

Background: The mechanism of DSB end resection in yeast nuclease deficient mre11 mutants and Mre11 nuclease-independent ATM activation in mammalian cells remains unclear.
Results: Mre11 binds to DSB ends and also promotes end bridging. Rad50, Xrs2, and Sae2 potentiate Mre11-catalyzed DNA unwinding activity.
Conclusion: Mre11 nuclease activity is dispensable for DNA binding and unwinding activity.
Significance: These studies reveal a novel mechanism of processing of DSBs by MRX-Sae2 complex.
Keywords: DNA Damage Response, DNA Repair, DNA-Protein Interaction, Molecular Cell Biology, Yeast Genetics
Abstract
Saccharomyces cerevisiae RAD50, MRE11, and XRS2 genes are essential for telomere length maintenance, cell cycle checkpoint signaling, meiotic recombination, and DNA double-stranded break (DSB) repair via nonhomologous end joining and homologous recombination. The DSB repair pathways that draw upon Mre11-Rad50-Xrs2 subunits are complex, so their mechanistic features remain poorly understood. Moreover, the molecular basis of DSB end resection in yeast mre11-nuclease deficient mutants and Mre11 nuclease-independent activation of ATM in mammals remains unknown and adds a new dimension to many unanswered questions about the mechanism of DSB repair. Here, we demonstrate that S. cerevisiae Mre11 (ScMre11) exhibits higher binding affinity for single- over double-stranded DNA and intermediates of recombination and repair and catalyzes robust unwinding of substrates possessing a 3′ single-stranded DNA overhang but not of 5′ overhangs or blunt-ended DNA fragments. Additional evidence disclosed that ScMre11 nuclease activity is dispensable for its DNA binding and unwinding activity, thus uncovering the molecular basis underlying DSB end processing in mre11 nuclease deficient mutants. Significantly, Rad50, Xrs2, and Sae2 potentiate the DNA unwinding activity of Mre11, thus underscoring functional interaction among the components of DSB end repair machinery. Our results also show that ScMre11 by itself binds to DSB ends, then promotes end bridging of duplex DNA, and directly interacts with Sae2. We discuss the implications of these results in the context of an alternative mechanism for DSB end processing and the generation of single-stranded DNA for DNA repair and homologous recombination.
Introduction
Among the various types of DNA damage, DNA double-stranded breaks (DSBs)2 are the most lethal forms, which, if left unrepaired, will lead to genome instability and/or cell death (1–4). To maintain the genome integrity, organisms have evolved several mechanistically different pathways for DSB repair: nonhomologous end joining (NHEJ) and homologous recombination (HR) being the two major pathways (1–4). Both the pathways are essential for genome maintenance; however, stages of the cell cycle regulate the choice between the two DSB end repair pathways. NHEJ functions all through the cell cycle, whereas HR is active only postreplicative stages of the cell cycle, i.e., in the S and G2 phases, during which the sister chromatids are more easily available to serve as repair templates (5–9). A common step in recombinational DNA repair pathway is the resection of 5′ ends on each side of the DSB, leading to the formation of 3′ single-stranded tails, which are also required for activation of the ATM/ATR (ataxia telangiectasia and Rad3-related protein)-mediated checkpoint response (1–4).
In Saccharomyces cerevisiae, RAD50, MRE11, and XRS2 genes are essential for homologous recombination and DSB repair (via NHEJ and HR), telomere length maintenance, cell senescence, and cell cycle checkpoint signaling (10–21). Several lines of evidence indicate that Mre11-Rad50-Xrs2 subunits (henceforth called MRX complex) are the essential early components of DSB end repair (1–4). S. cerevisiae mutants harboring deletion of any one of the MRX complex genes are as sensitive as a triple gene deletion mutants to DNA-damaging agents (10–16, 18–22). Moreover, a number of studies have shown that the MRX(N) complex is a part of a larger surveillance machine that contains several repair and DNA damage checkpoint proteins (1–4). For example, MRX complex requires Sae2 for the removal of covalently bound Spo11 to the DSBs, as well as for efficient resection of the DSB to generate extended 3′-ended ssDNA tails (23–31). Although Sae2 has intrinsic nuclease activity (30), its relevance and role in DSB end processing remains unclear. In S. cerevisiae, other studies have shown the existence of two functionally redundant pathways of DSB end resection downstream of MRX and Sae2 (32–34). The first involves Exo1, whereas the second is a protein complex comprising of Sgs1-Rmi1-Top3 and Dna2 nuclease (1–4, 35).
The formation of DSBs trigger the recruitment of MRX(N)-Sae2/CtIP complex to the DSB ends, which then exert both structural and catalytic functions (1–4, 35). Genetic studies in S. cerevisiae have shown that both MRX complex and Sae2 are required for the repair of DSBs associated with repetitive DNA and the processing of DNA secondary structures (36–39). Several studies have demonstrated that Mre11 exhibits Mn2+-dependent 3′ to 5′ exonuclease activity on DNA substrates, including nicked substrates, possessing blunt-ended or 5′ protruding ends and endonuclease activity on single- and noncanonical DNA substrates (18, 21, 22, 40–46), whereas Sae2 exhibits ssDNA endonuclease activity (30). Also, Rad50 negatively regulates Mre11 nucleolytic activity, and repression was alleviated upon ATP hydrolysis by Rad50, indicating that ATP and ADP function as allosteric effectors (46, 47). In agreement with these findings, structural studies show that Rad50 negatively regulates Mre11 nuclease activity by blocking the active site of Mre11, and hydrolysis of ATP disengages Rad50 subunit, thus unmasking the active site of Mre11 (48). Furthermore, the human Mre11-Rad50-Nbs1 complex has been shown to possess weak ATP-stimulated DNA unwinding and does not need ends for DNA binding activity (49, 50), and hMre11 by itself can bind both ssDNA and dsDNA and catalyze the annealing of complementary ssDNA molecules (51). Intriguingly, Mre11 nuclease activity is dispensable for the resection of DSBs in vegetatively growing yeast cells, although it is essential for the processing of meiotic DSBs, which are covalently bound on the 5′ strand to the Spo11 protein (21, 22, 28, 34, 36, 41). Further complicating the picture, Mre11 nuclease activities are not required for the activation of ATM after DNA damage in mammals (52).
The crucial step that determines the choice of DNA repair pathways and the efficiency of HR is the resection of DSB ends (1–4). Although studies have provided important insights into our understanding of the biochemical and structural aspects of the MRX(N) complex, the relevant mechanistic roles of individual subunits remain unclear. Recent genetic data define a two-step mechanism for DSB end resection. First, the nuclease activity of MRX-Sae2 complex generates 50–100 nucleotide 3′ overhangs (1, 4, 22, 35). The second step involves two pathways and two sets of components that promote bulk resection (1, 4, 22, 35). Despite advances in our understanding of how organisms and cells respond to DSBs, questions remain. For example, the molecular basis underlying DSB end resection in S. cerevisiae mre11 nuclease deficient mutants and Mre11 nuclease-independent activation of ATM in mammalian cells remains unknown (21, 27, 37, 40, 41, 52). In this study, we have performed a comprehensive analysis of the role of S. cerevisiae Mre11 in the processing of DSB ends using a variety of DNA substrates. Our results show that ScMre11, by itself, binds to double-stranded DNA ends and catalyzes significant unwinding of DNA structures possessing only 3′ ssDNA overhangs, and its cognates Sae2, Rad50, and Xrs2 stimulate the DNA unwinding activity of ScMre11. Interestingly, ScMre11 nuclease activity is dispensable for its intrinsic DNA unwinding activity. Altogether, these results reveal important insights into the mechanism of DSB end processing and support a model in which Sae2, Rad50, and Xrs2 positively regulate the ScMre11-mediated DNA unwinding activity via direct interactions or through allosteric effects on the DNA or cofactors.
EXPERIMENTAL PROCEDURES
Expression and Purification of MRX and Sae2 Proteins
S. cerevisiae Mre11 was overexpressed in and purified from Escherichia coli BL21 (DE3) pLysS as described previously (46). mre11 alleles (D16A and D56N) were generated by one-step PCR-mediated strategy using primers 1 and 2 for D16A allele and primers 3 and 4 for D56N allele: primer 1, D16A (5′-ATAAGGATTTTAATTACTACAGCGAATCATGTGGGTTAC-3′); primer 2, D16A (5′-GTAACCCACATGATTCGCTGTAGTAATTAAAATCCTTAT-3′); primer 3, D56N (5′-GACATGGTTGTACAGTCCGGTAATCTTTTTCACGTGATAAG-3′); and primer 4, D56N (5′-CTTATTCACGTGAAAAAGATTACCGGACTGTACAACCATGTC-3′). The mutant proteins were overexpressed in and purified from E. coli BL21 (DE3) pLysS to homogeneity using the same purification protocol as was used for wild-type Mre11 protein (46). S. cerevisiae Rad50 was overexpressed in strain BJ5464 and purified to homogeneity as described previously (43). S. cerevisiae Xrs2 was overexpressed in E. coli BL21 (DE3) pLysS and purified to homogeneity as described previously (45). S. cerevisiae SAE2 construct was kindly provided by Dr. Tanya Paull (University of Texas, Austin, TX). S. cerevisiae SAE2 gene was subcloned into pET21a, with forward primer (5′-TACTTCCAGCATATGCTGACTGGTGAA-3′) harboring a NdeI site and reverse primer (5′-GTACAAGAAAGCGGCCGCACATCTAGCATA-3′) with a NotI site. Further, pET21a-SAE2 construct was transformed into the expression host E. coli C43 (DE3) strain (Novagen). The cells were grown in LB broth until A600 = 0.4 at 37 °C, and then isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1 mm to induce the expression of Sae2 protein. The cells were further grown at 13 °C for 18 h, harvested by centrifugation at 5000 × g for 10 min. The pellet was resuspended in buffer A (10 mm Tris-HCl, pH 8, 150 mm NaCl, 5 mm 2-mercaptoethanol, and 10% (v/v) glycerol). The cells were lysed by an Ultrasonic processor set to 60% amplitude for 5 min and centrifuged at 72,000 × g for 90 min. The supernatant was applied onto a pre-equilibrated cobalt affinity column (5-ml bed volume) by gravity. The column was washed with buffer A containing 10 mm imidazole. The bound protein was eluted with a linear gradient of 20 to 800 mm imidazole in buffer A. Fractions from the column were analyzed by SDS-PAGE (10% (w/v) polyacrylamide gel), and peak fractions containing Sae2 were pooled and dialyzed against buffer B (10 mm Tris-HCl, pH 8, 10% glycerol, 1 m NaCl, and 5 mm 2-mercaptoethanol). The dialysate was applied onto a pre-equilibrated Superdex 75 gel filtration column (120-ml bed volume), and peak fractions containing Sae2 were pooled. The resulting protein was 99% pure as ascertained by SDS-PAGE and Western blotting with custom made anti-ScSae2 antibody (Imgenex). The fractions containing Sae2 were combined and dialyzed against storage buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 10% glycerol), and aliquots were stored at −80 °C.
Construction of DNA Substrates
Synthetic ODNs used in this study are depicted in supplemental Table S1. DNA substrates used in this study are shown schematically in supplemental Table S2. The ODNs were labeled at the 5′ end by using [γ-32P]ATP as described (53). DNA substrates were generated by annealing the appropriate combinations of ODNs, as indicated in Table 2. For each substrate, stoichiometric amounts of purified ODNs were added to 100 μl of 0.3 m sodium citrate buffer (pH 7) containing 3 m NaCl. The ODN mixtures annealed by incubation at 95 °C for 5 min and then by slow cooling to 4 °C over a period of 2 h. Annealed substrates were gel-purified by electrophoresis on a 6% (w/v) polyacrylamide gel in 44.5 mm Tris borate buffer (pH 8.3) containing 0.5 mm EDTA. We excised the bands corresponding to the DNA substrates from the gel, and DNA was eluted into TE buffer (10 mm Tris-HCl, pH 7.5, 1 mm EDTA) precipitated with 0.3 m sodium acetate (pH 5.2) and 95% (v/v) ethanol. The pellet was washed with 70% ethanol, dried, and resuspended in 20 μl of TE buffer.
TABLE 2.
Dissociation rate constants for binding of ScMre11 to recombinational DNA repair intermediates

Electrophoretic Mobility Shift Assays
DNA binding activity of ScMre11 was assayed in a 20-μl reaction mixture containing 20 mm Tris-HCl (pH 7.5), 0.1 mm DTT, and 100 μg/ml BSA with a fixed amount (3 nm) of 5′ 32P-labeled DNA substrate and indicated concentrations of ScMre11. After incubation at 37 °C for 60 min, the reaction was stopped by the addition of 2 μl of loading dye (0.1% (w/v) of bromphenol blue and 0.1% (w/v) xylene cyanol in 20% glycerol). Reaction mixtures were separated on a 6% native PAGE in 89 mm Tris borate buffer (pH 8.3) at 80 V for 3 h at 4 °C. The gels were dried and exposed to the phosphorimaging screen, and images were acquired using Fuji FLA-5000 phosphorimaging device and subsequently by autoradiography. The bands were quantified in UVI-Tech gel documentation station using UVI-Band Map software (version 97.04), and the data were plotted using GraphPad Prism (version 5.0). The results were analyzed by nonlinear regression equation. The slope of the curves yielded the indicated Kd values.
DNA Unwinding Assay
The indicated amounts of ScMre11 or other proteins (where specified) were incubated with 3 nm 5′ 32P-labeled DNA in 20 μl of assay buffer containing 20 mm Tris-HCl (pH 7.5), 0.1 mm DTT, and 100 μg/ml BSA. After incubation at 37 °C for 30 min, the reaction was stopped by the addition of EDTA, SDS, and proteinase K to final concentrations of 10 mm, 1%, and 10 μg/ml, respectively, and 5 nm ssDNA complementary to the unlabeled strand. However, we note that addition of unlabeled strand is not essential for Mre11-catalyzed unwinding reaction. The reaction mixtures were further incubated for an additional 15 min at 37 °C. Reaction mixtures were separated on a 12% nondenaturing polyacrylamide gel in 89 mm Tris borate buffer (pH 8.3) at 80 V for 14 h at 4 °C. The gels were dried and exposed to a phosphor screen, and images were acquired using phosphorimaging with a Fuji FLA-5000 device or autoradiography. The bands were quantified in UVI-Tech gel documentation station using UVI-Band Map software (version 97.04), and the data were plotted using GraphPad Prism (version 5.0).
Surface Plasmon Resonance Measurements
All of the measurements were performed on BIAcore 2000 optical biosensor (GE Healthcare) at 30 °C. Immobilization of proteins, binding experiments, and data analysis were performed according to the manufacturer's protocol (GE Healthcare). In a typical experiment, 1200 and 1300 response units/flow cell of ScMre11 and ScSae2, respectively, were immobilized on a CM5 chip using the amine coupling method. Flow cell 1 was used as a control. Proteins were diluted in running buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, and 0.005% NP20) at concentrations ranging from 2 to 400 nm and injected at a flow rate of 30 μl/min. Regeneration was carried out by 4 m MgCl2. The kinetic fitting was carried out with Biacore evaluation software using 1:1 Langmuir binding model to obtain the equilibrium and kinetic constants.
Far Western Analysis
Increasing amounts of BSA or ScMre11 in buffer consisting of 20 mm Tris-HCl (pH 7.5) and 0.1 mm DTT was applied to a nitrocellulose membrane. The membrane was blocked with buffer A (10 mm HEPES, pH 7.4, 100 mm NaCl, and 1 mm EDTA) containing 5% (w/v) nonfat milk. After incubation at 24 °C for 2 h, the membranes were incubated with Mre11, Xrs2, or Sae2 in buffer A for 12 h at 4 °C and washed six times at 10-min intervals with buffer A containing 0.001% Nonidet P-40. The membranes were then incubated with anti-Mre11, -Xrs2, or -Sae2 antibodies, rinsed, and incubated with horseradish peroxidase-conjugated anti-rabbit antibody. The signals were visualized using chemiluminescence detection. Anti-Mre11 antibody was obtained from BD Biosciences. Anti-Xrs2 and anti-Sae2 antibodies (custom made by Imgenex India Pvt. Ltd., Bhubaneswar, India) in rabbits using the synthetic epitope CSKQSRHSRSATSRSRGS (corresponding to amino acid residues 811–828 of ScXrs2) and epitope CSDTVIHEKDNDKENKTR (corresponding to ScSae2 amino acid residues 148–165 of Sae2) and characterized as described (53).
DNA Bridging Assay
Reaction mixtures (20 μl) containing 75 ng (4.2 kb) of linear DNA (generated by digestion of pET21a circular plasmid DNA with HincII), 40 mm Tris-HCl (pH 7.5), 10 mm DTT, and 10 mm MgCl2 where specified were incubated in the absence or presence of ScMre11. After incubation for 30 min at 37 °C, we added 10 mm MgCl2, 0.5 mm ATP, and 10 units of T4 DNA ligase (Fermentas), and incubation was extended for 10 min at 37 °C. In experiments with ExoIII, samples were further incubated at 37 °C for 60 min in the presence of 5 units of ExoIII. Except in case of Fig. 3, lane 3, the reactions were terminated by the addition of 1% SDS and 0.5 μg/μl proteinase K and incubated for 30 min at 37 °C. The samples were analyzed by electrophoresis through 1.2% agarose in 45 mm Tris borate buffer (pH 8.3) containing 1 mm EDTA at 60 V for 5 h and visualized by staining with ethidium bromide.
FIGURE 3.

ScMre11 promotes bridging between linear DNA molecules to form concatemers. The assay was performed as described under “Experimental Procedures. ” Lane 1, 1-kb DNA ladder; lanes 2 and 7, linear double-stranded DNA alone; lane 3, ScMre11 incubated with linear dsDNA in the absence of MgCl2 and ATP; lane 4, linear dsDNA in the presence of DNA ligase; lanes 5, 6, 8, and 9, incubated with 400 and 800 nm ScMre11, respectively, and then with T4 DNA ligase. Lanes 8 and 9, reaction mixtures were digested by ExoIII. The arrows on the right-hand side indicate products formed by bridging between linear DNA molecules. The asterisk denotes a product generated by ScMre11 nuclease activity in the presence of MgCl2.
AFM Imaging
The samples were imaged in air using a silicon tip on nitride lever AFM probe (Agilent Technologies; force constant, 21–98 N/m) and Agilent AFM controller operated in the tapping mode. Binding reactions were carried out at 37 °C for 20 in a buffer (10 μl) containing 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 10 μg/ml linear pVZ77 plasmid DNA (5.5 kb) and with indicated concentrations of ScMre11, before the sample was deposited on a mica surface. A 5-μl aliquot of the reaction mixture was deposited on freshly cleaved mica for 90 s, rinsed with sterile Milli-Q water, and air-dried. Imaging was done at a resolution of 512 × 512 pixels. Raw data were selected with the Picoimage software, and the same was used to “flatten” AFM images with second order polynomial fitting. The scan frequency was typically 1.5 Hz per line, and the modulation amplitude was a few nanometers.
RESULTS
Purification of S. cerevisiae Wild-type Sae2 and Mre11 and Its Mutant Proteins
S. cerevisiae wild-type Sae2 and Mre11 were expressed in and purified from E. coli as described under “Experimental Procedures.” To investigate the role of conserved amino acid residues in the phosphoesterase motif I and II in DNA binding (Fig. 1, a and b), we constructed two mre11 alleles (D16A and D56N) by site-directed mutagenesis. Mre11 mutant proteins were expressed in and purified from E. coli. SDS-PAGE analysis of wild-type ScSae2 and ScMre11 and its variants indicates that they are homogenous (Fig. 1, c and d). Further confirmation of the identity of the purified recombinant proteins was ascertained by determining the sequences of the N-terminal 10 amino acid residues and by Western blotting analysis with antibodies (BD Biosciences) specific for ScMre11 and ScSae2 (data not shown). In agreement with previous studies (36, 54), ScMre11 mutant proteins were devoid of nuclease activity (data not shown).
FIGURE 1.

Proteins used in this study. a and b, schematic linear representation of Mre11 domain organization. Alignment of S. cerevisiae Mre11 phosphoesterase motifs I (a) and II (b) with E. coli SbcD and human Mre11. An arrow indicates the site of point mutations mre11D16A (a) and mre11D56N (b) within the conserved phosphoesterase motifs. Purified protein preparations were analyzed by electrophoresis through 7.5% polyacrylamide gel and Coomassie Blue staining. c, wild-type ScMre11 and mre11D16A and mre11D56N mutant proteins. d, ScSae2. Lanes indicated by Marker contain molecular size markers. BPB, bromophenol blue.
ScMre11 Binds More Tightly to 70-mer ssDNA and Beyond than to Shorter ssDNA
The idea that MRX/N is recruited to the DSB ends to generate duplex DNA with a single-strand overhang is supported by numerous observations in many model organisms and mammalian cell lines (1–4). Consequently, a comprehensive understanding of how Mre11 binds to multiple divergent target DNA substrates is crucial for reconstructing the fidelity and efficiency of DSB end resection networks in vivo. Using the assay conditions defined previously (45, 46), we investigated the of ability of ScMre11 to bind to varying lengths of 32P-labeled ssDNA, ranging from 20- to 100-mer length of random sequence. Their ability to fold into DNA secondary structural motifs was minimized as described previously (55). The results show that ScMre11 formed a distinct complex with 20-mer ssDNA, which increased in a dose-dependent manner (supplemental Fig. S1a). However, even at the highest concentration tested (250 nm), all of the DNA molecules were not bound to the protein. Similarly, binding to 30–50-mer ssDNA revealed that the abundance of Mre11-DNA complexes increased concurrent with increase in the length of ssDNA (supplemental Fig. S1, b–i). Again, all the DNA molecules were not bound to the protein. Beyond a length of 70-mer ssDNA, ScMre11 exhibited very rapid and robust binding and reached saturation at relatively low protein concentrations compared with shorter ssDNA substrates.
Quantification of these results revealed that the formation of ScMre11-ssDNA complexes was higher with substrates in the range of 70–100-mer ssDNA, whereas the extent of complex formation with other ssDNA substrates was relatively weak (supplemental Fig. S1j). We determined the binding affinity of ScMre11 to varying lengths of ssDNA (Kd). We found that ScMre11p showed the strongest binding affinity for 100-mer ssDNA (Kd ≈ 11 nm), compared with ssDNA substrates of shorter length (Table 1). We conclude that ScMre11, like hMre11 (51), exhibits high affinity for ssDNA, and its binding is length-dependent, and a minimum length of 90–100-mer ssDNA is sufficient to form a high affinity complex.
TABLE 1.
Dissociation rate constants for binding of ScMre11 to varying lengths of single- and double-stranded DNA
| Kd | |
|---|---|
| nm | |
| Single-stranded DNA | |
| 20-mer | 176 |
| 30-mer | 147 |
| 40-mer | 97 |
| 50-mer | 75 |
| 60-mer | 57 |
| 70-mer | 42 |
| 80-mer | 32 |
| 90-mer | 22 |
| 100-mer | 11 |
| Duplex DNA | |
| 20 bp | 641 |
| 30 bp | 577 |
| 40 bp | 537 |
| 50 bp | 396 |
| 60 bp | 222 |
| 70 bp | 176 |
| 80 bp | 105 |
| 90 bp | 85 |
| 100 bp | 36 |
Length-dependent Binding of ScMre11 to Double-stranded DNA
To further characterize the DNA length dependence, we investigated the binding affinity of ScMre11 for dsDNA containing random sequence. We reasoned that the binding behavior of ScMre11 to dsDNA may be similar; therefore, we examined its binding to 20–100-bp fragments using gel mobility shift assays. The reactions were performed as described above, except that the indicated double-stranded DNA substrate was replaced by ssDNA of similar length. Interestingly, ScMre11 exhibited similar dsDNA length-dependent binding affinity (supplemental Fig. S2, a–i). Quantification of these results indicated that the extent of binding was 5–15-fold higher to substrates beyond a length of 80 bp (supplemental Fig. S2j). The Kd values for binding of ScMre11 to dsDNA substrates were determined as described above (Table 1). These results, in agreement with hMre11 (51), indicate that ScMre11 exhibited higher binding affinity for ssDNA over dsDNA and that the affinity was strongly influenced by the length of DNA. We note however that ScMre11 binding to shorter lengths of DNA substrates was not saturable. We speculate that it may be due to a lack of stable binding of ScMre11 to DNA, which is in close agreement with relatively high Kdvalues (see below). To validate the relative affinities of binding to different lengths of single- and double-stranded DNA, we carried out competition assays to determine substrate preference. Consistent with the above results, competition experiments showed that ScMre11 had much higher binding affinity for the longer than for the shorter lengths of either single-stranded or duplex DNA substrates (data not shown).
ScMre11 Binds to Intermediates of DNA Recombination and Repair
Repetitive DNA motifs in the genome, which can fold into putative physiological DNA substrates such as cruciforms, triplexes, slipped structures, G-quadruplexes, and Z-DNA, are thought to function as regulatory elements and mutation hot spots and also serve as potential sources of genome instability (56, 57). Furthermore, genetic studies in S. cerevisiae have implicated a role for Mre11 nuclease activity in the resolution of recombinational DNA repair intermediates (36, 37). However, the molecular basis of their resolution has remained elusive. To this end, synthetic ODNs were used to construct a variety of intermediates in recombination and repair (58, 59). ScMre11 exhibited higher binding affinity for the replication fork, cruciform DNA over that of its structural analog, the Holliday junction, among the substrates tested (supplemental Fig. S3, compare b with a and f). Similarly, ScMre11 showed relatively high affinity for the flayed duplex over the 5′ flap and partial duplex with a 5′ overhang (supplemental Fig. S3, compare c with d and g). Under these conditions, we obtained sigmoidal dose-response curves, indicative of apparent positive cooperativity in the binding ScMre11 to these substrates (supplemental Fig. S3h).
The quantitative data from these studies indicate that ScMre11 exhibits high affinity to replication fork, cruciform DNA, and flayed duplex (Table 2). Although the sizes of flap duplex substrates are identical, ScMre11 exhibits ∼2-fold higher affinity for the 3′ flap structure compared with that of the 5′ flap structure, thereby excluding the possibility that the differences in affinities are not due to differences in the size of the substrates. ScMre11 showed significantly lower affinity (8–10-fold) for the Holliday junction with an apparent Kd value of 275 nm, compared with those previously reported for a number of Holliday junction binding and/or resolvases (60).
ScMre11 Catalyzes Unwinding of Duplex DNA with 3′ Overhangs, but Not of 5′ Overhangs or Blunt-ended DNA Fragments
DNA end resection is a multistep process involving multiple components that can be divided into distinct steps: an initial resection step during which a short oligonucleotide tract is removed from the 5′ strand, followed by a second step of long range end resection to generate extensive tracts of ssDNA. The MRX-Sae2 complex catalyzes the initial resection step, whereas the long range end resection comprises of two distinct pathways: the first step is promoted by ExoI, and the second employs Sgs1-Top3-Rmi1 complex together with Dna2 (1–4). Additional evidences suggest that Dna2 nuclease is redundant with Mre11 nuclease; however, Dna2 does not replace the complete absence of Mre11 (34, 61). Mre11 exhibits 3′ to 5′ exonuclease activity on double-stranded DNA (31, 42–46). In addition, hMre11 stimulated by Nbs1 catalyzes a modest amount (∼6%) of unwinding of blunt-ended duplex DNA or duplex DNA containing a 3′ ssDNA overhang in the absence of ATP in a divalent cation-dependent manner (49). Because the unwinding activity has not been studied further, we sought to characterize the unwinding activity of MRX complex, in the absence as well as the presence of Sae2, using short duplexes and a wide variety of replication/recombination intermediates.
In the first set of experiments, which were motivated by observations by Paull and Gellert (49), we investigated the requirement for 3′ or 5′ ssDNA overhangs for the unwinding activity of ScMre11. To this end, we used three different substrates: blunt-ended 20-bp duplex and 20-bp duplexes with a 20-mer ssDNA overhang either at the 3′ or 5′ end, and monitored DNA unwinding as described under “Experimental Procedures.” We observed that ScMre11 was unable to unwind either blunt-ended duplex or the same substrate containing a 5′ overhang (Fig. 2, panels (i) and (ii)). On the other hand, ScMre11 catalyzed efficient unwinding of duplex DNA containing a 3′ overhang in a dose-dependent manner (Fig. 2, panel (iii)). These results also indicated that the unwinding reaction was essentially complete within 30 min at 37 °C. ScMre11 binds both the splayed substrates to a similar extent (supplemental Fig. S3c), thereby negating the possibility of thermally induced DNA strand separation of duplex DNA containing a 3′ ssDNA overhang. Although the basis for this difference is unclear, it is likely that ScMre11 requires a 3′ end for loading onto and translocation in a 3′ → 5′ polarity while it unwinds duplex DNA.
FIGURE 2.
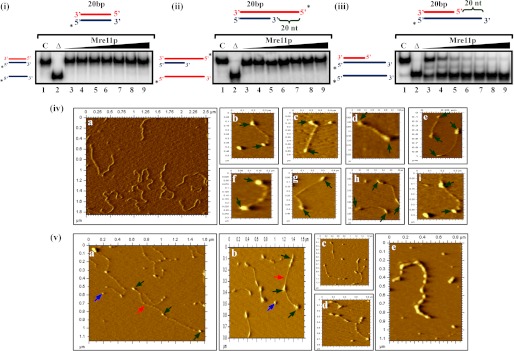
ScMre11 binds to DSB ends, promotes end bridging, and catalyzes unwinding of duplex DNA. Radiolabeled DNA substrate (3 nm) (depicted at the top of each gel image) was incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm DTT, 100 μg/ml BSA,0.1 mm EDTA, and the reaction products were separated and visualized as described under “Experimental Procedures.” Lane 1 (marked C), DNA substrate alone; lane 2 (marked Δ), heat-denatured DNA substrate; lanes 3–9, reaction mixtures contained 100, 200, 300, 400, 500, 600, and 700 nm ScMre11, respectively. The filled triangle on top of the gel image denotes the increasing concentration of ScMre11. Panel (i), 20-bp blunt duplex. Panel (ii), 20-bp duplex with 20-mer 5′ ssDNA overhang. Panel (iii), 20-bp duplex with 20-mer 3′ ssDNA overhang. Panel (iv), frame a, a typical AFM image of linear plasmid DNA (5.5 kb) in the absence of ScMre11. Frames b–i, linear plasmid DNA (10 μg/ml) was incubated with 2 nm ScMre11 as described under “Experimental Procedures.” Representative AFM images showing ScMre11 binding to both ends of linear dsDNA. Panel (v), frames a–d, same conditions as in panel (iv). Shown are randomly selected AFM images showing the coexistence of ScMre11 promoted end bridging of linear dsDNA molecules and ScMre11 binding to both the ends. Frame e, an AFM image of a linear dsDNA molecule apparently completely bound by ScMre11. Here, we incubated DNA under conditions as in panel (iv), but in the presence of 14 nm ScMre11. Green arrows indicate ScMre11 binding, and naked DNA is indicated by red arrows. DNA fragment lengths were measured as described previously (62). Blue arrows indicate unbound ScMre11.
ScMre11 Binds to DSB Ends and Promotes End Bridging between Linear Duplex DNA Molecules
Previous AFM studies have reported that S. cerevisiae Rad50-Mre11 complex, or its human counterpart, promotes bridging of linear DNA molecules (62–64). The fact that ScMre11 alone was able to bind and catalyze robust unwinding of DSB ends (Fig. 2, panel (iii)) prompted us to test whether it can directly bind to the DSB ends. We formed ScMre11-DNA complexes under conditions as described above, except that Mn2+ was omitted in the reaction buffer to inhibit its nuclease activity. Aliquots of the reaction mixtures were deposited onto the mica surface and visualized in the AFM. In agreement with DNA end binding activity shown for Rad50-Mre11 or MRX complexes (62–64), we observed that ScMre11 by itself binds to both ends of dsDNA (Fig. 2, panel (iv)) and, more importantly, promotes end bridging of unit length molecules to form concatemers (Fig. 2, panel (v)). At higher concentrations where all of the DNA molecules were bound by ScMre11 as ascertained in EMSA, we observed binding of ScMre11 to internal sites of the DNA fragment (Fig. 2, panel (v), frame e). Nevertheless, both the visualization of Mre11 binding to the DSB ends and its ability to promote intermolecular joining of linear DNA molecules are consistent with its role in the NHEJ pathway.
To ascertain DNA bridging activity of ScMre11, we performed in vitro ligation assay with linear duplex DNA. In the absence of ScMre11, incubation with DNA ligase alone resulted in the formation of concatemers (Fig. 3, lane 3). Upon the addition of ScMre11, concatemer formation was significantly enhanced (Fig. 3, lanes 5 and 6), although the extent may be underestimated because the presence of both unwinding and end bridging activities within the same protein would tend to work against one another. In control reactions where DNA ligase was omitted or in the presence of ExoIII, we did not observe concatemer formation (Fig. 3, lanes 2, 7, 8, and 9).
ScMre11-catalyzed DNA Unwinding as a Function of Length
To correlate the duplex DNA length to ScMre11 catalyzed unwinding activity, we constructed a series of molecules with different lengths of duplex region (ranging from 20 to 80 bp), all of them having a 20-mer 3′ ssDNA overhang. In particular, we wanted to examine the ability of ScMre11 to unwind duplex DNA substrates to generate extended ssDNA regions. Using increasing concentrations of ScMre11 with a fixed amount each of the substrate, we monitored the efficiency of the unwinding reaction. As shown in Fig. 4, ScMre11 catalyzed unwinding was robust on 20- and 30-bp duplex DNA substrates and then progressively decreased as the length of the duplex region was increased to 70 bp (Fig. 4, compare f with a and b). The unwinding activity was essentially undetectable with 80-bp duplex DNA under identical conditions (Fig. 4, compare g with h). We conclude first that ScMre11 catalyzes efficient unwinding of duplex DNA containing a 3′ ssDNA overhang but not of blunt-ended or duplex DNA containing a 5′ ssDNA overhang and second that ScMre11 binds to duplex DNA, unwinds and then terminates its unwinding activity after 80 bp, unless an end is reached before the limit.
FIGURE 4.

ScMre11 catalyzes DNA unwinding and separation of complementary single strands. The indicated 32P-labeled DNA substrate (3 nm) containing duplex regions of different lengths and ends possessing 20-mer 3′ overhangs (depicted at the top of each gel image) were incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm DTT, 100 μg/ml BSA, 0.1 mm EDTA, and the reaction products were separated and visualized as described under “Experimental Procedures. ” Lane 1 (marked C), DNA substrate alone; lane 2 (marked Δ), heat-denatured DNA substrate; lanes 3–9, reaction mixtures contained 100, 200, 300, 400, 500, 600, and 700 nm ScMre11, respectively. The filled triangle on top of the gel image denotes the increasing concentration of ScMre11. a, 20-bp duplex DNA with 20-mer 3′ ssDNA overhang; b, 30-bp duplex DNA with 20-mer 3′ ssDNA overhang; c, 40 bp duplex DNA with 20-mer 3′ ssDNA overhang; d, 50-bp duplex DNA with 20-mer 3′ ssDNA overhang; e, 60-bp duplex DNA with 20-mer 3′ ssDNA overhang; f, 70-bp duplex DNA with 20-mer 3′ ssDNA overhang; g, 80-bp duplex DNA with 20-mer 3′ ssDNA overhang; h, graphical representation of percentage of displaced single strand with increasing concentration ScMre11. Each data point in the graph is the average of three independent experiments. The error bars indicate S.E. The data were subjected to nonlinear regression analysis in GraphPad PRISM (version 5.00), using the equation for one site-specific binding with Hill slope.
ScMre11 Unwinds Intermediates of Recombination/Repair
The Mre11 complex is thought to play a central role in both homology-directed repair and NHEJ (1–4, 35). Moreover, recent studies have shown that Mre11 complex, specifically its nuclease activity, plays an essential role in processing of the stalled replication forks, thereby helping to maintain genome integrity during replication stress (65–70). To understand the mechanism of processing of stalled replication forks and other intermediates by MRX complex better, we considered whether ScMre11 could unwind replication and recombination intermediates, such as the Holliday junction and flayed duplex substrates. To this end, we performed unwinding experiments as described above, except that the ssDNA-containing substrates were replaced by Holiday junction or flayed duplex substrates, containing 20-mer ssDNA overhangs either at the 3′ or 5′ ends. We note that tailed Holliday junction intermediates exist in the cell during damage to replication forks (64). We found that ScMre11 was able catalyze the unwinding of both the Holliday junction and flayed duplex substrates containing 3′ ssDNA overhangs in a dose-dependent manner (Fig. 5, b and d). However, Mre11 was unable to catalyze the unwinding of the Holliday junction to its constituent ssDNA under these conditions. By contrast, ScMre11 failed to catalyze the unwinding of DNA substrates that contained 5′ ssDNA overhangs (Fig. 5, a and c).
FIGURE 5.

ScMre11 catalyzes unwinding of flayed duplex DNA and Holliday junction containing 3′ ssDNA overhangs. The indicated 32P-labeled DNA substrate possessing 20-mer ssDNA overhangs (3 nm) either at the 3′ or 5′ ends (depicted in the gel image at the top) was incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm DTT, 100 μg/ml BSA, 0.1 mm EDTA, and the reaction products were separated and visualized as described under “Experimental Procedures. ” Lane 1 (marked C), DNA substrate alone; lane 2 (marked Δ), heat-denatured DNA substrate; lanes 3–9, reaction mixtures contained 100, 200, 300, 400, 500, 600, and 700 nm ScMre11, respectively. The filled triangle on top of the gel image denotes the increasing concentration of ScMre11. Reaction products were separated and visualized as described under “Experimental Procedures.” a, flayed duplex DNA with 20-mer 5′ ssDNA overhang; b, flayed duplex with 20-mer 3′ ssDNA overhang; c, Holliday junction with 20-mer 5′ ssDNA overhang; d, Holliday junction with 20-mer 3′ ssDNA overhang.
Mre11 Catalyzed DNA Unwinding Is Independent of Its Nuclease Activity
To investigate whether ScMre11 nuclease activity is important for its DNA unwinding activity, we generated two mre11 alleles with substitutions of conserved residues in phosphoesterase motifs I (D16A) and II (D56N) of its nuclease domain (Fig. 1, a and b). These point mutations in ScMre11 have been shown individually to abolish its endo- and exonuclease activity (18, 20, 36). Purified S. cerevisiae mre11D16A and mre11D56N proteins were then examined for their ability to bind ssDNA and exhibit ssDNA nuclease activity. As shown in Fig. 6 (a and b), both the mutant proteins retained the ssDNA binding activity and behaved similar to the wild-type protein. We determined the DNA unwinding activity on a 20-bp partial duplex tethered to 20-mer 3′ ssDNA overhang. As shown in Fig. 6 (c and d), both mre11D16A and mre11D56N mutant proteins exhibited rapid and efficient DNA unwinding activity. Indeed, the extent of DNA unwinding catalyzed by both mre11D16A and mre11D56N mutant proteins was similar to that of the wild-type ScMre11 (Figs. 2, panel (iii), and 4a).
FIGURE 6.

ScMre11 nuclease deficient mutant proteins bind ssDNA and promote unwinding of duplex DNA. The indicated 32P-labeled 30-mer ssDNA fragment or partial DNA duplex possessing 20-mer 3′ ssDNA overhang (3 nm) of the indicated length (depicted at the top of gel images) were incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm DTT, 100 μg/ml BSA, and 0.1 mm EDTA, and the reaction products were separated and visualized as described under “Experimental Procedures.” a and b, ssDNA binding activity of mutant Mre11 proteins. Lane 1 (marked C), DNA substrate alone; lanes 2–9, reaction mixtures contained 25, 50, 100, 125, 150,175, 200, and 250 nm of mre11D16A (a) or mre11D56N (b), respectively. c and d, DNA unwinding activity of mutant ScMre11 proteins. Lane 1 (marked C), DNA substrate alone; lane 2 (marked Δ), heat-denatured DNA; lanes 3–9, reaction mixtures contained 100, 200, 300, 400, 500, 600, and 700 nm of mre11D16A (c) or mre11D56N (d), respectively. The filled triangle on top of the gel image denotes the increasing concentration of ScMre11 nuclease deficient mutant protein.
Stimulation of ScMre11 Catalyzed DNA Unwinding by Rad50, Xrs2, and Sae2 Proteins
We showed previously that the ScMre11 nuclease activity is allosterically regulated by Rad50 and ATP (46), which has been corroborated by x-ray crystallographic studies (48). These observations prompted us to investigate whether the DNA unwinding activity of ScMre11 is also subjected to regulation by proteins that are known to interact with Mre11 during DSB end repair. For our purpose, we used an 80-bp DNA duplex having a 20-mer 3′ ssDNA overhang because ScMre11 alone was essentially inactive on this substrate (Fig. 4g). Fig. 7 shows the results of a typical experiment for each of these proteins. In one set of experiments, we examined the ability of DSB end repair proteins such as Rad50, Xrs2, and Sae2 individually on Mre11 to catalyze unwinding of 32P-labeled substrate as described above. We observed that none of these proteins by themselves, or ScRad50-Mre11, were able to catalyze DNA unwinding (Fig. 7, lanes 3–5, 7, and 10). We then tested the ability of Xrs2 and Sae2 to stimulate ScMre11 catalyzed DNA unwinding in a dose-dependent manner. As shown in Fig. 7 (lanes 8, 9, 11, and 12), the addition of either Xrs2 or Sae2 potentiated the extent of DNA unwinding activity. On the other hand, the ability of Rad50 to stimulate DNA unwinding activity of ScMre11 was dependent on ATP, implying that ATP serves as a key allosteric effector in the unwinding reaction (Fig. 7, compare lanes 5 and 6). Most important, Rad50 and Xrs2, which are devoid of nuclease activity, were able to stimulate the unwinding activity of Mre11. Because the exonuclease activity of Mre11 is of the opposite polarity to that expected for resection of DSBs, our results suggest that the DNA unwinding activity Mre11, in conjunction Rad50, Xrs2, and Sae2, might provide an alternate mechanism for the generation ssDNA intermediates for DSB end repair and HR.
FIGURE 7.

Rad50, Xrs2, and Sae2 proteins stimulate DNA unwinding activity of ScMre11. Radiolabeled 80-bp duplex DNA possessing 20-mer ssDNA overhang at the 3′ end (3 nm) (depicted in the gel image on the top) was incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm DTT, 100 μg/ml BSA, and 0.1 mm EDTA, and the reaction products were separated and visualized as described under “Experimental Procedures.” Lane 1, DNA substrate alone; lane 2, heat-denatured DNA substrate. The remaining lanes contained 300 nm ScMre11 alone (lane 3), 300 nm ScRad50 alone (lane 4), 300 nm ScRad50 + 300 nm ScMre11 (lane 5), 300 nm ScMre11 + 300 nm ScRad50 + 5 mm ATP (lane 6), 700 nm ScXrs2 alone (lane 7), 300 nm ScMre11 + 300 nm ScXrs2 (lane 8), 300 nm ScMre11 + 700 nm ScXrs2 (lane 9), 700 nm ScSae2 (lane 10), 300 nm ScMre11 + 300 nm ScSae2 (lane 11), and 300 nm ScMre11 and 700 nm ScSae2 (lane 12), respectively.
Direct Interaction between S. cerevisiae Mre11 and Sae2
The aforesaid findings posit that stimulation of ScMre11 unwinding activity by Rad50, Xrs2, and Sae2 might involve their direct interaction. Multiple studies have demonstrated strong interaction among the three MRX subunits (1–4); however, Sae2 has not yet been shown to bind directly with either MRX complex or with any of its subunits. Rather, Sae2 formed complexes with MRX in the context of DNA (71). Direct interaction between Mre11 and Sae2 may not have been detected for a variety of technical reasons including experimental conditions or insufficient sensitivity of the assay. We investigated the possibility of direct interaction between ScMre11 and ScSae2 using two fairly sensitive techniques. First, we used Far Western blotting, a technique that has been widely used to study protein-protein interactions under nondenaturing conditions (72). Purified ScMre11 or BSA was immobilized onto the nitrocellulose membranes. The membranes were treated with blocking buffer, washed, and then incubated with ScMre11, ScXrs2, or ScSae2. Following incubation, the membranes were washed and probed with anti-ScMre11, -ScXrs2, or -ScSae2 antibodies. The antigen-antibody complexes were detected by chemiluminescence reaction (72). We observed specific binding between Xrs2 and Mre11 (used as positive control) (Fig. 8a, middle panel). The same pattern was also seen for ScSae2, indicating direct physical interaction between Sae2 and Mre11 (Fig. 8a, bottom panel). Under similar conditions, we observed no measurable binding of Mre11 to BSA (Fig. 8a, top panel). The results support the notion of the existence of physical interaction between ScMre11 and its cognate Sae2. To corroborate the far Western blot results, Western blot analysis was performed on cell-free lysates to ascertain the specificity of complex formation. As shown in supplemental Fig. S4, we observed specific association between ScMre11 and ScXrs2 and between ScMre11 and ScSae2.
FIGURE 8.

S. cerevisiae Sae2 binds directly to its cognate Mre11. a, far Western analysis of interactions between ScMre11 and ScSae2. Increasing concentrations of homogeneous preparation of Mre11 or BSA (25–200 nm) were spotted (from left to right) on nitrocellulose membranes. After blocking with nonfat milk, the membranes were incubated with ScMre11 or ScSae2; probed with anti-Mre11, anti-Xrs2, or anti-Sae2 antibodies; and developed as described under “Experimental Procedures.” b–d, sensorgrams showing homotypic and heterotypic interactions between Mre11, Xrs2, and Sae2 proteins. b, sensorgrams showing homotypic association of ScMre11; c, sensorgrams showing heterotypic interactions between ScMre11 and Sae2; d, sensorgrams showing heterotypic interactions between ScMre11 and Xrs2. The arrows (from left to right) indicate the time points used for injection of protein and buffer, respectively.
Second, to determine whether ScMre11 and ScSae2 interact directly in real time, we used plasmon resonance spectroscopy, a sensitive technique for quantitative affinity measurements involving biomolecules (73). We immobilized ScMre11 and ScSae2 on CM5 sensor chips to a surface density of 1200RU and 1300RU, respectively, and then injected various concentrations of the indicated analyte into the fluid chamber. In control experiments, we observed no measurable binding of ScMre11, ScSae2, or ScXrs2 either to CM5 sensor chips or chips tethered with BSA (data not shown). On the other hand, we found that immobilized ScMre11 was able to interact with ScMre11 or ScXrs2 (Fig. 8, b and d), and the analyzed data, according to a simple 1:1 Langmuir equation, yielded KD values of 2.3 × 10−8 m and 0.91 × 10−8 m, respectively (Table 3). We have used the same approach to measure the interaction between ScMre11 and Sae2. The results indicated that ScMre11 binds directly to Sae2 (Fig. 8c) and appears to be specific, and the analyzed data, according to a simple 1:1 Langmuir equation, yielded a KD value of 1.92 × 10−8 m (Table 3), similar to that of ScMre11 binding to ScXrs2. We conclude that ScMre11 immobilized on CM5 sensor chip provides appropriate specificity and conformation for interaction with ScMre11 and ScXrs2 and, more importantly, reveals direct interaction between ScMre11 and ScSae2.
TABLE 3.
Kinetic rate constants for homotypic and heterotypic interaction between ScMre11, Xrs2, and Sae2
| Interaction | Ka | Kd | KA | KD |
|---|---|---|---|---|
| m−1 s−1 | s−1 | m−1 | m | |
| Mre11-Xrs2 | 4.94 × 105 | 3.3 × 10−3 | 29.9 × 107 | 0.91 × 10−8 |
| Mre11-Sae2 | 2.11 × 105 | 3.96 × 10−3 | 8.62 × 107 | 1.92 × 10−8 |
| Mre11-Mre11 | 2.76 × 105 | 2.64 × 10−3 | 22.7 × 107 | 2.37 × 10−8 |
DISCUSSION
In this study, we show that Mre11, which was thought to be simple in its DNA binding preferences, can bind multiple DNA structures that are physiologically relevant to DNA repair and HR. ScMre11 exhibits higher binding affinity for single- over double-stranded DNA and intermediates of recombination and repair and catalyzes robust unwinding of the substrates that have 3′ ssDNA overhangs but not of 5′ ssDNA overhangs or blunt-ended DNA. Additional evidence suggests that ScMre11 nuclease activity is dispensable for the DNA binding and unwinding activity. Importantly, Rad50, Xrs2, and Sae2 potentiated the unwinding activity of Mre11. Consistent with its pivotal role in DSB end processing, we show direct interaction between Sae2 and Mre11. Our results have broad implications for understanding the mechanism of DSB end processing and generation of ssDNA during DNA repair and HR, as well as activation of the ATM/ATR cell cycle checkpoint signaling pathway.
The results presented here, together with the literature data (22, 42, 45, 49, 51), point out the fact that ScMre11 possesses certain functional characteristics of ssDNA-binding protein. ScMre11has a 5-fold higher affinity for single- and double-stranded DNA as the length was increased from 40- to 70-mer ssDNA or 40- to 90-bp duplex DNA. These results indicate that a minimum length of 60-mer ssDNA or 90-bp duplex DNA is needed for the formation of a high affinity complex between ScMre11-DNA. Interestingly, ScMre11 showed the ability to interact with putative physiological DNA substrates that are known to arise during replication of palindromic sequences and DNA repair/recombination. In contrast, the unwinding activity catalyzed by hMre11 or hMRN complex was much weaker (49). This discrepancy may be attributed to the source of proteins and/or differences in the assay conditions.
Genetic studies in S. cerevisiae have shown that Mre11 nuclease activity is required for processing of meiotic DSBs and DNA hairpins but is not essential for resection of DSB ends generated by the HO endonuclease (35). Notably, mre11 nuclease defective mutants show only mild sensitivity to DNA-damaging agents and weak resection defects compared with the mre11Δ and other mre11 mutant cells (1–4). Our findings suggest that Mre11 indeed has a role in DSB end processing but is independent of its nuclease activity. What is the biological significance underlying the specific binding of ScMre11 to intermediates of recombination and repair? A large body of evidence supports the view that the Mre11 complex plays a key role in the process of DNA replication (65–70); however, the mechanism is less well characterized. The observation that Mre11 exhibits striking selectivity toward replication fork and flayed duplex is consistent with its demonstrated role during “normal” replication or under conditions of replicative stress (70). Similarly, genetic studies have shown that the E. coli SbcCD complex, the Rad50-Mre11 complex functional homolog, is essential for the processing of DNA secondary structures that arise during replication/recombination (37, 74, 75).
Biochemical and structural studies suggest that Rad50 and Mre11 subunits are allosterically coupled to each other, and Rad50 negatively regulates the nuclease activity of Mre11, which is alleviated by ATP (46, 47, 48, 76–80). Furthermore, the crystal structure data of archaeal Mre11 suggest that DNA molecules act as allosteric ligands, inducing conformational changes in the MRN complex (80), which may aid in the recruitment of other DSB end repair factors. Intriguingly, although ssDNA is a prerequisite for DSB end repair and HR, as well as ATM activation, Mre11 nuclease activity is not (52). Costanzo and co-workers (81) subjected this idea to a series of elegant experiments and found that the MRN complex was required for the generation of ssDNA and activation of checkpoint pathways.
Genetic studies in S. cerevisiae suggest that MRX complex in concert with Sae2 catalyze the removal of ∼50–100 nucleotide residues from the 5′ end of the DSBs (1–4). These intermediates are subjected to extensive resection by the concerted action either by Sgs1/Top3/RmiI, Dna2, and RPA or ExoI and RPA to generate long 3′ ssDNA (which ranges from ∼500 nucleotides to a few kilobases in size) (22, 33, 34, 61, 82–89). Recent studies have revealed that Sgs1, Dna2, and RPA constitute a minimal set of proteins needed for resection of DSBs in an ATP-dependent manner, and the MRX complex stimulates the end resection activity (61). The addition of MRX-Sae2 to such reactions elicited strong stimulation of ExoI activity (60–300-fold), and more importantly, MRX or Sae2 alone could stimulate degradation of the 5′ strand catalyzed by ExoI as well as synergistic stimulation when added simultaneously (71). These studies also discerned that resection was more efficient when the substrate was preresected by MRX-Sae2, indicating that stimulation may involve cooperative binding in the context of DNA substrates by ExoI, MRX, and Sae2 (70). These results are consistent with the notion that MRX complex, or its individual subunits, in concert create a processed intermediate that is stimulatory to the ExoI and Sgs1-Dna2-mediated resection (35). However, further work is required to elucidate the finer points of this speculation by direct assays.
CONCLUSIONS
Altogether, our findings support the idea that ScMre11, in addition to its intrinsic nuclease activity, possesses significant DNA unwinding activity. Moreover, our results shed light on the conundrum underlying DSB end resection in mre11 nuclease deficient mutants. Fig. 9 shows, in accordance with the hypothesis proposed by Paull and co-workers (71), an integrated model for DSB end binding and unwinding by the components of MRX-Sae2 complex. After the removal of Spo11-oligonucleotide complex from the 5′ end by the combined action of MRX-Sae2 complex, Mre11 alone or MRX-Sae2 nuclease resects the 5′-ended strand or unwinds the duplex DNA to generate long 3′ ssDNA tracks; this reaction is independent of Mre11 nuclease activity. Furthermore, these findings may help to explain why mre11 nuclease deficient mutants exhibit mild sensitivity to DNA-damaging agents and weak resection defects, compared with mre11Δ cells (21, 22, 27, 37, 40, 41). Together, these results support and expand our understanding of the mechanism(s) underlying MRX-Sae2 promoted DSB repair. Given the complexity of the DSB repair pathway, understanding of how it is put together and how it contributes to the maintenance of genome integrity will likely lead to the discovery of “emergent” properties.
FIGURE 9.

Model for processing of DSB ends by MRX-Sae2 complex. Each horizontal line represents a DNA strand with polarity as indicated. Our findings support the idea that Mre11 or its nuclease deficient mutant protein (oval), by itself, binds (potentially in a cooperative manner) to the DSB ends and catalyzes unwinding of duplex DNA leading to the separation of complementary strands, a process that is stimulated by Rad50 (light gray triangle), Xrs2 (pear shape), and Sae2 (pac man shape). See text for details.
Acknowledgments
We gratefully acknowledge S. Usha and N. S. Srilatha for assistance with AFM and SPR measurements, respectively.
This work was supported by a grant from the Council of Scientific and Industrial Research, New Delhi, and by a J. C. Bose National Fellowship from the Department of Science and Technology, Government of India, New Delhi (to K. M.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S4.
- DSB
- double-stranded DNA break
- AFM
- atomic force microscope
- ATM
- ataxia telangiectasia mutate
- HR
- homologous recombination
- MRX
- Mre11-Rad50-Xrs2 proteins
- NHEJ
- nonhomologous end-joining
- ODN
- oligonucleotide
- ssDNA
- single-stranded DNA.
REFERENCES
- 1. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 2. Raynard S., Niu H., Sung P. (2008) DNA double-strand break processing. The beginning of the end. Genes Dev. 22, 2903–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stracker T. H., Petrini J. H. (2011) The MRE11 complex. Starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forment J. V., Kaidi A., Jackson S. P. (2012) Chromothripsis and cancer. Causes and consequences of chromosome shattering. Nat. Rev. Cancer 12, 663–670 [DOI] [PubMed] [Google Scholar]
- 5. Kadyk L. C., Hartwell L. H. (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shrivastav M., De Haro L. P., Nickoloff J. A. (2008) Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147 [DOI] [PubMed] [Google Scholar]
- 7. Barlow J. H., Lisby M., Rothstein R. (2008) Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell 30, 73–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aylon Y., Liefshitz B., Kupiec M. (2004) The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 23, 4868–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karanam K., Kafri R., Loewer A., Lahav G. (2012) Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 47, 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johzuka K., Ogawa H. (1995) Interaction of Mre11 and Rad50. Two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics 139, 1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivanov E. L., Sugawara N., White C. I., Fabre F., Haber J. E. (1994) Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol. Cell Biol. 14, 3414–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsubouchi H., Ogawa H. (1998) A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol. Cell Biol. 18, 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nairz K., Klein F. (1997) mre11S. A yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev. 11, 2272–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moore J. K., Haber J. E. (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell Biol. 16, 2164–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kironmai K. M., Muniyappa K. (1997) Alteration of telomeric sequences and senescence caused by mutations in RAD50 of Saccharomyces cerevisiae. Genes Cells 2, 443–455 [DOI] [PubMed] [Google Scholar]
- 16. Boulton S. J., Jackson S. P. (1998) Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 17, 1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nugent C. I., Bosco G., Ross L. O., Evans S. K., Salinger A. P., Moore J. K., Haber J. E., Lundblad V. (1998) Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr. Biol. 8, 657–660 [DOI] [PubMed] [Google Scholar]
- 18. Furuse M., Nagase Y., Tsubouchi H., Murakami-Murofushi K., Shibata T., Ohta K. (1998) Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 17, 6412–6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Usui T., Ohta T., Oshiumi H., Tomizawa J., Ogawa H., Ogawa T. (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell 95, 705–716 [DOI] [PubMed] [Google Scholar]
- 20. Bressan D. A., Olivares H. A., Nelms B. E., Petrini J. H. (1998) Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics 150, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreau S., Ferguson J. R., Symington L. S. (1999) The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell Biol. 19, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia V., Phelps S. E., Gray S., Neale M. J. (2011) Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keeney S., Kleckner N. (1995) Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc. Natl. Acad. Sci. U.S.A. 92, 11274–11278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bergerat A., de Massy B., Gadelle D., Varoutas P. C., Nicolas A., Forterre P. (1997) An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386, 414–417 [DOI] [PubMed] [Google Scholar]
- 25. Keeney S., Giroux C. N., Kleckner N. (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 [DOI] [PubMed] [Google Scholar]
- 26. McKee A. H., Kleckner N. (1997) A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics 146, 797–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rattray A. J., McGill C. B., Shafer B. K., Strathern J. N. (2001) Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae. A role for SAE2/COM1. Genetics 158, 109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neale M. J., Pan J., Keeney S. (2005) Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clerici M., Mantiero D., Lucchini G., Longhese M. P. (2005) The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 280, 38631–38638 [DOI] [PubMed] [Google Scholar]
- 30. Lengsfeld B. M., Rattray A. J., Bhaskara V., Ghirlando R., Paull T. T. (2007) Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 28, 638–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hopkins B. B., Paull T. T. (2008) The P. furiosus Mre11/Rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell 135, 250–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mimitou E. P., Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Budd M. E., Campbell J. L. (2009) Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS ONE 4, e4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mimitou E. P., Symington L. S. (2011) DNA end resection. Unravelling the tail. DNA Repair 10, 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lewis L. K., Storici F., Van Komen S., Calero S., Sung P., Resnick M. A. (2004) Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics 166, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lobachev K. S., Gordenin D. A., Resnick M. A. (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108, 183–193 [DOI] [PubMed] [Google Scholar]
- 38. Kim H. S., Vijayakumar S., Reger M., Harrison J. C., Haber J. E., Weil C., Petrini J. H. (2008) Functional interactions between Sae2 and the Mre11 complex. Genetics 178, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Argueso J. L., Westmoreland J., Mieczkowski P. A., Gawel M., Petes T. D., Resnick M. A. (2008) Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. U.S.A. 105, 11845–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Llorente B., Symington L. S. (2004) The Mre11 nuclease is not required for 59 to 39 resection at multiple HO-induced double-strand breaks. Mol. Cell Biol. 24, 9682–9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bonetti D., Clerici M., Manfrini N., Lucchini G., Longhese M. P. (2010) The MRX complex plays multiple functions in resection of Yku- and Rif2-protected DNA ends. PLoS One 5, e14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paull T. T., Gellert M. (1998) The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell 1, 969–979 [DOI] [PubMed] [Google Scholar]
- 43. Trujillo K. M., Sung P. (2001) DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50-Mre11 complex. J. Biol. Chem. 276, 35458–35464 [DOI] [PubMed] [Google Scholar]
- 44. Williams R. S., Moncalian G., Williams J. S., Yamada Y., Limbo O., Shin D. S., Groocock L. M., Cahill D., Hitomi C., Guenther G., Moiani D., Carney J. P., Russell P., Tainer J. A. (2008) Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosal G., Muniyappa K. (2005) Saccharomyces cerevisiae Mre11 is a high-affinity G4 DNA-binding protein and a G-rich DNA-specific endonuclease. Implications for replication of telomeric DNA. Nucleic Acids Res. 33, 4692–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghosal G., Muniyappa K. (2007) The characterization of Saccharomyces cerevisiae Mre11/Rad50/Xrs2 complex reveals that Rad50 negatively regulates Mre11 endonucleolytic but not the exonucleolytic activity. J. Mol. Biol. 372, 864–882 [DOI] [PubMed] [Google Scholar]
- 47. Majka J., Alford B., Ausio J., Finn R. M., McMurray C. T. (2012) ATP hydrolysis by RAD50 protein switches MRE11 enzyme from endonuclease to exonuclease. J. Biol. Chem. 287, 2328–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lim H. S., Kim J. S., Park Y. B., Gwon G. H., Cho Y. (2011) Crystal structure of the Mre11-Rad50-ATPγS complex. Understanding the interplay between Mre11 and Rad50. Genes Dev. 25, 1091–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Paull T. T., Gellert M. (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50complex. Genes Dev. 13, 1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trujillo K. M., Yuan S. S., Lee E. Y., Sung P. (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 273, 21447–21450 [DOI] [PubMed] [Google Scholar]
- 51. de Jager M., Dronkert M. L., Modesti M., Beerens C. E., Kanaar R., van Gent D. C. (2001) DNA-binding and strand-annealing activities of human Mre11. Implications for its roles in DNA double-strand break repair pathways. Nucleic Acids Res. 29, 1317–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buis J., Wu Y., Deng Y., Leddon J., Westfield G., Eckersdorff M., Sekiguchi J. M., Chang S., Ferguson D. O. (2008) Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 54. Krogh B. O., Llorente B., Lam A., Symington L. S. (2005) Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics 171, 1561–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dong F., Allawi H. T., Anderson T., Neri B. P., Lyamichev V. I. (2001) Secondary structure prediction and structure-specific sequence analysis of single-stranded DNA. Nucleic Acids Res. 29, 3248–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bacolla A., Wells R. D. (2004) Non-B DNA conformations, genomic rearrangements, and human disease. J. Biol. Chem. 279, 47411–47414 [DOI] [PubMed] [Google Scholar]
- 57. Leach D. R. (1994) Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. BioEssays 16, 893–900 [DOI] [PubMed] [Google Scholar]
- 58. Khanduja J. S., Muniyappa K. (2012) Functional analysis of DNA replication fork reversal catalyzed by Mycobacterium tuberculosis RuvAB proteins. J. Biol. Chem. 287, 1345–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tripathi P., Anuradha S., Ghosal G., Muniyappa K. (2006) Selective binding of meiosis-specific yeast Hop1 protein to the Holliday junctions distorts the DNA structure and its implications for junction migration and resolution. J. Mol. Biol. 364, 599–611 [DOI] [PubMed] [Google Scholar]
- 60. Déclais A.-C., Lilley D. M. (2008) New insight into the recognition of branched DNA structure by junction-resolving enzymes. Curr. Opin. Struct. Biol. 18, 86–95 [DOI] [PubMed] [Google Scholar]
- 61. Shim E. Y., Chung W. H., Nicolette M. L., Zhang Y., Davis M., Zhu Z., Paull T. T., Ira G., Lee S. E. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moreno-Herrero F., de Jager M., Dekker N. H., Kanaar R., Wyman C., Dekker C. (2005) Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 437, 440–443 [DOI] [PubMed] [Google Scholar]
- 63. Chen L., Trujillo K., Ramos W., Sung P., Tomkinson A. E. (2001) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell 8, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 64. Trujillo K. M., Roh D. H., Chen L., Van Komen S., Tomkinson A., Sung P. (2003) Yeast Xrs2 binds DNA and helps target Rad50 and Mre11 to DNA ends. J. Biol. Chem. 278, 48957–48964 [DOI] [PubMed] [Google Scholar]
- 65. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Robison J. G., Lu L., Dixon K., Bissler J. J. (2005) DNA lesion-specific co-localization of the Mre11/Rad50/Nbs1 (MRN) complex and replication protein A (RPA) to repair foci. J. Biol. Chem. 280, 12927–12934 [DOI] [PubMed] [Google Scholar]
- 67. Ying S., Hamdy F. C., Helleday T. (2012) Mre11-dependent degradation of stalled (DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 72, 2814–2821 [DOI] [PubMed] [Google Scholar]
- 68. Trenz K., Smith E., Smith S., Costanzo V. (2006) ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 25, 1764–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Foster S. S., Balestrini A., Petrini J. H. (2011) Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol. Cell Biol. 31, 4379–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Borde V., Cobb J. (2009) Double functions for the Mre11 complex during DNA double-strand break repair and replication. Int. J. Biochem. Cell Biol. 41, 1249–1253 [DOI] [PubMed] [Google Scholar]
- 71. Nicolette M. L., Lee K., Guo Z., Rani M., Chow J. M., Lee S. E., Paull T. T. (2010) Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 17, 1478–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wu Y., Li Q., Chen X.-Z. (2007) Detecting protein-protein interactions by far western blotting. Nat. Protoc. 2, 3278–3284 [DOI] [PubMed] [Google Scholar]
- 73. Malmqvist M., Karlsson R. (1997) Biomolecular interaction analysis. Affinity biosensor technologies for functional analysis of proteins. Curr. Opin. Chem. Biol. 1, 378–383 [DOI] [PubMed] [Google Scholar]
- 74. Connelly J. C., Kirkham L. A., Leach D. R. (1998) The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. U.S.A. 95, 7969–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Petrini J. H. (2000) The Mre11 complex and ATM. Collaborating to navigate S phase. Curr. Opin. Cell Biol. 12, 293–296 [DOI] [PubMed] [Google Scholar]
- 76. Wyman C., Lebbink J., Kanaar R. (2011) Mre11-Rad50 complex crystals suggest molecular calisthenics. DNA Repair 10, 1066–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Möckel C., Lammens K., Schele A., Hopfner K. P. (2012) ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 40, 914–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lammens K., Bemeleit D. J., Möckel C., Clausing E., Schele A., Hartung S., Schiller C. B., Lucas M., Angermüller C., Söding J., Strässer K., Hopfner K. P. (2011) The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 145, 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. He J., Shi L. Z., Truong L. N., Lu C. S., Razavian N., Li Y., Negrete A., Shiloach J., Berns M. W., Wu X. (2012) Rad50 zinc hook is important for the Mre11 complex to bind chromosomal DNA double-stranded breaks and initiate various DNA damage responses. J. Biol. Chem. 287, 31747–31756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hopfner K. P., Craig L., Moncalian G., Zinkel R. A., Usui T., Owen B. A., Karcher A., Henderson B., Bodmer J. L., McMurray C. T., Carney J. P., Petrini J. H., Tainer J. A. (2002) The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418, 562–566 [DOI] [PubMed] [Google Scholar]
- 81. Jazayeri A., Balestrini A., Garner E., Haber J. E., Costanzo V. (2008) Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 27, 1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fiorentini P., Huang K. N., Tishkoff D. X., Kolodner R. D., Symington L. S. (1997) Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and. in vitro. Mol. Cell Biol. 17, 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tsubouchi H., Ogawa H. (2000) Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell 11, 2221–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gravel S., Chapman J. R., Magill C., Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNAdouble-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lydeard J. R., Lipkin-Moore Z., Jain S., Eapen V. V., Haber J. E. (2010) Sgs1 and exo1 redundantly inhibit break-induced replication and de novo telomere addition at broken chromosome ends. PLoS Genet. 6, e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., Kowalczykowski S. C. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Niu H., Chung W. H., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L., Ira G., Sung P. (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mimitou E. P., Symington L. S. (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]