

Abstract
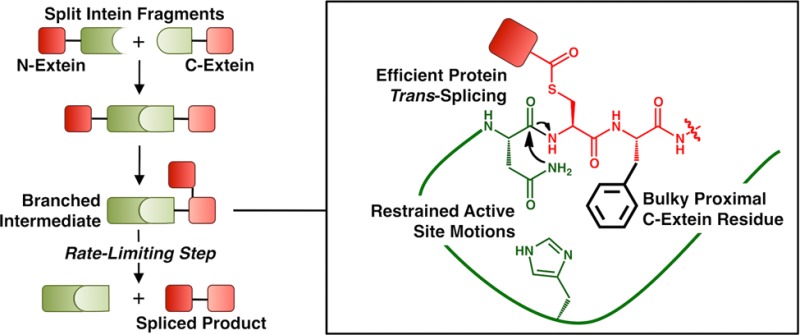
Split inteins play an important role in modern protein semisynthesis techniques. These naturally occurring protein splicing domains can be used for in vitro and in vivo protein modification, peptide and protein cyclization, segmental isotopic labeling, and the construction of biosensors. The most well-characterized family of split inteins, the cyanobacterial DnaE inteins, show particular promise, as many of these can splice proteins in less than 1 min. Despite this fact, the activity of these inteins is context-dependent: certain peptide sequences surrounding their ligation junction (called local N- and C-exteins) are strongly preferred, while other sequences cause a dramatic reduction in the splicing kinetics and yield. These sequence constraints limit the utility of inteins, and thus, a more detailed understanding of their participation in protein splicing is needed. Here we present a thorough kinetic analysis of the relationship between C-extein composition and split intein activity. The results of these experiments were used to guide structural and molecular dynamics studies, which revealed that the motions of catalytic residues are constrained by the second C-extein residue, likely forcing them into an active conformation that promotes rapid protein splicing. Together, our structural and functional studies also highlight a key region of the intein structure that can be re-engineered to increase intein promiscuity.
Introduction
Protein splicing is a post-translational autoprocessing event carried out by a class of proteins known as inteins.1 During this process, an intein domain excises itself from a larger precursor protein and ligates its N- and C-terminal flanking sequences (termed exteins) through a native peptide bond. Inteins naturally exist in two forms. Most are cis-splicing inteins that are expressed as single polypeptide chains embedded within their host proteins. By contrast, the far less abundant trans-splicing inteins are transcribed and translated as two separate protomers that associate and fold into the canonical intein domain structure.2 The association of naturally split inteins is rapid compared with the subsequent protein splicing reaction.3 Regardless of whether splicing occurs in cis or trans fashion, the mechanism of protein splicing is the same (Figure 1). First, the N-extein/intein peptide bond is activated through an N-to-S acyl shift to form a linear thioester intermediate. Next, this activated acyl group undergoes trans-thioesterification to form a branched thioester intermediate on the first residue of the C-extein, Cys+1. In the last chemoenzymatic step, the C-terminal Asn residue of the intein cyclizes, thereby resolving this branched intermediate (BI) into an excised intein and an N-extein/C-extein thioester adduct. Finally, this transient thioester spontaneously rearranges to a native peptide bond to yield the spliced product, and the excised intein succinimide hydrolyzes to yield a free carboxylate.
Figure 1.
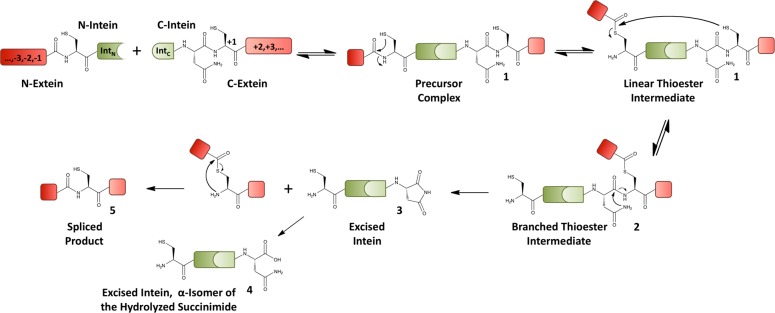
Mechanism of protein trans-splicing (PTS). Relevant species along the reaction coordinate are labeled. Numbers 1–4 refer to the chemically distinct C-intein adducts that can be observed in the splicing assays described in this report (see Figure 3 and Figure S8 in the Supporting Information). For simplicity, only the α-amino acid isomer of 4 is shown, but the ring in species 3 can open into an α- or β-amino acid form.
It is noteworthy that while different families of inteins utilize subtle variations on this general biochemical mechanism (such as Ser or Thr nucleophiles rather than Cys), the catalytic residues for protein splicing are always confined to the intein domain and the first C-extein residue.1 Despite this fact, a growing body of experimental evidence indicates that the intein splicing efficiency is highly dependent on the identity of two or three local extein residues on either side of the splice junction.4−10 For example, introduction of non-native residues at the −3, −2, and −1 positions, located on the N-extein (Figure 1), can alter the linear thioester formation efficiency or promote hydrolysis of this intermediate. Mutation of the +1, +2, and +3 residues, located on the C-extein (Figure 1), can abolish or greatly diminish the splicing activity and even lead to premature asparagine cyclization before formation of the BI. For each intein family, this context-dependent activity is dictated by evolutionary pressures, as inteins are naturally embedded between highly conserved residues in a number of different endogenous host proteins.11 As a result, different inteins are biased toward different sequences at their splice junctions.
The chemical synthesis of larger and more complex peptides and proteins is an ongoing challenge, and inteins are being widely used to facilitate such syntheses.12 Thus, the sensitivity of protein splicing to the local extein sequence (i.e., the residues immediately flanking the intein) has significant practical implications. All intein-based technologies are premised on a single notion: the chemical perturbations that an intein exerts on its endogenous host protein can be applied in a virtually traceless manner to any exogenous protein of interest. In reality, however, efficient and traceless synthesis of complex products is not always achieved. Rather, current technologies often require either the incorporation of non-native residues surrounding the splice junction in the target molecule or sacrifices in reaction kinetics and product yield to obtain the desired native sequence. An improved understanding of the general splicing mechanism and its sensitivity to local extein sequences thus remains of central concern.
Of particular interest as protein engineering tools are the split DnaE inteins, all of which endogenously generate the catalytic subunit of DNA polymerase III after protein trans-splicing (PTS).13 Until recently, many split intein-based technologies relied on the founding member of this family, termed Ssp, which derives its name from the model cyanobacterium that encodes it, Synechocystis species PCC6803.2 However, Ssp catalyzes PTS in hours, which is too slow for many practical applications.14 With the discovery and characterization of new split DnaE inteins, such as the now prevalent Nostoc punctiforme (Npu) intein, it is clear that several members of this family catalyze protein splicing with extraordinary efficiency (in minutes or less).5,9,15,16 Thus, many intein technologies are now being developed and improved with these new tools, including in vitro and in vivo protein semisynthesis,17−19 segmental isotopic labeling,20,21 peptide cyclization,22 and the construction of novel biosensors.23,24
The DnaE split intein family, however, is also plagued by poor tolerance for non-native local extein sequences. All split DnaE inteins are naturally embedded within the local N-extein sequence AEY (Figure 1, residues −3, −2, and −1) and the C-extein sequence CFN (Figure 1, residues +1, +2, and +3). Several reports indicate that DnaE inteins can tolerate significant deviation from this native N-extein sequence.6,10,16,19,25 Conversely, the presence of non-native C-extein residues can lead to dramatic reductions in splicing efficiency. For example, mutation of the canonical CFN sequence to SGV was shown to inhibit BI resolution for Ssp, although the contributions of each C-extein mutation were not individually assessed.7 Additionally, the identity of the +2 C-extein residue has a dramatic impact on the splicing activity for all members of the DnaE family, but it is not clear what step in the splicing pathway is modulated by this residue.5,9,10
Despite the fact that C-extein-dependent splicing activity is well-documented for DnaE inteins, little is known about the magnitude of this effect on the reaction kinetics or the physical basis of this phenomenon. We envisioned that a detailed understanding of how C-extein residues participate in the splicing reaction could help guide the practical use of split inteins and help lay the foundation for the design of more promiscuous engineered inteins. To this end, we performed a detailed structure–activity analysis on the Npu intein, employing semisynthesis to alter the C-extein moiety systematically, thereby providing the raw materials for a series of kinetic and structural analyses. This effort led to the finding that the +2 residue in the C-extein plays a critical role in constraining the active site of the intein during BI resolution. The work also draws attention to a loop region in the intein structure that appears to sense the C-extein composition and thus might be a productive focus of engineering efforts geared toward increasing intein promiscuity.
Results
Semisynthesis of Split Inteins with Varying C-Extein Composition
Our efforts began with the construction of a library of C-intein fragments (IntC) bearing a variety of model C-exteins ranging from a single Cys residue with different capping groups to tripeptides with unique sequences (see Table 1). For rapid generation of the desired constructs (17 proteins in all), we employed a semisynthetic approach utilizing expressed protein ligation (Figure 2).26 Specifically, the IntC fragments of Npu and Ssp (termed NpuC and SspC, respectively) were expressed in Escherichia coli fused to the cis-splicing His6-tagged GyrA intein and enriched over Ni columns (Figure S4). The crude fusion proteins were then reacted with either a large excess of a cysteine derivative (100 mM) to yield an IntC–Cys adduct directly, or they were thiolyzed with 100 mM 2-mercaptoethanesulfonate (MES) in the presence of a 1–5 mM di- or tripeptide to yield IntC–peptide adducts (Figure 2A,B). The desired product from each reaction was readily purified by reversed-phase high-performance liquid chromatography (RP-HPLC) (Figures 2C and S7), and its identity was confirmed by electrospray ionization mass spectrometry (ESI-MS) (Figure 2D and Table S2 in the Supporting Information). Importantly, this semisynthesis approach allowed for the modular assembly of constructs with natural amino acid mutations within the C-intein and effectively any functional groups in the C-extein side chains and backbone.
Table 1. Rate Constants for Individual Steps and the Overall Splicing Reactiona.
| reaction | intein | C-extein | k1 (s–1) | k2 (s–1) | k3 (s–1) | ksplice (s–1) |
|---|---|---|---|---|---|---|
| 1 | NpuWT | CFN(NH2) | (5.21 ± 0.28) × 10–2 | (1.77 ± 0.38) × 10–2 | (3.15 ± 0.04) × 10–2 | (1.36 ± 0.02) × 10–2 |
| 2 | SspWT | CFN(NH2) | (4.70 ± 0.26) × 10–3 | (7.03 ± 0.44) × 10–3 | (3.86 ± 0.17) × 10–4 | (1.46 ± 0.03) × 10–4 |
| 3b | NpuC1A | CFN(NH2) | – | – | (1.43 ± 0.03) × 10–4 | – |
| 4c | NpuN137A | CFN(NH2) | (1.70 ± 0.13) × 10–2 | (1.86 ± 0.11) × 10–3 | – | – |
| 5d | NpuWT | C(OH) | (2.41 ± 0.07) × 10–2 | (4.40 ± 0.14) × 10–3 | – | – |
| 6 | NpuWT | C(OCH3) | (6.59 ± 0.20) × 10–2 | (1.56 ± 0.06) × 10–2 | (4.76 ± 0.13) × 10–4 | (4.32 ± 0.16) × 10–4 |
| 7 | NpuWT | C(NH2) | (3.16 ± 0.09) × 10–2 | (6.59 ± 2.41) × 10–3 | (7.31 ± 0.26) × 10–5 | (6.30 ± 0.87) × 10–5 |
| 8 | NpuWT | C(NHCH3) | (4.40 ± 0.35) × 10–2 | (1.13 ± 0.10) × 10–2 | (1.33 ± 0.01) × 10–4 | (1.08 ± 0.03) × 10–4 |
| 9 | NpuWT | CF(OCH3) | (5.90 ± 0.85) × 10–2 | (1.20 ± 0.39) × 10–2 | (1.56 ± 0.16) × 10–3 | (1.28 ± 0.01) × 10–3 |
| 10 | NpuWT | CF(NH2) | (6.10 ± 1.10) × 10–2 | (1.13 ± 0.40) × 10–2 | (9.30 ± 0.42) × 10–3 | (6.32 ± 0.11) × 10–3 |
| 11 | NpuWT | CFA(NH2) | (6.05 ± 0.36) × 10–2 | (1.31 ± 0.27) × 10–2 | (2.57 ± 0.04) × 10–2 | (1.31 ± 0.02) × 10–2 |
| 12 | NpuWT | CAN(NH2) | (7.11 ± 2.11) × 10–2 | (2.74 ± 0.58) × 10–2 | (3.12 ± 0.28) × 10–4 | (2.39 ± 0.12) × 10–4 |
| 13b | NpuC1A | CAN(NH2) | – | – | (2.41 ± 0.02) × 10–6 | – |
| 14 | NpuH125N | CFN(NH2) | (4.21 ± 0.46) × 10–2 | (8.96 ± 3.22) × 10–3 | (5.53 ± 0.50) × 10–4 | (4.92 ± 0.13) × 10–4 |
| 15e | NpuH125N | CAN(NH2) | (7.81 ± 0.34) × 10–2 | (2.94 ± 0.01) × 10–2 | (3.23 ± 0.27) × 10–5 | (3.23 ± 0.27) × 10–5 |
| 16 | NpuD124Y | CFN(NH2) | (7.75 ± 0.55) × 10–2 | (2.06 ± 0.23) × 10–2 | (3.27 ± 0.11) × 10–2 | (1.74 ± 0.07) × 10–2 |
| 17 | NpuD124Y | CAN(NH2) | (1.06 ± 0.76) × 10–1 | (3.87 ± 0.47) × 10–2 | (4.43 ± 0.05) × 10–4 | (3.61 ± 0.21) × 10–4 |
k1, k2, and k3 were extracted from a global fit of all three normalized curves for one reaction to the analytical solutions for the differential rate equations that describe our kinetic model. ksplice was extracted by fitting the product formation curve to a standard first-order rate equation. The reported values are means ± standard deviations from three individually fit unique trials.
In reactions 3 and 13, the mutation of Cys1 precluded the first steps of the splicing pathway. k3 represents the rate of succinimide formation and thus C-extein cleavage in the absence of BI formation.
In reaction 4, mutation of the catalytic asparagine abolishes succinimide formation, thus the reaction does not progress past the BI.
In reaction 5, although all of the catalytic residues were present, no BI resolution was observed during the course of the assay.
The extremely slow BI resolution in reaction 15 led to roughly 10–20% N-extein hydrolysis as a side reaction, preventing a global fit to our kinetic model. For this reaction, k1 and k2 were extracted from a two-state equilibrium kinetic model using only the pre-equilibrium phase of the reaction (first 10 min). k3 was assumed to be identical to ksplice, which was determined by fitting the product formation curve to a first-order rate equation.
Figure 2.
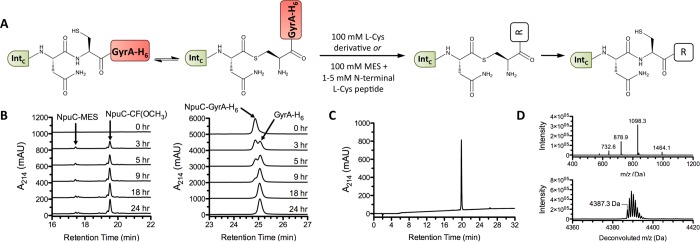
Semisynthesis of C-intein constructs. (A) Semisynthetic scheme (R = OH, OCH3, NH2, NHCH3, or an additional one or two amino acids, as indicated in Table 1). (B) RP-HPLC analysis of a one-pot MES-thiolysis/ligation sequence to synthesize NpuC–CF(OCH3). NpuC–MES and ligation product accumulation are shown in the left panel, and cleavage of the NpuC–GyrA-His6 fusion protein is shown in the right panel. (C) RP-HPLC and (D) ESI-MS analysis of NpuC–CF(OCH3) after purification. In (D), the raw and deconvoluted mass spectra are shown in the top and bottom panels, respectively (expected monoisotopic mass = 4387.28 Da).
Kinetic Assays To Monitor Formation and Resolution of the Branched Intermediate
To provide a rigorous assessment of C-extein effects on PTS, we developed two complementary analytical approaches that allowed us to distinguish various chemical species along the reaction coordinate in a time-resolved fashion. First, N-intein (IntN) proteins bearing a minimized N-extein tripeptide (AEY–IntN) were generated recombinantly and purified (Figures S5–S7 and Table S2). These constructs were mixed with their IntC counterparts at 30 °C, and aliquots were removed from the reaction solution at various time points and quenched by acidification to pH 1–2. Importantly, all of the reactions were carried out at pH 7.2 in the absence of thiol-based reducing agents to prevent any undesired hydrolysis or thiolysis reactions that would convolute the kinetic analyses. The time-point aliquots were analyzed by RP-HPLC, and for most of the reactions, the various IntC-related species (1–4 in Figure 1) and the spliced product (5 in Figure 1) could be readily separated (Figures 3A and S9). For reactions where sufficient separation between species 1–5 was not achieved by RP-HPLC, the quenched aliquots at various time points were desalted and analyzed as complex mixtures by ESI-MS (Figures 3B and S10). Because of the similarities in sequence composition, size, and net charge among species 1–4, the molecules showed similar levels of ionization, and thus, the RP-HPLC and ESI-MS analyses gave virtually identical results (compare panels A and B in Figure 3; for quantitative analysis of the error between the two assays, see Figure S13). Importantly, in both assay formats, the starting material and linear intermediate were indistinguishable, so the data were fit to a simplified kinetic model that collapsed the first two catalytic steps into a single equilibrium reaction (Figure 3C,D). The results of our kinetic analyses are summarized in Table 1 and Figure 4.
Figure 3.
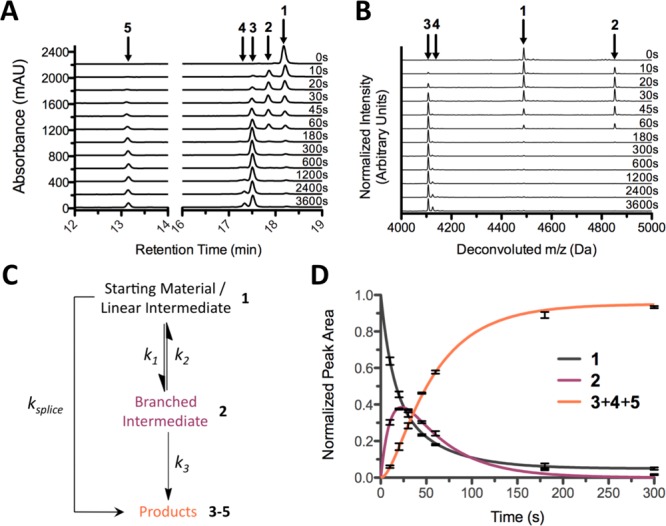
Splicing assays to analyze formation and resolution of the branched intermediate. (A, B) Time-dependent RP-HPLC (A) and ESI-MS (B) analyses of the reaction between AEY–NpuN and NpuC–CFN(NH2) (Table 1, reaction 1). The deconvoluted mass spectra in (B) are normalized to the intensity of the largest peak at each time point. (C) Simplified three-state kinetic model of protein splicing compatible with the analytical techniques presented herein. (D) Quantified reaction progress data for reaction 1 fit to the kinetic model in (C). The product curve represents the combined peak areas of all three products of the PTS process (the two excised intein species 3 and 4 and the spliced product 5). Error bars represent standard deviations from three independent trials. Numbering corresponds to that defined in Figures 1 and S8.
Figure 4.
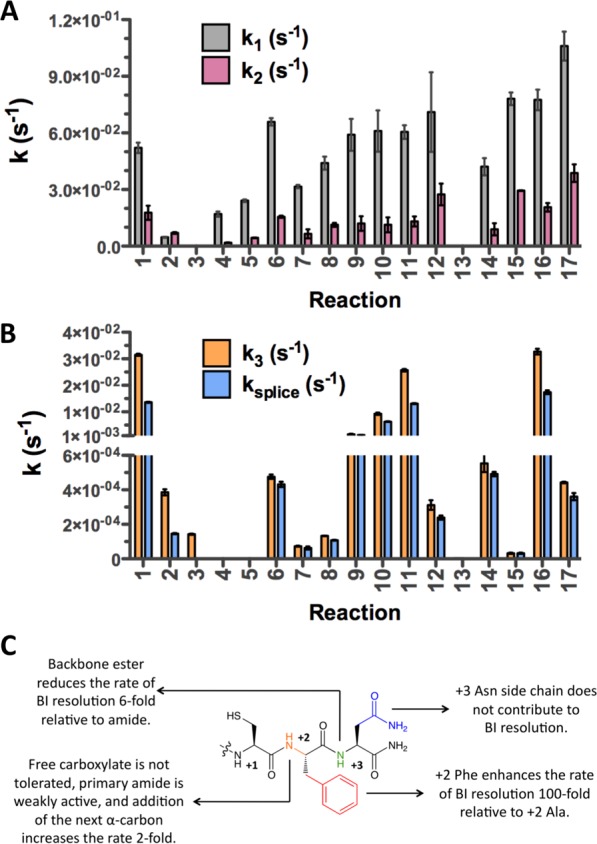
C-extein contributions to splicing activity. (A) Forward (k1) and reverse (k2) rate constants for BI formation from starting materials. (B) Rate constant for BI resolution (k3) and overall rate constant for trans-splicing (ksplice). (C) Scheme highlighting the key conclusions from the kinetic data.
We initially carried out a series of control reactions to validate our assays. The splicing kinetics of the wild-type Npu and Ssp inteins were assessed in their native N- and C-extein contexts (Table 1, reactions 1 and 2). The overall rate constants for spliced product formation (ksplice) were 1.36 × 10–2 and 1.46 × 10–4 s–1, respectively, consistent with the results of previous measurements using gel-based assays.9,14,16 These experiments also demonstrated that BI resolution (described by k3) is the slow step for Ssp, whereas for the faster Npu reaction, the initial and latter steps of PTS are kinetically coupled. As additional controls, we independently mutated the first catalytic cysteine (Cys1) and the C-terminal asparagine (Asn137) in Npu to alanine and analyzed the effect of these mutations on the splicing activity. As expected, the C1A mutation completely inhibited splicing, although basal levels of succinimide formation and thus C-extein cleavage were observed on a time scale of hours (Table 1, reaction 3). This result is consistent with the notion that cyclization of the C-terminal asparagine is stimulated by BI formation, as shown previously for the GyrA intein.27 Additionally, the N137A mutation abolished splicing and C-extein cleavage but only modestly reduced the kinetics of the initial steps (Table 1, reaction 4).
C-Extein Effects on BI Formation and Resolution
Next, we employed our kinetic assays to determine the effect of C-extein composition on individual steps in the PTS reaction (Table 1, reactions 5–12). These experiments revealed that variation of the C-extein had only a small effect on the kinetics of BI formation (k1 and k2) but profoundly affected the BI resolution step (k3) and thus the overall splicing rate constant (ksplice) (Figure 4A,B). A detailed comparison of these kinetic analyses revealed several important trends (Figure 4C). First, the C-extein chain length had a substantial effect on the activity. Cys+1 alone could not sustain BI resolution with an uncapped carboxylate, suggesting that a negative charge near the active site is undesirable (Table 1, reaction 5). Capping the +1 residue as an amide or ester restored a basal level of splicing activity (Table 1, reactions 6–8). Interestingly, Cys+1 capped with a methyl ester afforded a 4-fold rate increase over the methylamide analogue, possibly indicating an inhibitory role for this amide N–H moiety or an anomalous non-native effect of this subtle perturbation (Table 1, reactions 6 and 8). Ultimately, the effect of C-extein chain length on BI resolution was more pronounced once the entire Phe+2 residue was added (Table 1, reaction 10), but three C-extein residues were required to recapitulate the highest reported rates for Npu (Table 1, reaction 1).
Through our kinetic analyses, we also identified two specific functional groups that make major contributions to BI resolution. First, we found that the amide bond after Phe+2 provided a 6-fold rate enhancement relative to a methyl ester (compare reactions 9 and 10). This result suggests that the amide N–H group is involved in a hydrogen bond that facilitates BI resolution, perhaps by stabilizing a catalytically competent conformation. The second, more significant functional group is the Phe+2 phenyl ring. While this residue is known to be important, as discussed above, the extent of its contribution to BI resolution was not previously known. Our measurements indicate that the addition of the bulky Phe side chain enhances the BI resolution kinetics 100-fold relative to Ala (compare reactions 1 and 12). Interestingly, the presence of the Phe side chain also stimulated the basal rate of succiminide formation (i.e., C-extein cleavage) in the context of a C1A mutant of NpuN (compare reactions 3 and 13), implying that the Phe side chain forms favorable interactions even in the absence of the BI. By contrast, the side chain of Asn+3 does not contribute to the PTS reaction (compare reactions 1 and 11).
A Structural Role for the +2 C-Extein Residue
Given the significant contribution of the Phe+2 side chain to the splicing kinetics, we next sought to understand the structural origin of its involvement in split intein chemistry. Most high-resolution structures of inteins, including the only published structure of Npu,28 do not contain C-extein residues. One important exception to this is a crystal structure of Ssp bearing five native N-extein residues (KFAEY), three native C-extein residues (CFN), and mutations of the terminal intein residues (Cys and Asn) to Ala.29 In this structure, the Phe+2 side chain packs against a catalytic histidine that lies on a flexible loop (Figure 5A). This histidine (His125 in Npu) is completely conserved in the DnaE family and has been implicated as a general acid or base in the BI resolution step of many inteins.27,29 Mutation of His125 in Npu to Asn reduced the rate of BI resolution roughly 60-fold, similar to the F+2A mutation (Table 1, reactions 14 and 12, respectively). The Ssp structure suggests that Phe+2 participates in PTS by stabilizing His125 through a direct interaction. Indeed, mutating both residues in Npu had a nonadditive effect on the BI resolution kinetics (ΔΔGcoupling = 1.07 kcal mol–1), indicating some cooperativity between Phe+2 and His125 with respect to this step (Table 1, reaction 15; see Figure S14 for thermodynamic cycle analysis).
Figure 5.
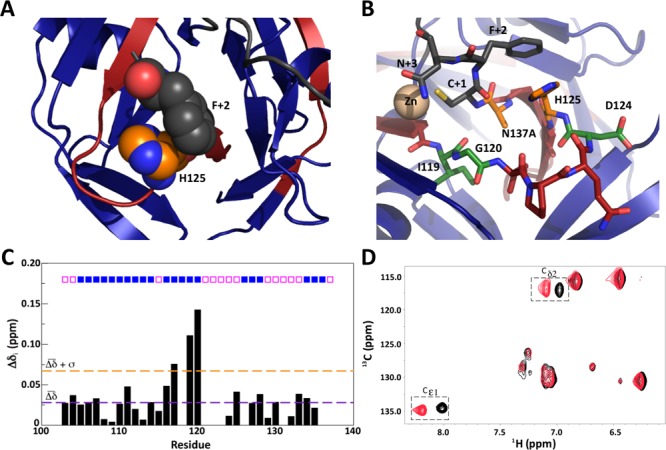
Structural effects of mutating the C-extein +2 residue. (A) Crystal structure of the Ssp DnaE intein (PDB entry 1ZDE), with the close packing of His125 and Phe+2 highlighted in spheres. The N-intein and C-intein are shown as blue and red ribbons, respectively. (B) The active site of Ssp bearing catalytic Cys and Asn mutations, native extein residues, and a coordinated zinc ion. Important residues surrounding the C-intein/C-extein junction (orange/black junction) are shown as sticks, and the C-extein is shown in gray. Key catalytic residues are shown in orange, and other noncatalytic residues highlighted in this study are shown in green. (C) Composite 1H and 15N backbone chemical shift perturbations (Δδi) for 13C,15N-labeled NpuC in complex with unlabeled NpuN upon changing the +2 C-extein residue from Phe to Ala (see the Supporting Information for calculations). The mean value is shown as a dashed purple line, and one standard deviation above the mean is shown as a dashed orange line. Residues in secondary structure elements are marked with boxes above the bars: solid blue boxes denote strands, and open pink boxes denote loops. (D) Overlay of the aromatic regions of the 1H–13C HSQC spectra of segmental labeled NpuN:NpuC complexes containing either Phe (black) or Ala (red) as the +2 C-extein residue. Chemical shift perturbations of the His125 imidazole ring 1H–13C correlations (Cε1 and Cδ2) are marked with dashed boxes.
To obtain a better understanding of the structural impact of the +2 residue, we carried out solution NMR analyses of Npu in CFN(NH2) and CAN(NH2) C-extein contexts. NMR constructs were prepared analogously to those used in the kinetic assays but with some additional provisions. Specifically, the NpuN protein contained the native N-extein sequence (AEY) and an inactivating C1A mutation but was neither 13C- nor 15N-labeled. The NpuC constructs, which bore the N137A mutation, were 13C- and 15N-enriched in the recombinant IntC portion but not in the synthetic C-extein region. The N- and C-inteins were mixed, and the complexes were purified to homogeneity by size-exclusion chromatography (Figures S15 and S16). The use of this segmental labeling scheme meant that only the NpuC residues (Ile103–Ala137), which had identical chemical composition in the two complexes, would be visible in heteronuclear correlation experiments. This was expected to simplify assignment while still allowing the putative interaction between the +2 residue and the catalytic His125 to be interrogated. The inactivating mutations (C1A and N137A) ensured that chemistry would not occur during data acquisition.
With the exception of several residues in the loop containing the catalytic His125 residue, we were able to assign the majority of the NpuC backbone resonances in the complexes using standard triple-resonance experiments (Figure S17). Most of the backbone resonances were unperturbed when the +2 C-extein residue was changed from Phe to Ala (Figure 5C). The only exceptions to this were the amide resonances from Ile119 and Gly120, which showed a modest perturbation. These residues are located at the beginning of the loop containing the catalytic His residue and, in the Ssp crystal structure, lie close to the C-intein/C-extein peptide bond that is ultimately attacked during BI resolution (Figure 5B). The His125 backbone amide resonance was not itself sensitive to the nature of the +2 C-extein residue. However, the aromatic side-chain protons of this residue did exhibit significant chemical shift perturbations upon mutation of the +2 residue, suggesting an altered chemical environment for this side chain in the absence of the +2 phenyl ring (Figure 5D). Together with our mutagenesis and kinetic data, these NMR studies lend support to the idea that the active-site conformation of Npu is coupled to the identity of the C-extein +2 residue.
The Phe+2 C-Extein Residue Constrains Active-Site Motions
To gain additional insight into the interplay between C-extein residues and the Npu active site, we carried out molecular dynamics (MD) simulations of two wild-type intein complexes bearing either CFN(NH2) or CAN(NH2) as C-exteins (identical to the constructs in Table 1, reactions 1 and 12). Simulations were carried out in explicit solvent in 1 fs steps for 0.5 μs. Comparison of the two simulation trajectories afforded a more detailed picture of the coupling between the +2 residue and the intein active site. One of the more striking results from the simulation was the effect of changing the +2 C-extein on the dynamics of the His125 side chain. In the presence of Phe+2 the His side chain primarily adopts a single rotameric state with only a brief excursion to an alternate rotamer (Figure 6A and black trajectory in Figure 6C). By contrast, with an Ala+2 residue, His125 frequently switches among three side-chain rotamers and favors a different conformation than the one found with Phe+2 (Figure 6B and red trajectory in Figure 6C). Interestingly, the backbone ϕ and ψ dihedral angles for His125 showed virtually no change as a function of C-extein composition (Figure S19). These data are consistent with the fact that there were chemical shift perturbations for the His125 side chain but not the backbone.
Figure 6.
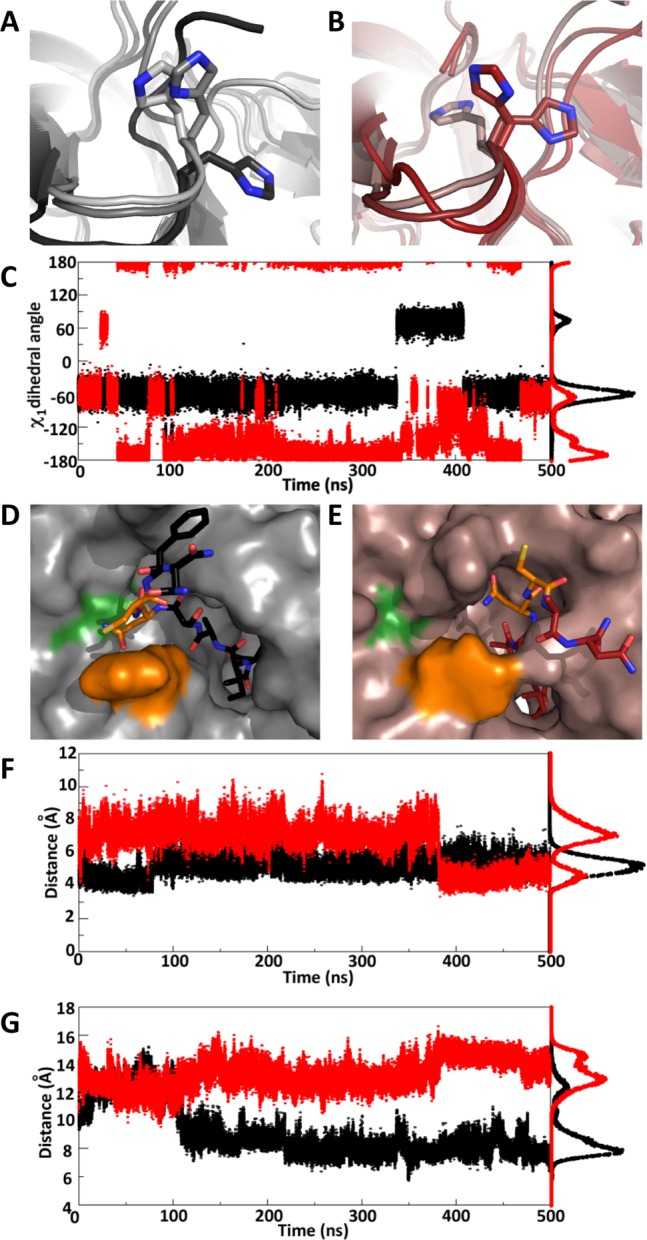
Results of MD simulations to probe +2 amino acid-dependent active-site dynamics. (A, B) Overlays of three representative frames from the MD trajectories for (A) AEY–NpuN + NpuC–CFN(NH2) and (B) AEY–NpuN + NpuC–CAN(NH2), highlighting His125 rotameric states. (C) Trajectory of the His125 side-chain χ1 dihedral angle during the simulations. (D, E) Representative frames highlighting the positioning of Asn137 relative to the His125 loop in the (D) CFN and (E) CAN simulations. Asn137 and Cys+1 are shown as orange sticks, His125 is shown as an orange surface, and Ile119 and Gly120 are shown as green surfaces. (F) Distance between the His125 and Asn137 Cβ atoms during the simulations. (G) Distance between the Ile119 and Cys+1 amide nitrogens during the simulations. In (C), (F), and (G), data from the CFN and CAN simulations are shown in black and red, respectively, and the traces to the right of the trajectory graphs are histograms indicating the distribution of angles or distances sampled throughout the simulation.
The second major consequence of the +2 residue mutation was the overall positioning of the C-intein/C-extein junction (i.e Asn137–Cys+1) relative to the His125 loop. In the simulation with CFN as the C-extein, Asn137 remained buried in the groove above this loop, similar to the Ssp structure (Figure 6D). By contrast, in the CAN simulation, the entire strand bearing Asn137 and the C-extein occupied space outside of this groove region (Figure 6E). An important consequence of this difference is that the distance between Asn137 and His125 (Figure 6F) and that between Ile119 and the scissile peptide bond (Figure 6G) were significantly shorter for the majority of the CFN simulation than for the CAN simulation. Overall, these MD simulations indicate that the presence of a sterically bulky amino acid at the +2 position in the C-extein acts to constrain the motions of key catalytic residues, leading to a more compacted arrangement around the scissile peptide bond.
In considering the mechanistic implications of these observations, it is important to emphasize that by necessity the simulations employed a linear precursor protein as the starting point. The use of a BI structure in the simulations would have been more desirable in view of the fact that our kinetic data revealed that formation of this intermediate stimulates cleavage of the peptide bond at the C-intein/C-extein junction (Table 1, compare reactions 1 and 3). Unfortunately, there is currently no high-resolution structural information on any intein in the BI state. Thus, we were forced to extrapolate from the structures available. Despite this caveat, the major conclusion from the simulation work is broadly consistent with our mutagenesis and kinetic data. In particular, we observed coupling between the +2 residue and the catalytic His125 in both the simulations and the studies of the BI resolution kinetics. We further note that the Phe side chain stimulates C-extein cleavage even in the absence of the BI (Table 1, compare reactions 3 and 13), which argues that this bulky side chain augments catalysis even in the linear precursor.
An Activating Point Mutation on the His125 Loop
Local C-extein residues appear to affect the structure and dynamics of residues surrounding the flexible His125 loop, thereby modulating the BI resolution kinetics. Thus, it is conceivable that point mutations within the intein that alter the loop conformation or flexibility could also modulate the splicing activity and even the tolerance to non-native extein residues. In a previous directed-evolution study on an NpuN–SspC chimera, we identified several mutations that make this intein more tolerant of the C-extein sequence SGV rather than CFN.7 Intriguingly, one of these mutations was an Asp-to-Tyr mutation adjacent to His125 (Asp124). We found that this mutation enhanced the rate of Npu splicing by 50% in the presence of Ala+2 (Figure 7A; compare reactions 12 and 17 in Table 1). Importantly, this mutation was still tolerated when Phe+2 was present, suggesting that it increases the overall promiscuity toward C-exteins (compare reactions 1 and 16). The Npu NMR structure28 and the Ssp crystal structures29,30 indicate that Asp124 packs against a β-turn from the N-intein. Because of this close packing, the bulky D124Y mutation would require conformational rearrangement and possibly also rigidification of the catalytic His125 loop, which could modulate the activity. As predicted, in a 100 ns MD simulation of NpuD124Y with a CAN(NH2) C-extein, the His125 loop conformation was altered and the His125 rotamer dynamics constrained, and Asn137 persistently remained above the His125 loop, similar to the NpuWT–CFN(NH2) simulation (Figures 7B–D, S21, and S22). This simulation suggests that the D124Y mutation reduces the C-extein dependence by recapitulating the constraints on the active-site dynamics typically applied by Phe+2, specifically the stabilization of His125 and the appropriate positioning of the C-intein/C-extein junction close to His125.
Figure 7.
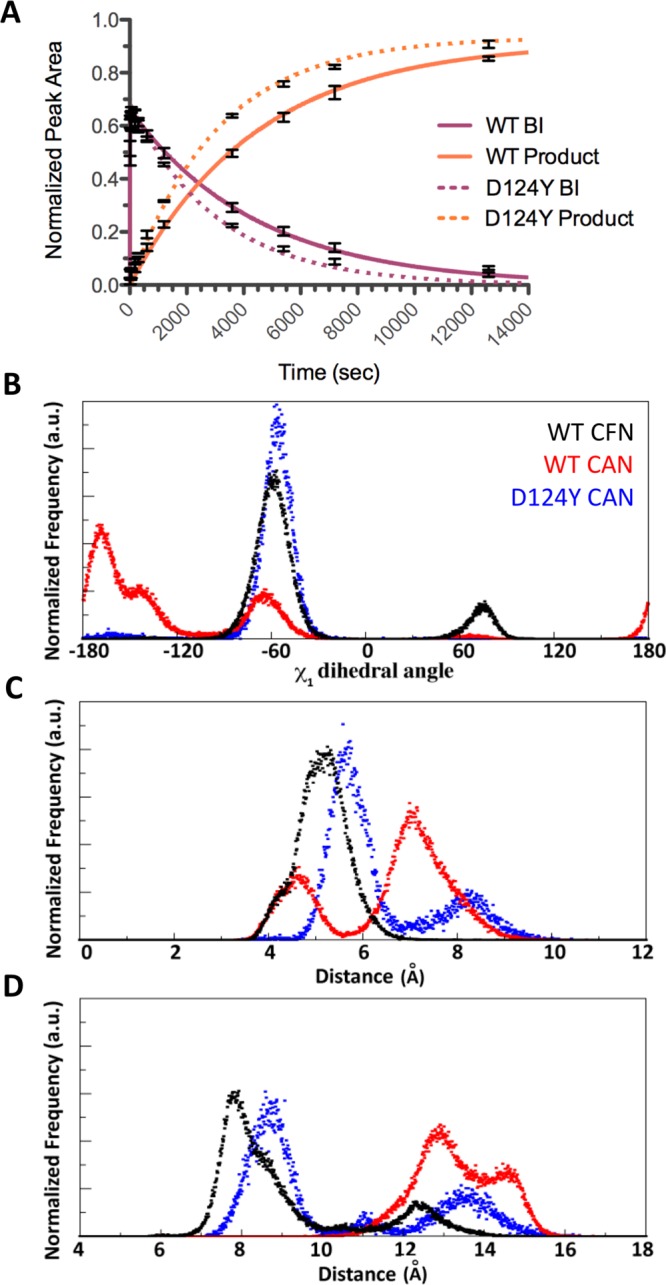
Structural and functional effect of the D124Y mutation in Npu. (A) Kinetic data showing rate enhancement for the BI resolution step with the D124Y mutation in the CAN(NH2) C-extein context. Solid lines correspond to best-fit kinetic curves for the wild-type intein (Table 1, reaction 12), and dotted lines correspond to the D124Y mutant (Table 1, reaction 17). Only the BI and product reaction curves are shown; the starting material curve has been omitted for clarity. Error bars represent standard deviations from three independent trials. (B–D) Histograms showing the distributions of (B) His125 χ1 dihedral angles, (C) distances between the His125 and Asn137 Cβ atoms, and (D) distances between the Ile119 and Cys+1 amide nitrogens during the wild-type CFN (black), wild-type CAN (red), and D124Y CAN (blue) simulations.
Discussion and Conclusions
In this work, we examined the molecular determinants for C-extein-dependent protein trans-splicing. This investigation was facilitated by the utilization of protein semisynthesis to generate inteins linked to a variety of C-exteins and the development of novel kinetic assays that provide information about individual steps along the PTS reaction coordinate. Through these studies, we not only extracted information on C-extein requirements but also gained additional mechanistic insights into split DnaE intein splicing. Specifically, our experiments confirmed that resolution of the branched intermediate is the slowest step for PTS (k3) and also provided evidence supporting the notion that some DnaE inteins have a highly activated N-terminal splice junction (k1/k2 > 2 for all Npu constructs), consistent with our previous report.9 Interestingly, this N-terminal activation appears to be roughly 10-fold slower and is significantly less efficient (k1/k2 = 0.67) for the Ssp intein. Additionally, we found that for Npu, the rate of Asn cyclization upon BI formation is 200-fold faster than its rate in the absence of the branched structure. Stimulation of Asn cyclization upon BI formation is also found in the cis-splicing GyrA intein.27 We propose that this kinetic stimulation is a common feature of inteins, in effect creating a trigger that helps ensure the proper fidelity of the reaction by minimizing premature cleavage of the C-extein. Lastly, it is particularly surprising that the H125N mutation does not completely abolish BI resolution but rather reduces its rate 60-fold. Indeed, the splicing rate of this mutant is still higher than that of wild-type Ssp. For many non-DnaE inteins, this step requires two histidine residues, one analogous to His125 and another immediately preceding the C-terminal Asn residue.27,31 In view of the lack of this penultimate histidine in the DnaE inteins, His125 has been implicated as the sole general acid/base for BI resolution.29 Our data suggest that while His125 is clearly important for BI resolution, other unidentified residues must also contribute to catalysis of this step.
The current study improves our understanding of the relationship between C-extein composition and trans-splicing efficiency. The kinetic data indicate that the C-extein almost exclusively affects the BI resolution step. Within the C-extein, we identified specific functional groups that contribute significantly to the splicing kinetics, in particular the Phe+2 side chain. Our NMR experiments and MD simulations illustrate that this bulky functional group constrains active-site motions, forcing catalytic histidine and asparagine residues and the scissile peptide bond into close proximity. The need for a bulky side chain at the +2 position is further highlighted by a recent genetic selection study on the Npu intein showing that Trp is also well-tolerated at this position.10 Collectively, these data paint a picture of the Npu active site that effectively extends beyond the intein domain itself to include the +2 C-extein residue.
During PTS, the N-extein is transferred from the N-terminus of the intein onto a C-extein side chain, which creates a unique branched protein structure. As BI resolution is the slowest and often the rate-limiting step for many inteins, this structure is the most relevant to the overall activity. To date, all of the published high-resolution structural data on inteins involve either a precursor or product form of the intein. While these studies (including this report) have provided substantial insights into the structural basis for protein splicing, they cannot examine interactions that are exclusively present in the BI. Indeed, our kinetic analyses revealed several important functional groups in the C-extein that affect BI resolution (Figure 4C), but only in the case of the Phe+2 side chain could we postulate any kind of structural basis for this. Thus, these results reinforce the need for high-resolution structural information on the BI in the PTS reaction.
The fullest deployment of split inteins in protein engineering ultimately requires a truly traceless trans-splicing system with no sequence requirements. While bulky hydrophobic residues other than phenylalanine are tolerated at the critical +2 position for DnaE inteins, thus alleviating some sequence constraints,5,10,25 these inteins are still only modestly promiscuous. Our results suggest that the interplay between the C-extein and the His125 active-site loop has direct implications for the rational design of improved, more extein-tolerant split inteins. Indeed, the D124Y point mutation on this flexible loop increases the tolerance of Npu for a +2 alanine residue without affecting its activity in a native context. In a recent directed-evolution endeavor on a DnaB family intein, a mutation at this position was also found to reduce C-extein sequence constraints.32 Furthermore, we previously demonstrated that mutating other residues on this loop can generally enhance the activity of Ssp9 and the NpuN–SspC chimera7 in a native C-extein context. These results collectively indicate that the conformational preferences of this loop are intimately linked with inadequate BI resolution both for intrinsically slow inteins and for efficient inteins in an exogenous C-extein context. Thus, this loop is a hot spot on the intein structure that should be explicitly targeted in future engineering efforts for the design of more high-activity, broad-specificity inteins.
Experimental Section
Semisynthesis of C-Intein Constructs
Semisynthetic IntC–extein proteins were generated through expressed protein ligation of a synthetic fragment, corresponding to the desired model C-extein, and a reactive recombinant fragment corresponding to the C-intein. Model C-exteins were synthesized using standard solution-based or solid-phase protocols (see the Supporting Information for details). Reactive recombinant IntC polypeptides were derived from the corresponding IntC–GyrA-His6 fusion proteins, which were expressed in E. coli and purified using standard methods (Figure S4). Ligation reactions involved treatment of the purified IntC–GyrA-His6 fusion protein with an excess of the model C-extein, usually in the presence of an additional thiol. Semisynthetic products were purified by preparative RP-HPLC and characterized by ESI-MS (Figure S7 and Table S2).
Expression and Purification of N-Intein Constructs
AEY–NpuN and AEY–SspN were expressed with an N-terminal His6-SUMO tag in E. coli BL21(DE3) cells from an IPTG-inducible protein expression vector. The cells were lysed by sonication, and the protein was enriched over Ni-NTA resin in a pH 8.0 phosphate-buffered saline solution. The proteins were eluted from the Ni column in the presence of 250 mM imidazole (Figure S5), and the elutions were dialyzed to reduce the imidazole concentration to 5 mM. The dialyzed solutions were treated for 12 h at room temperature with His6-tagged Ulp1, a SUMO-specific protease, to yield the desired products. The proteolysis reactions were passed over Ni-NTA resin to deplete unreacted starting material, the cleaved His6-SUMO tag, and Ulp1 (Figure S6). The proteins were further purified by size-exclusion chromatography on a Superdex 75 column in splicing assay buffer (100 mM sodium phosphates, 150 mM NaCl, 1 mM EDTA, pH 7.2) supplemented with 1 mM dithiothreitol. Product identities were confirmed by ESI-MS, and their purities were assessed by analytical RP-HPLC (Figure S7 and Table S2).
RP-HPLC and ESI-MS Analyses of Splicing Assays
Prior to any splicing assay, the N-intein solutions were dialyzed against splicing assay buffer (100 mM sodium phosphates, 150 mM NaCl, 1 mM EDTA, pH 7.2) overnight at 4 °C. It should be noted that thiols were omitted from this buffer since substantial N-extein cleavage was observed for reactions with a low k3. N-inteins and C-inteins were diluted to 15 and 10 μM, respectively, and tris(2-carboxyethyl)phosphine (TCEP) was added to each solution to a final concentration of 2 mM. Splicing reactions were initiated by mixing equal volumes of N-inteins and C-inteins at 30 °C. During the reaction, aliquots of the solution were removed and mixed 3:1 (v/v) with quenching solution (8 M guanidine hydrochloride and 4% trifluoroacetic acid). For RP-HPLC analysis, 100 μL of the quenched solutions were separated over a C18 analytical column, recording absorbance at 214 nm, and major peaks were collected and identified by ESI-MS (Figures 3A and S9 and Table S3). For direct ESI-MS analyses, 20 μL of the quenched solutions were desalted using Millipore C18 Zip-Tips, diluted, and loaded on the mass spectrometer by direct infusion. The complex mixture of multiply charged states of each species was deconvoluted into spectra depicting a well-defined mixture of singly charged species (Figure 3B and S10 and Table S3).
Kinetic Analyses
Peaks corresponding to species 1–5 in either the RP-HPLC chromatogram or ESI-MS spectrum were integrated and expressed as a fraction of total peak intensity for each time point. For the RP-HPLC analyses, the product was expressed as the sum of the integrated intensities for species 3–5 to account for changes in relative extinction coefficients. For ESI-MS analyses, the product was expressed as the sum of the integrated intensities of only species 3 and 4, since species 5 was not visible and the ionizabilities of 1–4 were assumed to be identical. The time-dependent reaction curves for all three states of the reaction (Figures S11 and S12), starting material (1), branched intermediate (2), and products (3 and 4 or 3–5), were collectively fit to the analytical solution for the coupled differential equations describing our kinetic model (Figure 3C; also see the Supporting Information). From this global fit, we extracted the values for k1, k2, and k3 for each individual reaction. The value of ksplice was determined by fitting the product formation curves (3 and 4 or 3–5) to a first-order rate equation. Reactions were repeated three or four times, and the means and standard deviations of all four kinetic parameters are reported in Table 1.
NMR Spectroscopy
NMR experiments were carried out on uniformly 15N,13C-labeled NpuCN137A ligated to an unlabeled C-extein [CFN(NH2) or CAN(NH2)] in complex with unlabeled AEY–NpuNC1A. Experiments were run on 600 MHz (Bruker or Varian Inova), 800 MHz, and 900 MHz Bruker spectrometers. Backbone resonance assignments of labeled NpuC in complex with NpuN were achieved using triple-resonance experiments with standard pulse sequences.33 The complex harbors one labeled histidine (His125). The side-chain carbons, Cδ2 and Cε1, of His125 were resolved with a standard 13C,1H aromatic heteronuclear single-quantum correlation (HSQC) experiment.34−36 Standard pulse sequences were used for the measurements of R1, R2, and 15N–1H nuclear Overhauser effect (NOE) rates.
Molecular Dynamics Simulations
All-atom MD simulations were performed on Npu constructs at constant temperature and pressure (300 K and 1 atm) using the MD suite AMBER 12.37,38 Simulations contained explicit water molecules, and the net charge of the system was neutralized with sodium ions. The constructs were generated from the first representative solution NMR structure of Npu (PDB entry 2KEQ).28 Prior to the simulations, this structure was modified in silico using UCSF Chimera39 to generate the constructs of interest, namely, (1) a wild-type split intein complex with canonical extein sequences [AEY–NpuN:NpuC–CFN(NH2)], (2) a wild-type split intein complex with a mutant C-extein [AEY–NpuN:NpuC–CAN(NH2)], and (3) a D124Y mutant with the same mutant C-extein sequence [AEY–NpuN:NpuCD124Y–CAN(NH2)]. For the wild-type CFN and CAN constructs, 500 ns long simulations were run, and a 100 ns long simulation was run for the D124Y mutant. Prior to the runs, a series of minimization, heating, and density equilibration steps were performed.
Acknowledgments
The authors thank the members of the Muir laboratory for valuable discussions. This work was supported by the U.S. National Institutes of Health (Grant GM086868). The program Chimera was supported by NIGMS Grant P41-GM103311. NMR resources at NYSBC were supported by NIGMS Grant P41-GM066354.
Supporting Information Available
Full methods and experimental data, including protein semisynthesis and purification protocols, characterization of proteins, details of kinetic analyses, NMR experiments, and MD simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Volkmann G.; Mootz H. D. Cell. Mol. Life Sci. 2013, 70, 1185–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. H.; Muir T. W. Isr. J. Chem. 2011, 51, 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Muir T. W. J. Am. Chem. Soc. 2005, 127, 6198–6206. [DOI] [PubMed] [Google Scholar]
- Southworth M.; Amaya K.; Evans T.; Xu M.; Perler F. Biotechniques 1999, 27, 110–120. [DOI] [PubMed] [Google Scholar]
- Iwai H.; Züger S.; Jin J.; Tam P.-H. FEBS Lett. 2006, 580, 1853–1858. [DOI] [PubMed] [Google Scholar]
- Amitai G.; Callahan B. P.; Stanger M. J.; Belfort G.; Belfort M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 11005–11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Muir T. W. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 10999–11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemella P. T.; Topilina N. I.; Soga I.; Pereira B.; Belfort G.; Belfort M.; Nayak S. K. Biophys. J. 2011, 100, 2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. H.; Dann G. P.; Vila-Perelló M.; Liu Z.; Muir T. W. J. Am. Chem. Soc. 2012, 134, 11338–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan M.; Pedamallu C. S.; Tori K.; Perler F. J. Biol. Chem. 2013, 288, 6202–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S. Trends Genet. 2001, 17, 465–472. [DOI] [PubMed] [Google Scholar]
- Vila-Perelló M.; Muir T. W. Cell 2010, 143, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi J.; Amitai G.; Belenkiy O.; Pietrokovski S. Mol. Microbiol. 2003, 50, 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. D.; Xu M. Q.; Evans T. C. Biochemistry 2001, 40, 1393–1402. [DOI] [PubMed] [Google Scholar]
- Dassa B.; Amitai G.; Caspi J.; Schueler-Furman O.; Pietrokovski S. Biochemistry 2007, 46, 322–330. [DOI] [PubMed] [Google Scholar]
- Zettler J.; Schütz V.; Mootz H. D. FEBS Lett. 2009, 583, 909–914. [DOI] [PubMed] [Google Scholar]
- Dhar T.; Mootz H. D. Chem. Commun. 2011, 47, 3063–3065. [DOI] [PubMed] [Google Scholar]
- Borra R.; Dong D.; Elnagar A. Y.; Woldemariam G. A.; Camarero J. A. J. Am. Chem. Soc. 2012, 134, 6344–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Perelló M.; Liu Z.; Shah N. H.; Willis J. A.; Idoyaga J.; Muir T. W. J. Am. Chem. Soc. 2013, 135, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche A. E. L.; Aranko A. S.; Talebzadeh-Farooji M.; Bernhard F.; Dötsch V.; Iwaï H. Angew. Chem., Int. Ed. 2009, 48, 6128–6131. [DOI] [PubMed] [Google Scholar]
- Muona M.; Aranko A. S.; Raulinaitis V.; Iwaï H. Nat. Protoc. 2010, 5, 574–587. [DOI] [PubMed] [Google Scholar]
- Jagadish K.; Borra R.; Lacey V.; Majumder S.; Shekhtman A.; Wang L.; Camarero J. A. Angew. Chem., Int. Ed. 2013, 52, 3126–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yang W.; Chen L.; Shi Y.; Li G.; Zhou N. Anal. Biochem. 2011, 417, 65–72. [DOI] [PubMed] [Google Scholar]
- Wong S.; Mills E.; Truong K. Protein Eng., Des. Sel. 2012, 26, 207–213. [DOI] [PubMed] [Google Scholar]
- Shah N. H.; Vila-Perelló M.; Muir T. W. Angew. Chem., Int. Ed. 2011, 50, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir T. W.; Sondhi D.; Cole P. A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frutos S.; Goger M.; Giovani B.; Cowburn D.; Muir T. W. Nat. Chem. Biol. 2010, 6, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeemig J. S.; Aranko A. S.; Djupsjöbacka J.; Heinämäki K.; Iwaï H. FEBS Lett. 2009, 583, 1451–1456. [DOI] [PubMed] [Google Scholar]
- Sun P.; Ye S.; Ferrandon S.; Evans T. C.; Xu M.-Q.; Rao Z. J. Mol. Biol. 2005, 353, 1093–1105. [DOI] [PubMed] [Google Scholar]
- Callahan B. P.; Topilina N. I.; Stanger M. J.; Van Roey P.; Belfort M. Nat. Struct. Mol. Biol. 2011, 18, 630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Benner J.; Perler F. B. J. Biol. Chem. 2000, 275, 20431–20435. [DOI] [PubMed] [Google Scholar]
- Appleby-Tagoe J. H.; Thiel I. V.; Wang Y.; Wang Y.; Mootz H. D.; Liu X.-Q. J. Biol. Chem. 2011, 286, 34440–34447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M.; Schleucher J.; Griesinger C. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar]
- Palmer A. G.; Cavanagh J.; Wright P. E.; Rance M. J. Magn. Reson. 1991, 93, 151–170. [Google Scholar]
- Kay L. E.; Keifer P.; Saarinen T. J. Am. Chem. Soc. 1992, 114, 10663–10665. [Google Scholar]
- Schleucher J.; Schwendinger M.; Sattler M.; Schmidt P.; Schedletzky O.; Glaser S. J.; Sørensen O. W.; Griesinger C. J. Biomol. NMR 1994, 4, 301–306. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E.; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 12; University of California: San Francisco, 2012.
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory. Comput. 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.