

Abstract
Since the discovery of the glutamate NMDA receptor subunit 3A (GluN3A), the functional role of this unique inhibitory subunit has been largely obscure. GluN3A expression is high in the neonatal brain but declines to a low level in the adult brain; it is thus commonly believed that GluN3A does not have a major functional impact in adulthood. Using wild-type (WT) and GluN3A knockout (KO) mice, we show here that deletion of GluN3A affected multiple behavioural functions in adult animals. GluN3A KO mice showed impaired locomotor activity on a variety of motor function tests, and increased sensitivity to acute and sub-acute inflammatory pain. GluN3A KO mice also showed enhanced recognition and spatial learning and memory functions. Hippocampal slices from juvenile and adult GluN3A KO mice showed greater long-term potentiation (LTP) compared with WT slices. GluN3A deletion resulted in increased expression of Ca2+/calmodulin-dependent kinase II (CaMKII) in the forebrain, and the phosphorylated CaMKII level upon LTP induction was significantly higher in the GluN3A KO hippocampus compared with WT controls. CaMKII inhibition abrogated the enhanced LTP in GluN3A KO slices. These data reveal for the first time that the presence of GluN3A may have profound impacts on several functional/behavioural activities in adult animals, and could be a therapeutic target for neurological disorders associated with NMDA receptor functions.
Key points
Adult glutamate NMDA receptor subunit 3A (GluN3A) knockout (KO) mice showed slow locomotor activity, motor deficits and increased pain sensation.
Hippocampal slices from juvenile and adult GluN3A KO mice showed greater long-term potentiation compared with wild-type slices.
Adult GluN3A KO mice showed enhanced abilities in learning and memory tasks.
GluN3A deletion resulted in increased expression and/or phosphorylation of Ca2+/calmodulin- dependent kinase II in the brain.
This is the first investigation showing that deletion of GluN3A in the embryonic stage has imperative impacts on multiple behaviours in adults.
Introduction
NMDA receptors (NMDARs) are key components in neuronal development and functional activities in the nervous system, including membrane excitability, synaptic plasticity, pain sensation and neuronal excitotoxicity (Barkus et al. 2010). NMDARs are hetero-multimers that are composed of at least one GluN1 (NR1) plus one or more GluN2A-D (NR2A-D) subunits (Sheng et al. 1994). GluN2 subunits, especially GluN2B, play important roles in the induction and maintenance of long-term potentiation (LTP) and long-term depression (LTD) that are likely the underlying mechanisms of learning and memory (Bliss & Collingridge, 1993). The lately discovered and characterized glutamate NMDAR subunit 3A (GluN3A) (NMDAR-L, χ−1 or NR3A) and GluN3B (NR3B) subunits are unique subunits of NMDARs. GluN3A is widely expressed in different brain regions, such as the cortex, hippocampus, thalamus, hypothalamus, brainstem and spinal cord (Sucher et al. 1995). Unlike GluN2B whose expression gradually decreases after birth but remains highly expressed in adulthood (Wenzel et al. 1997), GluN3A mRNA and protein levels peak at the end of the first postnatal week, but decrease from P12 to a low level (∼15% of GluN1) by adulthood in rodents (Ciabarra et al. 1995). This sharp reduction in GluN3A expression has led most investigations on GluN3A to focus on neonatal stages (Roberts et al. 2009).
GluN3A co-localizes with other subunits to form functional channel complexes (Perez-Otano et al. 2001). Co-expression of GluN3A with GluN1 and GluN2 reduces NMDAR-mediated Ca2+ influx and whole-cell currents, resulting in smaller unitary conductance of NMDAR channels. Because of this property, GluN3 subunits (GluN3A and GluN3B) have been regarded as inhibitory subunits in the NMDAR complex. The smaller conductance is also seen in cortical neurons isolated from wild-type (WT) P8 rodents when compared with age-matched neurons from GluN3A knockout (KO) mice (Sasaki et al. 2002). These small NMDAR inward currents are most likely due to the relatively reduced Ca2+ permeability and reduced Mg2+ sensitivity in the GluN3A-containing NMDARs (Matsuda et al. 2002). Mice lacking GluN3A have increased dendritic spine densities in cortical neurons. This difference is more pronounced in cortical neurons of P19 mice compared with the adults (Das et al. 1998). Supporting a role in early brain development, the elimination of GluN3A from active synapses in the first 2 weeks in rodents is required to increase NMDAR conductance, and to form stronger and larger synapses. Recent evidence also suggests that presynaptic GluN3A receptors may participate in regulation of glutamate release (Larsen et al. 2011). At the behavioural level, GluN3A downregulation is required to form long-term memories (Roberts et al. 2009). Thus, GluN3A downregulation appears critical for synapse maturation and memory formation during development. On the other hand, whether GluN3A plays a potential role in the adult brain has been largely ignored and remains poorly understood.
A previous investigation showed that enhanced LTP was only observed in hippocampal slices from P6–P8 GluN3A KO mice, but not in juvenile and adult slices (Roberts et al. 2009). These observations have enforced the general assumption that GluN3A is not a major player in NMDAR-dependent activities in the adult brain. Different from previous observations and assumptions, here we demonstrate for the first time that after embryonic deletion of GluN3A, markedly significant differences in locomotor functions, pain sensation, learning and memory and synaptic plasticity (LTP) can be detected between adult WT and GluN3A KO mice. These novel findings indicate that the GluN3A subunit, despite its relatively low expression, has a surprisingly imperative functional role in adult animals and could be a target for therapeutic interventions.
Methods
WT and GluN3A KO mice
Animal handling and all experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University. The GluN3A KO mice and WT counterparts were originally provided by Nobuki Nakanishi and Stuart A. Lipton at Sanford-Burnham Medical Research Institute (La Jolla, California, USA). Detailed information about the background of these mice has been described before (Das et al. 1998). In brief, embryonic stem cells derived from 129/SvJ were electroporated with DNA carrying disrupted GluN3A gene and then injected into blastocysts from C57BL/6 mice. The resulting chimeric males were crossed with BlackSwiss or 129SvEv females to produce F1 heterozygotes. GluN3A KO homozygote mice were then produced by cross-breeding F1 mice. In our lab, homozygote colonies of either WT or GluN3A KO mice were maintained under the same conditions with identical room temperature (22°C), same food and water supply, and animal care environment.
Genotyping
DNA for genotyping was extracted from tail snips. Two separate sets of primers were used for the GluN3A KO and WT mice, respectively. For the WT reaction, forward primer: 5′-CCACGGTGAGCTTGGGGAAG-3′ and reverse primer 5′-TTGGGGAGCGCCCTGCATGG-3′. For the KO reaction, forward primer: 5′-CCACGGTGAGCTTGGGGAAG-3′ and reverse primer 5′-GCCTGAAGAACGAGATCAGG-3′. DNA (2 μl) was amplified on a thermocycler (MJ Mini, Personal Thermal Cycler, Bio-Rad, CA, USA) for 40 cycles (2 min, 94°C; 30 s, 60°C; 10 min, 72°C for the WT reaction; and 2 min, 94°C; 30 s, 58°C; 10 min, 72°C for the KO reaction).
Behavioural tests
For all behavioural tests, mice were handled for 3 days and acclimated to the testing location at least 1 h before starting any test.
SHIRPA primary screen
The screen test was performed to identify gross phenotypic and behavioural characteristics. We followed similar protocol procedures as described online by the MRC Harwell centre for mouse genetics (http://empress.har.mrc.ac.uk/; Lalonde et al. 2005). Mice were placed inside a glass beaker and inspected for the following phenotypes: body position (0 = inactive, 1 = active, 2 = excessively active); tremor (0 = absent, 1 = present); palpebral closure (0 = eyes open, 1 = eyes closed); coat appearance (0 = tidy, well groomed, 1 = irregular); whiskers (0 = present, 1 = absent); and defecation (0 = present, 1 = absent). The mice were then transferred to a new non-familiar arena and observed for the following responses: transfer arousal (0 = extended freeze >5 s, 1 = brief freeze then move, 2 = immediate movement); gait (0 = fluid movement and about 3 mm pelvic elevation, 1 = lack of fluidity); and tail elevation (0 = dragging, 1 = horizontal extension, 2 = elevated). Finally, the mice were held by their tails and assessed for the following: positional passivity (0 = struggles when held by tail, 1 = struggles when held by neck, 2 = struggles when laid supine, 3 = does not struggle); limb grasping (0 = absent, 1 = present); corneal reflex (0 = present, 1 = absent); biting (0 = none, 1 = biting with handling); and vocalization (0 = none, 1 = vocal).
Locomotor and anxiety tests
The open field test was conducted to measure baseline locomotor activity. In the open field locomotion test, mice were allowed to freely explore an open field container (45 × 35 × 25 cm; divided into 12 equal squares) for 5 min. Activity was recorded on a video camera. The average distance and speed, and the total time spent in the central zone were recorded using TopScan CleverSys (CleverSys, Inc., Reston, VA, USA). The Rota-rod test was performed to assess the ability of mice to maintain balance and coordination on an accelerating rotating rod (UGO Bacile 7650). The Rota-rod was set to accelerate from 4 to 40 rpm in 300 s. In each session, mice were placed on the rotating drum and the time to fall off was recorded. Each session was stopped after 6 min even if the mouse was still on the drum. The timer was also stopped if the mouse clings to the rotating drum and completes a full rotation. Drums were cleaned with ethanol after each trial. On each testing day, mice were tested three times separated by at least 15 min inter-trial interval. The elevated plus maze test was performed to measure anxiety levels. The elevated plus maze was built in the lab using black Plexiglas material with the following dimensions: arm's length 30 cm; width 5 cm; and wall's height 15 cm. The central compartment is 5 × 5 cm, and the stage is elevated 45 cm above the floor. Each mouse was placed in the central platform facing an open arm, and its behaviour was videotaped for 5 min. The maze was cleaned with ethanol between trials. We recorded the number of entries and total time spent in the open and closed arms. The defensive withdrawal test was performed to measure exploratory behaviour. In the defensive withdrawal test, animals were placed in a dark chamber (10 × 15 × 10 cm high) placed along the wall of a large box (50 × 50 × 50 cm high). Animals were habituated to the open field 24 h prior to behavioural testing by allowing them to explore it for 15 min without access to the sheltered chamber. The ‘latency to exit’ was measured as the time it takes the animal to place his four paws outside the small chamber. The ‘latency to cross’ was measured as the time to cross a square drawn to represent the central zone of the box. The experiment was video-taped and analysis was performed by a researcher blinded to the identity of the groups.
Pain tests
To test acute pain, we measured the latency of pain responses on the hot-plate and tail-flick tests. For the hot-plate test, mice were placed on a hot plate (55°C), and the latency to forepaw licking or jumping was recorded for each mouse in three trials separated by at least 15 min. For the tail-flick test, the latency to withdraw the tail from water (50°C) was recorded for three trials separated by 15 min. To test inflammatory pain, the response to subcutaneous formalin injection was recorded. In brief, 10 μl of 5% formalin was subcutaneously injected in the plantar surface of a forepaw. Pain scores were collected as detailed before (Roussy et al. 2009). In brief, the total time spent licking or biting the injected forepaw was recorded over a 3 min interval for each mouse for 1 h.
Memory tests
The Novel Object Recognition and Morris water maze tests were performed and analysed as previously described (Lalonde et al. 2002; Bevins & Besheer, 2006). All trials were videotaped and performance was analysed using TopScan or MazeScan (CleverSys, Inc.).
The Novel Object Recognition test was performed during the dark cycle. On the 2 days preceding the training session, mice were individually habituated to the testing environment (30 cm × 30 cm × 20 cm; polycarbonate box) for 10 min. During the training session, mice were allowed to explore a box in which two identical objects were placed in adjacent corners for 5 min. During the retention sessions (1 h, 1 day and 7 days after training), one of the objects was replaced by a novel object and mice were allowed to explore for 5 min. The ratio of the time spent exploring any of the identical objects during the training session or the time spent exploring the novel object during the retention session to the time spent exploring the familiar object was calculated.
The Morris water maze test was performed by the animal behaviour core lab at Emory University. The water maze apparatus is a round, water-filled tub (52 inch or 132.1 cm diameter filled with white tempura paint) placed in a room rich with extra maze cues. Mice were placed in the maze starting from four different positions (N, S, E and W). An invisible escape platform was located in the same spatial location 1 cm below the water surface independent of the starting position on a particular trial. In this manner, subjects were able to utilize extra maze cues to determine the platform's location. Each subject was given four trials per day (N, S, E and W) for 5 days with a 15 min inter-trial interval. The maximum trial length was 60 s, and subjects were manually guided to the platform if they do not reach it in the allocated time. Upon reaching the invisible escape platform, subjects were kept on it for an additional 5 s to allow them to survey the spatial cues in the environment to guide future navigation to the platform. After 5 days of task acquisition, a probe trial was carried out during which the platform was removed. The time spent and distance travelled in the quadrant that previously contained the escape platform during task acquisition was measured over 60 s.
Hippocampal slice preparation and electrophysiological recording
WT and GluN3A KO mice were anaesthetized with isoflurane, decapitated, and their brains were dropped in ice-cold artificial cerebrospinal fluid (aCSF) containing (in mm): NaCl, 124; KCl, 3; NaH2PO4, 1.25; MgCl2, 6.0; NaHCO3, 26; CaCl2, 2.0; glucose, 10. The high Mg2+ concentration here was used to suppress NMDAR activation and associated excitotoxicity during the cutting procedure. The aCSF solution was saturated with 95% O2 and 5% CO2, at pH 7.4. The hippocampi were cut into 400 μm-thick transverse slices with a vibrating blade microtome (Vibratome 1000 Plus Sectioning System, St Louis, MO, USA). After incubation at room temperature (23–24°C) in another aCSF containing 1 mm MgCl2 for 60–90 min, slices were placed in a recording chamber (RC-22C, Warner Instruments, Hamden, USA) on a stage of an upright microscope (Olympus CX-31, Pittsburgh, USA) and perfused at a rate of 3 ml per min with aCSF (1 mm MgCl2) at 23–24°C. A 0.1 MΩ tungsten monopolar electrode was used to stimulate the Schaffer collaterals. The field (f)EPSPs were recorded in CA1 stratum radiatum by a glass microelectrode filled with aCSF with resistance of 3–4 MΩ. The stimulation output (Master-8; AMPI, Jerusalem) was controlled by the trigger function of an EPC9 amplifier (HEKA Elektronik, Lambrecht, Germany). fEPSPs were recorded in the current-clamp mode. Data were filtered at 3 kHz and digitized at sampling rates of 20 kHz using Pulse software (HEKA Elektronik). The stimulus intensity (0.2 ms duration, 16–30 μA) was set to evoke 40% of the maximum fEPSP initial slope, and the test pulse was applied at a rate of 0.033 Hz.
To induce LTP, high-frequency stimulation (HFS) was applied at Schaffer collaterals at 100 Hz for 1 s once or repeated three times with 10 s intervals. Schaffer collaterals are axonal collaterals extending from the CA3 pyramidal neurons in the hippocampus, and play a critical role in learning and memory. Paired-pulse facilitation (PPF) was examined by applying pairs of pulses that were separated by 10–500 ms intervals. Low-frequency stimulation (LFS; 1 Hz, 15 min) or NMDA (30 μm) application were used for the induction of LTD in both WT and KO slices. To perform the low Mg2+ experiments, no HFS was applied. Normal aCSF was changed to low Mg2+ aCSF (containing 0.2 mm Mg2+), slices were perfused and fEPSPs were recorded for 60 min (Townsend et al. 2006). The magnitudes of LTP and LTD were expressed as the mean percentage of baseline fEPSP initial slope.
To monitor membrane potential changes (depolarization) in response to stimulation at the Schaffer collaterals, we conducted blind whole-cell patch-clamp recording of pyramidal neurons in the CA1 region of the hippocampus. The recording glass pipettes (Sutter Instrument, USA) had a tip resistance between 6 and 8 MΩ when filled with an internal solution containing (in mm): KCl, 140; MgCl2, 2; CaCl2, 1; Na2ATP, 2; EGTA, 10; Hepes, 10; at pH 7.2. The stimulus intensity applied to the Schaffer collaterals was 16 μA for 0.2 ms. The distance between stimulation electrode and recording pipette was about 300 μm.
Western blot
Western blot analysis was performed to analyse protein expression in brain tissues from WT and GluN3A KO mice following previous procedures (Manzerra et al. 2001). In brief, brain tissue was lysed in lysis buffer containing (in mm): Tris–HCl pH 7.4, 25; NaCl, 150; EDTA, 5; 0.1% SDS; sodium orthovanadate, 2; NaF, 100; 1% Triton X-100, leupeptin, aprotinin and pepstatin with continuous manual homogenization. Forebrain or hippocampal tissue lysate was then spun at 14,000 × g for 15 min and supernatant was collected. Protein concentration was determined using BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein (30 μg) were resolved on SDS–PAGE using gradient gels (6–18%), and gels were blotted onto polyvinylidene fluoride (PVDF) membranes (Amersham, Buckinghamshire, UK) blocked with 5% BSA in TBST buffer (20 mm Tris, 137 mm NaCl and 0.1% tween) and incubated overnight with primary antibodies against rabbit polyclonal anti-GluN3A (1:1000; Millipore, Billerica, USA), rabbit anti-GluN3B (1:1000; Millipore), mouse monoclonal anti-GluR1 (1:1000; Millipore), rabbit anti-cAMP response-element binding (CREB; 1:1000; Cell Signaling, Danvers, USA), rabbit anti-phospho-CREB (1:1000; Cell Signaling), rabbit anti-Akt (1:1000; Cell Signaling), rabbit anti-phospho-Akt (1:1000; Cell Signaling), rabbit anti-protein kinase C (PKC)α (1:1000; Cell Signaling), rabbit anti-protein kinase A (PKA) C-α (1:1000; Cell Signaling), rabbit anti-Ca2+/calmodulin-dependent kinase II (CaMKII)α (1:1000; Cell Signaling), rabbit anti-phospho-CaMKII (Thr286; 1:1000; Cell Signaling) and mouse anti-actin (1:5000; Sigma, St Louis, MO, USA). After three washes with TBST, blots were incubated with HRP-conjugated secondary antibodies (anti-rabbit or anti-mouse, 1:2000; Bio-Rad) in 5% BSA for 1 h. Blots were developed using Pierce ECL Western Blotting Substrate (Thermo Scientific, IL, USA). The level of protein expression was normalized to β-actin controls.
For synaptosomes and postsynaptic density (PSD) preparations, we used a modification of previous protocols (Dunkley et al. 2008). In brief, total forebrains were homogenized in 0.32 m sucrose solution using Teflon-coated homogenizers. The homogenate was centrifuged to collect the supernatant S1 and the pellet P1. S1 was further centrifuged to collect S2 and P2. P2 was resuspended in Percoll, and the P2/Percoll solution was differentially centrifuged in a Percoll gradient. The band corresponding to the heavy synaptosomes was resuspended in Hepes/EDTA/Triton-X buffer and centrifuged (32,000 × g; 70.1 Ti rotor) to collect the first PSD fraction (PSD-1T). PSD-1T was resuspended in Hepes/EDTA/Triton-X buffer and centrifuged again (200,000 × g; 70.1 Ti rotor) to collect the second PSD fraction (PSD-2T). The following antibodies were used: mouse monoclonal anti-GluN1 (1:1000; Millipore), rabbit polyclonal anti-GluN2B (1:1000; Millipore), rabbit polyclonal anti-GluN2A (1:1000; Millipore) and rabbit anti-PSD-95 (1:1000; Cell Signalling). The level of protein expression was normalized to PSD-95 controls.
Statistical analysis
Results were expressed as the mean ± SEM. Statistical comparisons were made with unpaired two-tailed Student's t test and one- or two-way analysis of variance (ANOVA), with the Bonferroni's post hoc analysis to identify significant differences. A value of P < 0.05 was considered significant difference for all comparisons.
Results
General phenotypic and behavioural features of adult GluN3A KO mice
Adult GluN3A KO mice (2–4 months old) appear indistinguishable from WT mice either visually (Fig. 1K) or as shown by the SHIRPA test (Table 1), which is a primary screen for gross phenotypic and behavioural characteristics and neurological dysfunctions. Adult WT and GluN3A KO mice showed no increased seizures or convulsions.
Figure 1. Locomotor deficits without anxiety in adult glutamate NMDAR subunit 3A (GluN3A) knockout (KO) micek.
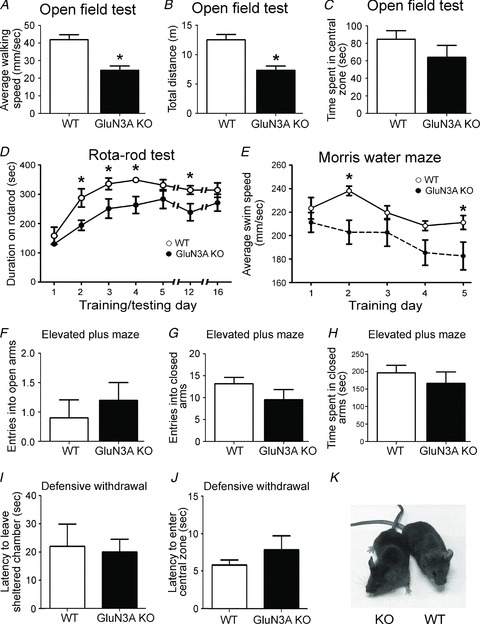
Adult wild-type (WT) and GluN3A KO mice were assessed for their motor function and anxiety level. A–C, in the open field test for locomotor activity, GluN3A KO mice walked slower (A) and covered less distance (B) during the 5 min period but spent a similar amount of time in the central zone (C) as the WT mice. These assays indicated that GluN3A KO mice had reduced locomotor abilities in novel environments. WT: n = 12; KO, n = 10 in all tests; *P < 0.05 vs. WT, Student's t test. D, in the Rota-rod test, animals were subjected to five training days, and then tested at days 12 and 16 (7 and 11 days after training). On day 1, WT and GluN3A KO mice performed similarly and, despite the difference during training, both WT and KO mice performed similarly at the end of a 5 day training session. After a long resting interval (from day 5 to 12), the performance of GluN3A KO mice dropped, but it soon recovered and remained similar to WT counterparts 4 days later (day 16). WT: n = 8; KO: n = 7; *P < 0.05 vs. WT, Student's t test at each time point separately; P > 0.05 using two-way ANOVA at all time points. E, morris water maze test demonstrated slower swim speeds of the GluN3A KO mice during the five consecutive training days. Both groups: n = 10; *P < 0.05 vs. WT, two-way ANOVA. F–H, stress/anxiety was tested in the elevated plus maze test. WT and GluN3A KO mice had a similar number of entries into the open (F) and closed (G) arms, and a similar total time spent in the closed arms (H). Both groups: n = 6; P > 0.05 by Student's t test. I and J, WT and GluN3A KO mice were evaluated for exploratory behaviour in the defensive withdrawal test. The latency to exit the sheltered chamber (I) and the latency to enter the central zone (J) were similar between the WT and KO mice, indicating unaltered exploratory behaviour between the two genotypes. WT: n = 11; KO: n = 9; P > 0.05 by Student's t test for both comparisons. K, adult WT and GluN3A KO mice are visually indistinguishable from each other.
Table 1.
Gross phenotypic and behavioural characteristics of adult WT and GluN3A KO mice using the SHIRPA primary screen
| Phenotype | WT mice | GluN3A KO mice | Phenotype | WT mice | GluN3A KO mice |
|---|---|---|---|---|---|
| Body position | 1 | 1 | Gait | 0 | 0 |
| Tremor | 0 | 0 | Tail elevation | 1 | 1 |
| Palpebral closure | 0 | 0 | Positional passivity | 2 | 2 |
| Coat appearance | 0 | 0 | Limb grasping | 1 | 1 |
| Whiskers | 0 | 0 | Corneal reflex | 0 | 0 |
| Defecation | 0 | 0 | Biting | 0 | 0 |
| Transfer arousal | 2 | 2 | Vocalization | 0 | 0 |
Each group: n = 5. For details on the scoring of each phenotype, please refer to the methods section. GluN3A, glutamate NMDA receptor subunit 3A; KO, knockout; WT, wild-type.
We noticed, however, that there were some body weight differences between WT and GluN3A KO pups (Table 2). While WT and GluN3A KO pups weighed similar at birth, GluN3A KO mice became heavier than WT animals starting day 10 of the first month after birth. The difference in weight, however, disappeared at early adulthood (1 month old). This observation suggested that GluN3A KO mice initially grew faster than the WTs. Given the published reports on the influence of maternal care on animal phenotypes (Kappeler & Meaney, 2010; Cameron et al. 2011), we measured the body weight of WT and GluN3A KO mice that were raised in the same cage. GluN3A KO mice still weighed more during development, and the difference in weight similarly disappeared in early adulthood.
Table 2.
Body weight changes of WT and GluN3A KO mice from postnatal stage to young adulthood
| Weight (g) of WT and KO litters separate | Weight (g) of WT and KO litters combined | |||||
|---|---|---|---|---|---|---|
| WT | KO | Significance | WT | KO | Significance | |
| Day 1 | 1.41 ± 0.08 n = 17 | 1.55 ± 0.17 n = 9 | NS; P = 0.42 | 1.23 ± 0.07 n = 13 | 1.50 ± 0.07 n = 10 | S; P = 0.01 |
| Day 5 | 3.31 ± 0.11 n = 16 | 3.64 ± 0.13 n = 14 | NS; P = 0.07 | 2.58 ± 0.14 n = 12 | 3.30 ± 0.15 n = 10 | S; P = 0.003 |
| Day 10 | 4.72 ± 0.19 n = 18 | 6.52 ± 0.14 n = 21 | S; P < 0.0001 | 4.58 ± 0.19 n = 12 | 5.10 ± 0.17 n = 10 | NS; P = 0.06 |
| Day 15 | 6.66 ± 0.16 n = 9 | 8.80 ± 0.20 n = 10 | S; P < 0.0001 | 6.25 ± 0.17 n = 12 | 7.30 ± 0.26 n = 10 | S; P = 0.02 |
| Day 20 | 7.91 ± 0.25 n = 24 | 11.90 ± 0.87 n = 10 | S; P < 0.0001 | 7.50 ± 0.28 n = 12 | 8.70 ± 0.33 n = 10 | S; P = 0.01 |
| Day 30 | 17.27 ± 0.62 n = 15 | 23.33 ± 0.61 n = 6 | S; P < 0.0001 | 13.64 ± 0.50 n = 11 | 15.10 ± 0.60 n = 10 | NS; P = 0.07 |
| Young adult | 24.38 ± 0.52 n = 13 | 25.14 ± 0.68 n = 14 | NS; P = 0.39 | 21.18 ± 0.62 n = 11 | 22.00 ± 1.75 n = 6 | NS; P = 0.59 |
KO, knockout; NS, not significant; S, significant difference at P < 0.05; WT, wild-type. Weight is expressed in grams. Unpaired Student's t test was used for all comparisons.
Impaired locomotor activity and normal stress/anxiety levels in adult GluN3A KO mice
The role of GluN3A in locomotion is completely unknown. We measured locomotion and locomotor learning using a variety of motor tests. We started with the open field test, which assesses the baseline locomotor activity. Adult GluN3A KO mice walked slower and travelled less distance than WT mice (Fig. 1A and B). However, the total time GluN3A KO mice spent in the central zone of the test box was similar to WT mice (Fig. 1C). Thus, GluN3A KO mice were less active in the open field test but did not show any sign of increased anxiety. In order to test for gross motor skills including balance, coordination and motor learning abilities, animals were trained everyday for 5 days on the Rota-rod test. On day 1, despite their reduced activity in the open field, both WT and GluN3A KO mice performed similarly on the Rota-rod, indicating that in forced movement, KO mice were able to perform as well as WT mice. With continuous training, adult WT mice improved their performance better (stayed longer on the rotating beam) than GluN3A KO mice. By day 5, however, GluN3A KO mice caught up with their WT counterparts. The animals were then rested for a week before being tested again at days 12 and 16. The performance of the KO mice declined at day 12, but improved again at day 16 to levels similar to the WTs (Fig. 1D). This test suggested that GluN3A KO mice had some motor deficits, as shown by their slowed ability to reach their plateau performance during the five training days and by the drop in performance at day 12. However, it was evident that this decreased motor ability was compensated for by prolonged training.
Consistent with the results from the Rota-rod test, in a 5 day Morris water maze testing, the swimming speed of adult GluN3A KO mice was similar to WT mice at day 1. In the following days, GluN3A KO mice swam slower than WT mice, and statistically significant difference was detected at days 2 and 5 (Fig. 1E). While the average swimming speed of WT mice remained about the same, the average speed of the KO mice during day 2–5 was significantly slower than that of WT mice (219.0 ± 6.7 mm s−1 and 193.0 ± 5.4 mm s−1 for WT and KO mice, respectively, n = 10, P < 0.05).
Because some of these locomotor phenotypes could alternatively be explained by increased anxiety levels, we specifically evaluated anxiety using the elevated plus maze (Takahashi et al. 1989). In this test, there was no difference in the number of entries to the open (Fig. 1F) or closed arms (Fig. 1G), or the total time spent in the closed arms (Fig. 1H). In testing exploratory behaviour using the defensive withdrawal test (Takahashi et al. 1989), GluN3A KO mice showed no difference in the time to leave the sheltered chamber and the time to cross into the central zone compared with WT mice (Fig. 1I and J). These data collectively indicated that GluN3A KO mice were not more stressed/anxious than their WT counterparts.
Enhanced pain sensation in adult GluN3A KO mice
NMDARs play a key regulatory role in pain sensation. Hot-plate and tail-flick tests were performed to elucidate whether adult GluN3A KO mice had altered acute nociception. The hot-plate test reflects supraspinal sensory integration (Rubinstein et al. 1996), while the tail-flick test is a measurement of spinal pathways of acute nociception (Tham et al. 2005). Adult GluN3A KO mice showed enhanced responses (shorter latency) on the hot-plate test (Fig. 2A). On the other hand, the tail-flick test showed no difference in the responses elicited in WT and GluN3A KO mice (Fig. 2B). Taken together, these results are consistent with a role of GluN3A in mediating an analgesic response in supraspinal but not spinal pain pathways.
Figure 2. Enhanced pain sensation in adult glutamate NMDAR subunit 3A (GluN3A) knockout (KO) mice.
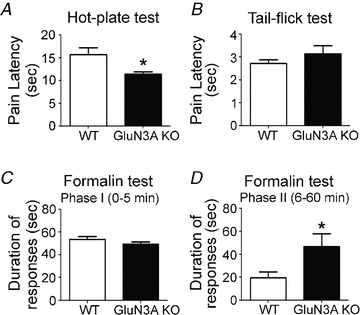
Adult wild-type (WT) and GluN3A KO mice were tested for their sensitivity to different pain stimuli. A and B, acute pain perceptions of supraspinal and spinal pain pathways were tested in WT and GluN3A KO mice in the hot-plate and tail-flick tests, respectively. Enhanced pain sensation (shorter pain latency) was seen in the hot-plate test (A) with KO mice (n = 8 per group; *P < 0.05, Student's t test), but not in the tail-flick test (n = 7 per group; B). C and D, acute and sub-acute pain perception (licking and biting the injected forepaw) triggered by formalin injection was evaluated in adult mice. Although there was no difference immediately (0–5 min) after the injection (C), significantly enhanced responses in GluN3A KO mice were seen 6–60 min after formalin injection (D). Both groups: n = 4; *P < 0.05 by Student's t test.
The established formalin inflammatory pain model was used to investigate the response to inflammatory nociception and tissue injuries. Formalin was subcutaneously injected into the plantar side of the right forepaw and the pain score was determined (Roussy et al. 2009). There was no obvious difference in the forepaw oedema between the WT and GluN3A KO genotypes 1 day after the formalin insult (data not shown). We analysed the duration of pain responses (licking and biting the injected forepaw) during the first hour, and pooled the data into two distinct phases: phase I (0–5 min); and phase II (6–60 min). In phase I, there was no difference in the responses between WT and KO mice (Fig. 2C). In phase II, GluN3A KO mice exhibited a more pronounced pain response compared with WT controls (Fig. 2D), indicating an enhanced response to inflammatory pain stimuli with GluN3A deletion.
Enhanced object recognition and spatial memory in adult GluN3A KO mice
Because learning and memory are intimately NMDAR dependent, adult WT and GluN3A KO mice were tested in the Novel Object Recognition and Morris water maze tests for assessing visual and spatial memory, respectively (Bevins & Besheer, 2006; Vorhees & Williams, 2006). In the Novel Object Recognition test, which requires acquisition, storage and recall of features of familiar objects, WT and GluN3A KO mice showed similar preference to the ‘familiar’ objects during the training session (Fig. 3A), indicating that these two genotypes had similar exploratory behaviours, curiosity and motivation. During the retention tests where one of the objects was replaced with a novel object, both WT and GluN3A KO mice showed more preference for the novel object 1 h after training. GluN3A KO mice, however, showed even more preference for novelty compared with WT controls. Different animal groups were tested with the novel object 1 or 7 days after training, respectively. WT mice lost preference for novelty in these tests, while the GluN3A KO mice still showed preference for the novel object at day 1 after training (Fig. 3A), indicating better recognition memory compared with WT mice.
Figure 3. Enhanced recognition and spatial memory in adult glutamate NMDAR subunit 3A (GluN3A) knockout (KO) mice.
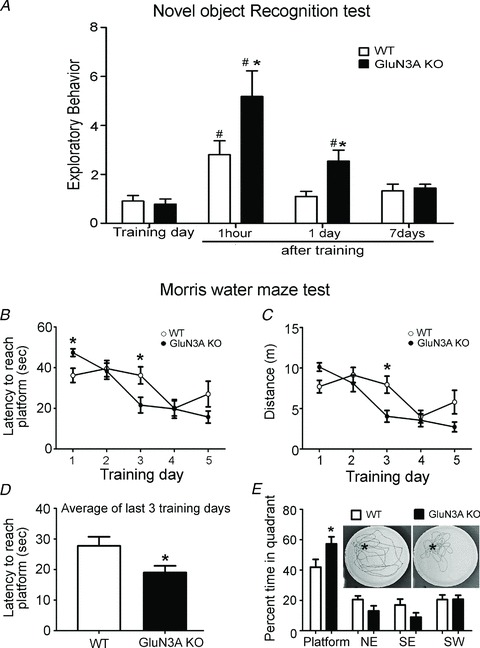
A, wild-type (WT) and GluN3A KO mice were evaluated for recognition memory in the Novel Object Recognition test. Both genotypes had equal exploratory preference during training as they spent similar amounts of time approaching, touching and smelling the similar objects. During the retention test (1 h after training), both WT and GluN3A KO mice showed enhanced preference for the novel object. This enhanced preference for novelty was more pronounced in KO animals. One and 7 days after training, WT mice lost preference for novelty, while GluN3A KO mice still showed preference for the novel object 1 day after training (but not at day 7). The exploratory behaviour on the y-axis represents the ratio of the time spent exploring the novel object to the time spent on the familiar object. All groups: n = 10; *P < 0.05 vs. WT; #P < 0.05 vs. training day controls using two-way ANOVA, Bonferroni post hoc analysis and Student's t test. B–E, WT and KO mice were evaluated for spatial memory in the Morris water maze test. B, latency to escape to platform during the 5 day training session. On day 1, GluN3A KO mice were slower to reach the platform, but their time improved at day 3 (*P < 0.05 at days 1 and 3, Student's t test; differences not significant using two-way ANOVA). C, swim distance to reach the platform during training. KO mice performed better at day 3 (*P < 0.05, two-way ANOVA with Bonferroni's analysis). D, average latency to reach the platform during the last three training days showing that KO mice reached the escape platform faster than WT mice (P = 0.022, Student's t test). E, the place preference test conducted at day 6 when the platform was removed after the last training day. GluN3A KO mice spent more time in the platform (marked as * in the inset photos) quadrant compared with WT mice, indicating enhanced spatial memory. The insets show representative traces of the navigation of both WT and GluN3A KO mice. Both groups: n = 10; *P < 0.05 vs. WT in the platform quadrant, two-way ANOVA and Bonferroni's post hoc analysis.
In the Morris water maze test in which animals use spatial cues to swim towards an escape platform (Vorhees & Williams, 2006), adult WT and GluN3A KO mice were trained to find the submerged platform four times a day for 5 days. On day 1, GluN3A KO mice needed more time to get to the platform (Fig. 3B) likely due to their locomotor deficit. The latency and distance travelled to escape to the platform decreased in both WT and GluN3A KO mice during the 5 day training (Fig. 3B and C). Analysis on the latency and distance covered by WT or KO mice during the 5 day training showed that there is a significant learning curve in both WT (day 3) and KO mice. By averaging the latency in the last three training days, GluN3A KO mice performed significantly better than WT controls (27.7 ± 3.0 s and 19.0 ± 2.2 s for WT and KO mice, respectively; n = 10, P < 0.05; Fig. 3D). When animals were tested after removal of the platform on day 6, both WT and GluN3A KO mice spent significantly more time in the target quadrant than in other quadrants (Fig. 3E; P < 0.0001 comparing the time spent in the target quadrant with all other quadrants in WT and GluN3A KO mice separately using one-way ANOVA). GluN3A KO mice, however, significantly spent even more time in the target quadrant compared with WT controls (41.9 ± 5.0% and 57.3 ± 4.6% for WT and KO mice, respectively; n = 10, P < 0.05), indicating better spatial memory of the KO mice.
Enhanced LTP in the juvenile GluN3A KO brain
LTP is a cellular and molecular event that is believed to be the electrophysiological basis of synaptic plasticity associated with learning and memory (Bliss & Collingridge, 1993). NMDARs are major regulators in the induction and maintenance of LTP (Coan et al. 1987). LTP was examined in the CA1 region of hippocampal slices from juvenile (postnatal day 10 and 20) WT and GluN3A KO mice. The fEPSPs were recorded before and after a single episode of HFS (100 Hz, 1 s) at the Schaffer collaterals. In this test, the average magnitude of HFS-induced LTP was similar between P10 WT and GluN3A KO mice (Fig. 4A), and between P20 WT and GluN3A KO animals (Fig. 4B). However, it is commonly known that the activation of AMPARs and NMDARs by glutamate released from presynapses contributes to postsynaptic membrane depolarization, and that LTP induction relies on sufficient activation of AMPARs that removes the voltage-dependent Mg2+ block on NMDARs (Premkumar & Auerbach, 1996). Because AMPAR expression in the developing brain is much lower than in adults (Pickard et al. 2000; see Fig. 6D) and NMDARs in the absence of GluN3A are more sensitive to the Mg2+ block than GluN3A-containing NMDARs (Chatterton et al. 2002), we hypothesized that LTP induction in P10/P20 mice might be greatly affected by the nature of HFS. Specifically, we proposed that a single HFS in GluN3A KO brain slices could not induce sufficient AMPAR-mediated membrane depolarization to remove the Mg2+ block as it can do in adult brains. Thus, sufficient postsynaptic depolarization might be needed to reveal the LTP capability in the P10 and P20 GluN3A KO brain.
Figure 4. Enhanced LTP in the neonatal and juvenile glutamate NMDAR subunit 3A (GluN3A) knockout (KO) brain.
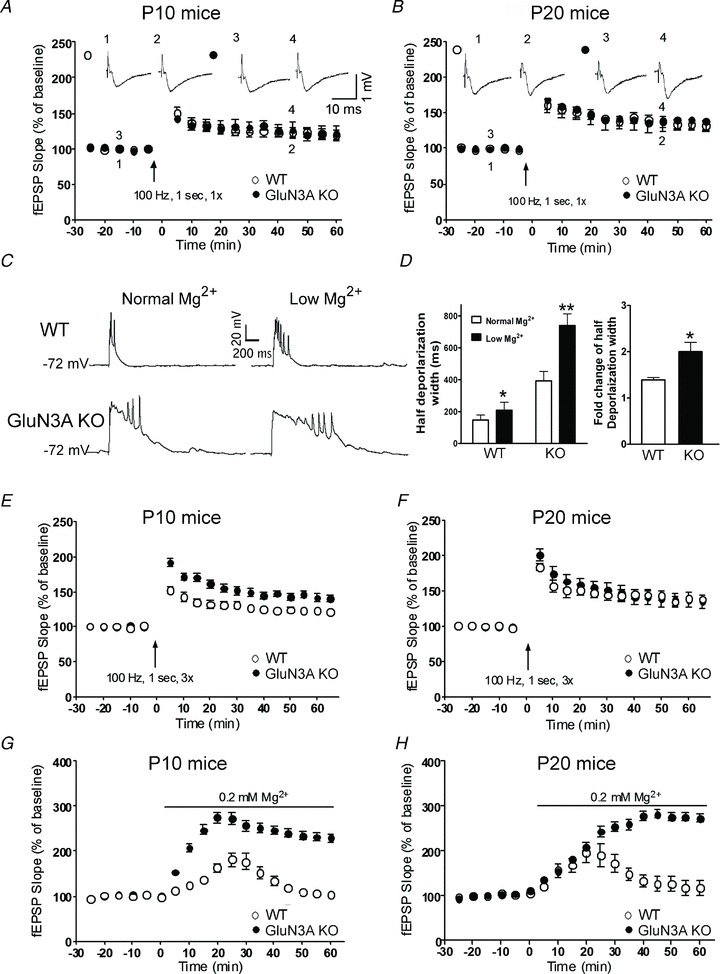
LTP induction in hippocampal slices from wild-type (WT) and GluN3A KO mice of different ages. A and B, LTP was induced by a single HFS episode (100 Hz, 1 s, 1×). The magnitude of LTP was similar between WT and GluN3A KO mice in P10 (A) and P20 (B) age groups. Representative field (f)EPSP traces recorded at the indicated time points are shown on top. All groups: n = 5 or 6. C and D, whole-cell current-clamp recording of membrane depolarization in response to stimulation at the Schaffer collaterals of P10 WT and GluN3A KO brain slices. C, membrane potential recordings in normal (left) and low Mg2+ (0.2 mm; right) from WT (upper) and GluN3A KO (lower) slices. D, summary of the widths (duration) measured at the half depolarization level in WT and GluN3A KO slices in both normal and low Mg2+ (*P < 0.05; **P < 0.01). The bar graph on the right shows low Mg2+ induced fold change of half depolarization width in WT and GluN3A KO mice (*P < 0.05; n = 5–6 per group). E and F, LTP was induced by HFS (100 Hz, 1 s) repeated three times (3×). The magnitude of LTP was greater in the postnatal day 10 KO mice (E). It was, however, similar between these two genotypes in the P20 (F) age group (n = 6 per group). G and H, after switching to 0.2 mm Mg2+ aCSF, fEPSP slopes were gradually and significantly increased in both WT and GluN3A KO slices of P10 (G) and P20 (H) mice. The LTP event was greater in the GluN3A KO slices in both age groups. *P < 0.05 between the average slopes of fEPSPs of WT and KO slices during time points 0–60 min. Each group in G, n = 6; each group in H, n = 5.
Figure 6. Expression of NMDARs and AMPARs in the neonatal and adult brains.
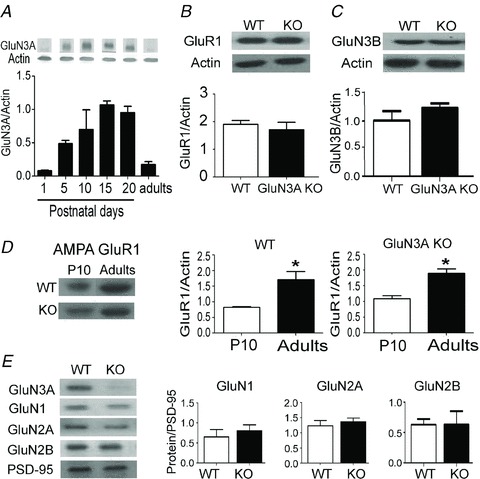
A, levels of glutamate NMDAR subunit 3A (GluN3A) protein in the developing and adult brain. GluN3A expression in the forebrain peaks early during development and then reaches a minimum level in adulthood. Each group: n = 3. B, the expression of the AMPAR subunit GluR1 in the adult brain is similar between wild-type (WT) and GluN3A knockout (KO) mice (n = 4 per group). C, the expression of GluN3B in the adult brain is similar between WT and GluN3A KO mice (n = 3 per group). D, expression of the AMPAR subunit GluR1 is higher in the adult forebrain of the WT and GluN3A KO mice as compared with P10 mice (each group: n = 4; *P < 0.05 between P10 and adult, Student's t test). Protein levels shown in A–D were determined in forebrain tissue lysates with β-actin as the loading control. E, the expression of the NMDAR subunits GluN1, GluN2A and GluN2B in the adult forebrain is similar between WT and GluN3A KO mice. Protein expression was determined in postsynaptic density fractions (PSD-2T), with PSD-95 as the loading control.
To examine our hypothesis, we then tested the effect of low Mg2+ (0.2 mm instead of 1 mm in aCSF) on membrane depolarization in hippocampal slices from P10 WT and GluN3A KO mice. Whole-cell current-clamp recording of pyramidal neurons in the CA1 region showed that stimulation at the Schaffer collaterals induced membrane depolarization that triggered action potential spikes (Fig. 4C and D). To quantify the depolarization response, we measured the width (duration) of the response at the half depolarization level. When brain slices from WT and GluN3A KO mice were perfused with the low Mg2+ aCSF, the depolarization response was much longer compared with the response in the normal aCSF solution (Fig. 4C and D). Moreover, the increased duration of membrane depolarization in low Mg2+ was greater in GluN3A KO than that in WT mice (Fig. 4D). This is consistent with the previous observation that NMDARs lacking GluN3A are more sensitive to Mg2+ block (Chatterton et al. 2002; Matsuda et al. 2002). The resting membrane potential did not change significantly in the low Mg2+ solution.
To ensure that sufficient membrane depolarization can overcome the voltage-dependent Mg2+ block of NMDARs, LTP was tested using either a 3× HFS protocol or the 0.2 mm Mg2+ external solution without HFS in young brain slices of P10 and P20 mice (Townsend et al. 2006). In the 3× HFS protocol, fEPSP recordings showed significantly greater LTP in P10 but not in P20 GluN3A KO slices (Fig. 4E and F). In the low Mg2+ solution, fEPSPs were gradually potentiated in P10 and P20 slices from both WT and KO animals. However, the increases were greater in GluN3A KO slices (Fig. 4G and H). Therefore, using protocols that provoke additional glutamate release and/or sufficient postsynaptic depolarization that can compensate for insufficient AMPAR expression/activation or the difference in Mg2+ sensitivity, we demonstrate that GluN3A can be a key regulator of synaptic plasticity in the developing brain.
Enhanced LTP in the adult GluN3A KO brain
In adult brain slices from 2- to 4-month-old mice, the role of GluN3A in LTP regulation was studied using normal aCSF (1 mm Mg2+) because a higher level of AMPARs exists in the adult brain (see Fig. 6D) and HFS should induce sufficient postsynaptic depolarization. Using the previously reported HFS protocol (100 Hz, 1 s, 3×; Roberts et al. 2009), we similarly observed that there was no significant difference in LTP between adult WT and GluN3A KO mice (Fig. 5A; P > 0.05, n = 6 in each group). However, a single HFS episode (100 Hz, 1 s, 1×) of a smaller depolarizing action revealed significantly greater LTP in adult GluN3A KO slices compared with WT slices (Fig. 5B). We then measured PPF, which is a specific indicator of presynaptic plasticity. PPF induced by various inter-stimulus intervals (ranging between 10 and 500 ms) was similar between WT and GluN3A KO hippocampal slices (Fig. 5C), indicating that the presynaptic function of GluN3A KO neurons was not different from that of WT neurons. As a control, we checked whether the enhanced LTP in the adult KO mice was dependent on NMDAR activation. The NMDAR antagonist d(−)2-amino-5-phosphonovalerate (AP-5; 100 μm) was bath applied 20 min prior to HFS. LTP induction was almost fully blocked in both WT and GluN3A KO slices (Fig. 5D). On the other hand, LTP induction was not affected when AP-5 was applied after HFS (Fig. 5E).
Figure 5. Enhancement of LTP in the adult glutamate NMDAR subunit 3A (GluN3A) knockout (KO) brain.
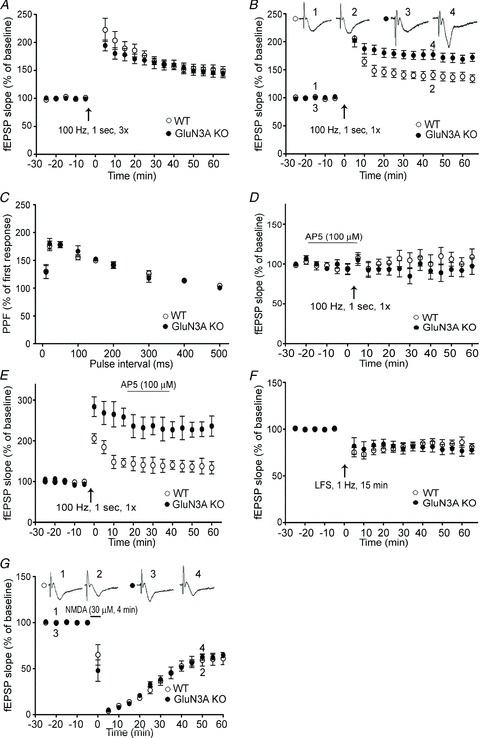
LTP induction in hippocampal slices from adult wild-type (WT) and GluN3A KO mice. A, LTP was induced by HFS (100 Hz, 1 s) repeated three times (3×). The magnitude of LTP was similar between WT and GluN3A KO slices. Each group: n = 6; B, LTP was induced by a single episode of HFS (100 Hz, 1 s, 1×). Although the initial elevated level of fEPSPs was similar, the magnitude of LTP in GluN3A KO slices stayed significantly higher than that in WT slices up to 60 min. WT and KO groups: n = 15 and 17, respectively; P < 0.05 between the averaged slopes of fEPSPs of WT and KO slices. C, paired-pulse facilitation (PPF) of fEPSP induced by various inter-pulse intervals was similar between adult WT and GluN3A KO slices. Both groups: n = 4. D, LTP was recorded in WT and GluN3A KO slices from adult animals in the presence of the NMDAR antagonist d(−)2-amino-5-phosphonovalerate (AP-5; 100 μm). LTP was similarly inhibited in both WT and GluN3A KO slices when AP-5 was bath applied 20 min prior to HFS (100 Hz, 1 s). Each group: n = 5. E, AP-5 (100 μm) application after the HFS (100 Hz, 1 s) stimulation could not block LTP induction in both WT and GluN3A KO slices. Each group: n = 5. F, similar LTD induced by LFS (1 Hz for 15 min) was recorded in WT and GluN3A KO slices from adult mice. Each group: n = 6. G, similar LTD was induced by 4 min exposure to 30 μm NMDA in WT and GluN3A KO slices from adult mice. Each group: n = 6.
LTD is another form of synaptic plasticity. We observed that LFS (1 Hz, 15 min) induced similar LTD in hippocampal slices of adult WT or GluN3A KO mice (Fig. 5F). We also tested a chemical induction protocol to induce LTD in the CA1 region of the hippocampus (Lee et al. 1998). A brief (4 min) bath application of NMDA (30 μm) transiently abolished the evoked field responses followed by stable responses that were depressed below the baseline. The magnitude of this chemically induced LTD in KO slices was also not different from that of the WT controls (Fig. 5G). Thus, LTD induced by two distinct inducers was not affected by deletion of GluN3A, suggesting that some GluN3A-independent mechanisms might regulate the induction of LTD.
Expression profile of glutamate receptors in the GluN3A KO brain
In Western blot examination, the expression of GluN3A in the forebrain of WT mice increased from postnatal day 1 to day 10, reached a maximal level between day 10 and 20, and then significantly dropped in adulthood (Fig. 6A; Wong et al. 2002). We evaluated the expression levels of major ionotropic glutamate receptor subunits in adult GluN3A KO mice. The expression levels of the AMPAR subunit GluR1 and NMDAR subunit GluN3B were similar in the adult brains of WT and GluN3A KO mice (Fig. 6B and C). As shown previously (Petralia et al. 1999), expression of GluR1 was low in postnatal day 10, and increased significantly in the adult brain for both WT and GluN3A KO mice (Fig. 6D). There was no difference in the expression of other NMDAR subunits (GluN1, GluN2A and GluN2B) measured in PSD-2Ts of adult WT and KO forebrains (Fig. 6E).
Enhanced CaMKII activity in the adult GluN3A KO mice
Western blot analysis of forebrain tissue lysates showed similar expression levels of different LTP regulators, including Akt, CREB, PKA and PKC (Fig. 7A and B). Moreover, there was no difference in the expression of phosphorylated Akt (p-Akt) and phosphorylated CREB (p-CREB; Fig. 7C).
Figure 7. Expression of LTP-related regulators in the forebrain of adult wild-type (WT) and glutamate NMDAR subunit 3A (GluN3A) knockout (KO) mice.
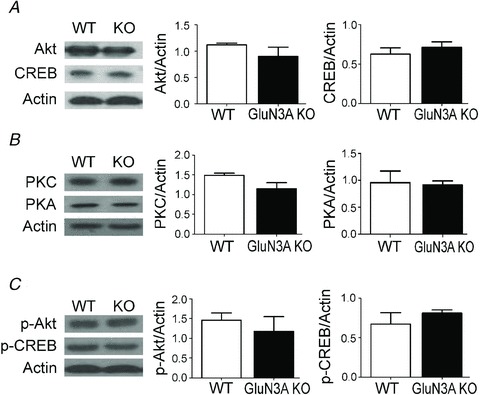
Western blotting was used to measure the protein levels of several mediators and effectors associated with LTP induction. A–C, there is no difference in the forebrain expression of Akt (A), cAMP response element-binding (CREB) (A), protein kinase C (PKC) (B), protein kinase A (PKA) (B), phospho-Akt (p-Akt) (C) and phospho-CREB (p-CREB) (C) between adult WT and GluN3A KO mice. Each group: n = 3 or 4. Protein expression was determined in forebrain tissue lysates with β-actin as the loading control.
The NMDA-dependent LTP induction requires NMDAR-mediated Ca2+ influx and rapid rise of intracellular Ca2+ concentration, which serves to activate CaMKII that acts as a major mediator in the induction of LTP (Barria et al. 1997; Giese et al. 1998). The expression of CaMKII was significantly higher in the GluN3A KO brain than in the WT brain, while no significant difference was seen in the level of phosphorylated CaMKII (p-CaMKII; Fig. 8A). To more closely assess this LTP key regulator in the brain region associated with LTP induction, CaMKII as well as the activated form of p-CaMKII were measured in the hippocampal region of adult animals before and 10 min after the LTP-inducing stimuli (1× HFS). We first calculated the ratio of p-CaMKII against CaMKII (p-CaMKII/CaMKII), and observed an expected trend of increased conversion to p-CaMKII after HFS in both WT and KO mice (Fig. 8B). Comparisons of the basal level of CaMKII and p-CaMKII, respectively, showed no difference before HFS between WT and GluN3A KO mice (Fig. 8C). However, and importantly, 10 min after the HFS, the level of p-CaMKII in the GluN3A KO hippocampus was significantly higher compared with that in the GluN3A KO slices (Fig. 8D). To understand whether the CaMKII signalling in the adult GluN3A KO mice could explain the differences in LTP and the increased memory performance, the selective CaMKII inhibitor, 1-[NO-bis-1,5-isoquinolinesulphonyl]-N-methyl-l-tyro-syl-4-phenylpiperazine (KN-62; 5 μm; Sigma), was applied into the bathing solution 15 min prior to and during the first 5 min after HFS. KN-62 suppressed LTP induction in hippocampal slices from both WT and GluN3A KO mice, and abrogated the LTP difference between them (Fig. 8E).
Figure 8. The key role of Ca2+/calmodulin-dependent kinase II (CaMKII) in the enhancement of LTP in adult glutamate NMDAR subunit 3A (GluN3A) knockout (KO) mice.
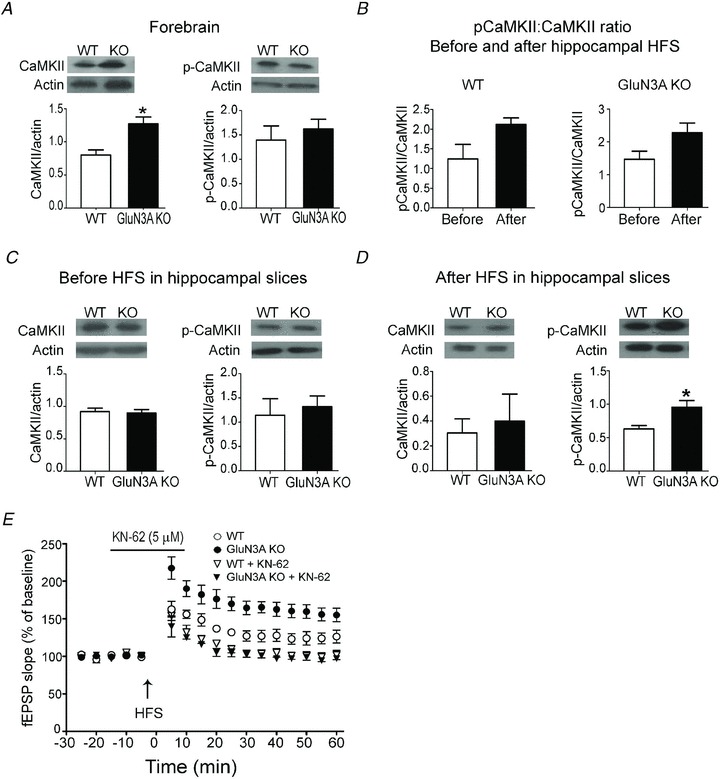
The expression of CaMKII and phosphorylated CaMKII (p-CaMKII) in the forebrain and hippocampus was measured using Western blotting. A, the basal CaMKII expression is higher in the forebrain cortex of adult GluN3A KO mice compared with wild-type (WT) mice, although there was no significant difference in the p-CaMKII level. *P < 0.05, Student's t test; n = 16 for CaMKII and n = 6 for p-CaMKII assay. B, the ratio of p-CaMKII versus total CaMKII in the hippocampus was compared before and 10 min after the LTP inducing high-frequency stimulation (HFS; 100 Hz, 1 s, 1×). The average values of the ratio appeared higher after the stimuli in both WT and KO slices (but P = 0.1 for WT and 0.06 for KO mice, respectively; n = 6 per group). C, the basal levels of CaMKII and p-CaMKII were similar in the unstimulated hippocampus of adult WT and GluN3A KO mice. For CaMKII, n = 13 per group; for p-CaMKII, n = 6. D, the level of CaMKII and p-CaMKII in the hippocampus 10 min after 1× HFS. The LTP stimulation significantly enhanced the p-CaMKII level in the adult GluN3A KO hippocampus compared with WT mice after HFS. *P < 0.05 vs. WT, Student's t test for both comparisons; n = 4 for both groups. Protein expression in A–D was determined in tissue lysates with β-actin as the loading control. E, 1-[NO-bis-1,5-isoquinolinesulphonyl]-N-methyl-l-tyrosyl-4-phenylpiperazine (KN-62; 5 μm), a specific inhibitor of CaMKII, suppressed the induction of LTP in WT and KO slices (downward triangles), and abrogated the difference between them. Each group: n = 3–4. Open and filled circles show LTP induction in WT and KO slices without KN-62 incubation. fEPSP, field EPSP.
Discussion
The present investigation reveals a potential novel role for the NMDAR subunit GluN3A in locomotion, pain sensation and learning/memory in adult animals. Previous investigations on the functional role of the GluN3A subunit have focused on neonates due to the high expression level of this subunit during the brain development. The marked impacts of GluN3A deletion on functional/behavioural activities of adult animals were previously unknown and unexpected. This is a surprising discovery in adult animals considering previous reports on adult GluN3A KO mice and that only low levels of GluN3A expression exist in the adult brain (Wong et al. 2002). The results from our investigation suggest that, even at relatively low expression levels, the functional impact of GluN3A in the adult brain should not be underestimated.
Adult GluN3A KO mice were morphologically and phenotypically indistinguishable from adult WTs. The KO animals grew faster than WT mice during the first month. The difference in body weight, however, disappeared in early adulthood as GluN3A expression declined. This is an interesting and previously unknown phenomenon. We suggest that the role of GluN3A in affecting body weight deserves a further detailed investigation. GluN3A KO mice were generally hypoactive in the open field test compared with the WT counterparts. Under forced circumstances, as in the Rota-rod or the swim test, the locomotor deficits were abrogated in the first trial day. Additionally, GluN3A KO mice showed reduced motor ability and motor skills/coordination on the Rota-rod test, but this deficit was compensated by training. At the brain structural and molecular level, GluN3A is moderately expressed in the striatum and the cerebellum (Wong et al. 2002); both regions heavily implicated in motor learning and coordination (Paulin, 1993; Dang et al. 2006).
Western blot analysis of different LTP-related regulators revealed that GluN3A KO mice have increased basal expression of CaMKII in the forebrain, and increased phosphorylation of CaMKII after HFS in the hippocampus. It is possible that GluN3A-deficient neurons with higher NMDAR Ca2+ permeability permit a facilitated intracellular Ca2+ accumulation that leads to a constitutively higher expression as well as activation of CaMKII. Transgenic mice overexpressing CaMKII in the forebrain show reduced locomotor activity in novel environments (Hasegawa et al. 2009). Thus, higher CaMKII expression in adult GluN3A KO mice might explain the reduction in locomotor activity. In contrast to the CaMKII overexpressing mice that display increased aggression and anxiety-like behaviours (Hasegawa et al. 2009), GluN3A KO mice exhibit unaltered anxiety levels as shown by the elevated plus maze. CaMKII is a major constituent of the PSDs, and plays an important role in initiating a cascade of events that regulate LTP and potentiate synaptic transmission (Lisman et al. 2002). Pharmacological inhibition of CaMKII blocks hippocampal LTP (Stanton & Gage, 1996), and impairs hippocampus-dependent memory and learning (Tan & Liang, 1996). Similar results were also found in transgenic mice with deletion of CaMKII (Silva et al. 1992). CaMKII also regulates nociception. Acute pharmacological inhibition of CaMKII with KN-62 reversed complete Freund's adjuvant (CFA)-induced inflammatory pain responses (Luo et al. 2008). On the other hand, forebrain overexpression of CaMKII reduces CFA-induced allodynia and hyperalgesia without affecting pain responses to heat, mechanical pressure and formalin injection (Wei et al. 2006). While the exact role of CaMKII in mediating LTP and pain sensation may be affected by multiple co-factors, the enhanced expression of CaMKII in the forebrain of the GluN3A KO mice is probably a molecular mediator in enhanced LTP and nociceptive responses.
The enhancement of learning and memory in GluN3A KO mice is similar to that seen in the ‘smart mice’ overexpressing GluN2B (NR2B; Tang et al. 1999). GluN2B, however, has been identified as a pro-death NMDAR subunit (Liu et al. 2007), whereas deletion or downregulation of GluN3A in the adult brain does not increase the risk of excitotoxicity or ischaemic injury (Nakanishi et al. 2009). In this respect, GluN3A appears to be a more realistic target for learning and memory modulation than GluN2B. To this end, the role of GluN3A in adult behaviour should be confirmed with conditional GluN3A manipulation studies. Supporting our observations, prolonged GluN3A expression in the forebrains of adult mice attenuates LTP and disrupts long-term memory (Roberts et al. 2009). Both memory tests used (Novel Object Recognition and Morris water maze) in this paper require locomotion and, given the difference in locomotor abilities between the two genotypes, it is possible that the difference seen in memory tests may not be due to memory and learning abilities alone. However, we were not able to objectively study emotional memory using contextual and cued fear conditioning, which does not require the measurement of movement because of the difference in acute pain responses. On the other hand, it is possible that the actual memory enhancement is much greater than what we observed in these tests because of the reduced locomotion in GluN3A KO mice. For example, despite their significantly reduced swim speed, GluN3A KO mice still managed to reach the platform in the water maze test faster than WT controls. Moreover, the expression of GluN3A in adult non-human primates is higher in contrast to the rodent brain (Mueller & Meador-Woodruff, 2005), suggesting that the GluN3A role observed in mice could be more prominent in primates.
Our data demonstrate that the GluN3A regulation of LTP is present in immature as well as adult brains. The observed facilitation in LTP is similar to what is seen in PSD-95 KO mice, except that GluN3A KO mice do not show any disruption in LTD (Carlisle et al. 2008). A previous study using the 3× HFS protocol could not detect any LTP difference between adult WT and GluN3A KO mice (Roberts et al. 2009). As we hypothesized in the current investigation, the strong stimulation protocol (3× HFS) used in the earlier study likely overcame the inhibitory effect of GluN3A in the WT mice, and thus prevented the detection of LTP differences between the two genotypes. That same study detected a difference in LTP between P6–P8 WT and GluN3A KO mice with a 3× HFS protocol. This is expected because 3× HFSs are strong enough to stimulate/depolarize the postnatal slices despite the low GluR1 expression at this developing stage. It is also important to mention that, as shown in Fig. 5A, upon more intensive stimuli (3× HFS), LTP in WT slices increased to the same level of KO slices that abrogated the LTP difference between WT and GluN3A KO mice seen with 1× HFS. Similar to our work and as shown in a study published while this work was in revision, there was no difference in the expression of AMPARs and other NMDARs between the WT and GluN3A KO adult brain (Henson et al. 2012). While it is possible that the behavioural differences in adults are due to the difference in GluN3A expression level between the WT and KO mice, a possibility that requires further investigation is that the phenotypic differences found in this study are inherited from the early knocking out of GluN3A during the embryonic and neonatal stages. There is a possibility that GluN3A deletion during brain development may cause subsequent gene regulation and/or synaptic modification that could have impacts on brain functions and behaviour in adulthood. To address the question whether early deletion of GluN3A is necessary for the observed changes in adults, the best approach is a conditional KO or shRNA knockdown in adult animals. As far as we know, successful knockdown or conditional KO of GluN3A in adult animals has not been reported by any group. This issue, therefore, remains to be specifically tested in a future investigation.
Patients suffering from schizophrenia exhibit impaired memory (Aleman et al. 1999), pain insensitivity (Dworkin, 1994) and disrupted motor functions (Marsden & Jenner, 1980). Animal models of schizophrenia also show decreased expression of CaMKII (Matsuo et al. 2009; Novak & Seeman, 2010). Noticeably, these symptoms and CaMKII expression are more or less the opposite phenotypes to what we observed in GluN3A KO mice. Indeed, GluN3A transcription level is elevated by 32% in schizophrenic patients and has been implicated in the pathogenesis of schizophrenia (Mueller & Meador-Woodruff, 2004). However, the association between the GluN3A expression level or polymorphisms and schizophrenia is still controversial (Gulli et al. 2007; Henson et al. 2008; Shen et al. 2009). The present work supports the merit for further investigations on the relationship between GluN3A and schizophrenia. It may additionally indicate a novel target whose modulation in adults may reverse phenotypes in disorders that involve impaired synaptic structures and dysfunctions such as those found in patients with schizophrenia.
Acknowledgments
This work was supported by NIH research grants NS057255, NS058710, NS062097 and the AHA Pre-doctoral Fellowship 10 PRE4430032. We thank G. Neigh for providing the behavioural analysis program CleverSys; J. Larimore, V. Faundez, J. McCoy III, Y. Zhu and D. Lambeth for helping with subcellular fractionation; the Emory Rodent Behavioural Core for performing the Morris water maze test; and K. Ressler for advice and comments.
Glossary
- AP-5
d(−)2-amino-5-phosphonovalerate
- CaMKII
Ca2+/calmodulin-dependent kinase II
- CFA
complete Freund's adjuvant
- CREB
cAMP response element-binding
- fEPSP
field EPSP
- GluN3A
glutamate NMDA receptor subunit 3A or NR3A
- HFS
high-frequency stimulation
- KN-62
1-[NO-bis-1,5-isoquinoli-nesulphonyl]-N-methyl-l-tyrosyl-4-phenylpiperazine
- KO
knockout
- LFS
low-frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- NMDAR
NMDA receptor
- PKA
protein kinase A
- PKC
protein kinase C
- PPF
paired-pulse facilitation
- PSD
postsynaptic density
- WT
wild-type
Author contributions
O.M.: design and perform experiments, data analysis and manuscript preparation/revision. M.S.: design and perform experiments, data analysis and manuscript preparation/revision. L.W.: concept development, data analysis and manuscript preparation, research grant support. S.P.Y.: concept development, design experiments, data analysis and manuscript preparation/revision, research grant support.
References
- Aleman A, Hijman R, de Haan EH, Kahn RS. Memory impairment in schizophrenia: a meta-analysis. Am J Psychiatry. 1999;156:1358–1366. doi: 10.1176/ajp.156.9.1358. [DOI] [PubMed] [Google Scholar]
- Barkus C, McHugh SB, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM. Hippocampal NMDA receptors and anxiety: at the interface between cognition and emotion. Eur J Pharmacol. 2010;626:49–56. doi: 10.1016/j.ejphar.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Cameron NM, Soehngen E, Meaney MJ. Variation in maternal care influences ventromedial hypothalamus activation in the rat. J Neuroendocrinol. 2011;23:393–400. doi: 10.1111/j.1365-2826.2011.02124.x. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, O’Dell TJ. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J Physiol. 2008;586:5885–5900. doi: 10.1113/jphysiol.2008.163469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415:793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- Ciabarra AM, Sullivan JM, Gahn LG, Pecht G, Heinemann S, Sevarino KA. Cloning and characterization of chi-1: a developmentally regulated member of a novel class of the ionotropic glutamate receptor family. J Neurosci. 1995;15:6498–6508. doi: 10.1523/JNEUROSCI.15-10-06498.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan EJ, Saywood W, Collingridge GL. MK-801 blocks NMDA receptor-mediated synaptic transmission and long term potentiation in rat hippocampal slices. Neurosci Lett. 1987;80:111–114. doi: 10.1016/0304-3940(87)90505-2. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Sasaki YF, Rothe T, Premkumar LS, Takasu M, Crandall JE, Dikkes P, Conner DA, Rayudu PV, Cheung W, Chen HS, Lipton SA, Nakanishi N. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393:377–381. doi: 10.1038/30748. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Dworkin RH. Pain insensitivity in schizophrenia: a neglected phenomenon and some implications. Schizophr Bull. 1994;20:235–248. doi: 10.1093/schbul/20.2.235. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Gulli R, Masnata B, Bonvicini C, Tura GB, Manglaviti L, Vaggi M, Mollica M, Bellone E, Mandich P, Gennarelli M, Di Maria E. A putative regulatory subunit (NR3A) of the NMDA receptor complex as candidate gene for susceptibility to schizophrenia: a case-control study. Psychiatr Genet. 2007;17:355–356. doi: 10.1097/YPG.0b013e328133f74f. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Furuichi T, Yoshida T, Endoh K, Kato K, Sado M, Maeda R, Kitamoto A, Miyao T, Suzuki R, Homma S, Masushige S, Kajii Y, Kida S. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol Brain. 2009;2:6. doi: 10.1186/1756-6606-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Larsen RS, Lawson SN, Perez-Otano I, Nakanishi N, Lipton SA, Philpot BD. Genetic deletion of NR3A accelerates glutamatergic synapse maturation. PLoS One. 2012;7:e42327. doi: 10.1371/journal.pone.0042327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Salimi K, Vadlamudi S, Hamer RM, Gilmore JH, Jarskog LF, Philpot BD. Developmental regulation of the NMDA receptor subunits, NR3A and NR1, in human prefrontal cortex. Cereb Cortex. 2008;18:2560–2573. doi: 10.1093/cercor/bhn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler L, Meaney MJ. Epigenetics and parental effects. Bioessays. 2010;32:818–827. doi: 10.1002/bies.201000015. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Sturchler-Pierrat C, Strazielle C. Spatial learning, exploration, anxiety, and motor coordination in female APP23 transgenic mice with the Swedish mutation. Brain Res. 2002;956:36–44. doi: 10.1016/s0006-8993(02)03476-5. [DOI] [PubMed] [Google Scholar]
- Larsen RS, Corlew RJ, Henson MA, Roberts AC, Mishina M, Watanabe M, Lipton SA, Nakanishi N, Perez-Otano I, Weinberg RJ, Philpot BD. NR3A-containing NMDARs promote neurotransmitter release and spike timing-dependent plasticity. Nat Neurosci. 2011;14:338–344. doi: 10.1038/nn.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Yang C, Chen Y, Shukla P, Tang L, Wang LX, Wang ZJ. Reversal of chronic inflammatory pain by acute inhibition of Ca2+/calmodulin-dependent protein kinase II. J Pharmacol Exp Ther. 2008;325:267–275. doi: 10.1124/jpet.107.132167. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Behrens MM, Canzoniero LM, Wang XQ, Heidinger V, Ichinose T, Yu SP, Choi DW. Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci U S A. 2001;98:11055–11061. doi: 10.1073/pnas.191353598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, Jenner P. The pathophysiology of extrapyramidal side-effects of neuroleptic drugs. Psychol Med. 1980;10:55–72. doi: 10.1017/s003329170003960x. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Kamiya Y, Matsuda S, Yuzaki M. Cloning and characterization of a novel NMDA receptor subunit NR3B: a dominant subunit that reduces calcium permeability. Brain Res Mol Brain Res. 2002;100:43–52. doi: 10.1016/s0169-328x(02)00173-0. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Yamasaki N, Ohira K, Takao K, Toyama K, Eguchi M, Yamaguchi S, Miyakawa T. Neural activity changes underlying the working memory deficit in alpha-CaMKII heterozygous knockout mice. Front Behav Neurosci. 2009;3:20. doi: 10.3389/neuro.08.020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller HT, Meador-Woodruff JH. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr Res. 2004;71:361–370. doi: 10.1016/j.schres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Mueller HT, Meador-Woodruff JH. Distribution of the NMDA receptor NR3A subunit in the adult pig-tail macaque brain. J Chem Neuroanat. 2005;29:157–172. doi: 10.1016/j.jchemneu.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Nakanishi N, Tu S, Shin Y, Cui J, Kurokawa T, Zhang D, Chen HS, Tong G, Lipton SA. Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci. 2009;29:5260–5265. doi: 10.1523/JNEUROSCI.1067-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak G, Seeman P. Hyperactive mice show elevated D2(High) receptors, a model for schizophrenia: calcium/calmodulin-dependent kinase II alpha knockouts. Synapse. 2010;64:794–800. doi: 10.1002/syn.20786. [DOI] [PubMed] [Google Scholar]
- Paulin MG. The role of the cerebellum in motor control and perception. Brain Behav Evol. 1993;41:39–50. doi: 10.1159/000113822. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Schulteis CT, Contractor A, Lipton SA, Trimmer JS, Sucher NJ, Heinemann SF. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci. 2001;21:1228–1237. doi: 10.1523/JNEUROSCI.21-04-01228.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang YX, Partridge JG, Zhao HM, Wenthold RJ, Malinow R. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat Neurosci. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci. 2000;20:7922–7931. doi: 10.1523/JNEUROSCI.20-21-07922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Auerbach A. Identification of a high affinity divalent cation binding site near the entrance of the NMDA receptor channel. Neuron. 1996;16:869–880. doi: 10.1016/s0896-6273(00)80107-5. [DOI] [PubMed] [Google Scholar]
- Roberts AC, Diez-Garcia J, Rodriguiz RM, Lopez IP, Lujan R, Martinez-Turrillas R, Pico E, Henson MA, Bernardo DR, Jarrett TM, Clendeninn DJ, Lopez-Mascaraque L, Feng G, Lo DC, Wesseling JF, Wetsel WC, Philpot BD, Perez-Otano I. Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron. 2009;63:342–356. doi: 10.1016/j.neuron.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussy G, Dansereau MA, Baudisson S, Ezzoubaa F, Belleville K, Beaudet N, Martinez J, Richelson E, Sarret P. Evidence for a role of NTS2 receptors in the modulation of tonic pain sensitivity. Mol Pain. 2009;5:38. doi: 10.1186/1744-8069-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japon M, Chan EC, Allen RG, Low MJ. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci U S A. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YF, Rothe T, Premkumar LS, Das S, Cui J, Talantova MV, Wong HK, Gong X, Chan SF, Zhang D, Nakanishi N, Sucher NJ, Lipton SA. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol. 2002;87:2052–2063. doi: 10.1152/jn.00531.2001. [DOI] [PubMed] [Google Scholar]
- Shen YC, Liao DL, Chen JY, Wang YC, Lai IC, Liou YJ, Chen YJ, Luu SU, Chen CH. Exomic sequencing of the ionotropic glutamate receptor N-methyl-D-aspartate 3A gene (GRIN3A) reveals no association with schizophrenia. Schizophr Res. 2009;114:25–32. doi: 10.1016/j.schres.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Gage AT. Distinct synaptic loci of Ca2+/calmodulin-dependent protein kinase II necessary for long-term potentiation and depression. J Neurophysiol. 1996;76:2097–2101. doi: 10.1152/jn.1996.76.3.2097. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Akbarian S, Chi CL, Leclerc CL, Awobuluyi M, Deitcher DL, Wu MK, Yuan JP, Jones EG, Lipton SA. Developmental and regional expression pattern of a novel NMDA receptor-like subunit (NMDAR-L) in the rodent brain. J Neurosci. 1995;15:6509–6520. doi: 10.1523/JNEUROSCI.15-10-06509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi LK, Kalin NH, Vanden Burgt JA, Sherman JE. Corticotropin-releasing factor modulates defensive-withdrawal and exploratory behavior in rats. Behav Neurosci. 1989;103:648–654. doi: 10.1037//0735-7044.103.3.648. [DOI] [PubMed] [Google Scholar]
- Tan SE, Liang KC. Spatial learning alters hippocampal calcium/calmodulin-dependent protein kinase II activity in rats. Brain Res. 1996;711:234–240. doi: 10.1016/0006-8993(95)01411-x. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tham SM, Angus JA, Tudor EM, Wright CE. Synergistic and additive interactions of the cannabinoid agonist CP55,940 with mu opioid receptor and alpha2-adrenoceptor agonists in acute pain models in mice. Br J Pharmacol. 2005;144:875–884. doi: 10.1038/sj.bjp.0706045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Wang GD, Zhang C, Shokat KM, Wang H, Tsien JZ, Liauw J, Zhuo M. Forebrain overexpression of CaMKII abolishes cingulate long term depression and reduces mechanical allodynia and thermal hyperalgesia. Mol Pain. 2006;2:21. doi: 10.1186/1744-8069-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- Wong HK, Liu XB, Matos MF, Chan SF, Perez-Otano I, Boysen M, Cui J, Nakanishi N, Trimmer JS, Jones EG, Lipton SA, Sucher NJ. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J Comp Neurol. 2002;450:303–317. doi: 10.1002/cne.10314. [DOI] [PubMed] [Google Scholar]