

Abstract
Gastrointestinal stromal tumors (GISTs) are rare but treatable soft tissue sarcomas. Nearly all GISTs have somatic mutations in either the KIT or PDGFRA gene, but there are no known inherited genetic risk factors. We assessed the relationship between KIT/PDGFRA mutations and select deletions or single nucleotide polymorphisms (SNPs) in 279 participants from a clinical trial of adjuvant imatinib mesylate. Given previous evidence that certain susceptibility loci and carcinogens are associated with characteristic mutations, or “signatures” in other cancers, we hypothesized that the characteristic somatic mutations in the KIT and PDGFRA genes in GIST tumors may similarly be mutational signatures that are causally linked to specific mutagens or susceptibility loci. As previous epidemiologic studies suggest environmental risk factors such as dioxin and radiation exposure may be linked to sarcomas, we chose 208 variants in 39 candidate genes related to DNA repair and dioxin metabolism or response. We calculated adjusted odds ratios (ORs) and 95% confidence intervals (CIs) for the association between each variant and 7 categories of tumor mutation using logistic regression. We also evaluated gene-level effects using the sequence kernel association test (SKAT). Although none of the association p-values were statistically significant after adjustment for multiple comparisons, SNPs in CYP1B1 were strongly associated with KIT exon 11 codon 557-8 deletions (OR = 1.9, 95% CI: 1.3-2.9 for rs2855658 and OR = 1.8, 95% CI: 1.2-2.7 for rs1056836) and wild type GISTs (OR = 2.7, 95% CI: 1.5-4.8 for rs1800440 and OR = 0.5, 95% CI: 0.3-0.9 for rs1056836). CYP1B1 was also associated with these mutations categories in the SKAT analysis (p = 0.002 and p = 0.003, respectively). Other potential risk variants included GSTM1, RAD23B and ERCC2. This preliminary analysis of inherited genetic risk factors for GIST offers some clues about the disease's genetic origins and provides a starting point for future candidate gene or gene-environment research.
Introduction
Gastrointestinal stromal tumors (GISTs) are soft tissue sarcomas that develop primarily in the stomach (60–70%) and small intestines (20–30%), but also appear in the rectum, colon, esophagus or omentum [1], [2]. These tumors are quite rare, with an estimated annual incidence of 6.8 cases per million individuals in the US between 1992 and 2000 [3], and 3300 to 6000 new US cases predicted each year [4], though systematic under-ascertainment of GIST cases implies the true rate is slightly higher [3], [5], [6]. Data from the National Cancer Institute's Surveillance, Epidemiology and End Results (SEER) program suggest that GISTs are more common in African-Americans than Caucasians (8.9 versus 4.5 cases per 1 million individuals per year, 1992-2002) but equally common in men and women [5]. Median age at diagnosis in the SEER population is 63 years.
Unlike other gastrointestinal neoplasms, more than 90% of GISTs express the KIT proto-oncogene, as measured by immunohistochemical analysis of CD117, the stem cell factor receptor protein encoded by KIT [7], [8]. In approximately 75% of GISTs, this CD117 overexpression is attributable to a gain-in-function mutation in the tyrosine kinase domain of KIT. Once mutated, KIT may encode tyrosine kinase receptors in which the tyrosine kinase domain can be activated in the absence of stem cell factor signaling, thereby stimulating excess, unregulated proliferation of the host tumor cells [8]–[10]. Another 10-15% of GISTs have mutations in the PDGFRA gene, another tyrosine kinase receptor encoding gene [8], [11].
Primary GIST-related KIT and PDGFRA mutations have been well characterized. Results from 3 population-based studies in Switzerland [12], Norway [13] and France [14] suggest that 50-60% of all GISTs have mutations in KIT exon 11, 5–10% in KIT exon 9, 3% in KIT exon 13, 1% in KIT exon 17, 2–5% in PDGFRA exon 12 and 2–6% in PDGFRA exon 18. The proportions observed in hospital-based or convenience samples are generally consistent with these estimates, with some variability due to inclusion criteria and small sample sizes [15]–[18].
Most GISTs with primary KIT or PDGFRA mutations respond to treatment with imatinib mesylate (STI571, Gleevec™, Novartis Pharmaceuticals, Basel, Switzerland), an inhibitor of the KIT and PDGFRA tyrosine kinase. Imatinib is more effective in patients with mutations in KIT exon 11 than in patients with no tumor mutations (wild type) or exon 9 mutations [19], [20]. Unfortunately, roughly half of the patients who initially respond to imatinib develop drug-resistant disease after prolonged treatment. This acquired resistance may be attributable to the development of secondary KIT mutations in residual tumor tissue [21]–[23].
While some KIT and PDGFRA germline mutations have been identified among families with multiple GIST cases [24], [25] and a few studies have identified single nucleotide polymorphisms (SNPs) associated with soft tissue sarcoma incidence (MDM2 [26]), survival (AhR [27]), or specific translocations common in some types of soft tissue sarcoma (XPG/ERCC5 [28]), no studies have looked for inherited genetic risk factors for sporadic GISTs. Though such studies are necessary to advance our understanding of disease etiology, recruitment of cases and compatible controls is limited by the disease's rarity. A population-based study with rapid case ascertainment and collection of detailed information on non-genetic risk factors would be especially arduous, as GISTs are often misclassified in reports to cancer surveillance systems [3], [5], [6].
Given these constraints, we decided to investigate the role of inherited genetic polymorphisms in GIST development by conducting a case-only analysis of the association between tumor mutation type (mutations in KIT exon 11, KIT exon 9, PDGFRA, or wild type) and 225 variants in 39 candidate genes using tumor and blood samples collected during a phase III clinical trial of adjuvant imatinib [29]. In previous studies, certain susceptibility loci have been linked to characteristic tumor mutations, or mutational “signatures”. These include associations between GSTM1-null genotype and TP53 transversion mutations among bladder cancer patients [30], and certain functional polymorphisms in XPD and G:C→T:A TP53 mutations among lung cancer patients [31]. Similarly, we hypothesized that the characteristic somatic mutations in the KIT and PDGFRA genes in GIST tumors may be mutational signatures that are causally linked to specific mutagens or susceptibility loci. To address this hypothesis, we selected candidate genes previously linked to soft tissue sarcoma or to environmental risk factors for soft tissue sarcoma. We included genes related to dioxin, phenoxyherbicide, insecticide, vinyl chloride, and radiation response, as well as variants in the previously identified AhR, MDM2, and ERCC5 genes [32]–[38]. We also looked at polymorphisms in genes encoding proteins on the AhR/ARNT dioxin-response pathway (CYP1A2, CYP1B1, HIF1A, NQO1, and G6PC/G6PT) [39]–[41], other related metabolizing pathways (ADH1A, ADH1B, ADH1C, ALDH18A1, ALDH1A1, ALDH1A2, ALDH1A3, ALDH1B1, ALDH1L1, ALDH1L2, ALDH2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2E1, CYP3A4, GSTM1, GSTT1, GSTP1, HNF4A, NAT2, NFE2L2, NOS2A, PTGS2/COX2, and SULT1A1) [42]–[45] and TP53, a tumor suppressor and cell cycle regulation gene closely related to MDM2 [26]. Additionally, we selected several DNA repair genes (ERCC2, RAD23B, XPA, and XPC) in the same DNA repair pathway as ERCC5, as polymorphisms in these nucleotide excision repair genes can affect individual sensitivity to carcinogen-induced DNA damage [46]. As our main objective was to conduct a preliminary assessment of these candidate genes rather than any specific variants, we conducted gene-level as well as SNP-level association analyses.
Materials and Methods
Study population
In total, 713 individuals participated in American College of Surgeons Oncology Group (ACOSOG) Z9001, a multicenter, phase III, randomized, double-blind study of adjuvant imatinib (Gleevec™) versus placebo for patients with resected, primary GISTs conducted between July 1, 2002 and April 18, 2007. Cases were eligible if they had a localized tumor of at least 3 cm that tested positive for CD117 by immunohistochemical analysis with the Dako antibody (DakoCytomation, Copenhagen, Denmark). Additional information on the Z9001 trial is published elsewhere [29].
This genetic ancillary study includes the first 333 Z9001 participants who provided a blood sample and consented in writing to unspecified future research using their blood and tumor tissue samples. After removing individuals missing mutation data (n = 52) or more than 10% of their genotype data (n = 2), 279 participants remained. Information on each participant's race, age, sex, and tumor size, site, stage, grade and mitotic rate was available from the parent study. The study protocol was approved by the Institutional Review Boards of Memorial Sloan-Kettering Cancer Center and the University of North Carolina. All participants provided written informed consent.
Variant selection
Once we selected our candidate genes, we identified single nucleotide polymorphisms (SNPs) or deletions within those genes that potentially affected function and had minor allele frequencies (MAF) equal to or greater than 10% in the HapMap CEU population [47]. This included nonsense, missense and splice site mutations, as well as mutations in seed microRNA regions or transcription binding sites. All selected nonsense, missense or splice site mutations were in or near coding regions (within 2000 and 500 base pairs of the 5′ and 3′ ends of the region, respectively). SNPs that did not pass the design phase (designability score <1 or final score <0.7) were replaced with surrogate SNPs in high linkage disequilibrium with the original candidate SNP.
Laboratory Analysis
During Z9001 enrollment, all tumor and blood specimens were banked with the ACOSOG Central Specimen Bank at Washington University School of Medicine in St. Louis, Missouri, then DNA extracted from these blood samples was sent to Memorial Sloan-Kettering Cancer Center (MSKCC) for storage at −80°C until analysis. Each sample was genotyped using the GoldenGate genotyping assay (Illumina Inc., San Diego, CA) [48], which consisted of allele-specific extension/ligation methodology followed by universal primer polymerase chain reaction (PCR) amplification regions for the candidate SNPs. Allele-specific oligos and locus-specific oligos hybridized directly to the genomic DNA, upstream and downstream from the targeted SNP before the universal PCR reaction took place [49]. For internal quality control purposes, twenty-seven participants underwent duplicate genotype analysis. Concordance for duplicate samples was 99.9%. SNPs were excluded if they were mono-allelic (n = 3), had a MAF less than 5% in our study samples (n = 6), showed poor clustering (n = 7), or had no individuals homozygous for the minor allele at some levels of the outcome (n = 1), leaving 208 SNPs in the final analysis.
Deletions in GSTM1 and GSTT1 were detected using multiplex PCR utilizing sets of target specific and housekeeping gene specific primers [50]. Here, individuals with no copies of the polymorphism of interest (null genotype) were differentiated from those who had one or two copies (wild type).
DNA for mutation analysis was extracted from tumor tissue that was snap-frozen and then analyzed as previously described [15], [51]. Briefly, all cases were first tested for KIT exon 11 mutations via PCR analysis using Platinum TaqDNA Polymerase High Fidelity (Life Technologies, Inc., Gaithersburg, MD). Tumors without exon 11 mutations were then subjected to PCR analysis using primers for KIT exon 9, 13, 14 and 17 and PDGFRA exon 12 and 18.
Statistical Analysis
Participants were categorized dichotomously based on the presence or absence of a specific mutation type. The following outcomes were considered: i) a deletion of KIT exon 11 codons 557–558, ii) any other (i.e. non-codon 557-8) KIT exon 11 deletion, iii) a KIT exon 11 insertion, iv) A KIT exon 11 point mutation, v) a KIT exon 9, exon 13, exon 14, or exon 17 mutation, vi) a PDGFRA exon 18 or 12 mutation, and vii) no KIT or PDGFRA mutation (wild type). Although differentiation by non-exon 11 KIT mutations would have been preferable, the prevalence of exon 9, 13, 14 and 17 mutations was too low for independent outcome assessment.
We conducted descriptive analyses of selected demographic variables and tumor characteristics, both overall and stratified by gender and race (white vs. non-white). We also compared the covariate distributions of our study population with the remaining Z9001 trial participants to look for possible indications of bias. For each variant, we calculated the race-specific MAF and Pearson χ2 p-value for the association between genotype and race. We used Fisher's exact test when one or more cells had less than 5 observations. Additionally, we conducted a crude case-control analysis by comparing the genotype distributions among the white participants (n = 273) to the genotype distributions among individuals of European descent using the HapMap database [47]. Individuals with missing mutation data were included in these descriptive analyses.
The association between germline polymorphisms and somatic mutations was analyzed using logistic regression. We obtained odds ratios (ORs), 95% confidence intervals (CIs) and p-values for each SNP-mutation combination, adjusting for race, sex, and age at diagnosis. We coded genotypes as ordinal variables (0 = homozygous for the major allele, 1 = heterozygous, 2 = homozygous for the minor allele) and estimated per-allele ORs and 1 df trend tests. All p-values were corrected for multiple testing by controlling for the false discovery rate [52].
Gene-level association tests were conducted using the sequence kernel association test (SKAT) developed by Wu et al [53], [54]. Here, SNPs are grouped based on prior biological knowledge, in this case occurrence in the same gene, and analyzed using a logistic kernel-machine-based multi-locus test. This method requires fewer hypothesis tests than standard techniques and improves power to detect the effect of an untyped, causal locus by incorporating data from several correlated surrogate SNPs. This method also allows for covariate adjustment, nonlinear effects, and epistasis.
Briefly, this method uses a modified version of the variance component score test to assess whether the variance of subject-specific random effects differs from 0. The subject-specific model for each of n individuals takes the form:
where yi is the outcome for individual i, x
i1 to x
im are the covariate values for individual i, α0 to αm are the regression parameters, z
i1 to z
ip are the genotypes for individual i at genotypes 1 to p, and h
i = h(z
i1, z
12,…z
ip) = h(Z
i) = 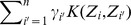 is a function for the subject-specific random effect defined by a positive, definite kernel function of the form K(•,•) and some γi, …, γn. Assuming h follows an arbitrary distribution with a mean of 0 and variance τK, testing the null hypothesis H0: h(Z) = 0 is equivalent to testing H0: τ = 0, which can be accomplished using a variance-component score statistic [55]:
is a function for the subject-specific random effect defined by a positive, definite kernel function of the form K(•,•) and some γi, …, γn. Assuming h follows an arbitrary distribution with a mean of 0 and variance τK, testing the null hypothesis H0: h(Z) = 0 is equivalent to testing H0: τ = 0, which can be accomplished using a variance-component score statistic [55]:
where logit  . To obtain a p-value, we can compare Q to a scaled χ2 distribution with scale parameter κ and degrees of freedom ν, which are modified to account for correlation between SNPs in the same SNP-set (for further explanation, see Appendix A in Wu et al [54]). In this analysis, we opted to use a kernel that models identity-by-state (IBS), or the number of alleles shared by a pair of individuals. This kernel is the most powerful option when epistatic effects may be present.
. To obtain a p-value, we can compare Q to a scaled χ2 distribution with scale parameter κ and degrees of freedom ν, which are modified to account for correlation between SNPs in the same SNP-set (for further explanation, see Appendix A in Wu et al [54]). In this analysis, we opted to use a kernel that models identity-by-state (IBS), or the number of alleles shared by a pair of individuals. This kernel is the most powerful option when epistatic effects may be present.
Results
Descriptive analyses are shown in Table 1. The median age for included participants was 58.0 years (range 18–85). Approximately half of the population was male (51%) and the majority were white (82%). Most tumors were located in the stomach (66%) or small intestines (31%) and were between 5 and 10 cm in diameter.
Table 1. Demographic information and tumor characteristics of patients included in genotyping ancillary study.
| Overall Sample | Sex Stratified | Race Stratified | |||
| N = 279 | Male (n = 142) | Female (n = 137) | White (n = 229) | Other (n = 50) | |
| Age: Median (range) | 58.0 (18–85) | 57.0 (18–85) | 58.0 (18–81) | 59.0 (18–85) | 53.0 (27–78) |
| Sex: N (%) | |||||
| Male | 142 (51) | --- | --- | --- | --- |
| Female | 137 (49) | --- | --- | --- | --- |
| Race: N (%) | |||||
| White | 229 (82) | 122 (86) | 107 (78) | --- | --- |
| Other | 50 (18) | 20 (14) | 30 (22) | --- | --- |
| Tumor Size: Median (range) | 6.5 (3.0–37.0) | 6.0 (3.0–37.0) | 6.5 (3.0–28.0) | 6.5 (3.0–37.0) | 6.0 (3.1–30.0) |
| Tumor Size: N(%) | |||||
| <5 cm | 79 (28) | 41 (29) | 38 (28) | 65 (28) | 14 (28) |
| 5-10 cm | 146 (52) | 72 (51) | 74 (54) | 119 (52) | 27 (54) |
| >10 cm | 54 (19) | 29 (20) | 25 (18) | 45 (20) | 9 (18) |
| Mitotic Rate: Median (range) | 3 (0–351) | 3 (0–351) | 3 (0–207) | 3 (0–351) | 4.5 (0–81) |
| Mitotic Rate: N(%) | |||||
| <5 | 156 (60) | 77 (58) | 79 (63) | 132 (62) | 24 (50) |
| ≥5 | 104 (40) | 57 (42) | 47 (37) | 80 (38) | 24 (50) |
| Missing | 19 | 8 | 11 | 17 | 2 |
| Tumor Location: N(%) | |||||
| Stomach | 182 (66) | 97 (69) | 85 (63) | 146 (64) | 36 (74) |
| Small Intestine | 85 (31) | 39 (28) | 46 (34) | 77 (34) | 8 (16) |
| Rectum | 2 (1) | 1 (1) | 1 (1) | 1 (0) | 1 (2) |
| Other | 8 (3) | 4 (3) | 4 (3) | 4 (2) | 4 (8) |
| Missing | 2 | 1 | 1 | 1 | 1 |
| Mutation Type: N(%) | |||||
| Exon 9 | 15 (5) | 9 (6) | 6 (4) | 15 (7) | 0 (0) |
| Exon 11 | 195 (70) | 95 (67) | 100 (73) | 153 (67) | 42 (84) |
| Exon 13 | 3 (1) | 0 (0) | 3 (2) | 2 (1) | 1 (2) |
| Exon 14 | 1 (0) | 1 (1) | 0 (0) | 1 (0) | 0 (0) |
| Exon 17 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| PDGFRA | 29 (10) | 21 (15) | 8 (6) | 25 (11) | 4 (8) |
| Wild type | 36 (13) | 16 (11) | 20 (15) | 33 (14) | 3 (6) |
| Exon 11 mutation type: N(%) | |||||
| 557-8 deletion | 66 (34) | 33 (35) | 33 (33) | 51 (33) | 15 (36) |
| Other deletion | 45 (23) | 25 (26) | 20 (20) | 34 (22) | 11 (26) |
| Insertion | 28 (14) | 14 (15) | 14 (14) | 23 (15) | 5 (12) |
| Point Mutation | 56 (29) | 23 (24) | 33 (33) | 45 (29) | 11 (26) |
| PDGFRA mutation type: N(%) | |||||
| D842V | 12 (41) | 10 (48) | 2 (25) | 10 (40) | 2 (50) |
| Other | 17 (59) | 11 (52) | 6 (75) | 15 (60) | 2 (50) |
70% of evaluated tumors had exon 11 KIT mutations, 10% had PDGFRA mutations and 13% had no identified KIT or PDGFRA mutations. Non-white participants were younger, on average (53.0 years vs. 59.0 years), and more likely to have stomach tumors (74% vs. 64%) and exon 11 KIT mutations (84% vs. 67%). The most common exon 11 KIT mutation was a deletion at codons 557–558 (34%).
Compared with other ACOSOG Z9001 participants, the individuals included in this genotyping substudy have similar demographic and tumor characteristics (Table 2). A somewhat higher proportion of participants in this ancillary study were white (82% versus 76%), but our subpopulation had nearly identical age, gender, tumor size, mitotic rate, tumor location, and tumor mutation type distributions to the full patient pool.
Table 2. Comparison of patients included in the genetic ancillary study to the remainder of the Z9001 clinical trial patients.
| Ancillary study (n = 279) | Remaining Z9001 patients (n = 436) | |
| Age: Median (range) | 58.0 (18–85) | 59.0 (21–91) |
| Sex: N (%) | ||
| Male | 142 (51) | 219 (50) |
| Female | 137 (49) | 217 (50) |
| Race: N (%) | ||
| White | 229 (82) | 332 (76) |
| Other | 50 (18) | 104 (24) |
| Tumor Size: Median (range) | 6.5 (3.0–37.0) | 6.6 (3.0–43.0) |
| Tumor Size: N(%) | ||
| <5 cm | 79 (28) | 118 (27) |
| 5–10 cm | 146 (52) | 112 (49) |
| >10 cm | 54 (19) | 105 (24) |
| Mitotic Rate: Median (range) | 3 (0–351) | 3 (0–289) |
| Mitotic Rate: N(%) | ||
| <5 | 156 (60) | 235 (65) |
| ≥5 | 104 (40) | 126 (35) |
| Missing | 19 | 75 |
| Tumor Location: N(%) | ||
| Stomach | 182 (66) | 263 (61) |
| Small Intestine | 85 (31) | 142 (33) |
| Rectum | 2 (1) | 8 (2) |
| Other | 8 (3) | 22 (5) |
| Missing | 1 | 2 |
| Mutation Type: N(%) | ||
| Exon 9 | 15 (5) | 20 (9) |
| Exon 11 | 195 (70) | 148 (64) |
| Exon 13 | 3 (1) | 6 (3) |
| Exon 14 | 1 (0) | 0 (0) |
| Exon 17 | 0 (0) | 1 (0) |
| PDGFRA | 29 (10) | 27 (12) |
| Wild type | 36 (13) | 28 (12) |
| Missing | 0 | 206 |
| Exon 11 mutation type: N(%) | ||
| 557-8 deletion | 66 (34) | 41 (28) |
| Other deletion | 45 (23) | 34 (23) |
| Insertion | 28 (14) | 18 (12) |
| Point Mutation | 56 (29) | 55 (37) |
| PDGFRA mutation type: N(%) | ||
| D842V | 12 (41) | 15 (56) |
| Other | 17 (59) | 12 (44) |
Genotype distributions of the 208 variants varied substantially by race (Table S1), but genotype frequencies among whites in our study population were very similar to the HapMap CEU sample for the 204 SNPs available in both populations. Notable discrepancies included SNPs on several aldehyde dehydrogenase genes, ALDH1A3, ALDH1A2, ALDH1L1 and ALDH1L2, and two DNA repair genes, ERCC2 and XPC.
The associations between each genetic variant and possible outcome are depicted in Figure 1, with the strength of the association quantified by the inverse of the log of the p-value. While no SNPs were statistically significant after controlling for an FDR level of 25%, some interesting patterns emerged. Most notably, minor alleles at CYP1B1 rs1056836 and rs2855658 were positively associated with a deletion at KIT exon 11 codons 557-8 (OR = 1.81, 95% CI: 1.21–2.71 and OR = 1.91, 95% CI: 1.27–2.86, respectively), while variation in another CYP1B1 SNP, rs1800440, was positively associated with wild type tumors (OR = 2.65, 95% CI: 1.48–4.76). Having a rare variant at rs1056836 was inversely associated with wild type tumors (OR = 0.54, 95% CI: 0.32–0.92).
Figure 1. Log p-values for individual variant (left) and SKAT (right) analyses by functional group and tumor mutation type.

Minor alleles in two RAD23B SNPs, rs7041137 and rs1805329, were more common among tumors with KIT exon 9, 13, or 14 mutations (ORrs7041136 = 3.05, 95% CI: 1.52–6.12 and ORrs1805329 = 3.24, 95% CI: 1.48–7.11) than tumors without such mutations. The rare form of a third RAD23B SNP, rs1805334, was also positively associated with non- exon 11 KIT mutations (OR = 2.45, 95% CI: 1.16–5.14).
rs50872 in ERCC2 was the strongest risk factor for KIT exon 11 insertion mutations (OR = 2.68, 95% CI: 1.43–5.04) and the rare variant of rs3815029 in GSTM1 was inversely associated with non-codon 557-8 KIT exon 11 deletions (OR = 0.43, 95% CI: 0.25,0.75). Based on the above evidence that at least one variant in CYP1B1, RAD23B, ERCC2, or GSTM1 was associated with one or more GIST mutation types at p<0.005, we provided a detailed evaluation of the estimated effects for all of the variants in these four key genes (Table 3). Effect estimates and p-values for the remaining variants were included in Table S2. This table includes results for rs4646755 in ALDH1L1 and rs3731149 in XPC, the strongest risk factors for PDGFRA mutations and KIT exon 11 point mutations, respectively, both of which had p-values of 0.02.
Table 3. Minor allele frequencies (MAF), Odds Ratios (ORs) and association p-values for SNPs in CYP1B1, ERCC2, GSTM1, and RAD23B by mutation type.
| Gene | SNP/ variant | KIT exon 11 codon 557-8 deletion | KIT exon 11 insertion | KIT exon 11 other deletion | KIT exon 11 point mutation | Other KIT mutation | PDGFRA mutation | Wild type | |
| CYP1B1 | rs1056836 | MAFa | 0.41/0.57 | 0.54/0.53 | 0.50/0.54 | 0.57/0.52 | 0.64/0.53 | 0.53/0.53 | 0.68/0.51 |
| OR (95% CI) | 1.81 (1.21, 2.71) | 1.01 (0.58, 1.75) | 1.11 (0.71, 1.73) | 0.81 (0.53, 1.22) | 0.71 (0.35, 1.42) | 0.93 (0.54, 1.61) | 0.54 (0.32, 0.92) | ||
| p-value | 0.004 | 1.0 | 0.6 | 0.3 | 0.3 | 0.8 | 0.02 | ||
| CYP1B1 | rs1800440 | MAFa | 0.11/0.20 | 0.16/0.18 | 0.14/0.19 | 0.13/0.19 | 0.31/0.17 | 0.24/0.17 | 0.32/0.16 |
| OR (95% CI) | 0.52 (0.28, 0.94) | 0.79 (0.37, 1.67) | 0.78 (0.42, 1.46) | 0.66 (0.36, 1.20) | 1.88 (0.91, 3.86) | 0.75 (0.39, 1.42) | 2.65 (1.48, 4.76) | ||
| p-value | 0.03 | 0.5 | 0.4 | 0.2 | 0.1 | 0.4 | 0.001 | ||
| CYP1B1 | rs2855658 | MAFa | 0.40/0.58 | 0.55/0.53 | 0.50/0.54 | 0.58/0.52 | 0.64/0.53 | 0.53/0.53 | 0.67/0.51 |
| OR (95% CI) | 1.91 (1.27, 2.86) | 0.94 (0.54, 1.63) | 1.12 (0.71, 1.75) | 0.77 (0.51, 1.17) | 0.71 (0.35, 1.42) | 0.93 (0.54, 1.61) | 0.56 (0.32, 0.96) | ||
| p-value | 0.002 | 0.8 | 0.6 | 0.2 | 0.3 | 0.8 | 0.04 | ||
| ERCC2 | rs13181 | MAFa | 0.33/0.33 | 0.41/0.32 | 0.36/0.32 | 0.32/0.33 | 0.39/0.32 | 0.33/0.33 | 0.22/0.34 |
| OR (95% CI) | 1.03 (0.68, 1.55) | 1.53 (0.87, 2.68) | 1.24 (0.78, 1.98) | 0.96 (0.62, 1.50) | 1.20 (0.60, 2.38) | 0.96 (0.54, 1.69) | 0.47 (0.26, 0.88) | ||
| p-value | 0.9 | 0.1 | 0.4 | 0.9 | 0.6 | 0.9 | 0.02 | ||
| ERCC2 | rs171140 | MAFa | 0.44/0.43 | 0.39/0.43 | 0.33/0.45 | 0.42/0.43 | 0.44/0.43 | 0.38/0.44 | 0.60/0.41 |
| OR (95% CI) | 1.16 (0.77, 1.76) | 0.81 (0.45, 1.45) | 0.67 (0.41, 1.10) | 0.94 (0.61, 1.45) | 0.88 (0.44, 1.76) | 1.33 (0.74, 2.39) | 1.98 (1.15, 3.42) | ||
| p-value | 0.5 | 0.5 | 0.1 | 0.8 | 0.7 | 0.3 | 0.01 | ||
| ERCC2 | rs1799787 | MAFa | 0.23/0.25 | 0.30/0.24 | 0.26/0.25 | 0.24/0.25 | 0.33/0.24 | 0.26/0.25 | 0.19/0.26 |
| OR (95% CI) | 0.95 (0.60, 1.51) | 1.40 (0.76, 2.57) | 1.15 (0.68, 1.93) | 0.95 (0.58, 1.55) | 1.36 (0.66, 2.79) | 0.92 (0.49, 1.71) | 0.58 (0.30, 1.10) | ||
| p-value | 0.8 | 0.3 | 0.6 | 0.8 | 0.4 | 0.8 | 0.1 | ||
| ERCC2 | rs3916874 | MAFa | 0.24/0.22 | 0.18/0.23 | 0.21/0.23 | 0.21/0.23 | 0.19/0.23 | 0.24/0.22 | 0.26/0.22 |
| OR (95% CI) | 1.20 (0.76, 1.91) | 0.70 (0.34, 1.46) | 0.94 (0.54, 1.64) | 0.95 (0.57, 1.59) | 0.77 (0.33, 1.80) | 0.97 (0.51, 1.85) | 1.28 (0.71, 2.28) | ||
| p-value | 0.4 | 0.3 | 0.8 | 0.8 | 0.5 | 0.9 | 0.4 | ||
| ERCC2 | rs50871 | MAFa | 0.36/0.45 | 0.45/0.42 | 0.33/0.44 | 0.43/0.43 | 0.42/0.43 | 0.53/0.41 | 0.57/0.41 |
| OR (95% CI) | 0.74 (0.49, 1.11) | 1.08 (0.62, 1.89) | 0.69 (0.43, 1.11) | 1.04 (0.68, 1.58) | 0.80 (0.41, 1.57) | 0.64 (0.37, 1.13) | 1.73 (1.03, 2.93) | ||
| p-value | 0.1 | 0.8 | 0.1 | 0.9 | 0.5 | 0.1 | 0.04 | ||
| ERCC2 | rs50872 | MAFa | 0.24/0.21 | 0.39/0.20 | 0.13/0.24 | 0.21/0.22 | 0.33/0.21 | 0.14/0.23 | 0.18/0.23 |
| OR (95% CI) | 1.21 (0.75, 1.95) | 2.68 (1.43, 5.04) | 0.47 (0.24, 0.91) | 0.88 (0.52, 1.49) | 1.91 (0.90, 4.04) | 2.09 (0.94, 4.66) | 0.79 (0.41, 1.54) | ||
| p-value | 0.4 | 0.002 | 0.03 | 0.6 | 0.1 | 0.1 | 0.5 | ||
| GSTM1 | deletion | MAFa | 0.38/0.55 | 0.50/0.51 | 0.47/0.52 | 0.56/0.50 | 0.76/0.49 | 0.59/0.50 | 0.56/0.50 |
| OR (95% CI) | 1.91 (1.07, 3.39) | 1.07 (0.48, 2.36) | 1.17 (0.61, 2.25) | 0.75 (0.41, 1.38) | 0.33 (0.10, 1.04) | 1.38 (0.62, 3.04) | 0.89 (0.43, 1.86) | ||
| p-value | 0.03 | 0.9 | 0.6 | 0.4 | 0.1 | 0.4 | 0.8 | ||
| GSTM1 | rs3815029 | MAFa | 0.42/0.37 | 0.34/0.39 | 0.24/0.41 | 0.45/0.37 | 0.39/0.38 | 0.40/0.38 | 0.40/0.38 |
| OR (95% CI) | 1.32 (0.87, 2.00) | 0.76 (0.40, 1.41) | 0.43 (0.25, 0.75) | 1.46 (0.93, 2.28) | 0.98 (0.48, 2.04) | 1.00 (0.56, 1.81) | 1.16 (0.68, 1.99) | ||
| p-value | 0.2 | 0.4 | 0.003 | 0.1 | 1.0 | 1.0 | 0.6 | ||
| RAD23B | rs10868 | MAFa | 0.10/0.10 | 0.09/0.10 | 0.09/0.10 | 0.07/0.11 | 0.14/0.10 | 0.12/0.10 | 0.14/0.10 |
| OR (95% CI) | 1.02 (0.51, 2.04) | 0.79 (0.28, 2.19) | 0.90 (0.39, 2.05) | 0.58 (0.26, 1.33) | 1.34 (0.46, 3.86) | 0.82 (0.33, 2.02) | 1.49 (0.66, 3.37) | ||
| p-value | 1.0 | 0.6 | 0.8 | 0.2 | 0.6 | 0.7 | 0.3 | ||
| RAD23B | rs1805329 | MAFa | 0.16/0.18 | 0.13/0.18 | 0.16/0.18 | 0.17/0.17 | 0.39/0.16 | 0.16/0.17 | 0.17/0.17 |
| OR (95% CI) | 0.95 (0.55, 1.64) | 0.69 (0.29, 1.61) | 0.97 (0.51, 1.86) | 1.03 (0.58, 1.82) | 3.24 (1.48, 7.11) | 1.19 (0.54, 2.61) | 0.77 (0.38, 1.55) | ||
| p-value | 0.8 | 0.4 | 0.9 | 0.9 | 0.003 | 0.7 | 0.5 | ||
| RAD23B | rs1805330 | MAFa | 0.06/0.09 | 0.07/0.08 | 0.11/0.08 | 0.08/0.08 | 0.14/0.08 | 0.07/0.08 | 0.08/0.08 |
| OR (95% CI) | 0.65 (0.30, 1.38) | 0.80 (0.29, 2.20) | 1.29 (0.64, 2.61) | 0.94 (0.45, 1.94) | 2.22 (0.83, 5.95) | 1.26 (0.45, 3.51) | 1.36 (0.55, 3.36) | ||
| p-value | 0.3 | 0.7 | 0.5 | 0.9 | 0.1 | 0.7 | 0.5 | ||
| RAD23B | rs1805334 | MAFa | 0.23/0.23 | 0.18/0.23 | 0.24/0.22 | 0.22/0.23 | 0.42/0.21 | 0.16/0.23 | 0.21/0.23 |
| OR (95% CI) | 1.10 (0.68, 1.79) | 0.75 (0.36, 1.56) | 1.23 (0.70, 2.16) | 1.01 (0.60, 1.69) | 2.45 (1.16, 5.14) | 1.72 (0.79, 3.75) | 0.74 (0.39, 1.41) | ||
| p-value | 0.7 | 0.4 | 0.5 | 1.0 | 0.02 | 0.2 | 0.4 | ||
| RAD23B | rs7041137 | MAFa | 0.27/0.29 | 0.21/0.29 | 0.30/0.28 | 0.28/0.28 | 0.53/0.27 | 0.24/0.29 | 0.26/0.29 |
| OR (95% CI) | 0.93 (0.61, 1.42) | 0.70 (0.37, 1.34) | 1.09 (0.68, 1.74) | 0.98 (0.63, 1.54) | 3.05 (1.52, 6.12) | 1.27 (0.69, 2.34) | 0.90 (0.52, 1.59) | ||
| p-value | 0.7 | 0.3 | 0.7 | 0.9 | 0.002 | 0.4 | 0.7 | ||
| aMAF among those with mutation/MAF among those without mutation. | |||||||||
These patterns were preserved in the gene-level SKAT analysis (Figure 1, Table 4, and Table S3), with CYP1B1 again associated with KIT exon 11 codon 557-8 deletions and wild type tumors (p = 0.002 and 0.003, respectively); strong associations between RAD23B and KIT exon 9, 13 or 14 mutations (p = 0.002); and GSTM1 and non-codon 557-8 KIT exon 11 deletions (p = 0.01). ALDH1L2 was also strongly associated with wild type tumors (p = 0.01). As for the other three possible tumor subtypes, ALDH2 was associated with KIT exon 11 insertions (p = 0.03) and the null GSTT1 genotype was associated with PDGFRA-mutated tumors (p = 0.04). No genes were associated with KIT exon 11 point mutations (p<0.05).
Table 4. P-values for sequence kernel association test (SKAT) for CYP1B1, ERCC2, GSTM1, and RAD23B, by mutation type.
| Gene | KIT exon 11 codon 557-8 deletion | KIT exon 11 insertion | KIT exon 11 other deletion | KIT exon 11 point mutation | Other KIT mutation | PDGFRA mutation | Wild type |
| CYP1B1 | 0.002 | 0.8 | 0.8 | 0.3 | 0.1 | 0.9 | 0.003 |
| ERCC2 | 0.6 | 0.1 | 0.1 | 0.9 | 0.6 | 0.4 | 0.02 |
| GSTM1 | 0.05 | 0.7 | 0.01 | 0.1 | 0.1 | 0.9 | 0.8 |
| RAD23B | 0.9 | 0.6 | 1.0 | 0.8 | 0.002 | 0.2 | 0.8 |
Although the effect estimates were very imprecise, the associations between the rare alleles of CYP1B1 SNPs rs1056836 and rs2855658 and KIT exon 11 codon 557-8 deletions were even stronger when the analysis was limited to small intestinal tumors (ORrs1056836 = 5.18, 95% CI: 2.07, 12.95 and ORrs2855658 = 5.17, 95% CI: 2.05, 13.03). Neither SNP was associated with the outcome in stomach GISTs. No other clear patterns emerged in site-specific subanalyses (data not shown).
Discussion
In this preliminary investigation of genetic risk factors for GIST tumor subtypes we identified several genes and SNPs worthy of further investigation. This included SNPs on two xenobiotic metabolizing genes, CYP1B1 and GSTM1, and two DNA repair genes, RAD23B and ERCC2. Further exploration of the relationship between GISTs and aldehyde dehydrogenase genes or other DNA repair genes (e.g. XPC), may also be warranted.
CYP1B1 encodes a cytochrome P450 enzyme that is involved with phase I metabolism of PAHs, dioxin, and other chemicals [43]. Two of the CYP1B1 SNPs we assessed have previously been linked to cancer. This included the rare variant at rs1056836, a missense mutation, which has been linked to increased risk of lung cancer [56], [57], multiple myeloma [39] and head and neck cancer [58], [59], with a possible inverse association with pancreatic cancer [60]. Previous evaluations of the SNP's association with breast, colorectal, endometrial and prostate cancer have produced mostly null findings [61]–[65]. The rare allele of rs1800440, another missense mutation, was also associated with lung and head and neck cancer [56], [59], with no reported association with breast or colorectal cancer [62], [66]. However, this SNP did exhibit an inverse association with endometrial cancer [61], [65]. The remaining CYP1B1 SNP, rs2855658, is located in a seed microRNA region, but has no previously established links to cancer.
Although there is little evidence of a link between cancer and the specific RAD23B, ERCC2, and GSTM1 variants identified here, previous studies have observed associations between one or more types of cancer and other variants on these three genes. For example, SNPs in RAD23B have been linked to esophageal [67] and bladder [68] cancers and one SNP near RAD23B was strongly associated with breast cancer in a genome-wide association study [69]. ERCC2 has also been linked to bladder cancer [68] and a large meta-analysis completed in 2006 reported statistically significant associations between ERCC2 SNPs and skin, breast and lung cancer [70]. Neither RAD23B nor ERCC2 have been linked to any type of sarcoma. Like the seed microRNA and missense mutation SNPs in CYP1B1 that were strongly associated with tumor mutations in the present study, some of the identified RAD23B and ERCC2 SNPs also have potentially functional roles. For example, rs13181 on ERCC2 is a missense mutation, as is rs1805329 on RAD23B. Additionally, RAD23B's rs1805330 is a splice site mutation and rs10868 and rs1805334 are located on transcription binding sites. As previously discussed, both RAD23B and ERCC2 are nucleotide excision repair genes. Polymorphisms in these and other DNA repair genes could impair an individual's DNA damage response and affect their carcinogen sensitivity [46].
GSTM1 is one of several genes encoding glutathione S-transferases, which are phase II xenobiotic metabolizing enzymes responsible for carcinogen activation or detoxification [45]. In previous studies, GSTM1 deletions have been linked to osteosarcoma incidence [71] and recurrence [72], with a non-statistically significant positive association with soft tissue sarcoma mortality [73]. Other studies of GSTM1 deletions have identified positive associations between the null genotype and a variety of cancers, included oral [45], colorectal [74], cervical [75], and bladder [76].
None of the association p-values were statistically significant after adjustment for multiple comparisons, whether we applied a false discovery rate correction of 25% or even 50%. While this implies that the observed associations may be due to chance, it should be noted that this was the first investigation of inherited risk factors for GISTs and our main study purpose was to identify variants worthy of further exploration. This study may also have limited generalizability. Study subjects were drawn from a predominantly white clinical trial population, and our findings may not be applicable to other racial groups or to all socioeconomic groups. As the HapMap CEU population is made up of 60 parent-child trios, it may not be an adequate comparison group for our population, especially since we were unable to adjust for unequal distributions of age, gender or other potential confounders.
Outcome misclassification is also a potential concern, as tumors with KIT exon 11 mutations were not assessed for other KIT or PDGFRA mutations and we did not test for PDGFRA exon 14 mutations in any tumors. However, previous reports suggest that GISTs with 2 or more mutations are rare (<5%) [12], [14], as are PDGFRA exon 14 mutations (<1%) [11], [77]. Thus, outcome misclassification is unlikely to be a substantial source of bias. While we have only limited evidence that our outcome classification system corresponds to distinct carcinogenic processes in GISTs, linking genetic polymorphisms to tumor phenotypes is valuable for generating etiologic hypotheses [37], [38].
In this small, yet novel, case-only study of genetic risk factors for GIST tumor subtypes we identified several variants in CYP1B1, RAD23B, GSTM1, and ERCC2 that we believe are worthy of further investigation. We hope that this exploratory analysis serves as a starting point for future research on genetic and environmental causes of these rare and understudied tumors.
Supporting Information
Minor allele frequencies (MAF) and p-values for comparison of genotype frequencies: Z9001 genotyped whites (n = 273) versus non-whites (n = 58) and genotyped Z9001 whites versus the HapMap CEU population (n = 180).
(PDF)
Minor allele frequencies (MAF), Odds Ratios (ORs) and association p-values by mutation type.
(PDF)
P-values for sequence kernel association test (SKAT) for remaining genes, by mutation type.
(PDF)
Acknowledgments
We thank Aliaksandra Samoila and Kenneth Cheung for their technical assistance during the preparation of DNA specimens for genotyping and direct sequencing. The authors also wish to thank Michael Wu for his statistical advice.
Funding Statement
This work was supported by Novartis Pharmaceuticals Corp. agreement #CSTI571BUS249. The funders had no role in study design, data collection and analysis, or decision to publish the manuscript. The funders did review the manuscript prior to submission, but their contributions were entirely cosmetic and did not alter the interpretation of the results.
References
- 1. Miettinen M, Lasota J (2001) Gastrointestinal stromal tumors--definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. . 438: 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Gastrointestinal Stromal Tumors Treatment (PDQ®) - National Cancer Institute. Available: http://www.cancer.gov/cancertopics/pdq/treatment/gist/HealthProfessional.Accessed 2012 Oct 24 .
- 3. Tran T, Davila JA, El-Serag HB (2005) The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol. . 100: 162–168. [DOI] [PubMed] [Google Scholar]
- 4. Corless CL, Heinrich MC (2008) Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. . 3: 557–586. [DOI] [PubMed] [Google Scholar]
- 5. Perez EA, Livingstone AS, Franceschi D, Rocha-Lima C, Lee DJ, et al. (2006) Current incidence and outcomes of gastrointestinal mesenchymal tumors including gastrointestinal stromal tumors. J Am Coll Surg. . 202: 623–629. [DOI] [PubMed] [Google Scholar]
- 6. Cheung MC, Zhuge Y, Yang R, Koniaris LG (2009) Disappearance of racial disparities in gastrointestinal stromal tumor outcomes. J Am Coll Surg. . 209: 7–16. [DOI] [PubMed] [Google Scholar]
- 7. Miettinen M, Lasota J (2006) Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. . 130: 1466–1478. [DOI] [PubMed] [Google Scholar]
- 8. Rubin BP, Heinrich MC, Corless CL (2007) Gastrointestinal stromal tumour. Lancet. . 369: 1731–1741. [DOI] [PubMed] [Google Scholar]
- 9. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, et al. (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. . 279: 577–580. [DOI] [PubMed] [Google Scholar]
- 10. Berman J, OLeary TJ (2001) Gastrointestinal stromal tumor workshop. Hum Pathol. . 32: 578–582. [DOI] [PubMed] [Google Scholar]
- 11. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, et al. (2005) PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. . 23: 5357–5364. [DOI] [PubMed] [Google Scholar]
- 12. Mazzola P, Spitale A, Banfi S, Mazzucchelli L, Frattini M, et al. (2008) Epidemiology and molecular biology of gastrointestinal stromal tumors (GISTs): a population-based study in the South of Switzerland, 1999–2005. Histol Histopathol. . 23: 1379–1386. [DOI] [PubMed] [Google Scholar]
- 13. Steigen SE, Eide TJ (2006) Trends in incidence and survival of mesenchymal neoplasm of the digestive tract within a defined population of northern Norway. APMIS. . 114: 192–200. [DOI] [PubMed] [Google Scholar]
- 14. Cassier PA, Ducimetière F, Lurkin A, Ranchère-Vince D, Scoazec J, et al. (2010) A prospective epidemiological study of new incident GISTs during two consecutive years in Rhône Alpes region: incidence and molecular distribution of GIST in a European region. Br J Cancer. . 103: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antonescu CR, Sommer G, Sarran L, Tschernyavsky SJ, Riedel E, et al. (2003) Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. . 9: 3329–3337. [PubMed] [Google Scholar]
- 16. Lasota J, Kuban W, Wardelmann E, Debiec-Rychter M, Merkelbach-Bruse S, et al. (2008) KIT codon 558 insertions in gastrointestinal stromal tumors. Analysis of 17 rare KIT mutants. Hum Pathol. . 39: 1728–1736. [DOI] [PubMed] [Google Scholar]
- 17. Miettinen M, Sobin LH, Lasota J (2005) Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. . 29: 52–68. [DOI] [PubMed] [Google Scholar]
- 18. Cassier PA, Fumagalli E, Rutkowski P, Schoffski P, van Glabbeke M, et al. (2012) Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. . 18: 4458–4464. [DOI] [PubMed] [Google Scholar]
- 19. Chen P, Zong L, Zhao W, Shi L (2010) Efficacy evaluation of imatinib treatment in patients with gastrointestinal stromal tumors: a meta-analysis. World J Gastroenterol. . 16: 4227–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao J, Dang Y, Sun N, Li J, Shen L (2012) C-KIT mutations were closely associated with the response to Imatinib in Chinese advanced gastrointestinal stromal tumor patients. Med Oncol. . 29: 3039–3045. [DOI] [PubMed] [Google Scholar]
- 21. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, et al. (2005) Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. . 11: 4182–4190. [DOI] [PubMed] [Google Scholar]
- 22. Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, et al. (2004) A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. . 64: 5913–5919. [DOI] [PubMed] [Google Scholar]
- 23. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, et al. (2008) Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. . 216: 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, et al. (2002) Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology. . 122: 1493–1499. [DOI] [PubMed] [Google Scholar]
- 25. Thalheimer A, Schlemmer M, Bueter M, Merkelbach-Bruse S, Schildhaus H, et al. (2008) Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutation N822Y. Am J Surg Pathol. . 32: 1560–5. [DOI] [PubMed] [Google Scholar]
- 26. Taubert H, Kappler M, Meye A, Bartel F, Schlott T, et al. (2000) A MboII polymorphism in exon 11 of the human MDM2 gene occuring in normal blood donors and in soft tissue sarcoma patients: an indication for an increased cancer susceptibility? Mutat Res. . 456: 39–44. [DOI] [PubMed] [Google Scholar]
- 27. Berwick M, Matullo G, Song YS, Guarrera S, Dominguez G, et al. (2004) Association between aryl hydrocarbon receptor genotype and survival in soft tissue sarcoma. J Clin Oncol. . 22: 3997–4001. [DOI] [PubMed] [Google Scholar]
- 28. Le Morvan V, Longy M, Bonaïti-Pellié C, Bui B, Houédé N, et al. (2006) Genetic polymorphisms of the XPG and XPD nucleotide excision repair genes in sarcoma patients. Int J Cancer. . 119: 1732–1735. [DOI] [PubMed] [Google Scholar]
- 29. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PWT, et al. (2009) Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. . 373: 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryk C, Berggren P, Kumar R, Hemminki K, Larsson P, et al. (2005) Influence of GSTM1, GSTT1, GSTP1 and NAT2 genotypes on the p53 mutational spectrum in bladder tumours. Int J Cancer. . 113: 761–768. [DOI] [PubMed] [Google Scholar]
- 31. Mechanic LE, Marrogi AJ, Welsh JA, Bowman ED, Khan MA, et al. (2005) Polymorphisms in XPD and TP53 and mutation in human lung cancer. Carcinogenesis. . 26: 597–604. [DOI] [PubMed] [Google Scholar]
- 32. Kogevinas M, Becher H, Benn T, Bertazzi PA, Boffetta P, et al. (1997) Cancer mortality in workers exposed to phenoxy herbicides, chlorophenols, and dioxins. An expanded and updated international cohort study. Am J Epidemiol. . 145: 1061–1075. [DOI] [PubMed] [Google Scholar]
- 33. Berwick M, Song Y, Jordan R, Brady MS, Orlow I (2001) Mutagen sensitivity as an indicator of soft tissue sarcoma risk. Environ Mol Mutagen. . 38: 223–226. [DOI] [PubMed] [Google Scholar]
- 34. Boffetta P, Matisane L, Mundt KA, Dell LD (2003) Meta-analysis of studies of occupational exposure to vinyl chloride in relation to cancer mortality. Scand J Work Environ Health. . 29: 220–229. [DOI] [PubMed] [Google Scholar]
- 35. Hardell L (2008) Pesticides, soft-tissue sarcoma and non-Hodgkin lymphoma--historical aspects on the precautionary principle in cancer prevention. Acta Oncol. . 47: 347–354. [DOI] [PubMed] [Google Scholar]
- 36. Brady MS, Gaynor JJ, Brennan MF (1992) Radiation-associated sarcoma of bone and soft tissue. Arch Surg. . 127: 1379–1385. [DOI] [PubMed] [Google Scholar]
- 37. Dixon K, Kopras E (2004) Genetic alterations and DNA repair in human carcinogenesis. Semin Cancer Biol. . 14: 441–448. [DOI] [PubMed] [Google Scholar]
- 38. Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC (2004) TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci Publ. . 157: 247–270. [PubMed] [Google Scholar]
- 39. Gold LS, De Roos AJ, Brown EE, Lan Q, Milliken K, et al. (2009) Associations of common variants in genes involved in metabolism and response to exogenous chemicals with risk of multiple myeloma. Cancer Epidemiol. . 33: 276–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fujii-Kuriyama Y (2010) Kawajiri (2010) Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci. . 86: 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lord-Dufour S, Copland IB, Levros L Jr, Post M, Das A, et al. (2009) Evidence for transcriptional regulation of the glucose-6-phosphate transporter by HIF-1alpha: Targeting G6PT with mumbaistatin analogs in hypoxic mesenchymal stromal cells. Stem Cells. . 27: 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoshimura K, Hanaoka T, Ohnami S, Ohnami S, Kohno T, et al. (2003) Allele frequencies of single nucleotide polymorphisms (SNPs) in 40 candidate genes for gene-environment studies on cancer: data from population-based Japanese random samples. J Hum Genet. . 48: 654–658. [DOI] [PubMed] [Google Scholar]
- 43. De Roos AJ, Gold LS, Wang S, Hartge P, Cerhan JR, et al. (2006) Metabolic gene variants and risk of non-Hodgkin's lymphoma. Cancer Epidemiol Biomarkers Prev. . 15: 1647–1653. [DOI] [PubMed] [Google Scholar]
- 44. Kiyohara C (2000) Genetic polymorphism of enzymes involved in xenobiotic metabolism and the risk of colorectal cancer. J Epidemiol. . 10: 349–360. [DOI] [PubMed] [Google Scholar]
- 45. Zhang Z, Hao K, Shi R, Zhao G, Jiang G, et al. (2011) Glutathione S-transferase M1 (GSTM1) and glutathione S-transferase T1 (GSTT1) null polymorphisms, smoking, and their interaction in oral cancer: a HuGE review and meta-analysis. Am J Epidemiol. . 173: 847–857. [DOI] [PubMed] [Google Scholar]
- 46. Jalal S, Earley JN, Turchi JJ (2011) DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. . 17: 6973–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.HapMap Homepage. Available: http://hapmap.ncbi.nlm.nih.gov/. Accessed 2012 Nov 24.
- 48. Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, et al. (2008) Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci U S A. . 105: 4340–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shen R, Fan J, Campbell D, Chang W, Chen J, et al. (2005) High-throughput SNP genotyping on universal bead arrays. Mutat Res. . 573: 70–82. [DOI] [PubMed] [Google Scholar]
- 50. Steinberg ML, Hubbard K, Utti C, Clas B, Hwang B, et al. (2009) Patterns of persistent DNA damage associated with sun exposure and the glutathione S-transferase M1 genotype in melanoma patients. Photochem Photobiol. . 85: 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dematteo RP, Gold JS, Saran L, Gönen M, Liau KH, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer. . 112: 608–615. [DOI] [PubMed] [Google Scholar]
- 52. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res. . 125: 279–284. [DOI] [PubMed] [Google Scholar]
- 53. Wu MC, Lee S, Cai T, Li Y, Boehnke M, et al. (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. . 89: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu MC, Kraft P, Epstein MP, Taylor DM, Chanock SJ, et al. (2010) Powerful SNP-set analysis for case-control genome-wide association studies. Am J Hum Genet. . 86: 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang D, Lin X (2003) Hypothesis testing in semiparametric additive mixed models. Biostatistics. . 4: 57–74. [DOI] [PubMed] [Google Scholar]
- 56. Xu W, Zhou Y, Hang X, Shen D (2012) Current evidence on the relationship between CYP1B1 polymorphisms and lung cancer risk: a meta-analysis. Mol Biol Rep. . 39: 2821–2829. [DOI] [PubMed] [Google Scholar]
- 57. Chen B, Qiu L, Li Y, Xu W, Wang X, et al. (2010) The CYP1B1 Leu432Val polymorphism contributes to lung cancer risk: evidence from 6501 subjects. Lung Cancer. . 70: 247–252. [DOI] [PubMed] [Google Scholar]
- 58. Harth V, Schafer M, Abel J, Maintz L, Neuhaus T, et al. (2008) Head and neck squamous-cell cancer and its association with polymorphic enzymes of xenobiotic metabolism and repair. J Toxicol Environ Health A. . 71: 887–897. [DOI] [PubMed] [Google Scholar]
- 59. Singh AP, Shah PP, Mathur N, Buters JTM, Pant MC (2008) Genetic polymorphisms in cytochrome P4501B1 and susceptibility to head and neck cancer. Mutat Res. . 639: 11–19. [DOI] [PubMed] [Google Scholar]
- 60. Vrana D, Novotny J, Holcatova I, Hlavata I, Soucek P (2010) CYP1B1 gene polymorphism modifies pancreatic cancer risk but not survival. Neoplasma. . 57: 15–19. [DOI] [PubMed] [Google Scholar]
- 61. Ashton KA, Proietto A, Otton G, Symonds I, McEvoy M, et al. (2010) Polymorphisms in genes of the steroid hormone biosynthesis and metabolism pathways and endometrial cancer risk. Cancer Epidemiol. . 34: 328–337. [DOI] [PubMed] [Google Scholar]
- 62. Reding KW, Chen C, Lowe K, Doody DR, Carlson CS, et al. (2012) Estrogen-related genes and their contribution to racial differences in breast cancer risk. Cancer Causes Control. . 23: 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xie Y, Liu G, Miao X, Liu Y, Zhou W, Zhong D (2012) CYP1B1 Leu432Val polymorphism and colorectal cancer risk among Caucasians: a meta-analysis. Tumour Biol. . 33: 809–816. [DOI] [PubMed] [Google Scholar]
- 64. Cui L, Dillehay K, Chen W, Shen D, Dong Z, et al. (2012) Association of the CYP1B1 Leu432Val polymorphism with the risk of prostate cancer: a meta-analysis. Mol Biol Rep. . 39: 7465–7471. [DOI] [PubMed] [Google Scholar]
- 65. McGrath M, Hankinson SE, Arbeitman L, Colditz GA, Hunter DJ, et al. (2004) Cytochrome P450 1B1 and catechol-O-methyltransferase polymorphisms and endometrial cancer susceptibility. Carcinogenesis. . 25: 559–565. [DOI] [PubMed] [Google Scholar]
- 66. Mei Q, Zhou D, Han J, Lu H, Tang B (2012) CYP1B1 Asn453Ser polymorphism and colorectal cancer risk: a meta-analysis. Metabolism. . 61: 1321–1329. [DOI] [PubMed] [Google Scholar]
- 67. Pan J, Lin J, Izzo JG, Liu Y, Xing J, et al. (2009) Genetic susceptibility to esophageal cancer: the role of the nucleotide excision repair pathway. Carcinogenesis. . 30: 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. García-Closas M, Malats N, Real FX, Welch R, Kogevinas M, et al. (2006) Genetic variation in the nucleotide excision repair pathway and bladder cancer risk. Cancer Epidemiol Biomarkers Prev. . 15: 536–542. [DOI] [PubMed] [Google Scholar]
- 69. Fletcher O, Johnson N, Orr N, Hosking FJ, Gibson LJ, et al. (2011) Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study. J Natl Cancer Inst. . 103: 425–435. [DOI] [PubMed] [Google Scholar]
- 70. Manuguerra M, Saletta F, Karagas MR, Berwick M, Veglia F, et al. (2006) XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am J Epidemiol. . 164: 297–302. [DOI] [PubMed] [Google Scholar]
- 71. Lu X, Yang W, Wan Z, Li J, Bi Z (2011) Glutathione S-transferase polymorphisms and bone tumor risk in China. Asian Pac J Cancer Prev. . 12: 3357–3360. [PubMed] [Google Scholar]
- 72. Salinas-Souza C, Petrilli AS, de Toledo SRC (2010) Glutathione S-transferase polymorphisms in osteosarcoma patients. Pharmacogenet Genomics. . 20: 507–515. [DOI] [PubMed] [Google Scholar]
- 73. Berwick M, Matullo G, Song YS, Guarrera S, Dominguez G, et al. (2004) Association between aryl hydrocarbon receptor genotype and survival in soft tissue sarcoma. J Clin Oncol. . 22: 3997–4001. [DOI] [PubMed] [Google Scholar]
- 74. Economopoulos KP, Sergentanis TN (2010) GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: a comprehensive meta-analysis. Eur J Cancer. . 46: 1617–1631. [DOI] [PubMed] [Google Scholar]
- 75. Wang D, Wang B, Zhai JX, Liu DW, Sun GG (2011) Glutathione S-transferase M1 and T1 polymorphisms and cervical cancer risk: a meta-analysis. Neoplasma. . 58: 352–359. [DOI] [PubMed] [Google Scholar]
- 76. Engel LS, Taioli E, Pfeiffer R, Garcia-Closas M, Marcus PM, et al. (2002) Pooled analysis and meta-analysis of glutathione S-transferase M1 and bladder cancer: a HuGE review. Am J Epidemiol. . 156: 95–109. [DOI] [PubMed] [Google Scholar]
- 77. Wozniak A, Rutkowski P, Piskorz A, Ciwoniuk M, Osuch C, et al. (2012) Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol. . 23: 353–360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Minor allele frequencies (MAF) and p-values for comparison of genotype frequencies: Z9001 genotyped whites (n = 273) versus non-whites (n = 58) and genotyped Z9001 whites versus the HapMap CEU population (n = 180).
(PDF)
Minor allele frequencies (MAF), Odds Ratios (ORs) and association p-values by mutation type.
(PDF)
P-values for sequence kernel association test (SKAT) for remaining genes, by mutation type.
(PDF)