

Background: Ras mutation drives tumor initiation as well as invasion.
Results: The ZEB1 transcription factor is sequentially induced with mutation of Rb1 and Ras, and these inductions are required for Ras-mediated tumor initiation and then invasion.
Conclusion: ZEB1 plays a critical role in initiation and progression of Ras-mediated tumors.
Significance: Induction ZEB1 is important for tumor initiation and invasion in a model of Ras-initiated cancer.
Keywords: Cancer, Cancer biology, Cancer Tumor Promoter, Epithelial-to-Mesenchymal Transition, Ras
Abstract
Rb1 restricts cell cycle progression, and it imposes cell contact inhibition to suppress tumor outgrowth. It also triggers oncogene-induced senescence to block Ras mutation. Loss of the Rb1 pathway, which is a hallmark of cancer cells, then provides a permissive environment for Ras mutation, and Ras is sufficient for invasive tumor formation in Rb1 family mutant mouse embryo fibroblasts (MEFs). These results demonstrate that sequential mutation of the Rb1 and Ras pathways comprises a tumor initiation axis. Both Rb1 and Ras regulate expression of the transcription factor ZEB1, thereby linking tumor initiation to the subsequent invasion and metastasis, which is induced by ZEB1. ZEB1 acts in a negative feedback loop to block expression of miR-200, which is thought to facilitate tumor invasion and metastasis. However, ZEB1 also represses cyclin-dependent kinase (cdk) inhibitors to control the cell cycle; its mutation in MEFs leads to induction of these inhibitors and premature senescence. Here, we provide evidence for two sequential inductions of ZEB1 during Ras transformation of MEFs. Rb1 constitutively represses cdk inhibitors, and induction of ZEB1 when the Rb1 pathway is lost is required to maintain this repression, allowing for the classic immortalization and loss of cell contact inhibition seen when the Rb1 pathway is lost. In vivo, we show that this induction of ZEB1 is required for Ras-initiated tumor formation. ZEB1 is then further induced by Ras, beyond the level seen with Rb1 mutation, and this Ras superinduction is required to reach a threshold of ZEB1 sufficient for repression of miR-200 and tumor invasion.
Introduction
The Rb1 family comprises three members (Rb1, p107, and p130) with overlapping functions that classically block cell cycle progression (1, 2). Knockdown or mutation of Rb1 in wild-type mouse embryo fibroblasts (MEFs)3 can reverse senescence, demonstrating that Rb1 itself plays a key role in establishing the cell cycle block (3, 4). However, p107 becomes induced upon Rb1 mutation, and DNA damage results from Rb1 mutation (3, 4), and together this induction of p107 and the DNA damage goes on to trigger cell senescence despite the loss of Rb1. Mutation of all three Rb1 family members eliminates senescence (5, 6), but Rb1 itself is only frequently mutated in rare tumors such as retinoblastoma, and the family is not collectively mutated in tumors. Instead, in most tumors the Rb1 family is inactivated by hyperphosphorylation resulting from deregulated cyclin-dependent kinase (cdk) activity (1, 2), and such loss of the Rb1 pathway is a hallmark of cancer; it reverses cell contact inhibition to allow tumor outgrowth, and it leads to immortalization of MEFs (2–7). Induction of senescence by Rb1 blocks proliferation of cells harboring activating Ras mutations (oncogene-induced senescence); thus, inactivation of the Rb1 pathway removes a barrier to Ras mutation in vivo (5, 6, 8, 9). Ras mutation becomes sufficient for tumor initiation when the Rb1 pathway is mutated in MEFs (6), demonstrating that Ras and Rb1 comprise two opposing arms of a cancer initiation axis.
ZEB1 is an epithelial-to-mesenchymal transition transcription factor thought to facilitate carcinoma invasion and metastasis by causing a migratory mesenchymal phenotype in cancer cells (10–14). It is repressed by Rb1 and induced by Ras (15, 16), suggesting a mechanism for linking ZEB1 to this cancer initiation axis. Indeed, in Ras-initiated pancreatic adenocarcinoma, ZEB1 is induced, and invasive and metastatic cells are evident remarkably early, even before a frank tumor can be identified (17). Although expression of ZEB1 and invasion/metastasis are generally associated with later stage tumors, these results suggest that Ras may accelerate the appearance of ZEB1 and resulting invasion/metastasis in tumors where it is mutated. ZEB1 acts in a negative feedback loop to repress expression of the miR-200 family, which has a key role in tumor invasion and metastasis (14, 18, 19); however, ZEB1 is also required to maintain repression of cell cycle inhibitory cdk inhibitors, and its down-regulation in ZEB1+/− and ZEB1−/− MEFs leads to a ZEB1 concentration-dependent induction of these cdk inhibitors and premature senescence (11, 20). Ras also drives sarcoma formation, and ZEB1 expression is linked to invasion and metastasis in these tumors, where epithelial-to-mesenchymal transition would not appear to be a factor. Taken together, these findings raise the possibility that ZEB1 may have roles in tumor progression extending beyond the ZEB1/miR-200 loop thought to facilitate invasion and metastasis (21–25).
Here, we examined the role for ZEB1 in a tumor model where sequential mutations of the Rb1 family and Ras in primary cultures of MEFs lead to generation of invasive sarcoma in nude mice. We demonstrate two sequential inductions of ZEB1 in this model, resulting first from mutation of Rb1 and then from Ras mutation. The Rb1 pathway constitutively represses cdk inhibitors, and we show that induction of ZEB1 when this pathway is lost is required to maintain repression of these inhibitors, allowing for the classic immortalization and loss of cell contact inhibition seen with Rb1 pathway mutation in cultured MEFs. Importantly, we demonstrate that the block in senescence resulting from this induction of ZEB1 is required subsequently for Ras-initiated tumor formation in nude mice. Then, when Ras is mutated, ZEB1 is further induced, and this induction by Ras is required to reach a threshold of ZEB1 sufficient to cause repression of miR-200, which we link to Ras-mediated tumor invasion.
EXPERIMENTAL PROCEDURES
Cell Culture
Rb1 family mutant MEFs and control wild-type MEFs were from T. Jacks. Cells derived from four separate embryos were used with similar results. The cells were cultured in DMEM with 10% heat-inactivated fetal bovine serum (26). Ras-triple knock-out (TKO) MEFs were created by infection with a V12Ras retrovirus and characterized with regard to Ras expression and activation as described in detail previously (27).
Tumor Formation in Nude Mice
Cells were injected subcutaneously into the hind limb of BALB/c nude mice as described (26). Tumors were fixed in 10% buffered formalin, embedded in paraffin, and sectioned for H&E.
RNA Extraction and Real-time PCR
RNA was extracted using TRIzol, and cDNA was synthesized using the Invitrogen RT kit (Invitrogen). SYBR Green real-time PCR was performed using a Stratagene Mx3000P Real-time PCR system (26). PCR primers are shown in supplemental Table 1. Three independent samples, each in triplicate, were analyzed for each real-time PCR condition. The detection of miRNAs was as described (28). Briefly, polyadenylation of at least 5 μg of the total RNA was completed by a poly(A) polymerase kit (PAP; Ambion) in a 20-μl reaction volume according to the manufacturer's instructions. The polyadenylated RNA was thereafter utilized directly for cDNA preparation using a reverse transcription kit (M-MLV reverse transcriptase; Invitrogen) and an adaptor primer (5′-GCGAGCACAGAATTAATACGACTCACTATAGG(T)12VN*-3′) in a 40-μl reaction volume. Real-time quantitative PCR was performed using a universal primer (5′-GCGAGCACAGAATTAATACGAC-3′) and a miRNA-specific primer (supplemental Table 2). Chromatin immunoprecipitation assays were performed as described previously (11), and primers are shown in supplemental Table 3.
Knockdown of ZEB1
We have described lentiviral shRNA knockdown of ZEB1 protein and mRNA previously (26). An additional five lentivirus shRNA knockdown vectors obtained from Open Biosystems were also used for knockdown of ZEB1 with equivalent results. Lentivirus with scrambled shRNA sequences was used as a control in the experiments (26).
Immunostaining
Immunostaining for E-cadherin was performed as described previously (11).
RESULTS
Induction of ZEB1 Is Required for Cell Immortalization and Loss of Contact Inhibition When the Rb1 Pathway Is Mutated
Rb1-E2F binds and represses the ZEB1 promoter, thus ZEB1 is induced in Rb1−/− MEFs, and it is further induced when all three Rb1 family members are mutated to generate TKO MEFs (Fig. 1A) (5, 6, 16). ZEB1 expression remained induced as TKO MEFs were passaged in culture (supplemental Fig. 1). TKO MEFs are immortal in culture, and they display loss of cell contact inhibition and consequently continue to proliferate after they reach confluence (5, 6), leading to formation of foci that in turn progress to large mounds (Fig. 1, B–D).
FIGURE 1.

ZEB1 is induced with Rb1 family mutation in MEFs, and Rb1 family mutation leads to loss of cell contact inhibition. A, real-time PCR comparing ZEB1 mRNA expression in wild-type MEFs with MEFs with the indicated Rb1 family mutation. TKO indicates MEFs with all three Rb1 family members mutated (6). Actb is β-actin, included as a control. Error bars, S.D. B, subconfluent TKO MEFs. C, TKO MEFs at confluence. The arrow denotes initial foci formation resulting from loss of cell contact inhibition. D, TKO MEFs after 2 weeks at confluence. The arrow denotes initial foci that have progressed to form a large mound of cells.
To assess the functional significance of ZEB1 induction in TKO MEFs, we knocked its expression down using lentiviral shRNA. Five TKO ZEB1 shRNA clones expressing varying levels of ZEB1 mRNA were selected (Fig. 2A). The level of ZEB1 in wild-type and ZEB1+/− MEFs is shown for comparison. The most severe knockdown of ZEB1 mRNA (clone 2, Fig. 2A) had little effect initially on proliferation of the cells (Fig. 3A); however, with passage, proliferation diminished, and each of the clones became senescent in a ZEB1 mRNA level-dependent fashion (Fig. 3, B–D). It is of note that with TKO ZEB1 shRNA clone 1, where ZEB1 mRNA is knocked down by only ∼50%, the percentage of senescent cells at passage 6 is similar to that seen in wild-type MEFs at this same passage (Fig. 3B). We then concluded that induction of ZEB1 is required for immortalization of TKO MEFs.
FIGURE 2.
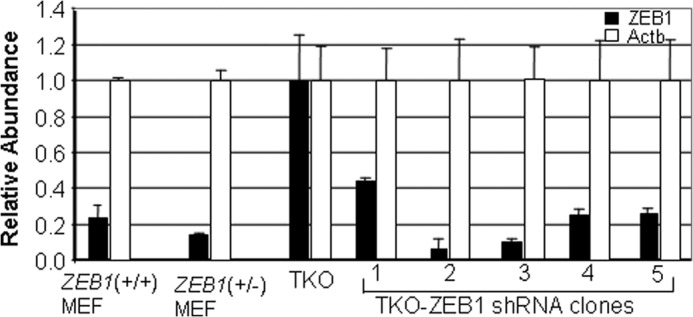
Real-time PCR comparing levels of ZEB1 mRNA in wild-type and ZEB1+/− MEFs with that in TKO MEFs and showing the effects of lentiviral ZEB1 shRNA knockdown in clones of TKO MEFs. TKO indicates TKO MEFs infected with a control lentiviral vector with a scrambled shRNA sequence (“Experimental Procedures”). Error bars, S.D.
FIGURE 3.

Knockdown of ZEB1 in TKO MEFs does not affect initial proliferation, but it leads to epithelial morphology and induction of E-cadherin, and it restores passage-dependent senescence and cell contact inhibition. A, initial proliferation of TKO ZEB1 shRNA clone 2 from Fig. 2 compared with uninfected TKO MEFs. B, passage of clones in Fig. 2 compared with parent TKO and wild-type MEFs. See B below for a description of Ras-TKO ZEB1 shRNA-2. C, example of senescent cells from B stained for senescent β-galactosidase (11). D, control proliferating TKO MEFs showing no β-galactosidase staining. E and F, TKO ZEB1 shRNA clone 5 from Fig. 2 shown after 3 weeks at confluence. G, immunostaining of cells from F for E-cadherin. Note transition to epithelial morphology and lack of foci formation. H and I, immunostaining of control TKO MEFs for E-cadherin. Error bars, S.D. Scale bars, 20 μm.
ZEB1 expression drives epithelial-to-mesenchymal transition, and we found previously that knockdown or mutation of ZEB1 led to an opposing mesenchymal-to-epithelial transition in wild-type MEFs, with induction of the epithelial specification factor E-cadherin (11). Moreover, E-cadherin became ectopically expression in ZEB1 mutant mice (11). As in wild-type MEFs, knockdown of ZEB1 in TKO MEFs led to epithelial morphology and induction of E-cadherin (Fig. 3, E–G). No E-cadherin was evident in the parent TKOs (Fig. 3, H and I).
Interestingly, knocking down ZEB1 in TKO MEFs to a level similar to that in wild-type MEFs (clone 5, Fig. 2) was sufficient to restore cell contact inhibition: TKO ZEB1 shRNA clone 5 at passage 1 was allowed to remain at confluence for 3 weeks, and no foci formation was evident (Fig. 3, E and F). We concluded that induction of ZEB1 in TKO MEFs is required for the loss of contact inhibition classically seen when the Rb1 pathway is mutated in MEFs.
Induction of ZEB1 Is Required to Maintain Repression of cdk Inhibitors after the Rb1 Pathway Is Mutated
Cell contact inhibition causes accumulation of cdk inhibitors such as p16 that block activity of cdk4/6, which hyperphosphorylates and inactivates Rb1. When cdk4/6 is inhibited, active hypophosphorylated Rb1 accumulates, leading to cell cycle arrest and eventually senescence (7, 8, 29–31). Senescence of wild-type MEFs with passage in culture is also the result of induction of cdk inhibitors. Chronic stimulation of the Ras pathway by serum growth factors in the culture media leads to activation of p53 and accumulation of cdk inhibitors that block activity cdk4/6, again leading to activation of Rb1 and senescence (32, 33). Beyond p16 and inhibition of cdk4/6, other cdk inhibitors including p21, p27, and p57 inhibit cdks important for Rb1-independent events in cell cycle progression. Interestingly, Rb1 is important in maintaining constitutive repression of these various cdk inhibitors: each of the inhibitors is modestly induced in TKO MEFs compared with wild-type cells (Fig. 4A) (5, 6), but the level of these inhibitors does not rise to a threshold sufficient to arrest the TKO MEFs. However, it is of note that proliferation of TKO MEFs is blocked if p21 and p27 become further elevated. When TKO MEFs are deprived of mitogens, they arrest in G2, and this arrest is linked to induction of p21 and p27 (34–36); mutation of p27 accelerates tumor initiation and progression in tissues where the Rb1 family is mutated, demonstrating that induction of p27 serves as a barrier in vivo that blocks progression of Rb1 family mutant tumors (37).
FIGURE 4.

Mutation of the Rb1 family leads to modest induction of mRNAs for cdk inhibitors in TKO MEFs, whereas knockdown of ZEB1 leads to rapid induction of E-cadherin mRNA (E-cad), and a more dramatic progressive induction of cdk inhibitors as cell are passaged. Senescence of the MEFs with ZEB1 knockdown does not require p53. A, real-time PCR comparing mRNA expression in wild-type and TKO MEFs at passage 4 (P4). B, real-time PCR showing that knockdown of ZEB1 in TKO ZEB1 shRNA clones from Fig. 2 leads to induction of mRNAs for E-Cad and cdk inhibitors. 5* indicates clone 5 cells at passage 1; other cells were passaged until they started to arrest (e.g. passage 2–3 for clones 2, 3, and 5; passage 6 for clone 1). C and D, infection of p53−/− MEFs with ZEB1 shRNA lentivirus leading to loss of proliferation and to large flat senescent-like cells, whereas a control virus with a scrambled shRNA sequence does not. GFP is expressed from the lentivirus, thus denoting infected cells. Note the proliferation of uninfected GFP− cells and lack of proliferation of infected GFP+ cells at day 9 in D. 60 ± 17 is the average percentage of infection at day 2. The boxed region at day 9 is shown at higher magnification on the left. Dotted lines outline the large flat GFP+ cell. Error bars, S.D. Scale bars, 20 μm.
ZEB1 binds the promoters of genes encoding p21, p27, and p57 and represses their transcription, and even heterozygous mutation of ZEB1 is sufficient for induction of these cdk inhibitors and premature senescence in MEFs (11, 20). Therefore, we wondered whether induction of ZEB1 in response to Rb1 family mutation might be important in preventing cdk inhibitors from rising to a threshold sufficient to cause cell cycle arrest. Indeed, when we knocked down ZEB1, these cdk inhibitors were dramatically induced in a ZEB1 concentration-dependent fashion as TKO MEFs were passaged (Fig. 4B). It is of note that knockdown of ZEB1 in TKO MEFs to a level similar to that in wild-type MEFs (e.g. clone 5, Fig. 1A) led to induction of the cdk inhibitors to levels well beyond those seen in wild-type MEFs, demonstrating that ZEB1 induction becomes required to maintain repression of these cdk inhibitors once the Rb1 pathway is lost (e.g. clone 5, Fig. 4B). By contrast to the progressive induction of cdk inhibitors with passage in ZEB1 knockdown TKO MEFs, E-cadherin mRNA induction occurred rapidly in TKO ZEB1 shRNA-5 MEFs and thus did not require multiple passages (Fig. 4B).
Because p21 is a target of p53 (33), we wondered whether p53 might be required for senescence of MEFs when ZEB1 is knocked down. We infected p53−/− MEFs with a ZEB1 shRNA lentivirus or a control virus with a scrambled shRNA sequence; as with wild-type or TKO MEFs the p53−/− MEFs arrested, and large flat senescent cells were evident (Fig. 4, C and D). These results demonstrate that senescence resulting from ZEB1 knockdown in MEFs does not require p53. It is of note that activation of p53 does not arrest TKO MEFs (5, 6). Taken together, our results suggest that induction of ZEB1 in response to Rb1 family mutation is required to maintain repression of cdk inhibitors, which in turn allows for immortalization and loss of cell contact inhibition when the Rb1 family is mutated in TKO MEFs.
Ras Causes Superinduction of ZEB1 in TKO MEFs
Introduction of V12Ras into TKO MEFs to create Ras-TKO MEFs has been described previously (6, 27). Loss of function of all Rb1 family members is required to prevent oncogene-induced senescence in response to Ras, and accordingly Ras-TKO MEFs are tumorigenic when injected into nude mice (6). We found that Ras superinduced ZEB1 in Ras-TKO MEFs to a level well beyond that seen in parent TKO MEFs (Fig. 5A). This induction of ZEB1 by Ras in primary culture MEFs is consistent with the previous finding that Ras/Erk2 induces expression of ZEB1 in an immortalized cell line (15). Because cdk inhibitor expression was already low in TKO MEFs, superinduction of ZEB1 in Ras-TKO MEFs had little noticeable addition effect on their expression (Fig. 5B).
FIGURE 5.

Mutant Ras causes superinduction of ZEB1 in Ras-TKO MEFs, but it represses ZEB1 in wild-type MEFs (Ras-MEF). Two levels of ZEB1 knockdown were used to assess the importance of ZEB1 induction in Ras-TKO MEFs. A, real-time PCR showing that ZEB1 is superinduced by Ras in Ras-TKO MEFs. Ras-TKO ZEB1 shRNA-1 and Ras-TKO ZEB1 shRNA-2 show two different levels of lentiviral ZEB1 shRNA knockdown of ZEB1 in Ras-TKO MEFs. B, real-time PCR showing that knockdown of ZEB1 in Ras-TKO MEFs to the level seen in WT MEFs (Ras-TKO ZEB1 shRNA-1) leads to rapid induction of E-cadherin (E-Cad) and cdk inhibitor mRNAs, whereas knockdown ZEB1 only to reverse Ras superinduction (Ras-TKO ZEB1 shRNA-2) does not. Ras-TKO ZEB1 shRNA-1 MEFs were prior to passage, and Ras-TKO ZEB1 shRNA-2 were at passage 2. C, real-time PCR showing Ras-mediated repression of ZEB1 and induction of p16 and p21 in wild-type (WT) MEFs. Error bars, S.D.
By contrast to TKO MEFs, introduction of mutant Ras into wild-type MEFs leads to oncogene-induced senescence, which as noted above requires accumulation of an active Rb1 family member. As opposed to TKO MEFs, Ras led to repression of ZEB1 in the wild-type MEFs along with induction of cdk inhibitors (Fig. 5C). We conclude that repression of ZEB1 by active Rb1 family members that accumulate when Ras is introduced into wild-type MEFs is dominant over its introduction by Ras. Thus, loss of the Rb1 pathway, which is a hallmark of cancer cells and is a prerequisite for Ras mutation, is required for Ras induction of ZEB1.
Ras-TKO MEFs Become Dependent upon Elevated ZEB1 to Block Senescence in Culture and for Tumor Formation in Nude Mice
Next, we used lentivirus shRNA to knock down ZEB1 in Ras-TKO MEFs. We chose cells with two different levels of ZEB1 knockdown: Ras-TKO ZEB1 shRNA-1 had ZEB1 knocked down to a level similar to that in wild-type MEFs, whereas in Ras-TKO ZEB1 shRNA-2 most of the superinduction of ZEB1 by Ras was reversed, but the level of ZEB1 was still above that in TKO MEFs (Fig. 5A). Not surprisingly, cdk inhibitors and E-cadherin were not significantly induced in Ras-TKO ZEB1 shRNA-2 compared with Ras-TKO or TKO MEFs because the level of ZEB1 was still beyond that seen in the parent TKO MEFs (Fig. 5B). Ras-TKO ZEB1 shRNA-2 MEFs were still immortal and showed a proliferation rate similar to Ras-TKO MEFs (Fig. 3B and 6A). However, knockdown of ZEB1 in Ras-TKO ZEB1 shRNA-1 to a level similar to that seen in wild-type MEFs led to rapid induction of cdk inhibitors and senescence of the cells before they could be passaged (Figs. 5B and 6, A–C). These results demonstrate that Ras causes TKO MEFs to become highly dependent upon or addicted to elevated ZEB1, which is highlighted by this rapid induction of cdk inhibitors and early senescence.
FIGURE 6.

Ras-TKO ZEB1 shRNA-2 MEFs show a rate of proliferation similar to parent TKO Ras MEFs, whereas Ras-TKO ZEB1 shRNA-1 MEFs undergo rapid senescence. A, cell proliferation assay. Note also in Fig. 3B that Ras-TKO ZEB1 shRNA-2 MEFs are immortal. B and C, Ras-TKO ZEB1 shRNA-1 MEFs at day 4 stained for senescent β-galactosidase. Error bars, S.D. Scale bars, 20 μm.
Ras Superinduction of ZEB1 Is Important for Repression of miR-200
Our results above suggested that superinduction of ZEB1 by Ras in TKO MEFs was not required for repression of cdk inhibitors and E-cadherin (they were already fully repressed in TKO MEFs); thus, we wondered what significance this superinduction might have. ZEB1 represses expression of miR-200 family members in the context of a negative feedback loop, and the collective induction of ZEB1 and loss of mir-200 within the context of this loop is thought to be critical for tumor invasion (14, 18, 19). Accordingly, miR-200 was induced in ZEB1−/− MEFs (supplemental Fig. 2). Although the level of miR-200 was reduced in TKO MEFs compared with wild-type MEFs, in contrast to E-cadherin and cdk inhibitors, miR-200 was still evident in TKO MEFs (Fig. 7A). miR-200 expression was further reduced in Ras-TKO MEFs (Fig. 7A), suggesting that superinduction of ZEB1 by Ras is required for full repression of mi-R200. Indeed, this Ras-dependent repression of miR-200 was largely reversed in Ras-TKO ZEB1 shRNA-2 MEFs (Fig. 7A). We conclude that repression of miR-200 requires a higher threshold of ZEB1 than repression of cdk inhibitors and E-cadherin and that superinduction of ZEB1 by Ras is required to generate a level of ZEB1 sufficient to repress miR-200. Consistent with a direct effect of ZEB1 on miR-200 expression, we found ZEB1 bound the promoters of miR-200 family members in chromatin immunoprecipitation assays (Fig. 7B).
FIGURE 7.

Ras superinduction of ZEB1 is required for repression of miR-200. A, real-time PCR demonstrating that Ras superinduction of ZEB1 (which is reversed in TKO ZEB1 shRNA-2 MEFs) is required for repression of miR-200. B, chromatin immunoprecipitation assays showing ZEB1 binding to the promoter regions of miR-200 family members in vivo. c1 and c2 indicate primers covering two different sets of E boxes (potential ZEB1 binding sites) in the miR-200c promoter (supplemental Table 3). IgG indicates control serum, and mock indicates no input chromatin. Gapdh is glyceraldehyde-3-phosphate dehydrogenase, a negative promoter control for ZEB1 binding. H3 and H4 show positive controls of histone H3 and histone H4 binding.
Superinduction of ZEB1 by Ras Is Required for Ras-initiated Invasion of Sarcomas in Nude Mice
It has been reported previously that Ras-TKO MEFs are tumorigenic in nude mice (6), and we found that these cells formed spindle cell sarcomas that were highly invasive into surround host tissues (Fig. 8). As noted above, it is thought that repression of miR-200 is important for tumor invasion and metastasis (38–42). Therefore, we wondered whether superinduction of ZEB1 by Ras and resulting repression of miR-200 might be critical for the Ras-initiated invasion of these tumors. To test this possibility, we injected TKO ZEB1 shRNA-2 MEFs into nude mice. These cells also generated spindle cell sarcomas of mass and histology similar to those formed with Ras-TKO MEFs, but these tumors were all noninvasive (Fig. 8). Consistent with their lack of proliferation, we did not observe tumors in nude mice with TKO ZEB1 shRNA-1 MEFs (Fig. 8). Taken together, these results demonstrate that ZEB1 induction to the level seen in TKO MEFs is required for Ras-dependent tumor initiation, and, Ras superinduction of ZEB1 beyond the level seen in TKO MEFs is then required for Ras-initiated transition to tumor invasion. We suggest that this Ras superinduction of ZEB1 is necessary to reach a threshold required for full repression of miR-200.
FIGURE 8.

Induction of ZEB1 in response to Rb1 family mutation is required for Ras to initiated sarcoma formation, whereas Ras superinduction of ZEB1 is required for transition to invasive sarcoma. 50,000 cells were injected into nude mice, and tumors were harvested after 31 days. Representative H&E sections of the tumor-host border are shown at the top, and sections of the tumor interior are shown below. The yellow arrow denotes the tumor-host border in a Ras-TKO ZEB1 shRNA-2 MEF tumor. Red arrows indicate sites of invasion into host muscle and the green arrow invasion into adipose tissue in a Ras-TKO MEF tumor. Scale bars, 50 μm.
DISCUSSION
Activation of the Rb1 pathway triggers oncogene-induced senescence to suppress Ras mutation, and sequential mutation of Rb1 and Ras in MEFs provides a model in which primary cells transition to invasive sarcoma. In this model, we demonstrate two sequential inductions of ZEB1, the first resulting from mutation of Rb1 and the second from Ras mutation. Although Rb1 classically represses genes in a cell cycle-dependent fashion to regulate proliferation, a number of other genes have been shown to be constitutively repressed by Rb1 in proliferating cells. Such genes include proapoptotic factors as well as cdk inhibitors (43). It has been suggested that release of constitutive repression of such genes can serve a tumor suppressor function that inhibits viability when the Rb1 pathway is lost. Although there is an increase in cdk inhibitors in TKO MEFs compared with wild-type MEFs, their expression does not rise to a level sufficient to block cell cycle progression. Our results suggest that induction of ZEB1 becomes critical to prevent an increase in expression of these cdk inhibitors leading to cell cycle arrest when the Rb1 pathway is lost. Evidence for this conclusion comes from our finding that knockdown of ZEB1 in TKO MEFs to the level seen in wild-type MEFs leads to induction of cdk inhibitors to a point well beyond that seen wild-type cells, where the Rb1 pathway is present to provide repression.
Knockdown of ZEB1 in TKO MEFs led to progressive induction of cdk inhibitors, with the inhibitors accumulating as the cells were passaged and the cells ultimately undergoing senescence in a ZEB1 concentration-dependent fashion. The reason for this delay in cdk inhibitor induction, which required multiple passages, is unclear, but a similar delay was also evident when ZEB1 mutant MEFs were placed in cell culture; several passages were required for accumulation of cdk inhibitors and premature senescence (11). One explanation for this delay is that it represents the time period for decay of ZEB1 or reversal of repressive epigenetic marks imposed by ZEB1 at the cdk inhibitor gene promoters, but, this delay was not evident in induction of E-cadherin. A second possibility is that derepression of the cdk inhibitors, resulting from knockdown of ZEB1, is not sufficient by itself for induction because an activation step, resulting for example from serum growth factors in the culture media, might be required once repression is removed. In this regard it is of note that in Ras-TKO MEFs, where Ras mutation causes constitutive activation of such growth factor pathways, knockdown of ZEB1 led to rapid induction of cdk inhibitors and senescence before the cells could be passaged.
Introduction of mutant Ras into TKO MEFs leads to superinduction of ZEB1, to a level well beyond that seen in TKO MEFs. Interestingly, this Ras induction of ZEB1 is dependent upon initial loss of the Rb1 pathway. Indeed, introduction of mutant Ras into wild-type MEFs led to severe repression of ZEB1. The explanation for this finding is that active Rb1, which accumulates in response to Ras in the wild-type cells, represses ZEB1, and this repression is dominant over the activation triggered by Ras, thereby causing loss of the Rb1 pathway to become a prerequisite for Ras induction of ZEB1. A partial knockdown of ZEB1 in Ras-TKO MEFs, to reverse most of the superinduction by Ras, did not affect cdk inhibitor expression or proliferation of the cells in culture; nor did it affect the type or size of tumors formed in nude mice. However, it blocked the ability of the tumors to invade surrounding soft tissues. We link this Ras superinduction of ZEB1 to repression of miR-200.
In summary, our results demonstrate two sequential inductions of ZEB1 during progression of primary culture MEFs to Ras-initiated invasive sarcoma. First, ZEB1 is derepressed with loss of the Rb1 pathway. We suggest that this initial induction is required to prevent expression of cell cycle inhibitory cdk inhibitors once their repression by Rb1 is lost. Importantly, this induction of ZEB1 arising from Rb1 pathway mutation becomes critical when Ras is mutated; reversal of the induction (e.g. by knockdown of ZEB1 in Ras-TKO MEFs back to the level seen in wild-type MEFs) triggered rapid senescence and blocked Ras-initiated tumor formation in nude mice. The second induction of ZEB1 is mediated by mutant Ras, and it is dependent on the first induction because it requires initial loss of the Rb1 pathway. This second induction is required for transition to invasive sarcoma and is linked to repression of miR-200, which is a well known regulator of invasion and metastasis. We propose that this second induction of ZEB1 by Ras is necessary for invasion because a higher threshold of ZEB1 is required for repression of miR-200.
Supplementary Material
Acknowledgments
We thank T. Jacks for Rb1 family mutant and wild-type MEFs and Guirong Liu for histological sections.
This work was supported, in whole or in part, by National Institutes of Health Grants EY019113 and EY018603 (to D. C. D.), National Institutes of Health Institutional Development Award P20GM103453 through the NIGMS (to Y. L.), and National Institutes of Health Core Grant EY015636 (to A. P.). This work was also supported by the Olga Torres Foundation (FOT), Spanish Association Against Cancer (AECC), Avon Breast Cancer Campaign (AVON-SAU), and Ministry of Economy and Competitiveness (MEC)/MICINN-BFU2010-15163 (to A. P.).

This article contains supplemental Figs. 1 and 2 and Tables 1–3.
- MEF
- mouse embryonic fibroblast
- cdk
- cyclin-dependent kinase
- Rb
- retinoblastoma
- TKO
- triple knock-out.
REFERENCES
- 1. Harbour J. W., Dean D. C. (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14, 2393–2409 [DOI] [PubMed] [Google Scholar]
- 2. Burkhart D. L., Sage J. (2008) Cellular mechanisms of tumor suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sage J., Miller A. L., Pérez-Mancera P. A., Wysocki J. M., Jacks T. (2003) Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424, 223–228 [DOI] [PubMed] [Google Scholar]
- 4. Chicas A., Wang X., Zhang C., McCurrach M., Zhao Z., Mert O., Dickins R. A., Narita M., Zhang M., Lowe S. W. (2010) Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dannenberg J. H., van Rossum A., Schuijff L., te Riele H. (2000). Ablation of the retinoblastoma gene family deregulates G1 control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14, 3051–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sage J., Mulligan G. J., Attardi L. D., Miller A., Chen S., Williams B., Theodorou E., Jacks T. (2000) Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 14, 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang H. S., Postigo A. A., Dean D. C. (1999) Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGF-β, and contact inhibition. Cell 97, 53–61 [DOI] [PubMed] [Google Scholar]
- 8. Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 9. Ho V. M., Schaffer B. E., Karnezis A. N., Park K. S., Sage J. (2009) The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene 28, 1393–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peinado H., Olmeda D., Cano A. (2007) Snail, Zeb and bHLH factors in tumor progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., El-Naggar S., Darling D. S., Higashi Y., Dean D. C. (2008) ZEB1 links epithelial-mesenchymal transition and cellular senescence. Development 135, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbons D. L., Lin W., Creighton C. J., Rizvi Z. H., Gregory P. A., Goodall G. J., Thilaganathan N., Du L., Zhang Y., Pertsemlidis A., Kurie J. M. (2009) Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 23, 2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olson P., Lu J., Zhang H., Shai A., Chun M. G., Wang Y., Libutti S. K., Nakakura E. K., Golub T. R., Hanahan D. (2009) MicroRNA dynamics in the stages of tumorigenesis correlates with hallmark capabilities of cancer. Genes Dev. 23, 2152–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brabletz S., Brabletz T. (2010) The ZEB/miR-200 feedback loop: a motor of cellular plasticity in development and cancer? EMBO Rep. 11, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin S., Dimitri C. A., Yoon S. O., Dowdle W., Blenis J. (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 38, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Constatino M. E., Montoya-Durango D., Higashi Y., Darling D. S., Dean D. C. (2007) The zinc finger transcription factor, ZFHX1A, is linked to cell proliferation by Rb/E2F1. Biochem. J. 408, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rhim A. D., Mirek E. T., Aiello N. M., Maitra A., Bailey J. M., McAllister F., Reichert M., Beatty G. L., Rustgi A. K., Vonderheide R. H., Leach S. D., Stanger B. Z. (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bracken C. P., Gregory P. A., Kolesnikoff N., Bert A. G., Wang J., Shannon M. F., Goodall G. J. (2008) A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 68, 7846–7854 [DOI] [PubMed] [Google Scholar]
- 19. Burk U., Schubert J., Wellner U., Schmalhofer O., Vincan E., Spaderna S., Brabletz T. (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Postigo A. (2003) Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J. 22, 2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoo J., Robinson R. A., Lee J. Y. (1999) H-Ras and K-Ras gene mutations in primary human soft tissue sarcoma: concomitant mutations if the ras genes. Mod. Pathol. 12, 775–780 [PubMed] [Google Scholar]
- 22. Young N. P., Crowley D., Jacks T. (2011) Uncoupling cancer mutations reveals critical timing of p53 loss in sarcomagenesis. Cancer Res. 71, 4040–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mora J., Rodríguez E., de Torres C., Cardesa T., Ríos J., Hernández T., Cardesa A., de Alava E. (2012) Activated growth signaling pathway expression in Ewing sarcoma and clinical outcome. Pediatr. Blood Cancer 58, 532–538 [DOI] [PubMed] [Google Scholar]
- 24. Geryk-Hall M., Hughes D. P. (2009) Critical signaling pathways in bone sarcoma: candidates for therapeutic interventions. Curr. Oncol. Rep. 11, 446–453 [DOI] [PubMed] [Google Scholar]
- 25. Shen A., Zhang Y., Yang H., Xu R., Huang G. (2012) Overexpression of ZEB1 relates to metastasis and invasion in osteosarcoma. J. Surg. Oncol. 105, 830–834 [DOI] [PubMed] [Google Scholar]
- 26. Liu Y., Clem B., Zuba-Surma E. K., El-Naggar S., Telang S., Jenson A. B., Wang Y., Shao H., Ratajczak M. Z., Chesney J., Dean D. C. (2009) Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 4, 336–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. El-Naggar S., Liu Y., Dean D. C. (2009) Mutation of the Rb1 pathway leads to overexpression of mTor, constitutive phosphorylation of Akt on serine 473, resistance to anoikis, and a block in c-Raf activation. Mol. Cell. Biol. 29, 5710–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi R., Chiang V. L. (2005) Facile means for quantifying microRNA expression by real-time PCR. BioTechniques 39, 519–525 [DOI] [PubMed] [Google Scholar]
- 29. Chassot A. A., Lossaint G., Turchi L., Meneguzzi G., Fisher D., Ponzio G., Dulic V. (2008) Confluence-induced cell cycle exit involves pre-mitotic cdk inhibition by p27kip1 and cyclin D1 down-regulation. Cell Cycle 7, 2038–2046 [DOI] [PubMed] [Google Scholar]
- 30. Prieur A., Peeper D. S. (2008) Cellular senescence in vivo: a barrier to tumorigenesis. Curr. Opin. Cell Biol. 20, 150–155 [DOI] [PubMed] [Google Scholar]
- 31. Malumbres M., Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 32. Ramirez R. D., Morales C. P., Herbert B. S., Rohde J. M., Passons C., Shay J. W., Wright W. E. (2001) Putative telomere-independent mechanism of replicative aging reflects inadequate growth conditions. Genes Dev. 15, 398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lowe S. W., Sherr C. J. (2003) Tumor suppression by Ink4a-Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13, 77–83 [DOI] [PubMed] [Google Scholar]
- 34. Foijer F., Wolthuis R. M., Doodeman V., Medema R. H., te Riele H. (2005) Mitogen requirement for cell cycle regulation in the absence of pocket protein activity. Cancer Cell 8, 455–466 [DOI] [PubMed] [Google Scholar]
- 35. van Harn T., Foijer F., van Vugt M., Banerjee R., Yang F., Oostra A., Joenje H., te Riele H. (2010). Loss of Rb proteins causes genomic instability in the absence of mitogen signaling. Genes Dev. 24, 1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li W., Kotoshiba S., Berthet C., Hilton M.B., Kaldis P. (2009) Rb/cdk2/cdk4 triple mutant mice elicit an alternative mechanism for regulation of the G1/S transition. Proc. Natl. Acad. Sci. U.S.A. 106, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Foijer F., Delzenne-Goette E., Dekker M., Te Riele H. (2007) In vivo significance of the G2 restriction point. Cancer Res. 67, 9244–9247 [DOI] [PubMed] [Google Scholar]
- 38. Shimono Y., Zabala M., Cho R. W., Lobo N., Dalerba P., Qian D., Diehn M., Liu H., Panula S. P., Chiao E., Dirbas F. M., Somlo G., Pera R. A., Lao K., Clarke M. F. (2009) Down-regulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138, 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dykxhoorn D. M., Wu Y., Xie H., Yu F., Lal A., Petrocca F., Martinvalet D., Song E., Lim B., Lieberman J. (2009) miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE 4, e7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Korpal M., Ell B. J., Buffa F. M., Ibrahim T., Blanco M. A., Celià-Terrassa T., Mercatali L., Khan Z., Goodarzi H., Hua Y., Wei Y., Hu G., Garcia B. A., Ragoussis J., Amadori D., Harris A. L., Kang Y. (2011) Direct targeting of Sec23 by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nature Med. 17, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park S. M., Gaur A. B., Lengyel E., Peter M. E. (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 22, 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wellner U., Schubert J., Burk U. C., Schmalhofer O., Zhu F., Sonntag A., Waldvogel B., Vannier C., Darling D., zur Hausen A., Brunton V. G., Morton J., Sansom O., Schüler J., Stemmler M. P., Herzberger C., Hopt U., Keck T., Brabletz S., Brabletz T. (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 [DOI] [PubMed] [Google Scholar]
- 43. van Oevelen C., Wang J., Asp P., Yan Q., Kaelin W.G., Jr., Kluger Y., Dynlacht B. D. (2008) A role for mammalian sin3a in permanent gene silencing. Mol. Cell 32, 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.