

Abstract
Background and Purpose
Hypoxia-mediated neovascularization plays an important role in age-related macular degeneration (AMD). There are few animal models or effective treatments for AMD. Here, we investigated the effects of the flavonoid silibinin on hypoxia-induced angiogenesis in a rat AMD model.
Experimental Approach
Retinal pigmented epithelial (RPE) cells were subjected to hypoxia in vitro and the effects of silibinin on activation of key hypoxia-induced pathways were examined by elucidating the hypoxia-inducible factor-1 alpha (HIF-1α) protein level by Western blot. A rat model of AMD was developed by intravitreal injection of VEGF in Brown Norway rats, with or without concomitant exposure of animals to hypoxia. Animals were treated with oral silibinin starting at day 7 post-VEGF injection and AMD changes were followed by fluorescein angiography on days 14 and 28 post-injection.
Key Results
Silibinin pretreatment of RPE cells increased proline hydroxylase-2 expression, inhibited HIF-1α subunit accumulation, and inhibited VEGF secretion. Silibinin-induced HIF-1α and VEGF down-regulation required suppression of hypoxia-induced phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (mTOR) pathway. In the rat model of AMD, silibinin administration prevented VEGF- and VEGF plus hypoxia-induced retinal oedema and neovascularization.
Conclusion and Implications
The effects of silibinin, both in vitro and in vivo, support its potential as a therapeutic for the prevention of neovascular AMD.
Keywords: silibinin, age-related macular degeneration, neovascularization, hypoxia, HIF-1α
Introduction
Age-related macular degeneration (AMD) is a chronic and multifactorial disease of the elderly, with an incidence of 17.5% in people aged >75 years (Klein et al., 2007). AMD patients experience reduced quality of life resulting from the irreversible retinal degeneration and blindness (Klein et al., 2011). Unfortunately, effective treatments remain scarce. Photodynamic therapy is an invasive method and has a number of systemic adverse side effects (Mozaffarieh et al., 2009). AMD pathogenesis occurs in four major steps; lipofuscinogenesis, drusogenesis, inflammation and neovascularization. In the dry form of AMD, drusen accumulates in the retinal pigment epithelium (RPE) and leads to atrophy of the epithelial layer. Over time, some patients with dry form AMD develop the wet form of the disease (neovascular AMD), which is characterized by abnormal retinal or choroidal angiogenesis (Bird, 2010).
Recent studies indicate that degeneration of retinal blood vessels in the elderly results in oxygen deprivation; thus, hypoxia-mediated signalling may play a critical role in AMD progression (Arjamaa et al., 2009). Hypoxic conditions are associated with rapid accumulation of the transcription factor hypoxia-inducible factor-1α (HIF-1α) and induction of hypoxia-inducible genes that participate in hematopoiesis (e.g. erythropoietin), glucose metabolism (e.g. glucose transporter 1), angiogenesis (e.g. VEGF), cell proliferation and apoptosis (DeClerck and Elble, 2010). Inactivation of HIF-1 is associated with defects in embryonic vascularization. HIFs are heterodimers composed of an O2-dependent α subunit and a constitutively expressed β subunit, the aryl hydrocarbon receptor nuclear translocator (ARNT). In essentially all cell types examined, HIF-1α subunits undergo oxygen-dependent hydroxylation on specific proline residues (catalysed by proline hydroxylase) and is then recognized by the von Hippel–Lindau E3 ubiquitin ligase (VHL) and targeted for proteasomal degradation. In low-oxygen conditions, the non-hydroxylated form of the HIF-1α subunit escapes degradation and binds to the constitutively expressed HIF-1β ARNT subunit through the PER-ARNT-SIM and helic-loop-helix domains. The HIF-1α/β heterodimers then bind to hypoxia-response elements (HREs) in the promoter regions of target genes, activating their transcription (Mole et al., 2009).
Accumulation of HIF-1α protein is induced by both cytokines and hypoxia and has been shown to involve the PI3K/Akt/mammalian target of rapamycin (mTOR) signalling pathway (Garcia-Maceira and Mateo, 2009). mTOR, a member of the PI3K-related kinase superfamily, appears to play a central role in signalling induced by depletion of oxygen, nutrients and energy. mTOR activation increases mRNA translation by activating the ribosomal protein S6 kinase (p70S6K), eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and eukaryotic elongation factor 2 kinase. RPE-associated neovascularization in dry form AMD is known to be strongly associated with hypoxia (Salminen et al., 2010). RPE damage stimulates the secretion of angiogenic factors through HIF-1α-dependent and -independent pathways (Kernt et al., 2010). VEGF is a critical angiogenic factor and has been implicated in the development of several ophthalmic diseases in humans such as retinopathy of prematurity, diabetic retinopathy, and AMD (Abu El-Asrar et al., 2011; Gonzalez Viejo et al., 2011), as well as in retinal neovascularization models in rats (Caldwell et al., 2005; Skondra et al., 2008; Sawada et al., 2010). It is likely that induced neovascularization triggers haemorrhage and oedema in the local macula, worsening the condition.
Silibinin is a bioactive polyphenolic flavonoid isolated from milk thistle (Silybum marianum) and has been used as a traditional medicine, especially for protection of liver function (Mayer et al., 2005; Beinhardt et al., 2011). In cancer models, silibinin treatment not only reduces free radical generation, but also inhibits endothelial and tumour cell proliferation, and thus shows anti-cancer and anti-angiogenic therapeutic potential (Zi and Agarwal, 1999; Varghese et al., 2005; Singh et al., 2008). In this study, we investigated the anti-neovascularization effects of silibinin in a Brown Norway (BN) rat model of AMD induced by intravitreal injection of VEGF, or VEGF injection followed by long-term exposure of animals to hypoxia. The molecular mechanisms of action of silibinin were studied in vitro using cultured RPE cells. We show that silibinin inhibits hypoxia-induced PI-3 K/Akt/mTOR/p70S6K signalling and reduces VEGF secretion in vitro, and rescues VEGF-mediated retinal neovascularization in vivo.
Methods
Chemicals
SP600125, LY294002, PD98059 and rapamycin were obtained from Calbiochem (San Diego, CA, USA). Silibinin, sodium fluorescein and all other chemicals used in this study were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and hypoxia treatment
Human RPE cells were obtained from American Type Culture Collection (Manassas, VA, USA). RPE cells were maintained in DMEM medium supplemented with 10% heat-inactivated FBS and were cultured in a humidified atmosphere of 5% CO2 at 37°C. For analysis of signalling pathways, RPE cells were preincubated with the appropriate inhibitors and/or silibinin for 30 min, and then conditioned in a hypoxia chamber with 95% N2 and 5% CO2 at 37°C (Anaerobic System ProOx model 110; BioSpherix, Lacona, NY, USA) for the indicated times.
Cell viability determination
Cell viability was assessed by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Carmichael et al., 1987). RPE cells were treated with different concentrations of silibinin for 24 h. After incubation, 50 μL·well–1 MTT (0.5 mg mL−1) was added and cells were incubated at 37°C for an additional 2 h. Finally, the medium was gently removed and the formazan product was dissolved in 100 μL dimethylsulfoxide. Absorption was measured at 570 nm using an ELISA plate reader (MRX-TC; Dynex Technology, Chantilly, VA, USA). The readings were corrected for background absorbance by subtracting the readings of a blank treatment. The data presented are from at least three independent assays.
Immunofluorescence
RPE cells were fixed with 4% paraformaldehyde in PBS for 30 min and stained for LC3A antibody (Cell Signaling, Beverly, MA, USA) and for nuclei with hoechst 33258 (Steinheim, Germany). Then the fluorescent changes were observed by Carl Zeiss Axio Imager a1 (Göttingen, Germany).
Western blot analysis
After incubation with silibinin, cells (seeded in 60-mm dishes) were washed twice with ice-cold PBS and lysed in RIPA lysis buffer [50 mM Tris-HCl (pH 7.4), 1% Nonidet P-40, 150 mM NaCl, 1 mM EGTA, 1 mM NaF, 1 mM Na3VO4 and protease inhibitor cocktail]. The whole-cell lysate was prepared by 14 000× g centrifugation, and the supernatant was collected. Protein concentration was determined using Braford reagent (Bio-Rad, Hercules, CA, USA). Aliquots of whole lysates were mixed with 4 × sample loading buffer (250 mM Tris-HCl, 8% SDS, 40% glycerin, β-mercaptoethanol, 0.5% bromophenol blue, pH 6.8), and separated by reducing 10% SDS-PAGE and electrotransferred onto PVDF membranes according to the manufacturer's procedures (PerkinElmer Life Sciences, Boston, MA, USA). The PVDF membrane was blocked in nonfat milk and incubated in TBST with antibodies specific to phospho-Akt, Akt, phospho-JNK, JNK and VEGF (Santa Cruz Biotechnology, Santa Cruz, CA, USA); Becilin-1, Atg-7, phosphor-p38, p38, phospho-mTOR, mTOR and phospho-p70S6 kinase (Cell Signaling); HIF-1α (BD Transduction, Erembodegem, Belgium); and β-actin (Sigma-Aldrich). For chemiluminescent detection, blots were incubated with HRP-conjugated secondary antibodies (1:5000 in TBST; Cayman Chemical, Ann Arbor, MI, USA) for 2 h at room temperature, followed by chemiluminescent HRP substrate detection according to the manufacturer's protocol (Millipore Corporation, Billerica, MA, USA).
Immunoprecipitation
Total cell lysates (1 mg protein mL−1 for each sample) were precleared by incubating for 2 h at 4°C with 20 μL of protein A magnetic beads (Millipore). Supernatants were collected, 1 μg mL−1 polyclonal rabbit anti-VHL antibody (Genetex Inc., Irvine, CA, USA) was added, and the samples were rotated overnight at 4°C. Protein A magnetic beads (20–30 μL) were then added and the samples were rotated for a further 2 h at 4°C. The bead-conjugated immune complexes were collected by centrifugation and washed three times with RIPA buffer before addition of 50–100 μL of 2 × SDS sample buffer and heating at 95°C for 5 min. The associated proteins were separated on 10% SDS-PAGE and immunodetection was performed as described later.
RNA isolation and reverse-transcriptase polymerase chain reaction (RT-PCR) assay
Total RNA was isolated by Tripure reagent (Roche Molecular Biochemicals, Basel, Switzerland) as described by the manufacturer's protocol. To analyse HIF-1α and VEGF messenger mRNA expression, an RT-PCR was performed and evaluated with a housekeeping gene, β-actin, as the internal control. First-strand cDNA was synthesized from 6 μg of total RNA at 42°C for 50 min. The cDNAs were amplified in a DNA thermal cycler (MJ Research, Watertown, MA, USA) using the following program: denaturation for 5 min at 95°C, followed by 35 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 52°C, and extension for 40 s at 72°C; with a final extension for 10 min at 72°C. The PCR primers were: HIF-1α sense: 5′-gctggccccagccgctggag-3′, antisense: 5′-gagtgcagggtcagcactac-3′; VEGF sense: 5′-aaggaggagggcagaatcat-3′, antisense: 5′-cacacaggatggcttgaaga-3′; β-actin sense: 5′-tgacccagatcatgtttgag-3′, antisense: 5′-tcatgaggtagtcagtcagg-3′. The PCR products were separated by 2% agarose gel electrophoresis and visualized by ethidium bromide staining.
VEGF quantification
VEGF secreted into the culture medium was measured by using an ELISA kit (R&D Systems, Minneapolis, MN, USA). RPE cells were plated in 60-mm dishes and cultured to 80–90% confluence. The growth medium was replaced with serum-free medium and cultures were treated as indicated in the text. VEGF present in 1-mL aliquots of culture medium was quantified at the indicated times, according to the ELISA manufacturer's instructions. Results were normalized to the amount of protein per well.
Animal husbandry and AMD model
BN rats (250–300 g body weight) were purchased from National Laboratory Animal Center (Taipei, Taiwan) and acclimated for 1 week in the housing room under controlled conditions of 12 h/12 h light/dark cycle, 23 ± 1°C temperature, and 39–43% relative humidity; water and food were available ad libitum. All procedures involving the use of animals complied with the Association for Research in Vision and Ophthalmology guidelines for the use of animals in ophthalmic and vision experimental research, and were approved by the Institutional Animal Care and Use Committee (approval number: NTUIACUC-20090024).
To establish the VEGF-induced retinal angiogenesis model, BN rats were intravitreally injected with 10 μg mL−1 VEGF recombinant protein (Pepro Tech, Rocky Hill, NJ, USA) in the right eye on day 1. The left eye was uninjected and served as a control. Silibinin (500 mg kg−1 body weight) was administered to the rats by oral gavage once daily starting on day 7. After five independent experiment, retinal angiogenesis were examined by fluorescein angiography on day 0 (prior to the VEGF injection), and again on days 14 and 28, as described later.
Choroidal neovascularization (CNV) evaluation by fluorescein angiography
Rats were anaesthetized with an intramuscular injection of ketamine (80 mg kg−1) and xylazine (10 mg kg−1). Before testing, the pupil of the right eye of each rat was dilated with atropine (0.125%, Sinphar, Ilan City, Taiwan). The eyes were covered with 2% Methocel gel (OmniVision, SA, Neuhausen, Switzerland) and the retinal and fluorescein angiography images were captured with a Micron III retinal imaging microscope (Phoenix Research Laboratories, San Ramon, CA, USA). For fluorescein angiography, 10% sodium fluorescein was injected intravenously and serial images were captured immediately (every 10 s for 10 min). The injected sodium fluorescein is completely excreted after 24 h. The images were quantified by Image-Pro software (Bethesda, MD, USA) to determine the extent of CNV formation and oedema area.
Statistical analysis
All data are expressed as the mean ± SD from at least three independent experiments (n ≥ 3). Statistically significant differences among groups were determined using one-way anova. A value of P < 0.05 was considered statistically significant.
Results
Silibinin treatment is not cytotoxic to RPE cells but inhibits hypoxia-induced autophagy
The viability of cultured RPE cells was assayed after 24 h incubation with silibinin (1, 5, 10, 50, 100 and 500 μM) by MTT. Silibinin treatment had no obvious toxic effect on RPE cell viability (Figure 1A) or morphology (Figure 1B) at any of the concentrations tested. Therefore, incubations of 24 h with 100 μM silibinin were used for the following experiments. To investigate whether silibinin could affect autophagy in RPE cells, The LC3A protein expression was detected by immunofluorescence assay (Figure 1C). Our data suggested silibinin inhibited hypoxia-induced autophagy in RPE cells.
Figure 1.
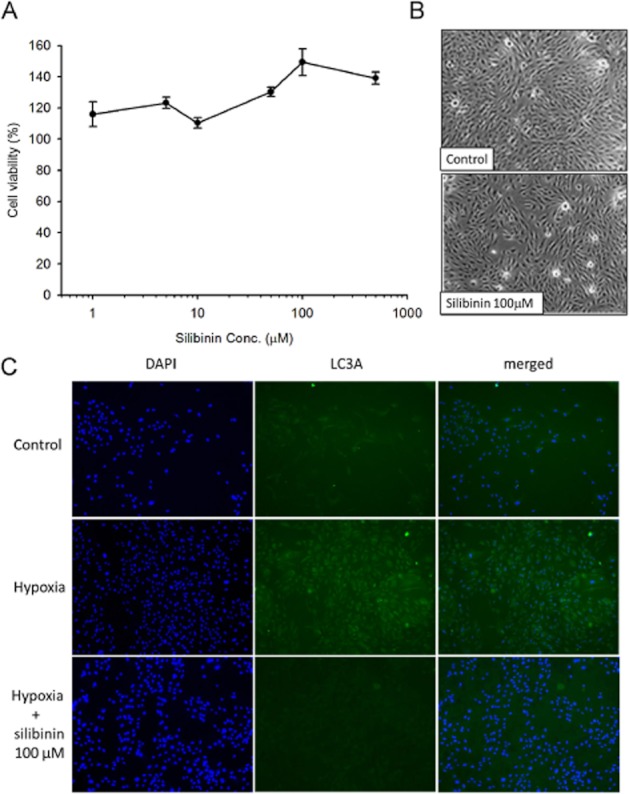
Silibinin is not cytotoxic to RPE cells. Cell viability (%) was measured by the MTT assay. Treatment of cells with silibinin (1–500 μM) for 24 h did not affect cell viability (A) or cell morphology (B) Autophagy was detected by immunofluorescence assay (C). Treatment of silibinin inhibited hypoxia-induced LC3A fluorescenceintensity. Data are mean ± SE from three independent experiments.
Silibinin inhibits HIF-1α protein accumulation, but does not affect hypoxia-induced VEGF mRNA expression
Incubation of RPE cells under hypoxic (1% O2) conditions resulted in a rapid and time-dependent accumulation of HIF-1α protein, which peaked at 6 h (3.1 ± 0.4-fold increase) and returned to basal levels at 12 h (Figure 2A). In comparison with the hypoxia-treated control cells, pretreatment of cells with silibinin inhibited hypoxia-induced HIF-1α accumulation, with complete inhibition being observed with 50 and 100 μM silibinin (Figure 2B). The expression of proline hydroxylase 2 (PHD2) was upregulated by silibinin pretreatment, whereas ARNT and VHL expression remained unaffected (Figure 2B). Incubation of cells under hypoxic conditions induced transactivation of an HRE luciferase reporter, which was also suppressed by pretreatment of the cells with silibinin (Figure 2C). Neither normoxia-conditioned (18% O2) nor hypoxia-conditioned RPE cells exhibited changes in HIF-1α or VEGF mRNA levels, even those pretreated and coincubated with silibinin (Figure 2D). These data suggest that silibinin reduces HIF-1α protein stability but does not alter their mRNA levels.
Figure 2.
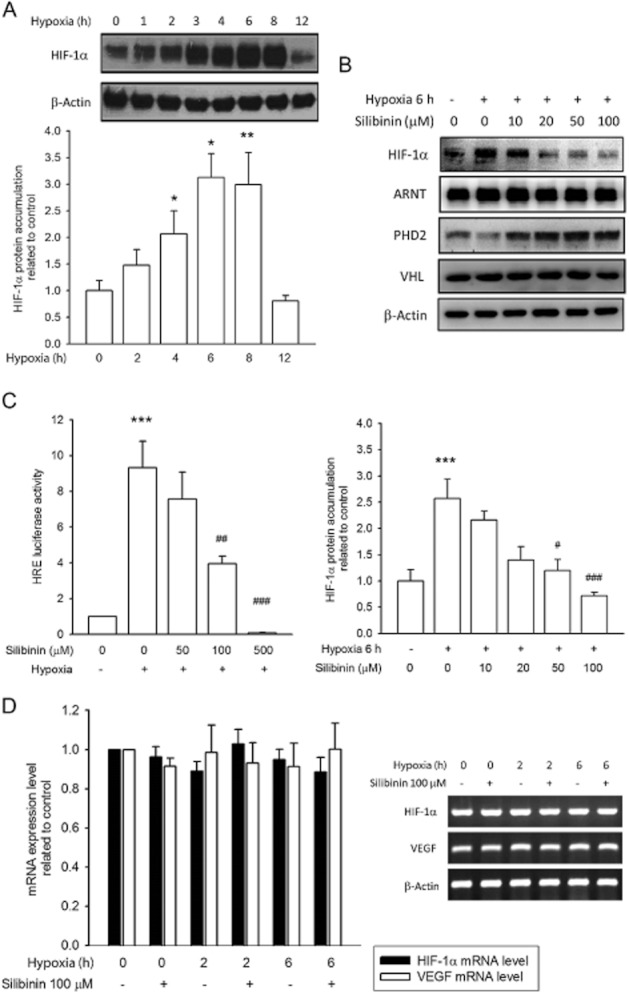
Silibinin inhibits HIF-1α protein accumulation, but not HIF-1α mRNA levels. (A) Images and quantification of Western blots show that 1% O2 hypoxia induces HIF-1α protein in RPE cells. (B) Hypoxia-induced HIF-1α protein accumulation, but not ARNT, PHD2, or VHL, was prevented by silibinin treatment. (C) Transactivation of an HRE reporter was increased in response to hypoxia, and this effect was inhibited by silibinin pretreatment. (D) HIF-1α and VEGF mRNA levels are unaffected by hypoxia or silibinin treatment. Data quantification was performed by densitometry and expressed as mean ± SE from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 compared with the hypoxia-treated control group.
HIF-1α and VHL protein–protein interactions are inhibited by silibinin
The interaction between HIF-1α and VHL proteins was evaluated by immunoprecipitation with anti-VHL antibodies followed by immunoblotting of the resolved complexes with anti-HIF-1α antibodies. We found that the HIF-1α/VHL association in RPE cells was increased by hypoxia (Figure 3A and B). However, the interaction was significantly reduced by treatment of cells with 100 μM silibinin, suggesting that silibinin treatment did not accelerate HIF-1α protein degradation through a proteasome-dependent pathway.
Figure 3.
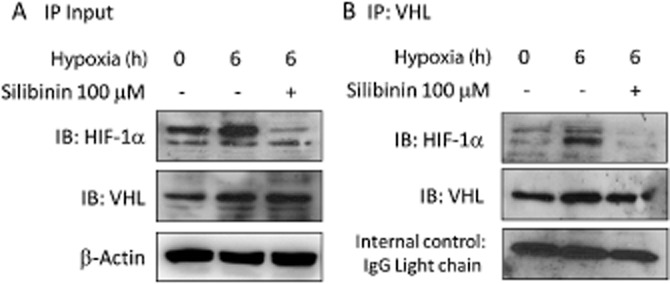
Association of HIF-1α and VHL proteins is prevented by silibinin. Formation of HIF-1α and VHL protein complexes was evaluated by immunoprecipitation and Western blotting. (A) RPE whole lysates used for immunoprecipitation were probed for their input HIF-1α and VHL levels. (B) Anti-VHL immune-complexes were analysed by blotting with anti-VHL and anti-HIF-1α antibodies. HIF-1α/VHL protein complex formation is increased by hypoxia and significantly reduced in the silibinin-pretreated group.
Silibinin inhibits HIF-1α protein accumulation by repressing signalling through the PI-3K/Akt/mTOR/p70S6K pathway
To identify the pathway involved in silibinin-induced down-regulation of HIF-1α, RPE cells were pretreated with specific signalling pathway inhibitors and then subjected to hypoxia. We found that hypoxia-induced HIF-1α protein accumulation was completely inhibited by treatment of cells with LY294002 (20 μM, PI-3K inhibitor) and rapamycin (20 nM, mTOR inhibitor), but was unaffected by treatment with PD98059 (20 μM, p42/p44 ERK inhibitor), SP600125 (20 μM, JNK inhibitor), or SB203580 (20 μM, p38-MAPK inhibitor) (Figure 4A). We also assessed the activation state of ERK, JNK, p38 Akt, mTOR and p70S6K in hypoxia-conditioned RPE cells with or without 100 μM silibinin by immunoblotting with antibodies against the phosphorylated forms of these enzymes. Akt and mTOR were clearly phosphorylated upon exposure to hypoxia (Figure 4B and C), with maximal phosphorylation of Akt (4.3-fold increase) occurring after a 3-h hypoxia and of mTOR (5.2-fold) after a 1-h hypoxia. Phosphorylation of both Akt and mTOR was inhibited to nearly basal levels by pretreatment of cells with 100 μM silibinin (Figure 4B and C). An earlier report linked mTOR to control of p70S6K activation in vivo and in vitro (Fenton and Gout, 2011). In this study, we observed asynchronous activation of mTOR and p70S6K in response to hypoxia, but silibinin treatment also effectively reduced p70S6K phosphorylation compared with the control group (Figure 4B and D). In contrast, neither ERK nor JNK and p38 phosphorylation was increased in hypoxia-treated RPE cells (Figure 4B). These data suggest that signalling through the PI3K/Akt/mTOR/p70S6K pathway is required for hypoxia-induced accumulation of HIF-1α protein and that silibinin treatment inhibited activation of this pathway and reduced HIF-1α protein levels.
Figure 4.
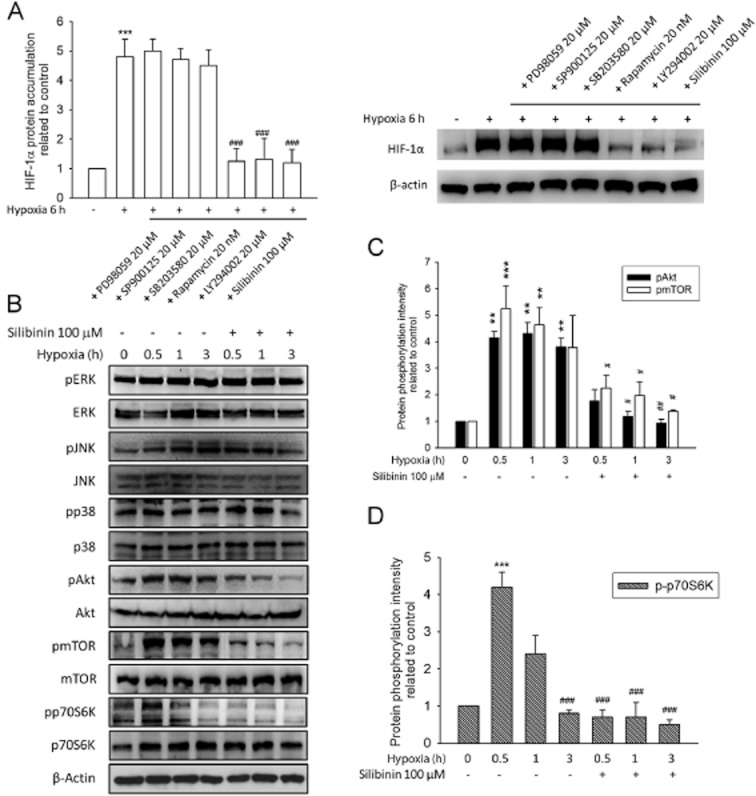
Silibinin inhibits HIF-1α protein accumulation by suppression of the PI3K/Akt/mTOR/p70S6K pathway. The expression of HIF-1α and phosphorylation of ERK, JNK, p38, Akt, mTOR, and p70S6K were evaluated by Western blotting. (A) Cultured RPE cells were pretreated with the inhibitors PD98059 (20 μM), SP600125 (20 μM), SB203580 (20 μM) LY294002 (20 μM), rapamycin (20 nM), and silibinin (100 μM) for 30 min and then exposed to hypoxic (1% O2) conditions for 6 h. The image and quantified data indicate that hypoxia-induced HIF-1α protein accumulation was prevented by LY294002, rapamycin, and silibinin pretreatment. (B) Western blot shows that silibinin pretreatment inhibited phosphorylation of Akt, mTOR, and p70S6K, but not that of ERK and JNK and p38. (C) Quantification of the data in (B) shows the phosphorylation intensity of Akt and mTOR. Both Akt and mTOR could be activated by hypoxia and silibinin reversed the hypoxia-mediated kinase phosphorylation (D) Quantification of the data in (B) showed that silibinin pretreatment inhibited the phosphorylation of p70S6K. Quantified data was analysed by densitometry and expressed as mean ± SE from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group. #P < 0.05, ##P < 0.01, and ###P < 0.001 compared with the hypoxia-treated control group.
Hypoxia-induced VEGF secretion is suppressed in silibinin-treated RPE cells
We next examined VEGF secretion by RPE cells, and found that hypoxia treatment increased VEGF secretion in a time-dependent manner (Figure 5A). Although silibinin treatment did not change VEGF mRNA levels (Figure 2D), quantities of secreted VEGF were significantly decreased by silibinin treatment (Figure 5B).
Figure 5.
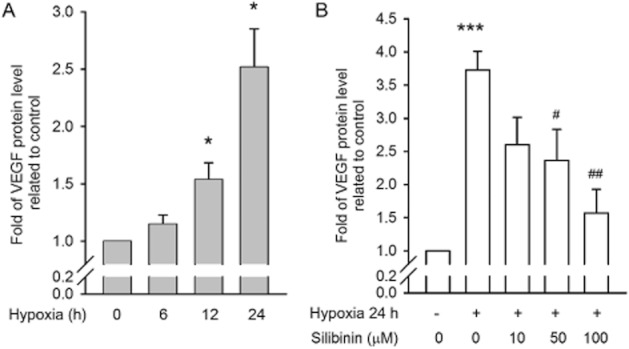
Hypoxia-induced VEGF secretion is suppressed by silibinin. VEGF protein secreted by cultured RPE cells was quantified by ELISA. (A) VEGF secretion was increased in a time-dependent manner in hypoxia-conditioned RPE cells. (B) Silibinin inhibits hypoxia-induced VEGF release. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group. #P < 0.05, ##P < 0.01, and ###P < 0.001 compared with the hypoxia-treated control group.
Silibinin prevents VEGF-induced and VEGF plus hypoxia-induced retinal oedema and neovascularization in a rat AMD model
Figure 6A shows a schematic of the VEGF-induced and VEGF plus hypoxia-induced rat model of retinal angiogenesis. Fluorescein angiography of VEGF-injected eyes was performed on days 14 and 28 post-injection. A significant increase in the permeability of retinal tissues was observed, as indicated by diffusion of fluorescein from the vessels, and resulted in retinal oedema and plexuses of microvascular angiogenesis (Figure 6B). Silibinin treatment of VEGF-injected animals rescued the oedema, retinal permeability and microvascular angiogenesis (Figure 6C). Additional groups of rats were injected with VEGF and then housed in a hypoxia chamber with 10% O2 for 28 days. This treatment synergized with VEGF to increase the retinal oedema and microvascular angiogenesis compared with that seen in response to the VEGF injection alone (Figure 6D). In addition, the diameter and length of the retinal vessels was obviously increased by hypoxia exposure. As was observed for the VEGF-induced retinal damage, treatment of animals with silibinin prevented the AMD-related microvascular angiogenesis and oedema in the VEGF plus hypoxia-induced model (Figure 6E). Figure 6F, G and H showed the quantification of the day 0, 14 and 28 in vessel length, vessel width and the oedema area. These data demonstrate that treatment of animals with silibinin delayed or protected the animals from AMD-related syndromes.
Figure 6.

Silibinin prevents VEGF- and VEGF plus hypoxia-induced retinal oedema and neovascularization in a rat AMD model. (A) Schematic of the VEGF-induced and VEGF plus hypoxia-induced retinal angiogenesis models in the BN rat. Fluorescein angiography was performed on day 0, 14 and 28. (B) In the group receiving intravitreal VEGF, the treated eye revealed diffusion of fluorescein from the vessel, representing increased permeability of retinal tissues (indicated by circle) and retinal oedema (indicated by arrow). (C) Silibinin treatment (500 mg kg−1 on days 7–28) decreased the VEGF-induced oedema, retinal permeability, and microvascular angiogenesis. (D) Animals injected with VEGF and exposed to hypoxia (10% O2) for 28 days show synergistic effects on retinal oedema (indicated by circle and arrow). The diameter and length of the retinal vessels were obviously increased. (E) Silibinin treatment of animals injected with VEGF and exposed to hypoxia (10% O2) for 28 days exhibit delayed and diminished AMD-related microvascular angiogenesis and oedema. The quantification of the day0, day14 and day 28 vessel length, vessel width and the oedema area were shown in Figure 6F, G and H. Data are expressed as mean ± SE from 5 independent experiments. ***P < 0.001 compared with the day 0 as control groups.
Discussion
The AMD Alliance International (AMDAI) has reported that the worldwide cost of vision loss (including people with low vision and blindness) was nearly USD $3 trillion in 2010. The cost due to AMD alone is estimated to be USD $343 billion, and is projected to rise dramatically through 2020 unless effective prevention and treatment modalities are adopted. Today, therapeutic strategies for AMD highlight the importance of early treatment to delay the progression of AMD. However, the development of therapies for AMD is restricted by the paucity of appropriate animal models. Laser-induced retinal choroidal angiogenesis and silver nitrite-induced corneal angiogenesis are popular in vivo models (Tobe et al., 1998; Edelman and Castro, 2000; Oh et al., 2009; Abdel-Rahman et al., 2010). However, both models are insufficient to elucidate the causes and factors involved in AMD development. In previous studies, exposure of mouse and zebrafish to hypoxia resulted in the formation of arterial sprouts and branches in the retina (Cao et al., 2008; Taylor et al., 2010). In the present study, we used a rat model in which intravitreal injection of VEGF combined with long-term exposure to hypoxia simulated the progression of AMD observed in humans. We found VEGF treatment alone increased the permeability of retinal tissue and oedema over 14–28 days, and housing in 10% O2 hypoxic conditions exacerbated the permeability and oedema and also increased vessel length and width. These data suggest that hypoxia might synergize with VEGF to induce microvascular angiogenesis in the eye. We also found that daily administration of silibinin prevented AMD-related symptoms in this rat model, supporting the potential therapeutic use of silibinin in AMD prevention.
We studied the molecular mechanism by which silibinin results in HIF-1α down-regulation and found that the protein accumulated rapidly in hypoxia-treated RPE cells, whereas HIF-1α mRNA was unaffected. HIF-1α is a key factor in cellular adaptation to low-oxygen conditions, and increases in HIF-1α protein stability and concentrations could enhance HIF-1α transactivation. In this study, silibinin treatment did not change expression of VHL in RPE cells, but did increase PDH2 levels. PHD2 is the most abundant isoform present in many cultured cells. The promoter region of PHD2 contains HREs that could be induced by hypoxia to serve as a negative feedback loop. Moreover, we found that silibinin reduced the association between HIF-1α and VHL in treated RPE cells, suggesting that silibinin-mediated HIF-1α down-regulation was unrelated to the proteasomal degradation pathway. The PI3K/Akt/mTOR pathway is a crucial regulator of cell proliferation and angiogenesis. This pathway contributes to HIF-1α protein accumulation and VEGF expression, in addition to its function in hypoxia-dependent HIF-1α up-regulation, activation of the epidermal growth factor receptor (EGFR) and PI3K, and inactivation of the tumour suppressor PTEN (Hudson et al., 2002; Thomas et al., 2006; Sudhagar et al., 2011; Befani et al., 2012). mTOR activation is sensitive to hypoxia, especially long-lasting hypoxic conditions. PI3K/mTOR activation is known to increase HIF-1α transactivation without altering its mRNA level (Jiang et al., 2010). In addition, mTOR activity is required for activation of p70S6K and modulates protein translation for a number of mRNAs that stimulate cell growth and angiogenesis. In this study, we found that the PI3K/Akt/mTOR/p70S6K pathway was activated in response to hypoxia in RPE cells. Pretreatment of cells with silibinin inhibited both the PI3K/Akt/mTOR/p70S6K pathway and HIF-1α accumulation. This is consistent with previous observations that silibinin-treated cancer cell lines exhibit reduced activation of the Akt/mTOR/p70S6K/4E-BP1 pathway, as well as increased instability of HIF-1α protein and decreased cell proliferation. These changes were reversed upon withdrawal of silibinin (Garcia-Maceira and Mateo, 2009). Together with previous reports, our study shows that suppression of the PI3K/Akt/mTOR/p70S6K pathway is essential for silibinin-induced HIF-1α down-regulation in RPE cells. In contrast, we saw no effect of silibinin on p42/p44 and p38 MAP kinases, which stimulate HIF-α activity without affecting its stability (Jing et al., 2012). Accordingly, inhibitors of these proteins did not inhibit hypoxia-induced HIF-1α protein accumulation.
It is reported that another mTOR-mediated autophage pathway leads to angiogenesis. In cancer cells, silibinin could induce autophagy through induction Becilin-1 and Atg-7 in HT1080, A431 and HeLa cells (Fan et al., 2011; Jiang et al., 2010). In our study, we found that a significant induction of autophagy was significantly induced in hypoxia-conditioned RPE cells, and data were detected by the LC3A fluorescence. In the autophagy-activated RPE cells, further administrated silibinin can reverse hypoxia-initiated autophagy induction. Although silibinin can inhibit mTOR pathway, which might lead us to predict the autophagy activation, but our data showed the regulation about mTOR and autophagy in hypoxia-conditioned RPE cells seems different. The detail mechanism needs further investigation.
In addition to its effect on VEGF, HIF-1α regulates numerous genes implicated in angiogenesis, such as placental growth factor, angiopoietin 1, angiopoietin 2, histone deacetylase 7, initiation factor 6 and β-arrestin 1 (Canales et al., 2006; Chen et al., 2009; Li et al., 2011; Shenoy et al., 2012). VEGF is one of the most important growth factors for tumourigenesis and angiogenesis. VEGF-A expression requires both PI3K and MAPK signalling, whereas VEGF-C expression is dependent on MAPK but not the PI3K/mTOR pathway (Luangdilok et al., 2011). In this study, we found that hypoxia-induced VEGF secretion in RPE cells, but did not affect VEGF mRNA levels. Inhibition of the PI3K/Akt/mTOR/p70S6K pathway by silibinin also reduced VEGF secretion in vitro, suggesting that silibinin might inhibit HIF-1α and VEGF mRNA translation. VEGF participates in many physiological and pathological events including vasculature formation, cell proliferation, inflammation and angiogenesis (Chung and Stadler, 2008; Halper, 2010; Kolar et al., 2011; Manalo et al., 2011). Early studies showed that VEGF rapidly induced phosphorylation of the tight-junction protein occludin, increased the permeability between endothelial cells, and resulted in retinal oedema, vascular haemorrhage and rhegmatogenous retinal detachment (Antonetti et al., 1999). In this study, silibinin administration to rats diminished VEGF-induced retinal vascular permeability and delayed retinal oedema until day 49 (data not shown). The data suggest that collapse of endothelial cell tight junctions might be involved in AMD pathogenesis, and this warrants investigation of the effects of silibinin on VEGF-stimulated endothelial cells.
Inhibition of hypoxia-induced PI3K/Akt/mTOR pathway has become a focus of anti-angiogenesis therapy. Rapamycin and NVP-BEZ235 (Novartis®), a dual PI3K/mTOR inhibitor, have been shown to inhibit VEGF production, proliferation, and VEGF-induced neovascularization in vivo (Zhong et al., 9006; Guba et al., 9003; Schnell et al., 9004). However, these drugs are generally cytotoxic, which limits their therapeutic application. In contrast, silibinin is widely used, has a long history in the treatment of liver diseases and is considered safer than other PI3K/mTOR inhibitors (Gu et al., 2007; Kaur and Agarwal, 2007; Cheung et al., 2010; Noh et al., 2011). Recently, silibinin has been investigated as an anti-cancer, anti-migration, anti-invasion and anti-angiogenesis agent (Singh et al., 2008). In this study, we firstly found that silibinin could inhibit hypoxia-induced VEGF secretion in RPE cells through suppression of the PI3K/Akt/mTOR/p70S6K pathway.
Innovatively, long-term hypoxic-conditioned animal experiments had been created up to 28 days, Micron III retinal microscope has been applied to observe the progression of AMD and a significant increased in the permeability of retinal tissues was observed. Comparing with physically laser-induced CNV, chronic whole body hypoxia could better mimic AMD progression.
We concluded that silibinin could protect against hypoxia- and VEGF-induced ocular neovascularization and retinal oedema in vivo. Collectively, our in vitro and in vivo data support the potential of silibinin for prevention of neovascular AMD.
Acknowledgments
This study was supported in part by a grant (NSC97-2320-B002-029-MY3) from the National Science Council, Taiwan.
Glossary
- AMD
age-related macular degeneration
- ARNT
aryl hydrocarbon receptor nuclear translocator
- HIF-1α
hypoxia-inducible factor-1α
- HRE
hypoxia-response element
- mTOR
mammalian target of rapamycin
- p70S6K
ribosomal protein S6 kinase
- PHD
proline hydroxylase
- RPE cell
retinal pigment epithelial cell
- VHL
von Hippel-Lindau protein
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- Abdel-Rahman MH, Yang Y, Salem MM, Meadows S, Massengill JB, Li PK, et al. Investigation of the potential utility of a linomide analogue for treatment of choroidal neovascularization. Exp Eye Res. 2010;91:837–843. doi: 10.1016/j.exer.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Abu El-Asrar AM, Nawaz MI, Kangave D, Siddiquei MM, Ola MS, Opdenakker G. Angiogenesis regulatory factors in the vitreous from patients with proliferative diabetic retinopathy. Acta Diabetol. 2011 doi: 10.1007/s00592-011-0330-9. (in press) [DOI] [PubMed] [Google Scholar]
- Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD) Ageing Res Rev. 2009;8:349–358. doi: 10.1016/j.arr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Befani CD, Vlachostergios PJ, Hatzidaki E, Patrikidou A, Bonanou S, Simos G, et al. Bortezomib represses HIF-1alpha protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med (Berl) 2012;90:45–54. doi: 10.1007/s00109-011-0805-8. [DOI] [PubMed] [Google Scholar]
- Beinhardt S, Rasoul-Rockenschaub S, Scherzer TM, Ferenci P. Silibinin monotherapy prevents graft infection after orthotopic liver transplantation in a patient with chronic hepatitis C. J Hepatol. 2011;54:591–592. doi: 10.1016/j.jhep.2010.09.009. author reply 592–593. [DOI] [PubMed] [Google Scholar]
- Bird AC. Therapeutic targets in age-related macular disease. J Clin Invest. 2010;120:3033–3041. doi: 10.1172/JCI42437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, et al. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6:511–524. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006;24:1115–1122. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- Cao R, Jensen LD, Soll I, Hauptmann G, Cao Y. Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PLoS ONE. 2008;3:e2748. doi: 10.1371/journal.pone.0002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- Chen L, Endler A, Shibasaki F. Hypoxia and angiogenesis: regulation of hypoxia-inducible factors via novel binding factors. Exp Mol Med. 2009;41:849–857. doi: 10.3858/emm.2009.41.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CW, Gibbons N, Johnson DW, Nicol DL. Silibinin – a promising new treatment for cancer. Anticancer Agents Med Chem. 2010;10:186–195. doi: 10.2174/1871520611009030186. [DOI] [PubMed] [Google Scholar]
- Chung EK, Stadler WM. Vascular endothelial growth factor pathway-targeted therapy as initial systemic treatment of patients with renal cancer. Clin Genitourin Cancer. 2008;6(Suppl. 1):S22–S28. doi: 10.3816/cgc.2008.s.004. [DOI] [PubMed] [Google Scholar]
- DeClerck K, Elble RC. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front Biosci. 2010;15:213–225. doi: 10.2741/3616. [DOI] [PubMed] [Google Scholar]
- Edelman JL, Castro MR. Quantitative image analysis of laser-induced choroidal neovascularization in rat. Exp Eye Res. 2000;71:523–533. doi: 10.1006/exer.2000.0907. [DOI] [PubMed] [Google Scholar]
- Fan S, Li L, Chen S, Yu Y, Qi M, Tashiro S, et al. Silibinin induced-autophagic and apoptotic death is associated with an increase in reactive oxygen and nitrogen species in HeLa cells. Free Radic Res. 2011;45:1307–1324. doi: 10.3109/10715762.2011.618186. [DOI] [PubMed] [Google Scholar]
- Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Garcia-Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene. 2009;28:313–324. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]
- Gonzalez Viejo I, Ferrer Novella C, Pueyo Royo V. [Use of anti-VEGF (anti-vascular endothelial growth factor) in retinopathy of prematurity (ROP)] Arch Soc Esp Oftalmol. 2011;86:207–208. doi: 10.1016/j.oftal.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67:3483–3491. doi: 10.1158/0008-5472.CAN-06-3955. [DOI] [PubMed] [Google Scholar]
- Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- Halper J. Growth factors as active participants in carcinogenesis: a perspective. Vet Pathol. 2010;47:77–97. doi: 10.1177/0300985809352981. [DOI] [PubMed] [Google Scholar]
- Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhu YS, Xu H, Sun Y, Li QF. Inflammatory stimulation and hypoxia cooperatively activate HIF-1α in bronchial epithelial cells: involvement of PI3K and NF-κB. Am J Physiol Lung Cell Mol Physiol. 2010;298:L660–L669. doi: 10.1152/ajplung.00394.2009. [DOI] [PubMed] [Google Scholar]
- Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, et al. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Agarwal R. Silymarin and epithelial cancer chemoprevention: how close we are to bedside? Toxicol Appl Pharmacol. 2007;224:350–359. doi: 10.1016/j.taap.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernt M, Neubauer AS, Liegl RG, Hirneiss C, Alge CS, Wolf A, et al. Sorafenib prevents human retinal pigment epithelium cells from light-induced overexpression of VEGF, PDGF and PlGF. Br J Ophthalmol. 2010;94:1533–1539. doi: 10.1136/bjo.2010.182162. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114:253–262. doi: 10.1016/j.ophtha.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Klein R, Chou CF, Klein BE, Zhang X, Meuer SM, Saaddine JB. Prevalence of age-related macular degeneration in the US population. Arch Ophthalmol. 2011;129:75–80. doi: 10.1001/archophthalmol.2010.318. [DOI] [PubMed] [Google Scholar]
- Kolar P, Gaber T, Perka C, Duda GN, Buttgereit F. Human early fracture hematoma is characterized by inflammation and hypoxia. Clin Orthop Relat Res. 2011;469:3118–3126. doi: 10.1007/s11999-011-1865-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ge C, Zhao F, Yan M, Hu C, Jia D, et al. Hypoxia-inducible factor 1 alpha-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin beta1 signaling in human hepatocellular carcinoma. Hepatology. 2011;54:910–919. doi: 10.1002/hep.24479. [DOI] [PubMed] [Google Scholar]
- Luangdilok S, Box C, Harrington K, Rhys-Evans P, Eccles S. MAPK and PI3K signalling differentially regulate angiogenic and lymphangiogenic cytokine secretion in squamous cell carcinoma of the head and neck. Eur J Cancer. 2011;47:520–529. doi: 10.1016/j.ejca.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Manalo KB, Choong PF, Becerra SP, Dass CR. Pigment epithelium-derived factor as an anticancer drug and new treatment methods following the discovery of its receptors: a patent perspective. Expert Opin Ther Pat. 2011;21:121–130. doi: 10.1517/13543776.2011.545347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer KE, Myers RP, Lee SS. Silymarin treatment of viral hepatitis: a systematic review. J Viral Hepat. 2005;12:559–567. doi: 10.1111/j.1365-2893.2005.00636.x. [DOI] [PubMed] [Google Scholar]
- Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarieh M, Schotzau A, Josifova T, Flammer J. The effect of ranibizumab versus photodynamic therapy on DNA damage in patients with exudative macular degeneration. Mol Vis. 2009;15:1194–1199. [PMC free article] [PubMed] [Google Scholar]
- Noh EM, Yi MS, Youn HJ, Lee BK, Lee YR, Han JH, et al. Silibinin enhances ultraviolet B-induced apoptosis in mcf-7 human breast cancer cells. J Breast Cancer. 2011;14:8–13. doi: 10.4048/jbc.2011.14.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EJ, Park K, Choi JS, Joo CK, Hahn SK. Synthesis, characterization, and preliminary assessment of anti-Flt1 peptide-hyaluronate conjugate for the treatment of corneal neovascularization. Biomaterials. 2009;30:6026–6034. doi: 10.1016/j.biomaterials.2009.07.024. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, Hyttinen JM, Toropainen E, Kaarniranta K. Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med. 2010;16:535–542. doi: 10.2119/molmed.2010.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada O, Miyake T, Kakinoki M, Sawada T, Kawamura H, Ohji M. Aqueous vascular endothelial growth factor after intravitreal injection of pegaptanib or ranibizumab in patients with age-related macular degeneration. Retina. 2010;30:1034–1038. doi: 10.1097/IAE.0b013e3181ce74c8. [DOI] [PubMed] [Google Scholar]
- Schnell CR, Stauffer F, Allegrini PR, O′Reilly T, McSheehy PM, Dartois C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68:6598–6607. doi: 10.1158/0008-5472.CAN-08-1044. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Han S, Zhao YL, Hara MR, Oliver T, Cao Y, et al. beta-arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene. 2012;31:282–292. doi: 10.1038/onc.2011.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RP, Gu M, Agarwal R. Silibinin inhibits colorectal cancer growth by inhibiting tumor cell proliferation and angiogenesis. Cancer Res. 2008;68:2043–2050. doi: 10.1158/0008-5472.CAN-07-6247. [DOI] [PubMed] [Google Scholar]
- Skondra D, Noda K, Almulki L, Tayyari F, Frimmel S, Nakazawa T, et al. Characterization of azurocidin as a permeability factor in the retina: involvement in VEGF-induced and early diabetic blood-retinal barrier breakdown. Invest Ophthalmol Vis Sci. 2008;49:726–731. doi: 10.1167/iovs.07-0405. [DOI] [PubMed] [Google Scholar]
- Sudhagar S, Sathya S, Lakshmi BS. Rapid non-genomic signalling by 17beta-oestradiol through c-Src involves mTOR-dependent expression of HIF-1alpha in breast cancer cells. Br J Cancer. 2011;105:953–960. doi: 10.1038/bjc.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AC, Seltz LM, Yates PA, Peirce SM. Chronic whole-body hypoxia induces intussusceptive angiogenesis and microvascular remodeling in the mouse retina. Microvasc Res. 2010;79:93–101. doi: 10.1016/j.mvr.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- Tobe T, Okamoto N, Vinores MA, Derevjanik NL, Vinores SA, Zack DJ, et al. Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci. 1998;39:180–188. [PubMed] [Google Scholar]
- Varghese L, Agarwal C, Tyagi A, Singh RP, Agarwal R. Silibinin efficacy against human hepatocellular carcinoma. Clin Cancer Res. 2005;11:8441–8448. doi: 10.1158/1078-0432.CCR-05-1646. [DOI] [PubMed] [Google Scholar]
- Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci U S A. 1999;96:7490–7495. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]