

Abstract
NMDA receptor (NMDAR)-mediated currents depend on membrane depolarization to relieve powerful voltage-dependent NMDAR channel block by external magnesium (Mgo2+). Mgo2+ unblock from native NMDARs exhibits a fast component that is consistent with rapid Mgo2+-unbinding kinetics and also a slower, millisecond time scale component (slow Mgo2+ unblock). In recombinant NMDARs, slow Mgo2+ unblock is prominent in GluN1/2A (an NMDAR subtype composed of GluN1 and GluN2A subunits) and GluN1/2B receptors, with slower kinetics observed for GluN1/2B receptors, but absent from GluN1/2C and GluN1/2D receptors. Slow Mgo2+ unblock from GluN1/2B receptors results from inherent voltage-dependent gating, which increases channel open probability with depolarization. Here we examine the mechanisms responsible for NMDAR subtype dependence of slow Mgo2+ unblock. We demonstrate that slow Mgo2+ unblock from GluN1/2A receptors, like GluN1/2B receptors, results from inherent voltage-dependent gating. Surprisingly, GluN1/2A and GluN1/2B receptors exhibited equal inherent voltage dependence; faster Mgo2+ unblock from GluN1/2A receptors can be explained by voltage-independent differences in gating kinetics. To investigate the absence of slow Mgo2+ unblock in GluN1/2C and GluN1/2D receptors, we examined the GluN2 S/L site, a site responsible for several NMDAR subtype-dependent channel properties. Mutating the GluN2 S/L site of GluN2A subunits from serine (found in GluN2A and GluN2B subunits) to leucine (found in GluN2C and GluN2D) greatly diminished both voltage-dependent gating and slow Mgo2+ unblock. Therefore, the residue at the GluN2 S/L site governs the expression of both slow Mgo2+ unblock and inherent voltage dependence.
Introduction
NMDA receptors (NMDARs) are ionotropic glutamate receptors critically involved in nervous system physiology and pathology and are tetramers usually composed of GluN1 and GluN2 subunits. NMDARs can be divided into four principal subtypes defined by the GluN2 subunit included in the receptor: GluN1/2A (composed of GluN1 and GluN2A subunits), GluN1/2B, GluN1/2C, and GluN1/2D receptors. NMDARs display diverse pharmacological and biophysical properties that vary among NMDAR subtypes (Monyer et al., 1992; Stern et al., 1992; Kuner and Schoepfer, 1996; Cull-Candy and Leszkiewicz, 2004; Erreger et al., 2005; Traynelis et al., 2010). The NMDAR subtype dependence of gating-related properties, including agonist affinity and channel open probability, is controlled by divergence among GluN2 subunits of the extracellular N-terminal domain (Gielen et al., 2009; Yuan et al., 2009). The NMDAR subtype dependence of channel-related properties, including inhibition by extracellular magnesium (Mgo2+), Ca2+ permeability, and single-channel conductance, is controlled by divergence among GluN2 subunits of the amino acid residue at a single site (Siegler Retchless et al., 2012). The site, termed the GluN2 S/L site, is located in the M3 transmembrane region.
Near typical resting potentials, endogenous Mgo2+ powerfully blocks the channels of NMDARs. GluN1/2A and GluN1/2B receptors are approximately fivefold more potently inhibited by Mgo2+ than GluN1/2C and GluN1/2D receptors (Monyer et al., 1992; Kuner and Schoepfer, 1996; Qian et al., 2005). Upon depolarization, Mgo2+ block of all NMDAR subtypes is relieved. The speed of depolarization-induced Mgo2+ unblock also varies strongly among NMDAR subtypes: Mgo2+ unblock from GluN1/2C and GluN1/2D receptors is very rapid, whereas Mgo2+ unblock from GluN1/2A and GluN1/2B receptors displays both a rapid component and a slow component that is slower for GluN1/2B receptors (Clarke and Johnson, 2006).
The slow component of Mgo2+ unblock from GluN1/2A and GluN1/2B receptors was unexpected based on the rapid kinetics of Mgo2+ unbinding from open channels (Nowak et al., 1984; Ascher and Nowak, 1988). Slow Mgo2+ unblock can be reproduced by models in which the rates of gating, desensitization, and/or agonist binding are altered when the channel is blocked by Mgo2+ (Kampa et al., 2004; Vargas-Caballero and Robinson, 2004). In contrast, we demonstrated previously that GluN1/2B receptors exhibit inherent (in the absence of Mgo2+) voltage-dependent gating and that slow Mgo2+ unblock from GluN1/2B receptors results from voltage-dependent gating (Clarke and Johnson, 2008). These data reconciled for GluN1/2B receptors the characteristics of slow Mgo2+ unblock with evidence that Mgo2+ block does not affect transitions among receptor states (Sobolevsky and Yelshansky, 2000; Qian et al., 2002; Blanpied et al., 2005).
Here we demonstrate that GluN1/2A receptors exhibit inherent voltage-dependent gating. We show that voltage-dependent gating leads to slow Mgo2+ unblock with glutamate concentration-dependent kinetics and to voltage dependence of synaptic response decay kinetics. We also determined the origins of NMDAR subtype dependence of voltage-dependent gating and slow Mgo2+ unblock. Our finding that mutating the GluN2 S/L site, which was shown to modify multiple other NMDAR channel properties, also attenuated slow Mgo2+ unblock and voltage-dependent gating, provides insight into mechanisms that may underlie both phenomena.
Materials and Methods
Cell culture and transfection.
Experiments were performed on human embryonic kidney (HEK) 293T cells, which were maintained as described previously (Qian et al., 2005), or on tsA cells, which were similarly maintained except that 10% (rather than 5%) fetal bovine serum was added to the culture medium. Cells were plated onto glass coverslips, either untreated or pretreated with poly d-lysine (0.1 mg/ml) and rat-tail collagen (0.1 mg/ml, BD Biosciences), in 35 mm culture dishes at 1–2 × 105 cells per dish. Eighteen to 24 h after plating the cells were transiently transfected with cDNAs encoding the GluN1–1a (GenBank X63255 in pcDM8) and GluN2A (GenBank M91561 in pcDNA1) or GluN2A(S632L) (in pcDNA1) (Siegler Retchless et al., 2012) subunit using a Ca2+ precipitation method (Qian et al., 2005) or FuGENE transfection reagents (Promega). cDNA for enhanced green fluorescent protein (eGFP) was cotransfected to identify transfected cells. The following amounts of cDNAs were added to each dish: 0.5–0.7 μg of eGFP, 0.5–1.3 μg of GluN1–1a, and 1–2 μg of GluN2A or GluN2A(S632L). Cells were incubated with the transfection solution for 6–8 h and then precipitates were washed off with culture medium containing 200–1000 μm APV and either 2 mm Mg2+ or 200 μm 7-chlorokynurentic acid. Experiments were performed 20–72 h after transfection.
Solutions.
Solutions were prepared from frozen stocks before experiments. Currents were activated by the indicated concentration of NMDA or glutamate. Glycine (10 μm) was added to all solutions. The external solution contained the following (in mm): NaCl 140, CaCl2 1, KCl 2.8, HEPES 10, and either 0, 1, or 5 Mgo2+, pH 7.2 adjusted with NaOH, osmolality 290 ± 10 mmol/kg. Other external solutions used are described in Results. The pipette solution contained the following (in mm): CsCl 125, EGTA 10, and HEPES 10, pH 7.2 adjusted with CsOH, osmolality 275 ± 10 mmol/kg. Sucrose was added if needed to adjust the osmolality of the external solution. All membrane voltages were corrected for the junction potential between the pipette and bath solution of 5 mV. Ultrapure salts were used if available. All chemicals were from Sigma.
Whole-cell recording.
Whole-cell recordings from transfected HEK 293T or tsA cells were performed as described previously (Qian et al., 2005). All experiments were performed at room temperature. Pipettes were pulled from borosilicate standard-walled glass with filaments (1.5 mm outer diameter; 0.86 mm inner diameter; Warner Instruments) and fire polished (resistance, 2–5 MΩ). Membrane current was recorded with an Axopatch 200 or 200B amplifier (Molecular Devices) in voltage-clamp mode. Series resistance correction and prediction circuitry were set to at least 80% in all experiments. Signals were low-pass filtered at a cutoff frequency of 2.5 or 5 kHz (8-pole Bessel; Warner Instruments), sampled at 10–50 kHz, and refiltered at 1 kHz for analysis and display.
Solutions were delivered using a fast perfusion system (Qian et al., 2002) connected to gravity-fed reservoirs (AutoMate Scientific). Solution exchange speed was estimated by recording whole-cell current from a transfected cell while moving between a solution with normal external solution plus 30 μm NMDA and 10 μm glycine and another solution that was identical except that the impermeant ion NMDG+ replaced Na+. The time course of current decrease upon movement into the NMDG+ extracellular solution was used to estimate the time course of solution exchanges, which were >90% complete within 20 ms.
Data analysis and kinetic modeling.
GluN1/2A receptor current responses were corrected for leak and capacitive currents by subtracting currents measured in 0 glutamate from currents measured in the presence of glutamate using identical voltage protocols. Examples of presubtraction and postsubtraction currents using identical procedures (except with GluN1/2B receptors) can be found in Figure 1 in Clarke and Johnson (2008). All current traces presented here are leak and capacitance subtracted. Current relaxations were fit with multiexponential equations as described previously (Clarke and Johnson, 2008). The number of exponential components was adjusted as necessary to provide high-quality fits as determined visually. Curve fitting was performed using Clampfit 9.2 or 10.3 (Molecular Devices) or Origin 7.0 (OriginLab) software.
Figure 1.

GluN1/2A receptor current relaxations after depolarizing steps display a slow component in 0 Mgo2+. A, Whole-cell current recording from an HEK 293T cell-expressing GluN1/2A receptors during application of 1 mm glutamate and 10 μm glycine in 0 Mgo2+. After the inward current response (bottom trace) reached a steady-state level, a depolarizing step from −100 to 190 mV (top trace) was applied. Leak and capacitive currents were subtracted in all figures. B, Voltage steps (top traces) and current responses (bottom traces) from the cell used for A with expanded time base during depolarizations from −100 to 80, 160, and 190 mV. C, Voltage step (top trace) and current response (bottom trace, thick line) replotted from A with a double-exponential fit (bottom trace, thin gray line) superimposed. Time constants of fast and slow components of the current relaxation are given; slow component of the current trace appears above the solid horizontal line. D, τslow did not depend significantly on the voltage during the depolarizing step (based on least-squares linear regression, p = 0.80) nor did τfast (p = 0.46; data not shown). E, Aslow as a percentage of the amplitude of the entire current relaxation induced by the depolarizing step increased with depolarization. F, Addition of 10 μm EDTA to normal external solution (No EDTA) did not affect Aslow significantly (p > 0.25 at each voltage; n = 3 for each condition) after depolarizing steps from −105 mV to 35, 95, or 185 mV.
Double-exponential fits to leak- and capacitance-subtracted whole-cell currents (Fig. 1C) were used to generate the data plotted in Figures 1D–F and 3, B and C. The fast component of the fits (time constant = τfast) resulted from voltage step-induced changes in driving force on current through open NMDARs. The value of τfast reflects technical limitations in patch-clamp experiments (Levis and Rae, 1992), including the speed of the voltage change during the step, and therefore does not provide physiologically relevant information. The mechanisms responsible for the slow component (amplitude as a percentage of entire current relaxation = Aslow; time constant = τslow) were investigated here.
Figure 3.
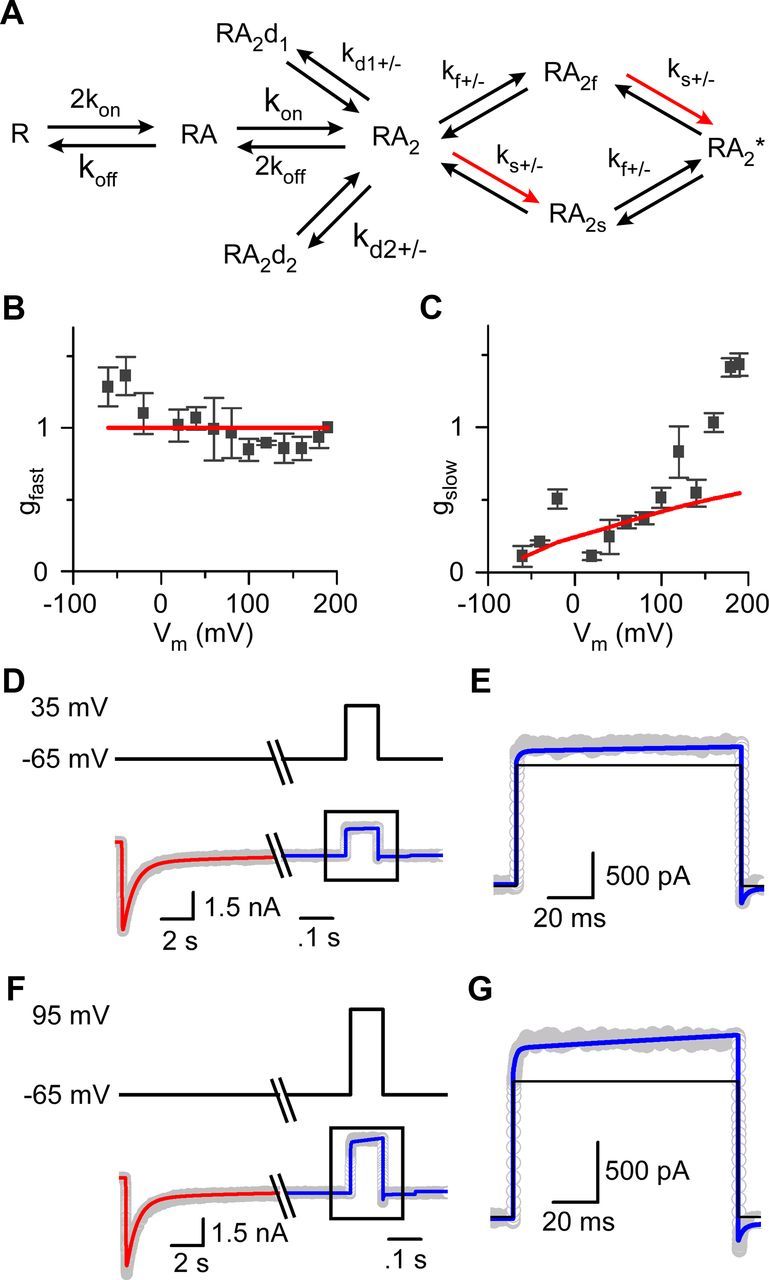
Depolarization-induced slow relaxations in 0 Mgo2+ can be reproduced by a GluN1/2A receptor model that incorporates voltage-dependent gating. A, Kinetic model used to simulate GluN1/2A receptor activation. Red arrows indicate rate that was altered to undergo an e-fold acceleration per 175 mV depolarization. B, C, Amplitudes of each component of double-exponential fits to current relaxations during depolarizing voltage steps (Fig. 1C) were converted to conductances (see Materials and Methods). gfast (B) and gslow (C) derived from fits to whole-cell data are plotted as a function of the voltage during the depolarizing voltage step (symbols). The corresponding values of gfast (B) and gslow (C) derived from GluN1/2AV-D model simulations also are plotted (red lines). D, Current trace (bottom, gray) during application of 1 mm glutamate in 10 μm glycine at −65 mV. Once a steady-state response was reached, the cell was depolarized to 35 mV. Results of fitting the GluN1/2AV-D model to determine desensitization rate constants and number of receptors (red line) and subsequent current simulations with all rates fixed (blue line) are overlaid. Voltage is shown by top trace. E, Enlarged view of the current trace (gray) and simulation by the GluN1/2AV-D model (blue line) in response to a depolarization from −65 to 35 mV. Current simulation from a model containing no voltage dependence is overlaid (black line); in this model, ks+ at all voltages equals the value measured by Erreger et al. (2005) at −100 mV. F, G, Same as D and E except the depolarization was from −65 to 95 mV.
We performed three tests to determine whether measurement procedures affected Aslow or τslow values. First, to determine whether, despite series resistance compensation, series resistance errors affected Aslow values, we investigated whether Aslow depended on the amplitude of the change in current (Total ΔI) during the largest depolarization used (−100 to 190 mV). A least-squares linear regression performed on a plot of Aslow (range, 45.0–55.4%) as a function of Total ΔI (range, 1.5–7.8 nA) for each of the 6 cells in which measurements were made revealed no correlation (p = 0.28). Therefore, it is unlikely that series resistance error affected our measurements of Aslow. Second, to determine whether low-pass filtering at 1 kHz affected Aslow or τslow, we compared Aslow and τslow values for three voltage steps (from −100 mV to 40, 100, and 190 mV) after low-pass filtering at 1 kHz to values after filtering at 2.5 kHz. Filter frequency did not significantly (p > 0.15 for each comparison) affect Aslow or τslow for any of the three voltage steps. Therefore, 1 kHz low-pass filtering does not appear to have affected quantification of Aslow or τslow. We also compared values of τfast after low-pass filtering at 1 kHz and at 2.5 kHz and found no significant differences for voltage steps to 40 or 100 mV, but τfast was significantly (p < 0.003; decrease of nearly twofold) faster after 2.5 kHz filtering for voltage steps to 190 mV. Therefore, 1 kHz low-pass filtering did in some cases affect τfast values (range, 0.089–0.593 ms), as expected for this fast component of current relaxations. Third, to determine whether use of double-exponential fits affected Aslow or τslow, we performed single-exponential fits to only the slow component of current relaxations for the same three voltage steps (from −100 mV to 40, 100, and 190 mV). To avoid interference from the fast component, fits were started 0.3 ms (twice the average τfast of ∼0.15 ms, a delay that excluded most of the fast component without missing a significant portion of the slow component) after completion of the voltage step. The values of Aslow and τslow based on single-exponential fits (to 2.5 kHz filtered currents) and based on double-exponential fits (to 1 kHz filtered currents) did not differ significantly for any of the three voltage steps (p > 0.12 for each comparison). Therefore, our standard measurement procedures (double-exponential fits to 1 kHz filtered currents) appear to have provided reliable measurements of Aslow and τslow.
To generate Figure 3, B and C, for voltage steps from −100 mV to each plotted ending voltage (Vm,e), the amplitudes for each Vm,e of the fast [Ifast(Vm,e)] and slow [Islow(Vm,e)] current components were first measured using double-exponential fits. Ifast(Vm,e) and Islow(Vm,e) were then converted to fast and slow conductances (Clarke and Johnson, 2008). To permit averaging of conductance values across cells, each conductance value was normalized to the fast conductance measured during the largest depolarization (−100 to 190 mV), which was performed in each cell. Because Ifast results from the change in driving force on current flow through NMDARs open at −100 mV, gfast was calculated as Ifast(Vm,e)/(Vm,e − (−100 mV)), which was then normalized by dividing by fast conductance for 190 mV (Ifast(190 mV))/(290 mV). Islow results from the change in NMDAR conductance that occurs after the voltage is stepped to Vm,e, so gslow was calculated as Islow(Vm,e)/(Vm,e − Vrev), which then was normalized by dividing by fast conductance for 190 mV. Vrev is the reversal potential for current flow through GluN1/2A receptors, which we measured as −7 mV.
Model fitting and current simulations were performed using SCoP 4.0 (Simulation Resources). The model of GluN1/2A receptors that we used (Fig. 3A) has the same structure as the model we used to investigate voltage-dependent gating of GluN1/2B receptors (Clarke and Johnson, 2008) and is based on previously developed GluN1/2B and GluN1/2A models (Banke and Traynelis, 2003; Erreger et al., 2005). State transition rates were fixed to the values for GluN1/2A receptors in Erreger et al. (2005) (Table 1), except for ks+ (the forward rate representing GluN2A subunit conformational change) and rates of entry into and recovery from desensitized states, which were set as described in Results. Because our GluN1/2A receptor model was based on the work of Erreger et al. (2005), it is important to compare experimental conditions. Most conditions were similar, including expression systems (HEK293 cells in Erreger et al., 2005; HEK293T cells here), temperature (room temperature in both cases), and pH (pH 7.3 external solution, pH 7.35 pipette solution in Erreger et al., 2005; pH 7.2 external and pipette solution here). Some conditions differed, but should not have affected the results substantially. Although the Ca2+ concentration was 0.5 mm in Erreger et al. (2005) but 1 mm here, we found that 0 Ca2+ + EGTA solution did not affect depolarization-induced slow relaxations significantly (Results). Although EDTA was used to chelate Zn2+ in Erreger et al., (2005), but was not used in most experiments here, we found that EDTA did not affect depolarization-induced slow relaxations (Fig. 1F).
Table 1.
Rates used for GluN1/2A receptor model fitting and current simulations
| Rate constant | Units | Value |
|---|---|---|
| kon | μm−1 s−1 | 31.6 |
| koff | s−1 | 1010 |
| ks+ | s−1 | 230a |
| ks− | s−1 | 178 |
| kf+ | s−1 | 3140 |
| kf-− | s−1 | 174 |
| kd1+ | s−1 | Determined by fitting |
| kd1+ | s−1 | Determined by fitting |
| kd1− | s−1 | Determined by fitting |
| kd2+ | s−1 | Determined by fitting |
| kd2− | s−1 | Determined by fitting |
“Determined by fitting” means these rates were determined by fitting the GluN1/2AV-D model to macroscopic currents as described in the text relevant to Figures 3 and 4. Other rate constants were taken directly from Erreger et al. (2005).
a This rate was set to the indicated value at −100 mV (the voltage at which rates were estimated in Erreger et al. (2005), but differed at other voltages by increasing exponentially with voltage (e-fold for 175 mV; Clarke and Johnson, 2008).
When modeling currents in 1 mm Mgo2+, corresponding rates in the unblocked and blocked arms of the model were set as equal. Voltage-dependent values of the Mgo2+ blocking rate and unblocking rate (Clarke and Johnson, 2008) were calculated from previously published equations (Antonov and Johnson, 1999).
Data are expressed as means ± SEM and statistical analysis was performed using 2-tailed Student's t tests unless otherwise noted.
Results
Voltage dependence of GluN1/2A receptor currents in 0 Mgo2+
To examine potential voltage-dependent gating of GluN1/2A receptors, currents were elicited from HEK 293T cells expressing GluN1/2A receptors by application of 1 mm glutamate in the continuous presence of 10 μm glycine in 0 Mgo2+. GluN1/2A receptor-mediated currents displayed marked desensitization (Fig. 3D,F) and reached a steady-state level after several seconds of continuous agonist application. Once a steady-state current was reached, depolarizing voltage steps were made. The current response to a voltage step from −100 to 190 mV and back to −100 mV is shown in Figure 1A and responses to three different depolarizing steps are shown at a faster time base in Figure 1B. Depolarizing steps caused a rapid outward jump in current, as expected due to the change in driving force, followed by a slower relaxation that increased in amplitude with larger depolarizations (Fig. 1B,E). Fitting of current relaxations in response to depolarizing voltage steps required double-exponential equations containing fast (τfast, <1 ms) and slow (τslow, several ms) components (Fig. 1C; see Materials and Methods). The value of τslow did not depend significantly on the amplitude of the depolarization (Fig. 1D). In contrast, Aslow depended significantly (ANOVA, p < 0.001) on the amplitude of the depolarization, becoming larger with increasing depolarization (Fig. 1E).
Zn2+ is a potent (IC50 ∼20 nm) inhibitor of GluN1/2A receptors. Even the very low Zn2+ concentrations that typically contaminate nominally Zn2+-free solutions may cause significant GluN1/2A receptor inhibition (Paoletti et al., 1997). To determine whether inhibition of NMDAR responses by Zn2+ contaminating our solutions influenced depolarization-induced slow relaxations, we compared Aslow in our normal external solution with an identical solution except for the addition of 10 μm EDTA, a very high-affinity Zn2+ chelator (Fig. 1F). EDTA had no effect on Aslow, suggesting that contaminating Zn2+ did not influence depolarization-induced slow relaxations.
We also investigated whether elimination of Ca2+, to which NMDARs are highly permeable, influenced depolarization-induced slow relaxations. We compared Aslow after depolarizations from −65 to 35 mV in normal external solution and in an identical solution except with 0 added CaCl2 and 1 mm EGTA. Aslow values in normal (1 mm Ca2+) external solution (9.0 ± 1.5%) and in 0 Ca2+/EGTA external solution (5.1 ± 0.9%) were not significantly different (p = 0.15), which is consistent with previous observations on GluN1/2B receptors (Clarke and Johnson, 2008). It is possible that the apparent trend toward a larger relaxation in Ca2+ reflects a moderate effect of 1 mm Ca2+ on depolarization-induced relaxations.
The results shown in Figure 1 bear a qualitative resemblance to recordings from GluN1/2B receptors that reflect voltage-dependent gating (Clarke and Johnson, 2008), suggesting that gating of GluN1/2A receptors may also depend on voltage. Voltage-dependent gating of GluN1/2B receptors results in a time-dependent increase in open probability (Popen) after depolarization, which is reflected by a slow component of relaxations after depolarizing voltage jumps (Clarke and Johnson, 2008). If the slow component of GluN1/2A receptor current relaxations results from a depolarization-induced increase in Popen, then steady-state current should increase supralinearly at positive voltages. Consistent with this prediction, the ratio of steady-state current at 35 to −65 mV (1.05 ± 0.06) was significantly (p = 0.0044) higher than predicted assuming a linear I–V curve with the measured reversal potential of −7 mV (0.72).
If GluN1/2A receptor Popen exhibits a time-dependent increase during depolarizations, a time-dependent decrease in Popen should be observed when membrane voltage is returned to a hyperpolarized value. Consistent with this prediction, immediately after repolarization to −100 mV, the inward current displayed an initial peak that exceeded the steady-state current level at −100 mV (Fig. 2A). The repolarization-induced inward current peak (Ipeak) depended significantly (ANOVA, p < 0.001) on the amplitude of the previous depolarization, becoming larger after repolarization from more positive voltages (Fig. 2B). After the Ipeak, the current relaxed to the baseline current level over several milliseconds and was well fit by a single-exponential equation. These data suggest that the Popen of GluN1/2A receptors is enhanced at depolarized voltages even in the absence of Mgo2+.
Figure 2.

Amplitude of tail currents after repolarizations in 0 Mgo2+ depend on voltage preceding the repolarization. A, Currents elicited by the application of 1 mm glutamate and 10 μm glycine in 0 Mgo2+ (bottom traces) during repolarization (top traces) from 90 mV (thick line) or 190 mV (thin line) to −100 mV. Immediately after repolarization to −100 mV, the current was transiently more negative than the steady-state current measured at −100 mV. B, Ipeak, the peak inward current following repolarization to −100 mV normalized to the steady-state current at −100 mV, plotted as a function of the voltage that preceded the repolarization. Ipeak depended significantly on the voltage during the preceding depolarization (one-way ANOVA; p < 0.0001).
Development and evaluation of GluN1/2A receptor voltage-dependent model in 0 Mgo2+
We next used a computational model to investigate the mechanism by which depolarization enhances GluN1/2A receptor currents. We started by testing the parsimonious hypothesis that the same underlying mechanism is responsible for voltage dependence of GluN1/2A and GluN1/2B receptor currents. To test this hypothesis, we developed a voltage-dependent model of GluN1/2A receptor function (the GluN1/2AV-D model) using as a template our previously described (Clarke and Johnson, 2008) voltage-dependent GluN1/2B receptor model (the GluN1/2BV-D model). In the GluN1/2AV-D model (Fig. 3A), “R” represents the NMDAR bound only to glycine (all experiments were performed in the continuous presence of 10 μm glycine) and “A” represents a single glutamate molecule. Once the NMDAR binds two glutamate molecules (state RA2), the NMDAR can enter one of two desensitized states, RA2d1 or RA2d2, or proceed toward the open state (RA2*). As described previously (Banke and Traynelis, 2003; Erreger et al., 2005), preopening conformational changes associated with the GluN1 (transition to RA2f) or GluN2 (transition to RA2s) subunits connect state RA2 with RA2*. We found (Clarke and Johnson, 2008) that this type of model reproduces GluN1/2B receptor current properties, including those that result from inherent voltage dependence, if ks+ (the forward rate associated with the GluN2 subunit preopening conformational change) exhibits weak exponential (Hille, 2001) dependence on membrane voltage. To determine whether GluN1/2A receptors express the same mechanism and strength of voltage dependence as GluN1/2B receptors, we imposed on ks+ in the GluN1/2AA-D model (Fig. 3A, red arrows) the same exponential voltage dependence (e-fold per 175 mV) found to model GluN1/2B receptors accurately (Clarke and Johnson, 2008). However, because of the kinetic differences between GluN1/2A and GluN1/2B receptors, we used rate constants equal to the values (Table 1) determined previously for GluN1/2A receptors (Erreger et al., 2005). Therefore, the value of ks+ at any membrane voltage (Vm) equaled ks+(−100 mV) * exp[−(Vm + 100 mV)/(175 mV)], where ks+(−100 mV) is the value measured by Erreger et al. (2005) at −100 mV. Importantly, GluN1/2A receptors undergo preopening conformational changes more rapidly than GluN1/2B receptors.
We investigated whether the GluN1/2AV-D model reproduces the depolarization-induced enhancement of GluN1/2A receptor currents, first by examining the voltage dependence of the rapid and slow components of current relaxations in response to voltage jumps. To compare data with model predictions, double-exponential equations were fit to current relaxations (Fig. 1B) measured during voltage jumps or simulated by the GluN1/2AV-D model. The amplitudes of the fast and slow components were then converted to normalized conductances (gfast and gslow; see Materials and Methods). The values derived from whole-cell recordings demonstrated that gfast is nearly voltage independent (Fig. 3B, symbols). Model simulations showed that the GluN1/2AV-D model similarly predicted gfast to be voltage independent (Fig. 3B, red line). Consistent with the data shown in Figure 1E, the voltage dependence of GluN1/2A receptor current slow relaxations was reflected by the increase with depolarization of gslow (Fig. 3C, symbols). Model simulations with the GluN1/2AV-D model predicted voltage dependence of gslow similar to measured values up to voltages of ∼150 mV. As was observed with GluN1/2B receptors (Clarke and Johnson, 2008), the voltage dependence of gslow appeared to increase sharply at extremely positive voltages, an observation that we did not explore further. The model predictions shown in Figure 3, B and C, were made with no adjustable parameters; the values of all parameters were derived from a previous GluN1/2A receptor model (Erreger et al., 2005) and from the voltage dependence of ks+ that we estimated previously for GluN1/2B receptors (Clarke and Johnson, 2008). Desensitization rates in the GluN1/2AV-D model were set to 0 for Figure 3, B and C, because desensitization had negligible effects on these rapid depolarization-induced current relaxations. Based on the good agreement between model and data, we further evaluated the ability of the GluN1/2AV-D model to reproduce voltage-dependent properties of GluN1/2A receptors.
We next compared recorded and simulated current waveforms during voltage steps. Before performing simulations, the desensitization rates and number of receptors, which vary substantially between cells, were determined separately for each cell. Values of these parameters were estimated by fitting the GluN1/2AV-D model to an application of 1 mm glutamate at −65 mV that preceded depolarizations to 35 mV (Fig. 3D) or 95 mV (Fig. 3F). During fitting, only the channel number and desensitization rates were allowed to vary (Table 1). Consistent with previous results (Erreger et al., 2005), GluN1/2A receptor current desensitization was biexponential and the fit required two desensitized states. Once excellent fits were obtained (Fig. 3D,F, red lines), all rates were fixed and the GluN1/2AV-D model was used to simulate currents in response to depolarization to either 35 or 95 mV. With no free parameters, the GluN1/2AV-D model accurately reproduced GluN1/2A receptor-mediated currents in response to depolarizations from −65 to 35 mV (Fig. 3D,E, blue line) and 95 mV (Fig. 3F,G, blue line). In contrast, a GluN1/2A receptor model identical to the GluN1/2AV-D model, except with no voltage dependence of ks+, predicted square depolarization-induced current responses and underestimated outward current levels (Fig. 3E,G, black lines). These data suggest that voltage-dependent gating of GluN1/2A receptors is sufficient to account for the depolarization-induced potentiation of GluN1/2A receptor-mediated currents in 0 Mgo2+.
An apparent disagreement between the GluN1/2AV-D model and our data involves voltage dependence of τslow. Although the model predicts τslow to be weakly voltage dependent, we did not observe significant voltage dependence of τslow (Fig. 1D). We do not believe that this disagreement is meaningful, however, because the large variability in our time constant measurements would not have allowed us to resolve weak voltage dependence of τslow.
Evaluation of the GluN1/2AV-D model in 1 mm Mgo2+
Voltage-dependent gating of GluN1/2B receptors results not only in inherent voltage dependence, but also in the slow component of Mgo2+ unblock from GluN1/2B receptors (Clarke and Johnson, 2008). We next determined whether the slow component of Mgo2+ unblock from GluN1/2A receptors also results from voltage-dependent gating. The slow component of Mgo2+ unblock is slower from GluN1/2B than from GluN1/2A receptors (Clarke and Johnson, 2006). Therefore, we initially found it unlikely, although nevertheless worth testing, that slow Mgo2+ unblock of both receptor subtypes could result from equal voltage dependence of the same single gating parameter (ks+). We performed comparisons of experimental data and model simulations similar to those shown in Figure 3D–G, but in 1 mm Mgo2+. To account for block by Mgo2+, we used a model identical to that shown in Figure 3A, except that a “blocked arm” was added to the GluN1/2AV-D model (Clarke and Johnson, 2008, their Fig. 4B). The blocked arm followed a trapping block scheme in which, after Mgo2+ binds, the NMDAR channel can close and glutamate can unbind, trapping the Mgo2+ ion in the pore. Similar to the GluN1/2BV-D model (Clarke and Johnson, 2008), we used a symmetric block model: corresponding rates in the blocked and unblocked arms were set as equal, in contrast to asymmetric models that have been developed to explain slow Mgo2+ unblock (Kampa et al., 2004; Vargas-Caballero and Robinson, 2004). Desensitization rates and channel number were again determined for each cell by fitting the model to whole-cell responses. However, because GluN1/2A receptor currents are strongly inhibited by 1 mm Mgo2+ at −65 mV, the model was fit to whole-cell currents elicited by glutamate application while the cell was held at 35 mV. Excellent fits were obtained with a model containing two desensitized states in the unblocked and in the blocked arms of the model (Fig. 4A, black line).
Figure 4.

Slow Mgo2+ unblock is reproduced by the GluN1/2AV-D model. A, Current trace (gray line) during application of 1 mm glutamate in 10 μm glycine and 1 mm Mgo2+ at 35 mV. The GluN1/2AV-D model, expanded to incorporate symmetric block by Mgo2+, was fit to the data with desensitization rates and channel number the only free parameters (black line). B, Experimental data (gray line) and current simulations (black line) during application of 1 mm glutamate in 10 μm glycine and 1 mm Mgo2+ at −65 mV. Once a steady-state response was reached, the cell was depolarized from −65 mV to 35, −25, and 95 mV. All model parameters were fixed during the simulation. C–E, Enlarged views of current traces (gray lines) and GluN1/2AV-D model simulations (black lines) in response to depolarizations from −65 to −25 mV (C), 35 mV (D), and 95 mV (E). F, Comparison of τslow values from whole-cell recordings (gray) and from fits to currents simulated by the GluN1/2AV-D model (black) during current relaxations activated by depolarizing steps from −65 mV to −25, 35, and 95 mV.
To compare model simulations and recordings of depolarization-induced slow Mgo2+ unblock, the membrane potential was returned to −65 mV after the desensitization rates and number of receptors were estimated and the values were fixed in the model. Glutamate was reapplied in the continued presence of 1 mm Mgo2+ and, once a steady-state current level was reached, the voltage was stepped from −65 mV to −25, 35, and 95 mV (Fig. 4B). With no free parameters, the GluN1/2AV-D model accurately simulated (Fig. 4B–E, black lines) the current waveforms recorded in 1 mm Mgo2+.
Agreement between model and data were assessed quantitatively by comparing the time constants of the slow component of depolarization-induced relaxations in whole-cell recordings and simulations. For whole-cell recordings, current relaxations were fit in most cases with a double-exponential equation and the slower exponential component was used to quantify the time constant (τslow; Fig. 4F, gray bars) of the slow component. In a few cases, a triple-exponential equation was used because of a small (<10%), slow (τ > 100 ms) component observed in current relaxations; in these cases, the intermediate time constant was used as τslow. In simulations used for Figure 4F, the GluN1/2AV-D model was used with no adjustable parameters; desensitization rates were fixed at average values determined from fits as shown in Figure 4A to three cells (average values ± SEM were as follows: kd1+, 107 ± 3 s−1; kd1−, 3.50 ± 0.84 s−1; kd2+, 46.7 ± 1.23 s−1; kd2−, 0.41 ± 0.09 s−1). Simulated currents were fit with multiexponential equations; accurate fits of the noiseless simulations required five-component exponential equations. The fastest three of the five components (τ1 − τ3) had values <1 ms and thus are likely to correspond to τfast of fits to data. The remaining two components had average values of ∼2 ms (τ4) and ∼220 ms (τ5). τ5 is likely to result from receptors leaving desensitized states (the only states with such slow kinetics), and so we referred to τ4 as the τslow of simulations and compared its value with τslow measured from the data. Relaxations observed in whole-cell recordings did not have an obvious component corresponding to the slowest component (τ5) of GluN1/2AV-D model relaxations, suggesting that modeling of desensitization could be refined (Gibb, 2004; Schorge et al., 2005). Good agreement was observed between the values of τslow at all three voltages (Fig. 4F). The ability of simulations based on the GluN1/2AV-D model to reproduce slow Mgo2+ unblock strongly supports the hypothesis that slow Mgo2+ unblock results from voltage-dependent gating of GluN1/2A receptors. This hypothesis is further supported by the observation that, after depolarizations from −65 to 95 mV, the τslow of current relaxations in 0 Mgo2+ (2.44 ± 0.94 ms) and of slow Mgo2+ unblock in 1 mm Mgo2+ (2.14 ± 0.25 ms) were not significantly different (p = 0.7).
The time constant of the slow component of Mgo2+ unblock is faster at high than at low NMDA concentrations (Clarke and Johnson, 2006). Whether the kinetics of Mgo2+ unblock depend on glutamate concentration and, if so, whether the concentration dependence of Mgo2+ unblock is reproduced by NMDAR models is not known. To further test the ability of the GluN1/2AV-D model to simulate characteristics of GluN1/2A receptor currents, we compared experimental results and model simulations of Mgo2+ unblock at low and high glutamate concentrations. The experimental conditions were similar to those used in the experiments shown in Figure 4. For each cell modeled, desensitization rates and number of receptors were determined using a glutamate application in 10 μm glycine and 1 mm Mgo2+ at 35 mV, after which time glutamate was removed and voltage was returned to −65 mV. Model simulations of subsequently recorded GluN1/2A receptor currents were made with no adjustable parameters. Recorded currents to be simulated were generated by glutamate reapplication, followed by a voltage step from −65 to 35 mV once a steady-state current level was reached. In these experiments, however, GluN1/2A receptor currents were activated by application of either 1 μm or 1 mm glutamate (Fig. 5A). In low agonist concentration (1 μm glutamate), the GluN1/2AV-D model predicted that Mgo2+ unblock would proceed more slowly than in high agonist concentration (1 mm glutamate; Fig. 5B, red lines). Experimental data confirmed this prediction, with depolarization-induced Mgo2+ unblock from GluN1/2A receptors proceeding more slowly when currents were activated by the low agonist concentration (Fig. 5B, black lines, C). The GluN1/2AV-D model also predicted that steady-state current at 35 mV, relative to steady-state current at −65 mV, should be larger in low than in high agonist concentrations (Fig. 5D, red lines). This prediction implies that GluN1/2A receptor responses should be slightly more voltage dependent at low than at high agonist concentrations. Again, the experimental data agreed well with model predictions (Fig. 5D, black lines, E). The overall agreement between GluN1/2AV-D model simulations and measurements of Mgo2+ unblock suggest that the slow component of Mgo2+ unblock from GluN1/2A receptors is a consequence of voltage-dependent receptor gating.
Figure 5.
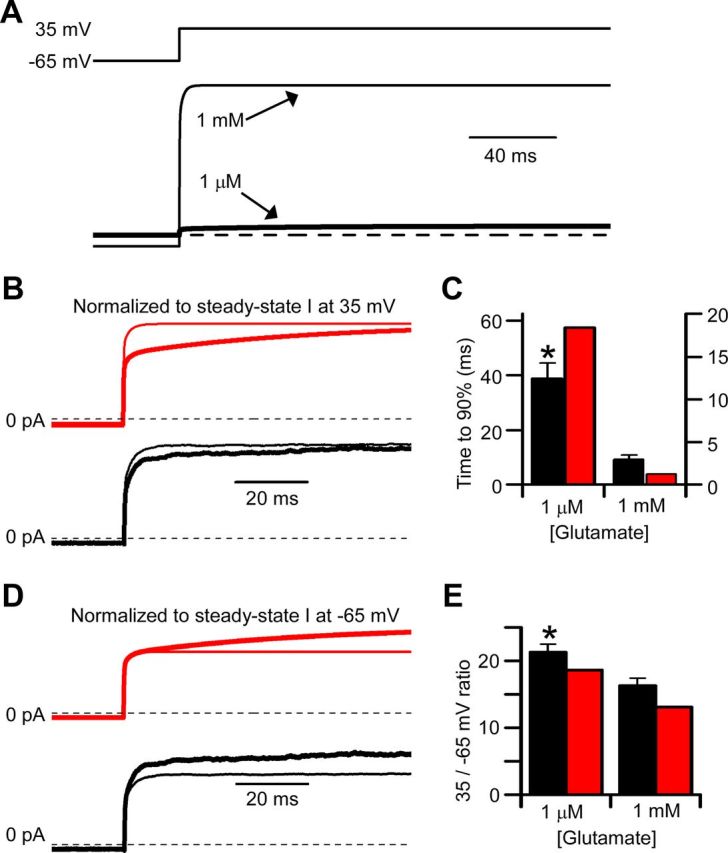
Agonist concentration dependence of slow Mgo2+ unblock is reproduced by the GluN1/2AV-D model. A, Currents (bottom traces) simulated with the GluN1/2AV-D model in 1 mm (thin line) or 1 μm (thick line) glutamate in response to a voltage jump from −65 to 35 mV (top trace) in 1 mm Mgo2+. B, D, Simulated (red lines) and recorded (black lines) currents in low (1 μm glutamate; thick line) and high (1 mm glutamate; thin lines) agonist concentration. B, Currents normalized so that the steady-state current at 35 mV in 1 μm and in 1 mm glutamate were equal; the currents at −65 mV differed slightly, although the differences are not visible in the figure. The slower approach to steady-state current at 35 mV in 1 μm glutamate reflects slower Mgo2+ unblock in low agonist concentration. C, Comparison of time required for recorded (black bars) and simulated (red bars) currents to reach 90% of their final value in low (left) and high (right) agonist concentrations. Left y-axis applies to data in 1 μm glutamate; right y-axis applies to data in 1 mm glutamate. D, Currents normalized so that the steady-state current at −65 mV (before the voltage step) in 1 μm and in 1 mm glutamate were equal. The greater outward current at 35 mV in 1 μm glutamate illustrates stronger voltage dependence of Mgo2+ inhibition in low agonist concentration. E, Comparison of absolute value of the ratio of steady-state current at 35 mV divided by steady-state current at −65 mV for recorded (black bars) and simulated (red bars) currents in low (left) and high (right) agonist concentrations. *Significantly (p < 0.05) different from corresponding value for recorded current in 1 mm glutamate.
Comparison of GluN1/2AV-D and GluN1/2BV-D model predictions and experimental results
The slow component of Mgo2+ unblock from GluN1/2A receptors is faster than the slow component of Mgo2+ unblock from GluN1/2B receptors (Clarke and Johnson, 2006). In the GluN1/2AV-D model developed here and in the previously developed GluN1/2BV-D model, inherent voltage-dependent gating resulted from voltage dependence of the same magnitude (e-fold for 175 mV) of the same single rate constant (ks+). It was important to determine whether, despite the identical voltage dependences of ks+ in the two models, the models reproduce the observed difference in kinetics of the slow component of Mgo2+ unblock. In Figure 6A, the predicted current relaxations of the GluN1/2AV-D model and GluN1/2BV-D model to the same depolarizing step (−65 to 35 mV) are compared. The slow component of Mgo2+ unblock appeared to be faster for the GluN1/2AV-D model. The slow components of simulated Mgo2+ unblock were quantified and compared with each other and with mean values measured in whole-cell experiments in Figure 6B. To optimize the validity of the comparisons, τslow was measured identically for simulations from the GluN1/2AV-D and GluN1/2BV-D models (as described for Fig. 4, the next-to slowest of a 5-component exponential fit was used as τslow). Similarly, values of τslow were measured identically for all whole-cell recordings from GluN1/2A and GluN1/2B receptors (because a very slow component of relaxations was sometimes observed, the intermediate time constant of a triple-exponential fit was used as τslow). The τslow value was substantially smaller (faster) from the GluN1/2AV-D model than from the GluN1/2BV-D model; the experimental τslow value was significantly smaller from GluN1/2A receptors than from GluN1/2B receptors; and the τslow value derived from each model was in excellent agreement with the corresponding experimental value (Fig. 6B). These results support the conclusion that GluN1/2A and GluN1/2B receptors exhibit little or no difference in their inherent voltage dependence of gating. Instead, differences in the kinetics of Mgo2+ unblock from GluN1/2A and GluN1/2B receptors (Clarke and Johnson, 2006) can be explained predominantly by differences between the gating kinetics of these receptor subtypes (Erreger et al., 2005).
Figure 6.

Differences in gating kinetics lead to differences in Mgo2+ unblocking kinetics of GluN1/2A and GluN1/2B receptors. A, Current simulations from the GluN1/2AV-D (black) and GluN1/2BV-D (gray) models in response to a voltage jump from −65 to 35 mV in 1 mm glutamate and 1 mm Mgo2+ (glycine sites are assumed always to be occupied in all simulations). Simulations are taken from Figure 4D for GluN1/2A and from Figure 6B of Clarke and Johnson (2008) for GluN1/2B. B, Values of τslow measured from current simulations based on the GluN1/2AV-D and GluN1/2BV-D models (triangles) were in excellent agreement with τslow values measured from experimental recordings from GluN1/2A and GluN1/2B receptors (open squares), respectively. Experimental values were compiled from all of our experiments in which voltage jumps from −65 to 35 mV were performed in 1 mm glutamate, 10 μm glycine, and 1 mm Mgo2+: GluN1/2A receptor values (n = 12) are derived from experiments performed in this study and from Clarke and Johnson (2006); GluN1/2B receptor values (n = 9) are derived from Clarke and Johnson (2006), (2008).
The residue at the GluN2 S/L site governs NMDAR voltage-dependent gating
A single divergent residue in GluN2 subunits is responsible for variation among NMDAR subtypes of multiple channel properties: single-channel conductance, Ca2+ selectivity, and inhibition by Mgo2+ (Siegler Retchless et al., 2012). The divergent residue, situated near the intracellular end of the M3 region, is a serine in the GluN2A and GluN2B subunits and a leucine in the GluN2C and GluN2D subunits (Fig. 7A). The site at which the residue is located is termed the GluN2 S/L site (Siegler Retchless et al., 2012). The slow component of Mgo2+ unblock and depolarization-induced potentiation of NMDARs followed a pattern consistent with a link to the GluN2 S/L site: both properties are expressed in GluN1/2A and GluN1/2B receptors, but not in GluN1/2C or GluN1/2D receptors (Clarke and Johnson, 2006, 2008; and data presented here). We therefore investigated the possibility that the GluN2 S/L site also influences NMDAR subtype dependence of depolarization-induced potentiation and the slow component of Mgo2+ unblock.
Figure 7.
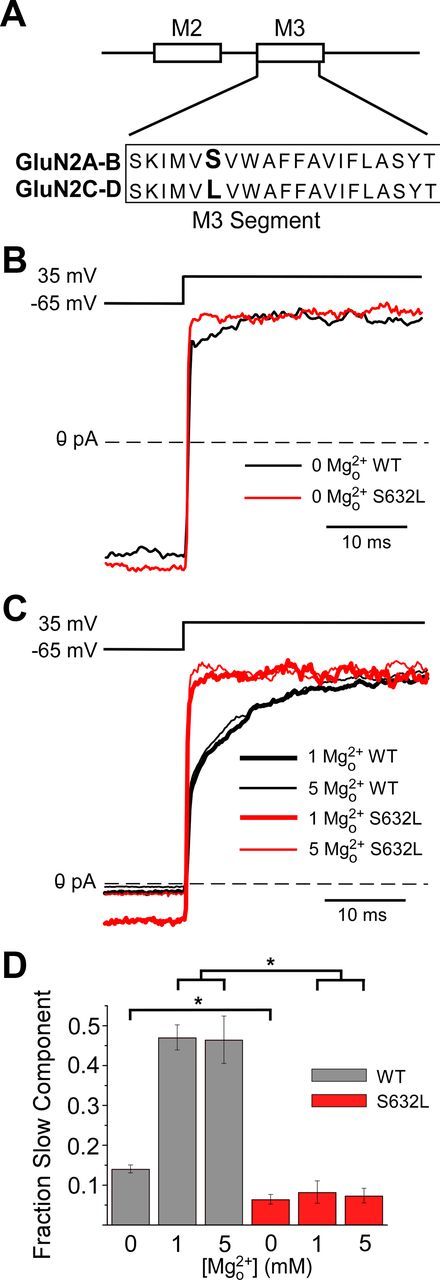
Control by a single divergent GluN2 subunit residue of voltage-dependent gating and slow Mgo2+ unblock. A, GluN2 S/L site (residues shown in bold) is located at the intracellular end of the M3 transmembrane region. Subunit residue numbering begins at the start methionine. The residues at the GluN2 S/L site are as follows: GluN2A(S632), GluN2B(S633), GluN2C(L643), and GluN2D(L657). B, Examples of GluN1/2A (WT; black line) and GluN1/2A(S632L; red line) current traces recorded from transfected tsA cells during depolarizing step from −65 to 35 mV in 30 μm NMDA and 10 μm glycine with 0 Mgo2+. C, Same as in B except in the presence of 1 or 5 mm Mgo2+. Currents in B and C were normalized to steady-state current at 35 mV (average current from 35–40 ms after the depolarizing step). D, Fractional amplitude of the slow component of current relaxations for GluN1/2A (gray) and GluN1/2A(S632L) (red) receptors in 0, 1, and 5 mm Mgo2+ (n = 5–7 in each condition). The amplitude of the slow component measured from double-exponential fits was normalized to the total amplitude of current relaxation in response to depolarizations from −65 to 35 mV. Slow components in 0 Mgo2+ were compared using the two-tailed Student's t test; slow components in 1 and 5 mm Mgo2+ were compared using one-way ANOVA followed by Tukey post hoc comparison. *Significantly different (p < 0.001).
We compared the properties of wild-type NMDARs and GluN1/2A(S632L) receptors in which the GluN2 S/L site of the GluN2A subunit was mutated from serine (normally in GluN2A and GluN2B subunits) to leucine (found in GluN2C and GluN2D subunits; Fig. 7A). In 0 Mgo2+, the slow component of GluN1/2A(S632L) receptor current relaxations in response to voltage steps from −65 to 35 mV was 48% as large as the slow component of wild-type GluN1/2A receptors (p < 0.001; Fig. 7B,D). Addition of Mgo2+ did not affect the small remaining slow component of current relaxations recorded from GluN1/2A(S632L) receptors (Fig. 7C,D), suggesting that it may be unrelated to inherent voltage dependence. In 1 mm Mgo2+, the slow component of GluN1/2A(S632L) receptor current relaxations was 15% as large as the slow component of GluN1/2A receptors (p < 0.001; Fig. 7C,D). However, because GluN1/2A(S632L) receptors are inhibited by Mgo2+ with an IC50 similar to that of GluN1/2C and GluN1/2D receptors (Siegler Retchless et al., 2012), 1 mm Mgo2+ inhibits GluN1/2A(S632L) receptors approximately fivefold less effectively than wild-type GluN1/2A receptors at −65 mV. It is possible that the reduction of the slow component of Mgo2+ unblock of GluN1/2A(S632L) receptors in 1 mm Mgo2+ results from weaker inhibition by Mgo2+. We therefore examined current relaxations in 5 mm Mgo2+ (Fig. 7C,D), a concentration that inhibits GluN1/2A(S632L) receptors as effectively as 1 mm Mgo2+ inhibits GluN1/2A receptors at −65 mV. We found that the slow component of GluN1/2A(S632L) receptor current relaxations in 5 mm Mgo2+ was 13% as large as the slow component of GluN1/2A receptors in 1 mm Mgo2+ (p < 0.001; Fig. 7C,D). Therefore, mutation of the divergent residue at the GluN2 S/L site substantially reduces both depolarization-induced potentiation and the slow component of Mgo2+ unblock. The concurrent reduction of both phenomena by a single mutation provides further compelling evidence that depolarization-induced potentiation and the slow component of Mgo2+ unblock are a consequence of voltage-dependent gating of NMDARs.
Discussion
We describe here a millisecond-timescale depolarization-induced potentiation of GluN1/2A receptor-mediated currents recorded in 0 Mgo2+ (Figs. 1, 2). A model of GluN1/2A receptor activation containing weak voltage dependence of the GluN2A subunit preopening conformational change accurately reproduced multiple properties of GluN1/2A receptor-mediated currents in response to depolarizations in both 0 Mgo2+ (Fig. 3) and 1 mm Mgo2+ (Figs. 4, 5). Models with identical voltage dependence accurately simulated the distinct kinetics of the slow component of Mgo2+ unblock from GluN1/2A and GluN1/2B receptors (Fig. 6). Remarkably, mutation of the single residue at the GluN2 S/L site in the GluN2A subunit strongly reduced both depolarization-induced potentiation and slow Mgo2+ unblock of GluN1/2A receptors (Fig. 7).
Inherent voltage-dependent gating
Models including inherent voltage-dependent gating have now been shown to accurately simulate currents mediated by both GluN1/2A (data presented here) and GluN1/2B (Clarke and Johnson, 2008) receptors. The form of these models was derived from previously developed kinetic models in which it was hypothesized that GluN1/2A receptors undergo preopening conformational changes more rapidly than GluN1/2B receptors (Banke and Traynelis, 2003; Erreger et al., 2005). Interestingly, incorporation of the same voltage dependence (e-fold per 175 mV) into the same gating transition (ks+) resulted in the GluN1/2AV-D model accurately reproducing more rapid depolarization-induced current relaxations than the GluN1/2BV-D model. Therefore, the differential responses of GluN1/2A and GluN1/2B receptors to depolarization arise predominantly from voltage-independent kinetic differences between the receptor subtypes. The similar underlying voltage dependence of GluN1/2A and GluN1/2B receptors suggests that the receptor structures responsible for voltage dependence do not differ between these NMDAR subtypes.
Numerous previous observations are consistent with the inherent voltage-dependent gating of NMDARs described here. A slow component of depolarization-induced current relaxation in 0 Mgo2+ was observed in recordings from native hippocampal NMDARs (Benveniste and Mayer, 1995; Spruston et al., 1995). In these studies, the slow current relaxation accounted for as much as 23% of the total response to a depolarization from −100 to 60 mV and was well fit by an exponential component with a τ of 2.5 ms (Benveniste and Mayer, 1995). In single-channel recordings from native hippocampal NMDARs, depolarization was observed to increase Popen in 0 Ca2+ (Gibb and Colquhoun, 1992), consistent with data presented here. Several studies of NMDAR inhibition by intracellular Mg2+ also included data consistent with inherent voltage-dependent gating. Measurements of I–V curves in 0 Mg2+ (both intracellular and extracellular) revealed outward rectification in several preparations: native NMDAR-mediated currents from cultured cortical neurons (Li-Smerin and Johnson, 1996); GluN1/2A and GluN1/2B receptors expressed in CHO cells (Li-Smerin et al., 2000); and GluN1/2A receptors expressed in Xenopus oocytes (Kupper et al., 1998). Interestingly, outward rectification was greater in whole-cell recordings from neurons than from CHO cells and was much greater in measurements of mean current from excised neuronal patches than in whole-cell experiments on neurons (Li-Smerin and Johnson, 1996; Li-Smerin et al., 2000). This observation suggests that the inherent voltage dependence of NMDARs may be subject to modulation.
Nowak and Wright (1992) described a voltage-dependent alteration in NMDAR Popen such that the Popen was significantly higher at positive than at negative membrane potentials. Such changes in Popen due to shifts in the frequency of NMDAR channel opening could explain the slow potentiation we report here. However, the change in Popen described by Nowak and Wright (1992) occurred on the time-scale of minutes. Therefore, this slower depolarization-induced potentiation may involve mechanisms distinct from the more rapid depolarization-induced current potentiation described here or perhaps may reflect a form of modulation of inherent voltage dependence.
A final voltage-dependent aspect of NMDAR function has been revealed by measurements of the decay kinetics of NMDAR-mediated EPSCs (NMDAR-EPSCs), which become slower with depolarization (Konnerth et al., 1990; Keller et al., 1991; D'Angelo et al., 1994; Lamotte d'Incamps and Ascher, 2008). The voltage-dependent decay was observed both in the absence and presence of Mgo2+, but was more pronounced in the presence of Mgo2+ (Konnerth et al., 1990; Keller et al., 1991). We simulated NMDAR-EPSCs in 1 mm Mgo2+ over a range of voltages using the GluN1/2AV-D model and found the model predicts modest slowing of synaptic currents with depolarization (Fig. 8). Additional mechanisms, such as voltage dependence of glutamate transport (Lamotte d'Incamps and Ascher, 2008), may contribute to the robust voltage dependence of NMDAR-EPSC kinetics observed in some preparations. Nevertheless, our results suggest that voltage-dependent gating of NMDARs contributes to the voltage dependence of NMDAR-EPSC decay kinetics.
Figure 8.

Voltage-dependent decay kinetics of synaptic currents in 1 mm Mgo2+ simulated by the GluN1/2AV-D model. A, Synaptic currents (top traces) were simulated with the GluN1/2AV-D model in 1 mm Mgo2+ at the indicated membrane voltages by applying a pulse of glutamate (bottom trace) that increased instantaneously from 0 to 1 mm and then decayed with a single-exponential time constant of 1 ms (Clements et al., 1992). B, Simulated synaptic currents were normalized to peak current to allow comparison of decay time course. Decay kinetics became slower with depolarization.
Slow Mgo2+ unblock from the channel of NMDARs
The GluN1/2AV-D model developed here reproduced accurately not only depolarization-induced current potentiation in 0 mm Mgo2+, but also the prominent slow component of Mgo2+ unblock. The GluN1/2AV-D model is a symmetric block model and thus does not require that the kinetics of NMDAR transitions be affected by Mgo2+ binding, which is consistent with previous conclusions (Sobolevsky and Yelshansky, 2000; Qian et al., 2002; Blanpied et al., 2005). In previous models developed to account for slow Mgo2+ unblock, it was proposed that block by Mgo2+ causes alterations in NMDAR gating, desensitization, and/or agonist unbinding (Kampa et al., 2004; Vargas-Caballero and Robinson, 2004). Although these models are able to predict accurately the slow component of Mgo2+ unblock, they do not reproduce the depolarization-induced current potentiation in 0 Mgo2+ described here and previously (Benveniste and Mayer, 1995; Spruston et al., 1995; Clarke and Johnson, 2008). The data presented here strongly support a common molecular mechanism for depolarization-induced current potentiation in 0 Mgo2+ and slow Mgo2+ unblock: adding voltage dependence to a single kinetic step in the GluN1/2AV-D model reproduces both phenomena; the kinetics of current potentiation and of slow Mgo2+ unblock do not differ; and a point mutation at the GluN2 S/L site reduces both phenomena (see below). Although it remains possible that Mgo2+ block does have limited effects on NMDAR gating, desensitization, and/or agonist binding, we conclude that slow Mgo2+ unblock results predominantly from voltage-dependent gating.
Dependence of voltage-dependent gating and slow Mgo2+ unblock on the GluN2 S/L site
The GluN2 S/L site is responsible for NMDAR subtype dependence of multiple channel properties (Siegler Retchless et al., 2012). Here we demonstrated that mutation from serine to leucine of the GluN2 S/L site in GluN2A subunits results in strong reduction of depolarization-induced potentiation in 0 Mgo2+ and of slow Mgo2+ unblock. Therefore, the GluN2 S/L site governs the NMDAR subtype dependence of voltage-dependent gating and slow Mgo2+ unblock of NMDARs in addition to inhibition by Mgo2+, selective permeability to Ca2+, and single-channel conductance. Identification of a single-site mutation that diminishes voltage-dependent gating will provide an important tool for further investigation of the mechanistic basis of NMDAR inherent voltage dependence.
Many types of ligand-gated ion channels in addition to NMDARs have been shown to be sensitive to voltage, including nicotinic acetylcholine receptors (Magleby and Stevens, 1972; Ascher et al., 1978), inhibitory glycine receptors (Legendre, 1999), and AMPA receptors (Raman and Trussell, 1995). Sensitivity of receptor gating to voltage has been most commonly proposed to result from either of two mechanisms: movement through the membrane field of charged amino acid residue(s) during gating (Magleby and Stevens, 1972; Auerbach et al., 1996) or voltage-dependent occupation of a permeant ion binding site that influences gating (Ascher et al., 1978). Reduction of voltage-dependent gating of NMDARs by the GluN2A(S632L) mutation favors the involvement of voltage-dependent occupation of a permeant ion-binding site in voltage dependence for at least two reasons. First, neither of the residues involved in the mutation, serine or leucine, is charged. This, however, is not a compelling observation because the GluN2 S/L site affects channel properties through interaction with a tryptophan residue on the adjacent GluN1 subunits, GluN1(W608) (Siegler Retchless et al., 2012). The mutation could also reduce allosterically the movement during gating of a charged residue within the membrane. Second, the GluN2A(S632L) mutation strongly affects interactions of ions (Mg2+, Ca2+, and permeant monovalent cations) with the NMDAR channel, but has limited effects on channel gating (Siegler Retchless et al., 2012), suggesting that it is more likely to affect voltage-dependent binding of an ion than movement of a charged residue during gating. Ion binding to NMDARs can influence channel gating (Li-Smerin and Johnson, 1996; Li-Smerin et al., 2001). However, elimination of Ca2+ and the use of symmetric KCl solutions have minimal effects on GluN1/2B receptor τslow (Clarke and Johnson, 2008) and we report here that elimination of Ca2+ did not affect GluN1/2A receptor Aslow significantly. These data suggest the following hypothesis: the GluN2A(S632L) mutation affects a nonspecific cation-binding site on NMDARs (Li-Smerin and Johnson, 1996; Antonov et al., 1998; Antonov and Johnson, 1999; Li-Smerin et al., 2001; Zhu and Auerbach, 2001) that is occupied in a voltage-dependent manner and the occupation of which can influence channel gating.
Footnotes
Support for this work was from National Institutes of Health Grants R01 MH045817 (to J.W.J.), T32 MH18273 to (R.J.C.), and T32 NS007433 and T32 NS073548 (to N.G.G.). We thank Karen Bouch and Christen Shiber for excellent technical assistance.
The authors declare no competing financial interests.
References
- Antonov SM, Johnson JW. Permeant ion regulation of N-methyl-D-aspartate receptor channel block by Mg2+ Proc Natl Acad Sci U S A. 1999;96:14571–14576. doi: 10.1073/pnas.96.25.14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov SM, Gmiro VE, Johnson JW. Binding sites for permeant ions in the channel of NMDA receptors and their effects on channel block. Nat Neurosci. 1998;1:451–461. doi: 10.1038/2167. [DOI] [PubMed] [Google Scholar]
- Ascher P, Nowak L. The role of divalent cations in the N-methyl-D-aspartate responses of mouse central neurones in culture. J Physiol. 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher P, Marty A, Neild TO. Life time and elementary conductance of the channels mediating the excitatory effects of acetylcholine in Aplysia neurones. J Physiol. 1978;278:177–206. doi: 10.1113/jphysiol.1978.sp012299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A, Sigurdson W, Chen J, Akk G. Voltage dependence of mouse acetylcholine receptor gating: different charge movements in di-, mono- and unliganded receptors. J Physiol. 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Traynelis SF. Activation of NR1/NR2B NMDA receptors. Nat Neurosci. 2003;6:144–152. doi: 10.1038/nn1000. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J Physiol. 1995;483:367–384. doi: 10.1113/jphysiol.1995.sp020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Clarke RJ, Johnson JW. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci. 2005;25:3312–3322. doi: 10.1523/JNEUROSCI.4262-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J Neurosci. 2006;26:5825–5834. doi: 10.1523/JNEUROSCI.0577-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. Voltage-dependent gating of NR1/2B NMDA receptors. J Physiol. 2008;586:5727–5741. doi: 10.1113/jphysiol.2008.160622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Rossi P, Taglietti V. Voltage-dependent kinetics of N-methyl-D-aspartate synaptic currents in rat cerebellar granule cells. Eur J Neurosci. 1994;6:640–645. doi: 10.1111/j.1460-9568.1994.tb00309.x. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AJ. NMDA receptor subunit gating-uncovered. Trends Neurosci. 2004;27:7–10. doi: 10.1016/j.tins.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Gibb AJ, Colquhoun D. Activation of N-methyl-D-aspartate receptors by L-glutamate in cells dissociated from adult rat hippocampus. J Physiol. 1992;456:143–179. doi: 10.1113/jphysiol.1992.sp019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion channels of excitable membranes. Ed 3. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller BU, Konnerth A, Yaari Y. Patch clamp analysis of excitatory synaptic currents in granule cells of rat hippocampus. J Physiol. 1991;435:275–293. doi: 10.1113/jphysiol.1991.sp018510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Keller BU, Ballanyi K, Yaari Y. Voltage sensitivity of NMDA-receptor mediated postsynaptic currents. Exp Brain Res. 1990;81:209–212. doi: 10.1007/BF00230117. [DOI] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper J, Ascher P, Neyton J. Internal Mg2+ block of recombinant NMDA channels mutated within the selectivity filter and expressed in Xenopus oocytes. J Physiol. 1998;507:1–12. doi: 10.1111/j.1469-7793.1998.001bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamotte d'Incamps B, Ascher P. Four excitatory postsynaptic ionotropic receptors coactivated at the motoneuron-Renshaw cell synapse. J Neurosci. 2008;28:14121–14131. doi: 10.1523/JNEUROSCI.3311-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P. Voltage dependence of the glycine receptor-channel kinetics in the zebrafish hindbrain. J Neurophysiol. 1999;82:2120–2129. doi: 10.1152/jn.1999.82.5.2120. [DOI] [PubMed] [Google Scholar]
- Levis RA, Rae JL. Constructing a patch clamp setup. Methods Enzymol. 1992;207:14–66. doi: 10.1016/0076-6879(92)07004-8. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y, Johnson JW. Effects of intracellular Mg2+ on channel gating and steady-state responses of the NMDA receptor in cultured rat neurons. J Physiol. 1996;491:137–150. doi: 10.1113/jphysiol.1996.sp021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y, Aizenman E, Johnson JW. Inhibition by intracellular Mg2+ of recombinant N-methyl-D-aspartate receptors expressed in Chinese hamster ovary cells. J Pharmacol Exp Ther. 2000;292:1104–1110. [PubMed] [Google Scholar]
- Li-Smerin Y, Levitan ES, Johnson JW. Free intracellular Mg2+ concentration and inhibition of NMDA responses in cultured rat neurons. J Physiol. 2001;533:729–743. doi: 10.1111/j.1469-7793.2001.t01-1-00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL, Stevens CF. A quantitative description of end-plate currents. J Physiol. 1972;223:173–197. doi: 10.1113/jphysiol.1972.sp009840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Nowak LM, Wright JM. Slow voltage-dependent changes in channel open-state probability underlie hysteresis of NMDA responses in Mg2+-free solutions. Neuron. 1992;8:181–187. doi: 10.1016/0896-6273(92)90119-x. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian A, Antonov SM, Johnson JW. Modulation by permeant ions of Mg2+ inhibition of NMDA-activated whole-cell currents in rat cortical neurons. J Physiol. 2002;538:65–77. doi: 10.1113/jphysiol.2001.012685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian A, Buller AL, Johnson JW. NR2 subunit-dependence of NMDA receptor channel block by external Mg2+ J Physiol. 2005;562:319–331. doi: 10.1113/jphysiol.2004.076737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Trussell LO. Concentration-jump analysis of voltage-dependent conductances activated by glutamate and kainate in neurons of the avian cochlear nucleus. Biophys J. 1995;69:1868–1879. doi: 10.1016/S0006-3495(95)80057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorge S, Elenes S, Colquhoun D. Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J Physiol. 2005;569:395–418. doi: 10.1113/jphysiol.2005.095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegler Retchless B, Gao W, Johnson JW. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat Neurosci. 2012;15:406-S2. doi: 10.1038/nn.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Yelshansky MV. The trapping block of NMDA receptor channels in acutely isolated rat hippocampal neurones. J Physiol. 2000;526:493–506. doi: 10.1111/j.1469-7793.2000.t01-2-00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol. 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern P, Béhé P, Schoepfer R, Colquhoun D. Single-channel conductances of NMDA receptors expressed from cloned cDNAs: comparison with native receptors. Proc Biol Sci. 1992;250:271–277. doi: 10.1098/rspb.1992.0159. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Caballero M, Robinson HP. Fast and slow voltage-dependent dynamics of magnesium block in the NMDA receptor: the asymmetric trapping block model. J Neurosci. 2004;24:6171–6180. doi: 10.1523/JNEUROSCI.1380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Auerbach A. K+ occupancy of the N-methyl-d-aspartate receptor channel probed by Mg2+ block. J Gen Physiol. 2001;117:287–298. doi: 10.1085/jgp.117.3.287. [DOI] [PMC free article] [PubMed] [Google Scholar]