley 2007). This reason is unlikely in the case of the SS/Jr-Ctr as there appears to be the complete absence of protein expression. The development of some cataracts in Cryg mutants has been attributed to the formation of large amyloid-like intranuclear inclusions and/or termination of the secondary lens fiber differentiation as indicated by the presence of remnants of cell nuclei (Sandilands, Hutcheson et al. 2002).
One motivation in elucidating the gene mutation that causes cataracts in the SS/Jr-Ctr strain was the possibility that the gene could also linked to kidney and/or cardiovascular disease exhibited by the strain. The kidney and eye share similar developmental pathways including PAX2 and WT1 (Dahl, Koseki et al. 1997; Izzedine, Bodaghi et al. 2003). WT1 is necessary for ureteric bud formation (Mrowka and Schedl 2000) as well as retinal ganglion cell differentiation (Wagner, Wagner et al. 2002). There are a number of retinal abnormalities found in inherited renal diseases (Savige, Ratnaike et al. 2011). However, γ-crystallins appear to only be expressed in the lens as opposed to α-crystallins and other eye proteins that are also expressed in other tissues and organs (Graw 1997). Thus, it is unlikely that the Crygd mutation would have any impact on cardiovascular or kidney disease and no further studies were performed to explore this possible association.
There are only a couple spontaneous cataract models in the rat with either mapped or identified genetic cause including an unidentified gene on chromosome 15 in the Ihara epileptic rat (Yokoyama, Amano et al. 2001), connexin 46 (Gja3) (Yoshida, Harada et al. 2005), and connexin 50 (Gja8) (Liska, Chylikova et al. 2008). The Gja8 point mutation (L7Q) in the SHR-Dca is different than that observed in the UPL rat (R340W) and appears to have significant impact on onset and severity of cataracts (Liska, Chylikova et al. 2008). Like the SS/Jr-Ctr model, the SHR-Dca is model of hypertension and while there appears no clear link between γ-crystallins and hypertension in the SS/Jr-Ctr model, there have been some studies that suggest a role of gap junctions in hypertension (Figueroa, Isakson et al. 2006; Lübkemeier, Machura et al. 2011).
In summary, we have identified a novel mutation in the start codon of the γ-crystallin D gene that leads to a complete knockout and the most likely cause of cataract formation in SS/Jr-Ctr rat. This finding could provide new utility to the model and allow investigation into eye development, cataract formation, and the function of crystallin proteins.
Acknowledgments
GRANTS
M.R.G is supported by NIH/NHLBI HL094446 and Robert M. Hearin Foundation. A.C.H is supported by 1T32HL105324. E.G.S is supported by Medical Research funds from the Department of Veterans Affairs and NIH Grants HL27255 and HL75321.
Footnotes
DISCLOSURE
The authors have nothing to disclose.
References
- Andley UP. Crystallins in the eye: Function and pathology. Progress in Retinal and Eye Research. 2007;26(1):78–98. doi: 10.1016/j.preteyeres.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Brady JP, Garland D, et al. Targeted disruption of the mouse αA-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein αB-crystallin. Proceedings of the National Academy of Sciences. 1997;94(3):884–889. doi: 10.1073/pnas.94.3.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady JP, Garland DL, et al. αB-Crystallin in Lens Development and Muscle Integrity: A Gene Knockout Approach. Investigative Ophthalmology & Visual Science. 2001;42(12):2924–2934. [PubMed] [Google Scholar]
- Chang B, Hawes NL, et al. Chromosomal Localization of a New Mouse Lens Opacity Gene (lop18) Genomics. 1996;36(1):171–173. doi: 10.1006/geno.1996.0439. [DOI] [PubMed] [Google Scholar]
- Churchill A, Graw J. Clinical and experimental advances in congenital and paediatric cataracts. Philos Trans R Soc Lond B Biol Sci. 2011;366(1568):1234–1249. doi: 10.1098/rstb.2010.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Koseki H, et al. Pax genes and organogenesis. Bioessays. 1997;19(9):755–765. doi: 10.1002/bies.950190905. [DOI] [PubMed] [Google Scholar]
- Duncan G, I, Wormstone M. Calcium cell signalling and cataract: Role of the endoplasmic reticulum. Eye. 1999;13(3b):480–483. doi: 10.1038/eye.1999.125. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Isakson BE, et al. Vascular Gap Junctions in Hypertension. Hypertension. 2006;48(5):804–811. doi: 10.1161/01.HYP.0000242483.03361.da. [DOI] [PubMed] [Google Scholar]
- Garrett MR, Dene H, et al. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J Am Soc Nephrol. 2003;14(5):1175–1187. doi: 10.1097/01.asn.0000060572.13794.58. [DOI] [PubMed] [Google Scholar]
- Garrett MR, Dene H, et al. Genome scan and congenic strains for blood pressure QTL using Dahl salt-sensitive rats. Genome Res. 1998;8(7):711–723. doi: 10.1101/gr.8.7.711. [DOI] [PubMed] [Google Scholar]
- Garrett MR, Gunning WT, et al. Dissection of a genetic locus influencing renal function in the rat and its concordance with kidney disease loci on human chromosome 1q21. Physiological Genomics. 2007;30(3):322–334. doi: 10.1152/physiolgenomics.00001.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett MR, Joe B, et al. Genetic linkage of urinary albumin excretion in Dahl salt-sensitive rats: influence of dietary salt and confirmation using congenic strains. Physiological Genomics. 2006;25(1):39–49. doi: 10.1152/physiolgenomics.00150.2005. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP, Gomez-Sanchez CM, et al. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol. 2010;95(1):120–130. doi: 10.1113/expphysiol.2009.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez EP, Zhou M, et al. Mineralocorticoids, salt and high blood pressure. Steroids. 1996;61(4):184–188. doi: 10.1016/0039-128x(96)00010-4. [DOI] [PubMed] [Google Scholar]
- Graw J. The crystallins: genes, proteins and diseases. Biol Chem. 1997;378(11):1331–1348. [PubMed] [Google Scholar]
- Graw J. Genetics of crystallins: Cataract and beyond. Experimental Eye Research. 2009;88(2):173–189. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Graw J. Mouse models of cataract. J Genet. 2009;88(4):469–486. doi: 10.1007/s12041-009-0066-2. [DOI] [PubMed] [Google Scholar]
- Graw J, Löster J, et al. V76D mutation in a conserved gD-crystallin region leads to dominant cataracts in mice. Mammalian Genome. 2002;13(8):452–455. doi: 10.1007/s00335-002-3021-6. [DOI] [PubMed] [Google Scholar]
- Huang B, He W. Molecular characteristics of inherited congenital cataracts. European Journal of Medical Genetics. 2010;53(6):347–357. doi: 10.1016/j.ejmg.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Hwang SR, Garza CZ, et al. Demonstration of GTG as an alternative initiation codon for the serpin endopin 2B-2. Biochemical and Biophysical Research Communications. 2005;327(3):837–844. doi: 10.1016/j.bbrc.2004.12.053. [DOI] [PubMed] [Google Scholar]
- Izzedine H, Bodaghi B, et al. Eye and Kidney: From Clinical Findings to Genetic Explanations. Journal of the American Society of Nephrology. 2003;14(2):516–529. doi: 10.1097/01.asn.0000051705.97966.ad. [DOI] [PubMed] [Google Scholar]
- Joe B, Saad Y, et al. Positional identification of variants of Adamts16 linked to inherited hypertension. Hum Mol Genet. 2009;18(15):2825–2838. doi: 10.1093/hmg/ddp218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang S, et al. Multiple-Interval Mapping for Ordinal Traits. Genetics. 2006;173(3):1649–1663. doi: 10.1534/genetics.105.054619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liska F, Chylikova B, et al. Microphthalmia and cataract in rats with a novel point mutation in connexin 50 - L7Q. Mol Vis. 2008;14:823–828. [PMC free article] [PubMed] [Google Scholar]
- Lübkemeier I, Machura K, et al. The Connexin40 A96S Mutation Causes Renin-Dependent Hypertension. Journal of the American Society of Nephrology. 2011;22(6):1031–1040. doi: 10.1681/ASN.2010101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margo CE, Gomez-Sanchez EP, et al. Hereditary cataracts in the John Rapp inbred strain of Dahl salt-sensitive rat. Invest Ophthalmol Vis Sci. 1987;28(8):1422–1428. [PubMed] [Google Scholar]
- Michael R, Bron AJ. The ageing lens and cataract: a model of normal and pathological ageing. Philosophical Transactions of the Royal Society B: Biological Sciences. 2011;366(1568):1278–1292. doi: 10.1098/rstb.2010.0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka C, Schedl A. Wilms’ Tumor Suppressor Gene WT1. Journal of the American Society of Nephrology. 2000;11(suppl 2):S106–S115. [PubMed] [Google Scholar]
- Nagai N, Ito Y, et al. Comparison of the Mechanisms of Cataract Development Involving Differences in Ca2+ Regulation in Lenses among Three Hereditary Cataract Model Rats. Biological and Pharmaceutical Bulletin. 2008;31(11):1990–1995. doi: 10.1248/bpb.31.1990. [DOI] [PubMed] [Google Scholar]
- Oki K, Gomez-Sanchez EP, et al. Role of mineralocorticoid action in the brain in salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2012;39(1):90–95. doi: 10.1111/j.1440-1681.2011.05538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp JP. Strain Designations for Inbred Dahl Salt Sensitive and Inbred Salt Resistant Rats. Rat New Letter. 1986;16:1. [Google Scholar]
- Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1985;7(3 Pt 1):340–349. [PubMed] [Google Scholar]
- Ryskulova A, Turczyn K, et al. Self-Reported Age-Related Eye Diseases and Visual Impairment in the United States: Results of the 2002 National Health Interview Survey. American Journal of Public Health. 2008;98(3):454–461. doi: 10.2105/AJPH.2006.098202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson J, Marcantonio JM, et al. A Human Lens Model of Cortical Cataract: Ca2+-Induced Protein Loss, Vimentin Cleavage and Opacification. Investigative Ophthalmology & Visual Science. 2000;41(8):2255–2261. [PubMed] [Google Scholar]
- Sandilands A, Hutcheson AM, et al. Altered aggregation properties of mutant gamma-crystallins cause inherited cataract. EMBO J. 2002;21(22):6005–6014. doi: 10.1093/emboj/cdf609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savige J, Ratnaike S, et al. Retinal Abnormalities Characteristic of Inherited Renal Disease. Journal of the American Society of Nephrology. 2011;22(8):1403–1415. doi: 10.1681/ASN.2010090965. [DOI] [PubMed] [Google Scholar]
- Smith RS, Hawes NL, et al. Lop12, a Mutation in Mouse Crygd Causing Lens Opacity Similar to Human Coppock Cataract. Genomics. 2000;63(3):314–320. doi: 10.1006/geno.1999.6054. [DOI] [PubMed] [Google Scholar]
- Suman SK, Mishra A, et al. Evolutionary Remodeling of βγ-Crystallins for Domain Stability at Cost of Ca2+ Binding. Journal of Biological Chemistry. 2011;286(51):43891–43901. doi: 10.1074/jbc.M111.247890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicart P, Caron A, et al. A missense mutation in the [agr]B-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20(1):92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- Wagner N, Wagner KD, et al. The Wilms’ Tumor Suppressor Wt1 Is Associated with the Differentiation of Retinoblastoma Cells. Cell Growth Differ. 2002;13(7):297–305. [PubMed] [Google Scholar]
- Wang K, Cheng C, et al. γD-Crystallin–Associated Protein Aggregation and Lens Fiber Cell Denucleation. Investigative Ophthalmology & Visual Science. 2007;48(8):3719–3728. doi: 10.1167/iovs.06-1487. [DOI] [PubMed] [Google Scholar]
- Wang S, Basten CJ, et al. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University; Raleigh, NC: 2012. ( http://statgen.ncsu.edu/qtlcart/WQTLCart.htm) [Google Scholar]
- West S. Epidemiology of cataract: accomplishments over 25 years and future directions. Ophthalmic Epidemiol. 2007;14(4):173–178. doi: 10.1080/09286580701423151. [DOI] [PubMed] [Google Scholar]
- Williams JM, Johnson AC, et al. Genetic Variants in Arhgef11 Are Associated With Kidney Injury in the Dahl Salt-Sensitive Rat. Hypertension. 2012 doi: 10.1161/HYPERTENSIONAHA.112.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C-h, Liu H, et al. Arginine 54 and Tyrosine 118 Residues of αA-Crystallin Are Crucial for Lens Formation and Transparency. Investigative Ophthalmology & Visual Science. 2006;47(7):3004–3010. doi: 10.1167/iovs.06-0178. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Amano S, et al. Genetic analysis of cataract in Ihara epileptic rat. Mamm Genome. 2001;12(3):207–211. doi: 10.1007/s003350010263. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Harada Y, et al. New genetic model rat for congenital cataracts due to a connexin 46 (Gja3) mutation. Pathology International. 2005;55(11):732–737. doi: 10.1111/j.1440-1827.2005.01896.x. [DOI] [PubMed] [Google Scholar]
- Zambelli-Weiner A, Crews JE, et al. Disparities in Adult Vision Health in the United States. American Journal of Ophthalmology. doi: 10.1016/j.ajo.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Zicha J, Dobesova Z, et al. Age-dependent salt hypertension in Dahl rats: fifty years of research. Physiol Res. 2012;61(Suppl 1):S35–87. doi: 10.33549/physiolres.932363. [DOI] [PubMed] [Google Scholar]