

Background: Energy conservation by Na+-translocating ferredoxin oxidation requires a low potential ferredoxin.
Results: Endergonic reduction of ferredoxin with NADH is driven by exergonic caffeyl-CoA reduction catalyzed by a soluble caffeyl-CoA reductase-electron transfer flavoprotein complex.
Conclusion: Caffeyl-CoA reductase uses electron bifurcation for energy coupling.
Significance: Ferredoxin reduction by electron bifurcation is essential for energy conservation in caffeate respiration.
Keywords: Bioenergetics, Electron Transfer, Enzyme Mechanisms, Iron-Sulfur Protein, Metalloenzymes, Acetogenesis, Electron Bifurcation, Energy Conservation
Abstract
A low potential electron carrier ferredoxin (E0′ ≈ −500 mV) is used to fuel the only bioenergetic coupling site, a sodium-motive ferredoxin:NAD+ oxidoreductase (Rnf) in the acetogenic bacterium Acetobacterium woodii. Because ferredoxin reduction with physiological electron donors is highly endergonic, it must be coupled to an exergonic reaction. One candidate is NADH-dependent caffeyl-CoA reduction. We have purified a complex from A. woodii that contains a caffeyl-CoA reductase and an electron transfer flavoprotein. The enzyme contains three subunits encoded by the carCDE genes and is predicted to have, in addition to FAD, two [4Fe-4S] clusters as cofactor, which is consistent with the experimental determination of 4 mol of FAD, 9 mol of iron, and 9 mol of acid-labile sulfur. The enzyme complex catalyzed caffeyl-CoA-dependent oxidation of reduced methyl viologen. With NADH as donor, it catalyzed caffeyl-CoA reduction, but this reaction was highly stimulated by the addition of ferredoxin. Spectroscopic analyses revealed that ferredoxin and caffeyl-CoA were reduced simultaneously, and a stoichiometry of 1.3:1 was determined. Apparently, the caffeyl-CoA reductase-Etf complex of A. woodii uses the novel mechanism of flavin-dependent electron bifurcation to drive the endergonic ferredoxin reduction with NADH as reductant by coupling it to the exergonic NADH-dependent reduction of caffeyl-CoA.
Introduction
Strictly anaerobic bacteria devoid of cytochromes are classically considered as fermenting bacteria without having the capability for ion gradient-driven ATP synthesis, as they lack ion-motive electron transfer systems (1, 2). This view has changed dramatically with the discovery of decarboxylation-driven (3) and methyltransfer-driven (4, 5) ATP synthesis. However, these types of reactions are limited to only a few species. Recently, an ion-motive electron transport system was discovered in the acetogenic model bacterium Acetobacterium woodii: a membrane-integral, sodium-motive ferredoxin:NAD+ oxidoreductase (Rnf complex) that couples the exergonic electron flow from reduced ferredoxin to NAD+ with the export of Na+ from the cytoplasm (6–11). The Rnf complex is the only coupling site in A. woodii, and the established transmembrane electrochemical Na+ gradient then drives the synthesis of ATP via a well characterized Na+ F1F0-ATP synthase (12–16). The Rnf complex is apparently widespread in the bacterial world and present also in a few archaea (17, 18).
A cellular bioenergetics based on just one coupling site that is fueled by reduced ferredoxin raises the question of how ferredoxin is reduced. The redox potential of the ferredoxin in A. woodii can be considered to be in the range of ≈−500 mV, and thus, electron transfer from hydrogen (E0′ = −414 mV) or NADH (E0′ = −320 mV) to ferredoxin is highly endergonic and thermodynamically impossible. Recently, a soluble hydrogenase was purified from A. woodii that drives the endergonic ferredoxin (Fd) reduction with H2 as reductant by coupling it to the exergonic electron transfer from hydrogen to NAD+ by electron bifurcation (19) according to Equation 1.
Electrons derived from NADH are then channeled by soluble cytoplasmic enzymes to the terminal electron acceptor carbon dioxide (carbonate respiration or acetogenesis) or caffeate (caffeate respiration) (7, 20–23).
Caffeate respiration involves activation of caffeate to caffeyl-CoA (22, 24) and a subsequent reduction of caffeyl-CoA to hydrocaffeyl-CoA. After a CoA is transferred to caffeate, hydrocaffeate is released into the ecosystem. In addition to the genes encoding the CoA transferase and an ATP-dependent caffeyl-CoA synthetase required for the initial activation, the car operon encodes a potential acyl-CoA dehydrogenase (CarC) and the two subunits of an electron transfer flavoprotein (CarDE). The reduction of caffeyl-CoA with NADH as reductant is exergonic, and the energy gained from this reaction may be used to drive the endergonic ferredoxin reduction with NADH as reductant by flavin-based electron bifurcation (23, 25, 26). To address this question, we purified the caffeyl-CoA reductase-Etf complex and present evidence for ferredoxin reduction coupled to caffeyl-CoA reduction with NADH as reductant. The energetics of caffeate reduction is discussed.
EXPERIMENTAL PROCEDURES
Growth of Cells and Purification of Caffeyl-CoA Reductase
A. woodii (DSM 1030) was grown at 30 °C under anaerobic conditions in 20-liter flasks (Glasgerätebau Ochs, Bovenden/Lenglern, Germany) using 20 mm fructose as the carbon and energy source as described previously (27, 28). At A600 ∼ 0.3, caffeate was added to a concentration of 5 mm. The medium and all buffers were prepared using the anaerobic techniques described previously (29, 30). All buffers contained 2 mm dithioerythritol (DTE)3 and 4 μm resazurin. 5 μm FAD was added to all purification buffers to avoid loss of activity. All purification steps were performed under strictly anaerobic conditions at room temperature in an anaerobic chamber (Coy Laboratory Products, Grass Lake Charter Township, MI) filled with 98–95% N2 and 2–5% H2 as described (27). Cells of A. woodii were harvested at A600 = 1.5 and washed twice with 25 mm Tris-HCl (pH 7.0) and 420 mm sucrose and resuspended in 150 ml of 25 mm Tris-HCl (pH 8.0) and 420 mm sucrose. 500 mg of lysozyme was added, and the suspension was incubated for 1 h at 37 °C. After centrifugation, the protoplasts were resuspended in 25 mm Tris-HCl (pH 7.5), 20 mm MgSO4, 20% glycerol, 0.5 mm PMSF, and 0.1 mg/ml DNase I and passed three times through a French pressure cell at 110 megapascals. Cell debris was removed by centrifugation at 24,000 × g for 40 min. Membranes were removed by centrifugation at 130,000 × g for 45 min. The supernatant containing the cytoplasmic fraction with ∼1600 mg of protein was applied to a Q Sepharose high performance column (2.6 × 5 cm) equilibrated with buffer A (50 mm Tris-HCl, 20 mm MgSO4, 5 μm FAD, and 20% glycerol (pH 7.5)). Protein was eluted with a 150-ml linear gradient of 0–0.35 m NaCl in buffer A. Methyl viologen-dependent caffeyl-CoA reductase activity eluted at ∼0.3 mm NaCl. After the addition of ammonium sulfate (1.25 m), the pooled fractions were loaded onto a phenyl-Sepharose high performance column (1.6 × 10 cm) equilibrated with 1.25 m (NH4)2SO4 in buffer A. Protein was eluted with a 250-ml linear gradient of 1.25 to 0 m (NH4)2SO4. Caffeyl-CoA reductase activity eluted at ∼0.7 m (NH4)2SO4. Pooled fractions were concentrated by ultrafiltration in 50-kDa Vivaspin tubes (Sartorius Stedim Biotech GmbH). For the following purification step with a HiTrap Blue Sepharose high performance column (5 ml), the removal of the ammonium sulfate in the sample was crucial to ensure binding to the column; this was done by a 10-fold dilution with buffer A. The sample was loaded onto the column equilibrated with buffer A, and protein was eluted with a linear gradient of 0–500 mm NaCl. Caffeyl-CoA reductase activity eluted in a broad peak at ∼170 mm NaCl. Pooled fractions were concentrated by ultrafiltration in 50-kDa Vivaspin tubes. The sample was applied to a Sephacryl S-300 high resolution column (1.6 × 60 cm) equilibrated with buffer A and eluted at a flow rate of 0.5 ml/min. Caffeyl-CoA reductase activity eluted as a single peak. Pooled fractions were concentrated to a minimum of 5 mg/ml and stored at 4 °C.
Assays of Caffeyl-CoA Reductase Activity
All measurements were performed at 30 °C in 1.8-ml anaerobic cuvettes (Glasgerätebau Ochs) sealed with rubber stoppers in a N2 atmosphere. A caffeyl-CoA regeneration system was used in all measurements (200 units of CoA transferase and 0.25 mm caffeate), and the CoA transferase was overproduced and purified as described (24). Measurements where done routinely using a methyl viologen oxidation assay with 5 mm methyl viologen in buffer B (50 mm Tris-HCl, 20 mm MgSO4, and 2 mm DTE, pH 7.5). The addition of 1 mm sodium dithionite resulted in partial reduction of the methyl viologen. The reaction was started by the addition of 5 μm caffeyl-CoA. Methyl viologen oxidation was monitored at 604 nm (ϵ = 13.8 mm−1 cm−1), and 1 unit is defined as the reduction of 1 μmol of caffeyl-CoA/min. Physiological caffeyl-CoA reductase activity was measured in buffer B with 0.5 mm NADH as electron donor. The reaction was started by the addition of at least one of the following electron acceptors: caffeyl-CoA (5 μm), ferredoxin (20 μm), FAD (0.25 mm), or FMN (0.25 mm). The reduction of caffeate was monitored at 312 nm (ϵ = 13.72 mm−1 cm−1), and the reduction of ferredoxin was monitored at 430 nm (ϵ = 13.1 mm−1 cm−1). Ferredoxin was purified from Clostridium pasteurianum as described (31). In initial experiments, caffeyl-CoA was synthesized chemically (24). For the experiments described here, caffeyl-CoA was synthesized enzymatically with the caffeyl-CoA synthetase from A. woodii (22). The synthesis of caffeyl-CoA in 50 mm Tris-HCl (pH 7.0), 20 mm MgSO4, 50 mm KCl, 1 mm ATP, 1 mm CoA, 0.25 mm caffeate, and 2 mg of enzyme was followed by ultrafiltration in 30-kDa Vivaspin tubes to remove the enzyme. The yellow flow-through was concentrated in a centrifugal evaporator to a final concentration of 1 mm caffeyl-CoA (ϵ346 = 18.0 mm−1 cm−1) and stored at 4 °C.
Analytical Methods
The concentration of proteins was measured according to Bradford (32). Proteins were separated on 12% polyacrylamide gels and stained with Coomassie Brilliant Blue G-250. The molecular mass of the purified caffeyl-CoA reductase was determined using a calibrated Sephacryl S-300 column equilibrated with buffer A. The iron and sulfur content of the purified enzyme was determined by colorimetric methods (33, 34). To determine the flavin content of the purified enzyme, 1 nmol of protein was precipitated with trichloroacetic acid to a final concentration of 500 mm. The nature of the flavin in the supernatant was identified by thin layer chromatography on Silica Gel 60 sheets (Macherey-Nagel GmbH & Co. KG) with 5% Na2HPO4 in H2O or 1-butanol/acetic acid/H2O (12:3:5) as the mobile phase. The amount of bound flavin was calculated by comparing the absorbance of the yellow supernatant at 450 nm with a calibration line with FAD treated the same way (35).
RESULTS
Assay for Caffeyl-CoA Reduction
The activity of the caffeyl-CoA reductase-Etf complex was measured using dithionite-reduced methyl viologen as electron donor and caffeyl-CoA as electron acceptor. Methyl viologen oxidation was followed spectroscopically at 604 nm. Because caffeyl-CoA is not commercially available, a caffeyl-CoA regeneration system was established. Caffeyl-CoA was reduced with the caffeyl-CoA reductase-Etf complex to hydrocaffeyl-CoA according to Equation 2,
 |
where MV is methyl viologen. Next, hydrocaffeyl-CoA was used as CoA donor by the hydrocaffeyl-CoA:caffeate-CoA transferase CarA from A. woodii (24) to activate caffeate to caffeyl-CoA (Equation 3).
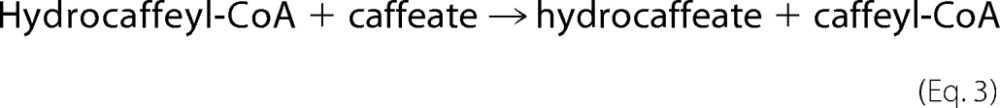 |
Thus, in sum, caffeate was reduced to hydrocaffeate according to Equation 4, and the rate of methyl viologen (MV) oxidation is proportional to caffeyl-CoA reduction (1 unit = 1 μmol of caffeyl-CoA/mg).
The dependence of methyl viologen oxidation on the caffeyl-CoA concentration showed saturation at 5 μm caffeyl-CoA with Km = 0.5 μm.
Purification of the Caffeyl-CoA Reductase-Etf Complex from A. woodii
To purify the caffeyl-CoA reductase-Etf complex, fructose/caffeate-grown cells were grown to the late exponential growth phase, harvested, and disrupted with a French pressure cell. Membranes were removed by ultracentrifugation, and the caffeyl-CoA reductase-Etf complex was purified from the cytoplasm by ion exchange chromatography on Q Sepharose, phenyl-Sepharose, and Blue Sepharose, followed by gel filtration on Sephacryl S-300. The enzyme was purified by 24-fold to apparent homogeneity with an average specific activity of 14 units/mg in the methyl viologen oxidation assay (Table 1).
TABLE 1.
Purification of the caffeyl-CoA reductase-Etf complex from A. woodii
| Purification step | Protein | Specific activitya | Yield | Purification |
|---|---|---|---|---|
| mg | units/mg | % | -fold | |
| Cell-free extract | 2579.2 | 0.57 | 100.0 | 1.0 |
| Cytoplasm | 2161.9 | 0.57 | 83.2 | 1.0 |
| Q Sepharose | 381.4 | 2.51 | 65.0 | 4.4 |
| Phenyl-Sepharose | 99.5 | 7.83 | 52.9 | 13.7 |
| Blue Sepharose | 46.5 | 12.93 | 40.8 | 22.6 |
| Gel filtration Sephacryl S-300 | 14.5 | 13.61 | 13.4 | 23.8 |
a The activity of the caffeyl-CoA reductase-Etf complex was determined by following the caffeyl-CoA-dependent oxidation of reduced methyl viologen at 604 nm. The assay buffer (buffer B) contained 5 mm reduced methyl viologen (reduced with 1 mm sodium dithionite) as electron donor, 5 μm caffeyl-CoA, and the caffeyl-CoA regeneration system (200 units of the CoA transferase CarA and 0.25 mm caffeate) under an atmosphere of 100% N2. Measurements were performed at 30 °C in anaerobic cuvettes.
The purified enzyme apparently has three subunits of 41, 39, and 29 kDa (Fig. 1). This fits well with the predicted masses of CarE (42.5 kDa), CarC (41.4 kDa), and CarD (28.2 kDa) and indeed, the proteins were identified by peptide mass fingerprinting as CarE, CarC, and CarD. CarDE is identical to EtfAB identified previously by a proteomics approach to be up-regulated during growth on caffeate (7). Analytical gel filtration, as well as separation on a native gel, revealed that the caffeyl-CoA reductase and the electron transfer flavoprotein formed a stable complex. Because the masses of the subunits of the complex are rather small, an accurate estimate of the subunit composition could not be made, but the estimated molecular mass of 350 kDa upon gel filtration is consistent with a homotrimer of CarCDE, which would have a mass of 336 kDa.
FIGURE 1.
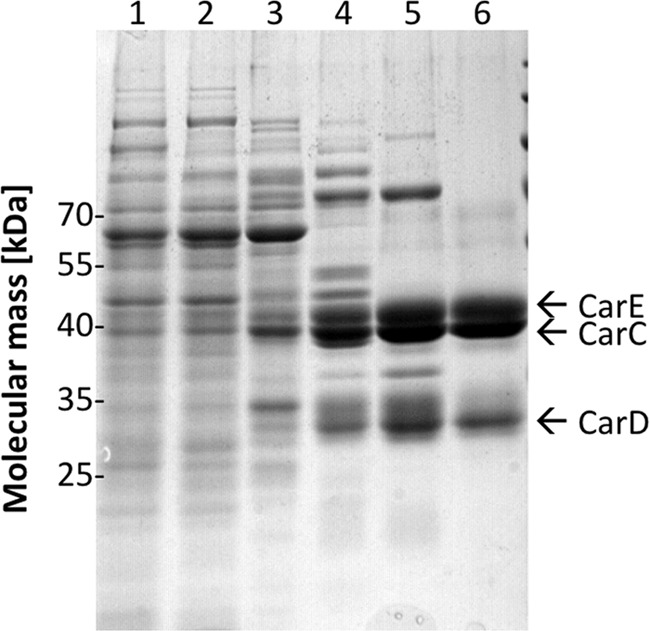
Purification of the caffeyl-CoA reductase-Etf complex. Samples from the different purification steps were separated by SDS-PAGE, and proteins were stained with Coomassie Brilliant Blue. Lane 1, cell extract; lane 2, cytoplasm; lane 3, pooled fractions from Q Sepharose; lane 4, pooled fractions from phenyl-Sepharose; lane 5, pooled fractions from Blue Sepharose; lane 6, pooled fractions from Sephacryl S-300. 10 μg of protein was applied to each lane.
Basic Biochemical Properties of the Caffeyl-CoA Reductase-Etf Complex of A. woodii
The spectrum of the yellow-brownish enzyme shows absorbance maxima at 372 and 445 nm (Fig. 2), which indicated oxidized flavin as the prosthetic group (35). The purified complex contained 4 ± 0.2 mol of flavin/mol of CarCDE. This flavin was tightly bound because it could not be removed by gel filtration. Thin layer chromatography identified the flavin as FAD. Iron and acid-labile sulfur were determined colorimetrically according to Fish (33) and Beinert (34). The enzyme contained 9 ± 0.5 mol of iron/mol of CarCDE and 9 ± 1.3 mol of acid-labile sulfide/mol of CarCDE. The C terminus of CarE contains eight conserved cysteine residues, which are homologous to the cysteine residues in the ferredoxins from C. pasteurianum and Clostridium acidi-urici harboring two [4Fe-4S] clusters (36, 37). With the determined content of iron and sulfur, two [4Fe-4S] clusters/CarCDE can be predicted.
FIGURE 2.
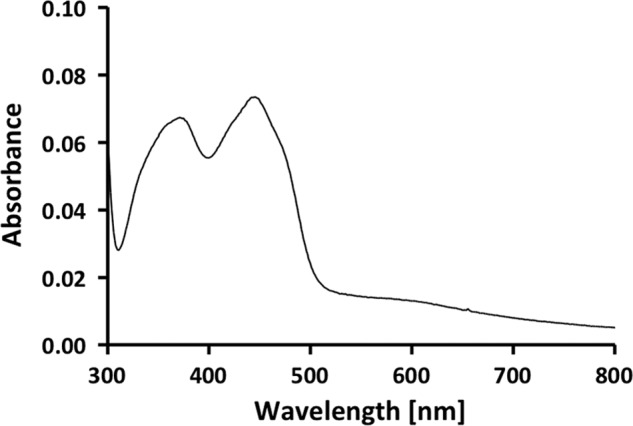
UV-visible spectrum of the purified caffeyl-CoA reductase-Etf complex. The spectrum of the enzyme (0.57 mg/ml) was recorded in buffer B.
The basic biochemical properties of the enzyme were determined using the caffeyl-CoA-dependent methyl viologen oxidation assay. The caffeyl-CoA reductase activity had an optimum at pH 7.5, and activity decreased to ∼30% at pH 6.0 or 9.0. The enzyme activity exhibited a rather broad temperature optimum, and maximum activity was detected at 30 °C, which is the optimum growth temperature of A. woodii. If caffeate was substituted with p-coumarate or ferulate, the corresponding CoA thioesters were reduced as well.
Caffeyl-CoA Reductase Uses NADH as Reductant and Catalyzes Flavin Reduction
The redox potential of caffeyl-CoA/hydrocaffeyl-CoA is not known, but considering that the reduction of the acrylate side chain of caffeate is thermodynamically equivalent to the reduction of fumarate (21), it can be considered to be in the same range as fumarate/succinate with +33 mV (38). We first determined whether the enzyme can catalyze the energetic “downhill” transport of electrons from NADH (E0′ = −320 mV) to caffeyl-CoA (E0′ = +33 mV). Thus, in the enzyme assay, reduced methyl viologen (Equation 4) was replaced with NADH, which is the physiological electron donor of caffeate reduction in A. woodii (7), and the decrease in caffeate was measured spectroscopically at 312 nm, which is proportional to the reduction of caffeyl-CoA (1 unit = 1 μmol of caffeyl-CoA/mg). With NADH (0.5 mm) as electron donor, caffeyl-CoA (5 μm) was reduced with an activity of 1 unit/mg.
To prevent loss of activity, 5 μm FAD had to be added to all buffers during purification. Thus, the CarCDE complex requires FAD for catalytic activity. Next, we tested whether flavins could be used as artificial electron acceptors. No activity could be detected when FAD or FMN (0.25 mm) was used alone as electron acceptor, but together with caffeyl-CoA, they were completely reduced with NADH as reductant. Caffeyl-CoA reduction was stimulated by 26- and 12-fold with FAD and FMN, respectively. The Km value for both FAD and FMN was 100 μm. Thus, caffeyl-CoA and flavins were reduced simultaneously.
Coupling of Caffeyl-CoA and Ferredoxin Reduction
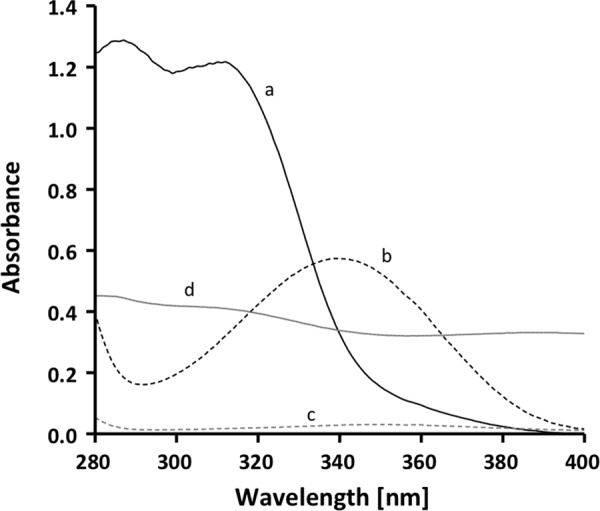
Next, we tested whether the addition of ferredoxin also stimulated NADH-dependent caffeyl-CoA reduction, and indeed, the reduction of caffeyl-CoA increased by 10-fold to 10 units/mg after the addition of 20 nmol of ferredoxin. The observed stimulation of caffeyl-CoA reduction by ferredoxin raised the question of whether ferredoxin was reduced alongside caffeyl-CoA. To test this, spectral analyses were performed (Fig. 3). The spectrum of oxidized ferredoxin reveals the characteristic shoulder of oxidized ferredoxin between 370 and 450 nm (trace a). Reduction with CO dehydrogenase purified from A. woodii with 100% CO in the gas phase led to the disappearance of the shoulder (trace b). The addition of NADH, the caffeyl-CoA regeneration system (caffeate and the CoA transferase CarA, with no caffeyl-CoA present), and the purified caffeyl-CoA reductase-Etf complex to oxidized ferredoxin led to the spectrum shown in trace c. The change in absorbance below 400 nm was due to NADH and caffeate, but above 400 nm, the spectrum was identical to that of oxidized ferredoxin. This spectrum did not change, indicating that ferredoxin alone was not reduced. The addition of caffeyl-CoA and incubation for 5 min gave rise to the spectrum shown in trace d. The change in the absorbance spectrum above 400 nm was similar to the change in the absorbance spectrum when ferredoxin was reduced with CO dehydrogenase and CO. This experiment clearly demonstrated that ferredoxin was reduced alongside caffeyl-CoA.
FIGURE 3.
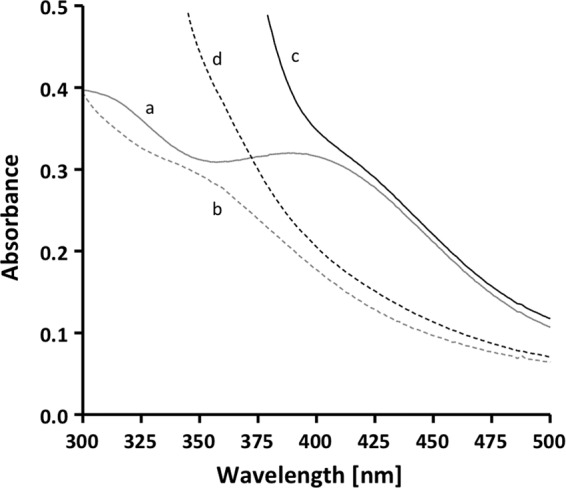
Caffeyl-CoA-dependent ferredoxin reduction. The assay buffer (buffer B) contained 0.2 mm NADH, 20 μm ferredoxin, 5 μm caffeyl-CoA, the caffeyl-CoA regeneration system (200 units of the CoA transferase CarA and 0.2 mm caffeate), and 8 μg of the purified CarCDE complex. Trace a, ferredoxin; trace b, ferredoxin reduced with CO dehydrogenase purified from A. woodii and 100% CO in the gas phase; trace c, ferredoxin, NADH, CarCDE, and the caffeyl-CoA regeneration system (with no caffeyl-CoA present); trace d, same as trace c after the addition of caffeyl-CoA and incubation of 5 min at 30 °C.
To determine the kinetic constants for this reaction, ferredoxin reduction was measured spectroscopically by following the decrease in absorbance at 430 nm. The caffeyl-CoA reductase-Etf complex reduced ferredoxin at a rate of 6 units/mg (1 unit = 1 μmol of ferredoxin/min). The dependence of the ferredoxin reduction on NADH (Fig. 4A) and ferredoxin (Fig. 4B) was hyperbolic. Saturation with NADH was at 500 μm with Km = 100 μm. Values in the same range were obtained when caffeyl-CoA reduction was followed or when FAD (Km for NADH = 125 μm) or FMN (Km for NADH = 150 μm) was used as oxidant. Saturation with ferredoxin was at 15 μm, but the Km value was below 5 μm and thus below the detection limit.
FIGURE 4.
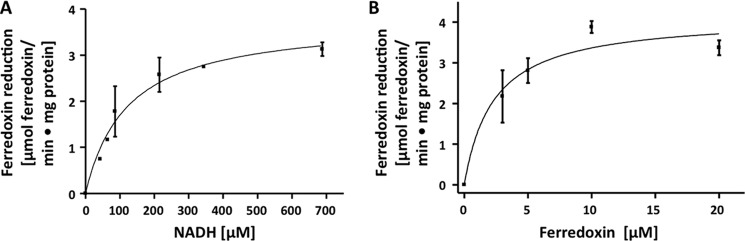
Dependence of ferredoxin reduction on NADH (A) and ferredoxin (B). Enzyme activity was measured in assay buffer (buffer B) containing the caffeyl-CoA regeneration system (200 units of the CoA transferase CarA and 0.2 mm caffeate), 8 μg of protein, and 5 μm caffeyl-CoA. All assays were done at 30 °C under an atmosphere of 100% nitrogen. For NADH dependence (A), NADH was added, and the reaction was started by the addition of 20 μm ferredoxin. For ferredoxin dependence (B), 0.5 mm NADH was added, and the reaction was started by the addition of ferredoxin.
To determine the ratio for the reduction of the two electron acceptors, the reduction of ferredoxin and caffeyl-CoA was monitored simultaneously at 430 and 312 nm, respectively (Fig. 5). The decrease in absorbance at 430 nm was used to calculate the amount of ferredoxin reduced, and the decrease in absorbance at 312 nm was used to calculate the amount of caffeyl-CoA reduced. From a number of experiments, a stoichiometry of 1:1.8 (ferredoxin:caffeyl-CoA) was calculated. However, NADH oxidation also led to a decrease in absorbance at 312 nm (ϵ = 3.6 mm−1 cm−1) (Fig. 6). All of the other substances involved did not have absorbance at 312 nm (hydrocaffeate and hydrocaffeyl-CoA), or the absorbance did not change after reduction (ferredoxin). Thus, the change in absorbance at 312 nm was the result of a combination of caffeyl-CoA reduction and NADH oxidation. Because one NADH is required to reduce one ferredoxin and the amount of ferredoxin reduced can be measured unequivocally at 430 nm, the amount of NADH used to reduce ferredoxin was subtracted. This gave a stoichiometry of 1:1.3 (ferredoxin:caffeyl-CoA) (Fig. 7). Considering that this ratio still reflects the amount of NADH required to reduce caffeyl-CoA and that reduction of caffeyl-CoA is also catalyzed in the absence of ferredoxin, under fully coupled conditions, the ratio should come close to 1:1. If FAD or FMN was used instead of ferredoxin as an additional electron acceptor, the stoichiometry without any corrections was 1:1.2 (flavin:caffeyl-CoA).
FIGURE 5.
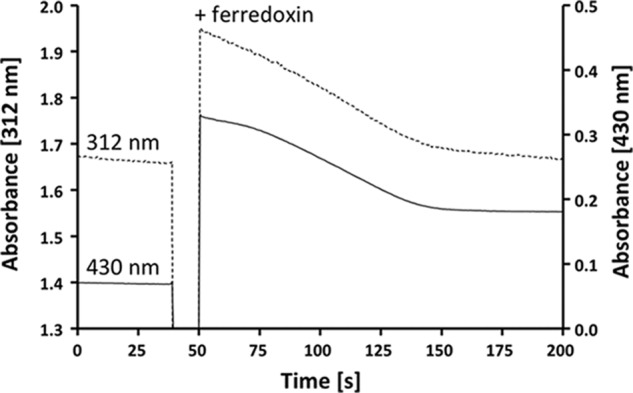
The purified CarCDE complex catalyzes reduction of ferredoxin and caffeyl-CoA simultaneously. The assay buffer (buffer B) contained 0.2 mm NADH, 5 μm caffeyl-CoA, 8 μg of the purified CarCDE complex, and the caffeyl-CoA regeneration system (200 units of the CoA transferase CarA and 0.2 mm caffeate). The reaction was started by the addition of 20 μm ferredoxin. Reduction of caffeyl-CoA (dashed line; monitored at 312 nm via decline of caffeate) and reduction of ferredoxin (solid line; monitored at 430 nm) were monitored simultaneously.
FIGURE 6.
Spectral properties of components used in the enzyme assay. Trace a, 0.2 mm caffeate; trace b, 0.2 mm NADH; trace c, 5 μm caffeyl-CoA; trace d, 20 μm ferredoxin (measured in buffer B).
FIGURE 7.
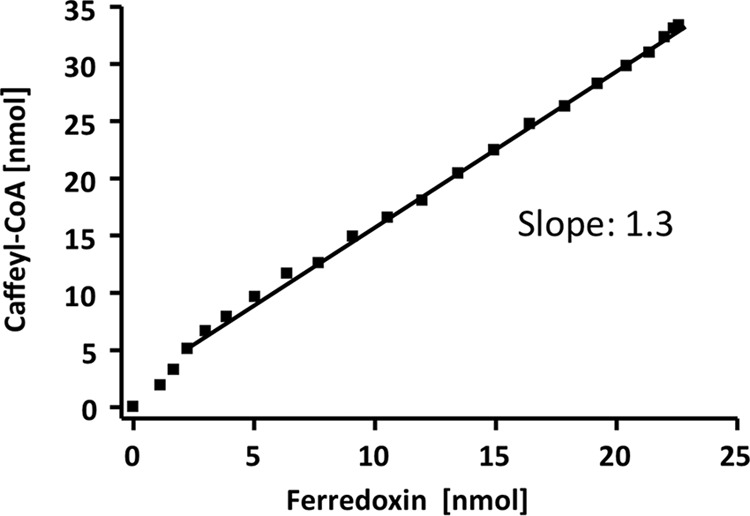
The purified CarCDE complex reduces ferredoxin and caffeyl-CoA in stoichiometric amounts. The reduction of caffeyl-CoA and ferredoxin was monitored simultaneously (see Fig. 5). Data points from 5-s intervals were taken to calculate the amount of reduced electron carrier from the absorbance difference and the molar extinction coefficient. The amount of reduced caffeyl-CoA is plotted against the amount of reduced ferredoxin.
DISCUSSION
We have shown that the gene carC encodes a caffeyl-CoA reductase and that the genes carD and carE encode the two subunits of the electron transfer flavoprotein. The Etf protein forms a stable complex with the caffeyl-CoA reductase, a homolog of the acyl-CoA dehydrogenase-Etf complexes from Clostridium propionicum (35) and Clostridium kluyveri (25). Electron transfer flavoproteins are found in all three domains of life (39) and consist of a large and a small subunit building a heterodimer containing noncovalently bound FAD. In general, they mediate the electron transfer between different donors and acceptors, e.g. between acyl-CoA dehydrogenases and the Etf:quinone oxidoreductase in the mitochondrial matrix in Mammalia (40, 41). Etf proteins usually contain, in addition to FAD, an AMP molecule buried in the small subunit, which may play a role in facilitating the correct structure of the protein (42, 43). In the Etf protein of Megasphaera elsdenii, this second nucleotide-binding site is occupied by a second FAD molecule (44), which seems to be the case also for the Etf proteins of other anaerobic bacteria characterized so far (25, 35). Besides these two FAD molecules bound to the Etf protein (CarDE), another one can be expected to bind to CarC (Fig. 8). This is in contrast to the four FAD molecules/CarCDE experimentally determined here. Because FAD can substitute ferredoxin and is also reduced in a stoichiometry close to 1:1 (FAD:caffeyl-CoA), a possible explanation is that an additional FAD molecule is tightly bound to the ferredoxin-binding site.
FIGURE 8.

Enzymology and bioenergetics of hydrogen-dependent caffeate respiration in A. woodii. The electron-bifurcating hydrogenase reduces ferredoxin (Fd) and NAD+ simultaneously. The Rnf complex couples the exergonic electron transfer from reduced ferredoxin to NAD+ with the translocation of Na+ ions. The electron acceptor caffeyl-CoA is reduced by the caffeyl-CoA reductase-Etf complex with NADH, which is coupled to the endergonic reduction of ferredoxin with NADH. The reduced ferredoxin is re-oxidized at the Rnf complex. The electrochemical Na+ gradient drives ATP synthesis by the Na+ F1F0-ATP synthase. The hydrocaffeyl-CoA:caffeate-CoA transferase CarA uses hydrocaffeyl-CoA to activate caffeate to caffeyl-CoA; this energy-saving CoA loop provides the regeneration of the acceptor under steady-state conditions. Initially, caffeate is activated by the caffeyl-CoA synthetase CarB at the expense of ATP (not shown). CM, cytoplasmic membrane.
In the Etf protein of A. woodii, in addition to FAD, 9 mol of iron/CarCDE and 9 mol of sulfur/CarCDE were determined. The large subunit of the Etf protein of A. woodii contains an N-terminal extension that contains two copies of the conserved consensus sequence (CX2CX2CX3CP) responsible for the binding of the [4Fe-4S] cluster in several ferredoxins (36, 37). This extension can be found in the large Etf subunit of other anaerobic bacteria such as Clostridium saccharolyticum and was proposed for several other organisms by comparing genomic sequences (41). In the mitochondrial electron transfer flavoprotein:ubiquinone oxidoreductase, a [4Fe-4S] cluster is the immediate acceptor for electrons derived from the Etf protein (45, 46). Thus, the iron-sulfur cluster in EtfA from A. woodii may be involved in electron transport, possibly in the intramolecular electron transfer from the Etf protein subunit to the caffeyl-CoA reductase subunit inside the complex.
The caffeyl-CoA reductase-Etf complex of A. woodii apparently couples the endergonic ferredoxin reduction with NADH as reductant to the exergonic reduction of caffeyl-CoA with the same reductant. A flavin is involved, and thus, the enzyme apparently uses the novel mechanism of electron bifurcation to overcome the steep energy barrier in ferredoxin reduction. The first electron-bifurcating enzyme discovered was the butyryl-CoA dehydrogenase-Etf complex from C. kluyveri (23, 25). This complex couples the exergonic reduction of crotonyl-CoA with NADH to the endergonic reduction of ferredoxin. For this complex, a similar stoichiometry of 1:1.4 (ferredoxin:butyryl-CoA) was determined because this reaction is also slightly uncoupled.
In C. kluyveri, the ferredoxin is used by the ferredoxin-dependent hydrogenase to reduce protons to molecular hydrogen. Part of the reduced ferredoxin can also be oxidized at the Rnf complex, improving the energy yield via electron transport phosphorylation. Additional electron-bifurcating enzymes have been described since then. The hydrogenase from Thermotoga maritima uses electron confurcation to drive hydrogen production from NADH by coupling to exergonic ferredoxin-dependent proton reduction (47). Transhydrogenases from C. kluyveri and Moorella thermoacetica couple the NADH-dependent reduction of NADP+ to the ferredoxin-dependent reduction of NADP+ (48, 49), and the heterodisulfide reductase complex from Methanothermobacter marburgensis couples the endergonic ferredoxin reduction with H2 to the exergonic reduction of a heterodisulfide with H2 (50). Recently, a hydrogen-activating, electron-bifurcating hydrogenase was purified and characterized from A. woodii (19). The enzyme oxidizes hydrogen only when NAD+ and ferredoxin are present, and both substrates are reduced in a stoichiometry of 1:1. This activity is strictly dependent on the presence of FMN. This hydrogenase couples the FMN-dependent exergonic reduction of NAD+ with H2 to the FMN-dependent endergonic reduction of ferredoxin with the same electron donor using flavin-based electron bifurcation. Here, we have described the caffeyl-CoA reductase-Etf complex, which couples the exergonic reduction of caffeyl-CoA with NADH to the endergonic reduction of ferredoxin with the same electron donor by flavin-based electron bifurcation.
The caffeyl-CoA reductase-Etf complex was the last missing link in our understanding of the enzymology and bioenergetics of caffeate respiration in A. woodii (Fig. 8). This remarkable example of microbial life under extreme energy limitations has only one coupling site, the sodium-motive ferredoxin:NAD+ oxidoreductase (Rnf) and two soluble enzymes that conserve energy in the form of reduced ferredoxin that fuels the ferredoxin:NAD+ oxidoreductase. Oxidation of 1 mol of hydrogen yields 0.5 mol of NADH and 0.5 mol of reduced ferredoxin. Oxidation of the latter via the ferredoxin:NAD+ oxidoreductase may be coupled to the translocation of one Na+ (Fig. 8) across the membrane. The NADH is then oxidized by the caffeyl-CoA reductase-Etf complex, and electrons are bifurcated to caffeyl-CoA and ferredoxin. The reduced ferredoxin is re-oxidized by the ferredoxin:NAD+ oxidoreductase, which gives rise to the translocation of two more Na+ ions. Thus, the internal Rnf loop accounts for up to 60% of the total amount of ions translocated during this respiration. In sum, 3 mol of Na+ are translocated (Equation 5).
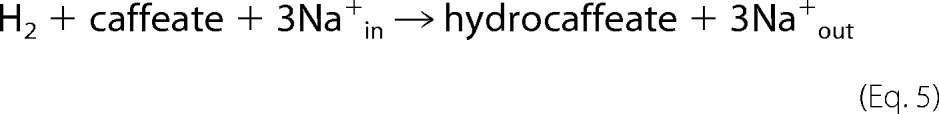 |
Considering that the Na+ F1F0-ATP synthase of A. woodii has 10 ion-binding sites (14, 16, 51), this makes 3.3 ions/ATP, and thus, caffeate respiration yields 0.9 mol of ATP/mol of caffeate. This is in good agreement with the experimental data (21).
Acknowledgments
We thank Dr. Julian Langer (Max Planck Institute of Biophysics, Frankfurt am Main, Germany) for performing the mass spectroscopy. CO dehydrogenase from A. woodii was a gift from K. Schuchmann.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft.
- DTE
- dithioerythritol.
REFERENCES
- 1. Müller V. (2008) Bacterial fermentation. in Encyclopedia of Life Sciences, John Wiley & Sons Ltd., Chichester, United Kingdom, DOI: 10.1002/9780470015902.a0001415.pub2 [DOI] [Google Scholar]
- 2. Schmitz R. A., Daniel R., Deppenmeier U., Gottschalk G. (2006) The anaerobic way of life. in The Prokaryotes (Dworkin M., Falkow S., Rosenberg E., Schleifer K. H., Stackebrandt E., eds) Vol. 2, pp. 86–101, Springer-Verlag, New York [Google Scholar]
- 3. Dimroth P., von Ballmoos C. (2008) ATP synthesis by decarboxylation phosphorylation. Results Probl. Cell Differ. 45, 153–184 [DOI] [PubMed] [Google Scholar]
- 4. Gottschalk G., Thauer R. K. (2001) The Na+-translocating methyltransferase complex from methanogenic archaea. Biochim. Biophys. Acta 1505, 28–36 [DOI] [PubMed] [Google Scholar]
- 5. Buckel W. (2001) Sodium ion-translocating decarboxylases. Biochim. Biophys. Acta 1505, 15–27 [DOI] [PubMed] [Google Scholar]
- 6. Boiangiu C. D., Jayamani E., Brügel D., Herrmann G., Kim J., Forzi L., Hedderich R., Vgenopoulou I., Pierik A. J., Steuber J., Buckel W. (2005) Sodium ion pumps and hydrogen production in glutamate fermenting anaerobic bacteria. J. Mol. Microbiol. Biotechnol. 10, 105–119 [DOI] [PubMed] [Google Scholar]
- 7. Imkamp F., Biegel E., Jayamani E., Buckel W., Müller V. (2007) Dissection of the caffeate respiratory chain in the acetogen Acetobacterium woodii: indications for a Rnf-type NADH dehydrogenase as coupling site. J. Bacteriol. 189, 8145–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biegel E., Schmidt S., Müller V. (2009) Genetic, immunological and biochemical evidence of a Rnf complex in the acetogen Acetobacterium woodii. Environ. Microbiol. 11, 1438–1443 [DOI] [PubMed] [Google Scholar]
- 9. Schmidt S., Biegel E., Müller V. (2009) The ins and outs of Na+ bioenergetics in Acetobacterium woodii. Biochim. Biophys. Acta 1787, 691–696 [DOI] [PubMed] [Google Scholar]
- 10. Biegel E., Müller V. (2010) Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poehlein A., Schmidt S., Kaster A.-K., Goenrich M., Vollmers J., Thürmer A., Bertsch J., Schuchmann K., Voigt B., Hecker M., Daniel R., Thauer R. K., Gottschalk G., Müller V. (2012) An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS ONE 7, e33439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heise R., Reidlinger J., Müller V., Gottschalk G. (1991) A sodium-stimulated ATP synthase in the acetogenic bacterium Acetobacterium woodii. FEBS Lett. 295, 119–122 [DOI] [PubMed] [Google Scholar]
- 13. Reidlinger J., Mayer F., Müller V. (1994) The molecular structure of the Na+-translocating F1F0-ATPase of Acetobacterium woodii, as revealed by electron microscopy, resembles that of H+-translocating ATPases. FEBS Lett. 356, 17–20 [DOI] [PubMed] [Google Scholar]
- 14. Aufurth S., Schägger H., Müller V. (2000) Identification of subunits a, b, and c1 from Acetobacterium woodii Na+-F1F0-ATPase. Subunits c1, c2, and c3 constitute a mixed c-oligomer. J. Biol. Chem. 275, 33297–33301 [DOI] [PubMed] [Google Scholar]
- 15. Fritz M., Müller V. (2007) An intermediate step in the evolution of ATPases–the F1F0-ATPase from Acetobacterium woodii contains F-type and V-type rotor subunits and is capable of ATP synthesis. FEBS J. 274, 3421–3428 [DOI] [PubMed] [Google Scholar]
- 16. Fritz M., Klyszejko A. L., Morgner N., Vonck J., Brutschy B., Muller D. J., Meier T., Müller V. (2008) An intermediate step in the evolution of ATPases: a hybrid F1-V0 rotor in a bacterial Na+ F1F0 ATP synthase. FEBS J. 275, 1999–2007 [DOI] [PubMed] [Google Scholar]
- 17. Biegel E., Schmidt S., González J. M., Müller V. (2011) Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68, 613–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schlegel K., Welte C., Deppenmeier U., Müller V. (2012) Electron transport during aceticlastic methanogenesis by Methanosarcina acetivorans involves a sodium-translocating Rnf complex. FEBS J. 279, 4444–4452 [DOI] [PubMed] [Google Scholar]
- 19. Schuchmann K., Müller V. (2012) A bacterial electron-bifurcating hydrogenase. J. Biol. Chem. 287, 31165–31171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bache R., Pfennig N. (1981) Selective isolation of Acetobacterium woodii on methoxylated aromatic acids and determination of growth yields. Arch. Microbiol. 130, 255–261 [Google Scholar]
- 21. Tschech A., Pfennig N. (1984) Growth yield increase linked to caffeate reduction in Acetobacterium woodii. Arch. Microbiol. 137, 163–167 [Google Scholar]
- 22. Hess V., Vitt S., Müller V. (2011) A caffeyl-coenzyme A synthetase initiates caffeate activation prior to caffeate reduction in the acetogenic bacterium Acetobacterium woodii. J. Bacteriol. 193, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herrmann G., Jayamani E., Mai G., Buckel W. (2008) Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hess V., González J. M., Parthasarathy A., Buckel W., Müller V. (2013) Caffeate respiration in the acetogenic bacterium Acetobacterium woodii: a CoA loop saves energy for caffeate activation. Appl. Environ. Microbiol. 79, 1942–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li F., Hinderberger J., Seedorf H., Zhang J., Buckel W., Thauer R. K. (2008) Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buckel W., Thauer R. K. (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113 [DOI] [PubMed] [Google Scholar]
- 27. Heise R., Müller V., Gottschalk G. (1992) Presence of a sodium-translocating ATPase in membrane vesicles of the homoacetogenic bacterium Acetobacterium woodii. Eur. J. Biochem. 206, 553–557 [DOI] [PubMed] [Google Scholar]
- 28. Heise R., Müller V., Gottschalk G. (1989) Sodium dependence of acetate formation by the acetogenic bacterium Acetobacterium woodii. J. Bacteriol. 171, 5473–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bryant M. P. (1972) Commentary on the Hungate technique for culture of anaerobic bacteria. Am. J. Clin. Nutr. 25, 1324–1328 [DOI] [PubMed] [Google Scholar]
- 30. Hungate R. E. (1969) A roll tube method for cultivation of strict anaerobes. in Methods in Microbiology (Norris J. R., Ribbons D. W., eds.) pp. 117–132, Academic Press, New York [Google Scholar]
- 31. Schönheit P., Wäscher C., Thauer R.K. (1978) A rapid procedure for the purification of ferredoxin from Clostridia using polyethylenimine. FEBS Lett. 89, 219–222 [DOI] [PubMed] [Google Scholar]
- 32. Bradford M. M. (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 33. Fish W. W. (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364 [DOI] [PubMed] [Google Scholar]
- 34. Beinert H. (1983) Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 35. Hetzel M., Brock M., Selmer T., Pierik A. J., Golding B. T., Buckel W. (2003) Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur. J. Biochem. 270, 902–910 [DOI] [PubMed] [Google Scholar]
- 36. Bertini I., Donaire A., Feinberg B. A., Luchinat C., Piccioli M., Yuan H. (1995) Solution structure of the oxidized 2[4Fe-4S] ferredoxin from Clostridium pasteurianum. Eur. J. Biochem. 232, 192–205 [DOI] [PubMed] [Google Scholar]
- 37. Duée E. D., Fanchon E., Vicat J., Sieker L. C., Meyer J., Moulis J. M. (1994) Refined crystal structure of the 2[4Fe-4S] ferredoxin from Clostridium acidurici at 1.84 Å resolution. J. Mol. Biol. 243, 683–695 [DOI] [PubMed] [Google Scholar]
- 38. Thauer R. K., Jungermann K., Decker K. (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41, 100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsai M. H., Saier M. H., Jr. (1995) Phylogenetic characterization of the ubiquitous electron transfer flavoprotein families ETF-α and ETF-β. Res. Microbiol. 146, 397–404 [DOI] [PubMed] [Google Scholar]
- 40. Crane F. L., Beinert H. (1956) On the mechanism of dehydrogenation of fatty acyl derivatives of coenzyme A. II. The electron-transferring flavoprotein. J. Biol. Chem. 218, 717–731 [PubMed] [Google Scholar]
- 41. Toogood H. S., Leys D., Scrutton N. S. (2007) Dynamics driving function: new insights from electron transferring flavoproteins and partner complexes. FEBS J. 274, 5481–5504 [DOI] [PubMed] [Google Scholar]
- 42. Roberts D. L., Frerman F. E., Kim J. J. (1996) Three-dimensional structure of human electron transfer flavoprotein to 2.1 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 93, 14355–14360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roberts D. L., Salazar D., Fulmer J. P., Frerman F. E., Kim J. J. (1999) Crystal structure of Paracoccus denitrificans electron transfer flavoprotein: structural and electrostatic analysis of a conserved flavin binding domain. Biochemistry 38, 1977–1989 [DOI] [PubMed] [Google Scholar]
- 44. Sato K., Nishina Y., Shiga K. (2003) Purification of electron-transferring flavoprotein from Megasphaera elsdenii and binding of additional FAD with an unusual absorption spectrum. J. Biochem. 134, 719–729 [DOI] [PubMed] [Google Scholar]
- 45. Swanson M. A., Usselman R. J., Frerman F. E., Eaton G. R., Eaton S. S. (2008) The iron-sulfur cluster of electron transfer flavoprotein-ubiquinone oxidoreductase is the electron acceptor for electron transfer flavoprotein. Biochemistry 47, 8894–8901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watmough N. J., Frerman F. E. (2010) The electron transfer flavoprotein:ubiquinone oxidoreductases. Biochim. Biophys. Acta 1797, 1910–1916 [DOI] [PubMed] [Google Scholar]
- 47. Schut G. J., Adams M. W. (2009) The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191, 4451–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang S., Huang H., Moll J., Thauer R. K. (2010) NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192, 5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang H., Wang S., Moll J., Thauer R. K. (2012) Electron bifurcation involved in the energy metabolism of the acetogenic bacterium Moorella thermoacetica growing on glucose or H2 plus CO2. J. Bacteriol. 194, 3689–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaster A. K., Moll J., Parey K., Thauer R. K. (2011) Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 108, 2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brandt K., Müller D. B., Hoffmann J., Hübert C., Brutschy B., Deckers-Hebestreit G., Müller V. (2013) Functional production of the Na+ F1F0 ATP synthase from Acetobacterium woodii in Escherichia coli requires the native AtpI. J. Bioenerg. Biomembr. 45, 15–23 [DOI] [PubMed] [Google Scholar]