

Abstract
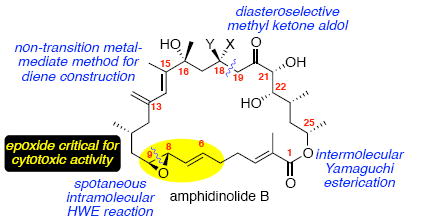
The total synthesis of amphidinolide B1 and the proposed structure of amphidinolide B2 has been accomplished. Key aspects of this work include the development of a practical, non-transition metal mediated method for the construction of the C13-C15 diene, the identification of α-chelation and dipole minimization models for diastereoselective methyl ketone aldol reactions, the discovery of a spontaneous Horner-Wadsworth-Emmons macrocyclization strategy and the development of a novel late stage method for construction of an allylic epoxide moiety. The originally proposed structure for amphidinolide B2 and diastereomers thereof display potent anti-tumor activities with IC50 values ranging from 3.3 nM to 94.5 nM against human solid and blood tumor cells. Of the different stereoisomers, the proposed structure of amphidinolide B2 is over 12-fold more potent than the C8,9-epimer and C18-epimer in human DU145 prostate cancer cells. These data suggest that the epoxide stereochemistry is a significant factor for anticancer activity.
Introduction
First reported in 1986, the amphidinolide family of natural products has long captured the attention of the scientific community.1 To date, over thirty members of this family have been isolated.2 Given their fascinating structures and diverse biological activity, these targets have attracted considerable attention in both the synthetic3,4,5 and biological communities.6
From this diverse broad collection of compounds, the amphidinolide B sub-family possesses some of the most intriguing structural features and biological activity. Kobayashi and co-workers reported the isolation of the 26-membered macrolide (amphidinolide B) from the dinoflagellate Amphidinium sp. in small amounts (Figure 1).2b The planar original structure was proposed as compound 1. Subsequent reisolation by Shimizu and co-workers as well as structure determination through X-ray crystallographic analysis by Clardy and co-workers provided the relative stereochemistry of amphidinolide B (which was renamed amphidinolide B1) as compound 2.2c In addition, the location of the methyl moiety of the dienyl system was reassigned to the C15 position. Absolute configuration of 2 was later established via degradation.2d Shimizu and co-workers also reported the isolation of two related members of this family – amphidinolide B2 (3) & B3 (4) and proposed their structures based on analogy to 2 and comparison on NMR spectra. More recently, Kobayashi and co-workers reported the isolation of additional members of this subfamily – amphidinolide B4 and B5 (5 and 6)2e as well as amphidinolides B6 and B7 (not shown).2f Structurally related amphidinolides G and H [e.g. amphidinolide H1 (7) and amphidinolide G1 (8)] have also been reported.2p-r In particular, amphidinolide B1 (2) has proven to be the most cytotoxic member of the amphidinolide family - demonstrating impressive potency in early cancer screening [IC50 levels: L1210 murine leukemia cell line (0.14 ng/mL),2b human colon tumor HCT 116 cell line (0.12 μg/mL)2c and KB cancer cell line (4.2 ng/mL)].1f
Figure 1.
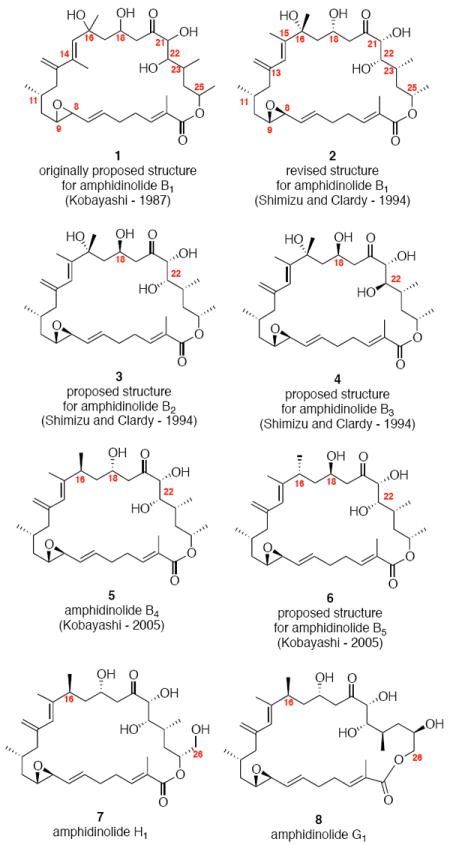
Amphidinolides B, G and H.
In addition to the intriguing biological activity, macrolide 2 has a compelling architecture – nine stereogenic centers embedded within a 26-membered macrocycle including a reactive allylic epoxide moiety at C6-C9 and a highly substituted diene moiety at C13-C15. Consequently, this target 2 has attracted considerable synthetic attention from numerous laboratories7 including our own.8,9,10 In 2008, we reported the first total synthesis of amphidinolide B1 (2) as well as the proposed structure of amphidinolide B2 (3) which we established to be incorrect.4 Subsequent to our efforts, Fürstner and coworkers published their synthesis of 2 in 20095 and more recently Nishiyama and co-workers completed their total synthesis in 2012.11 It should be noted that Fürstner reported the first syntheses of related amphidinolides G and H in 2007.3aa Herein, we disclose a full account of our work and the biological evaluation of synthesized analogues of amphidinolide B.
Results and Discussion
Our initial retrosynthetic strategy is shown in Scheme 1. We intended to install the fragile allylic epoxide moiety at a late stage from the corresponding C7-C9 enone motif. Macrolactonization of the corresponding seco acid 9 under Mitsunobu conditions12 could form the key 26-membered ring system. The C8-C9 bond would be accessible from a HWE olefination with the keto phosphonate 11 and aldehyde 10. The C18 stereogenic center could be constructed by a chelation-controlled aldol condensation between aldehyde 12 and α-benzyloxy ketone 13. The aldehyde 12 would be derived from the styrenyl compound 14 through a regioselective oxidative cleavage. The diene could be accessed from the coupling of an allylic metallo species such as 15 and the methyl ketone 16.
Scheme 1.
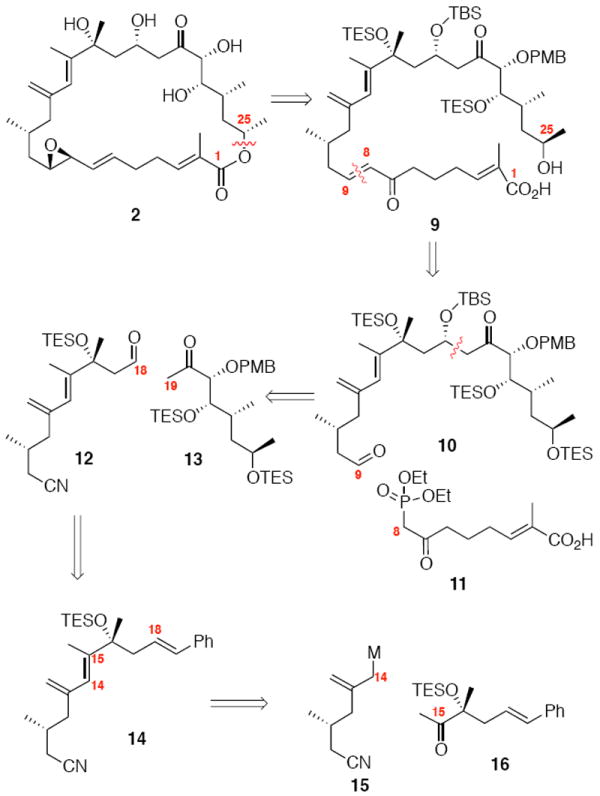
Initial Retrosynthetic Analysis of Amphidinolide B1.
Synthesis of the nucleophilic components 24-25 and styrenyl compound 16 are shown in Scheme 2. The methyl ketone 16 could be prepared in 4 steps from (L)-lactic acid (17). Using Seebach chemistry,13 stereochemistry originally contained in the hydroxyl acid 17 was transferred cleanly to the lactone 20 with high levels of diastereoselecitivity. Treatment of the lactone with MeLi yielded the α-hydroxy methyl ketone which was protected as its TES ether 16. For the nucelophilic component 15, we envisioned multiple options including phosphonate 25 and Peterson-type approaches (e.g. 24). Starting from the commercially available oxazolidinone 21 and known iodide 22,14 Evans alkylation followed by reduction and protection generated the allyl silane 24. Bromination at C14 using NBS followed by Arbuzov reaction generated the allyl phosphonate 25 in 55% yield over two steps.
Scheme 2.
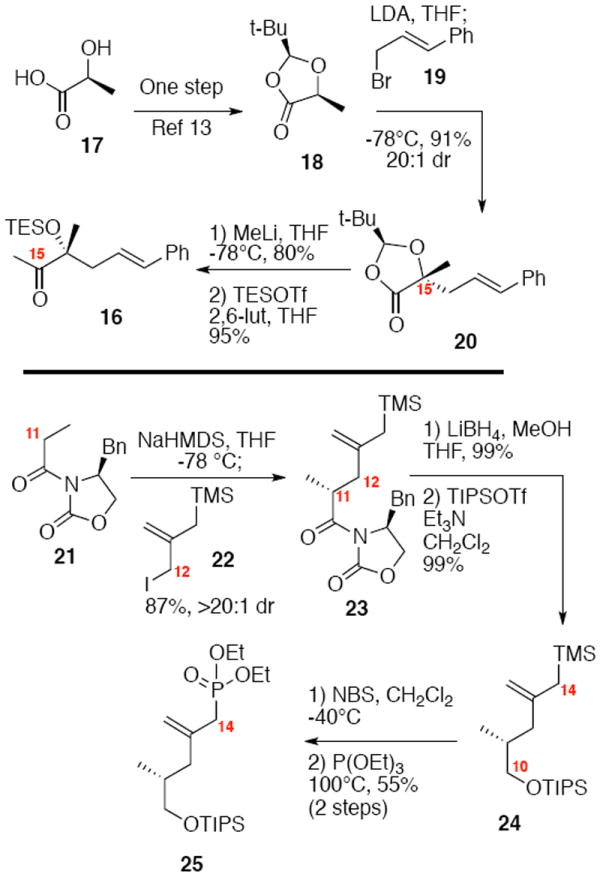
Synthesis of Diene Precursors.
With the coupling partners in hand, we envisioned multiple options for their combination (Scheme 3). One of the initial approaches we explored was a phosphonate olefination strategy. Unfortunately, using a range of bases (n-BuLi, t-BuLi, s-BuLi, KHMDS) we were unable to facilitate the olefination. An alternative strategy involving a Peterson olefination could arise at the same target 26 Anions derived from allyl trimethylsilanes can attack a carbonyl electrophile through either α-attack or γ-attack.15 Using conditions developed by Magnus and co-workers,16 we observed exclusive formation of the vinyl silane product 27 derived from reactivity in the γ position. A modified procedure using MgBr217 failed to override this preference. While the vinyl silane product 27 is in theory still a viable intermediate in the synthesis of the diene 26, we were unable to facilitate the subsequent conversion to the target diene.18 We also explored a Sakurai-type approach19 to C-C bond formation using allyl silane 24. Treatment of 24 and 16 under a range of Lewis acidic conditions (e.g. TMSOTf, BF3•Et2O, Me2AlCl, AlMe3, SnCl4) did not provide any of the desired coupled material 28. Fortunately, treatment with freshly distilled TiCl4 did facilitate the C14-C15 bond construction in up to 65% yield and 6:1 dr (stereochemistry at C15 undetermined). While effective, this reaction did prove to be scale dependent (typically providing 50-65% yield at 0.65 mmol scale, but 20-30% yield at 2.0 mmol scale). Despite considerable effort to ascertain the nature of the scale dependence, we were unable to fully address this shortcoming. Consequently, this transformation was often run in parallel batches to bring through sufficient amounts of material. Subsequent elimination of the 3° alcohol 28 was best facilitated using SOCl2 and pyridine in toluene at -78°C to yield the desired diene in near quantitative yield as a 2.2:1 ratio of diene regioisomers (rr) (26:29). The diene rr was not impacted by use of a single C15 stereoisomer of alcohol 28. Use of alternate conditions (MsCl, POCl3, Tf2O, Burgess reagent, Martin’s sulfurane) led to no product formation or inferior levels of regioselectivity. While these diene regioisomers 26 and 29 were not readily separable by column chromatrogrpahy, removal of the silyl protecting group provided the corresponding regioisomeric diols 30 and 31 that were separable.
Scheme 3.
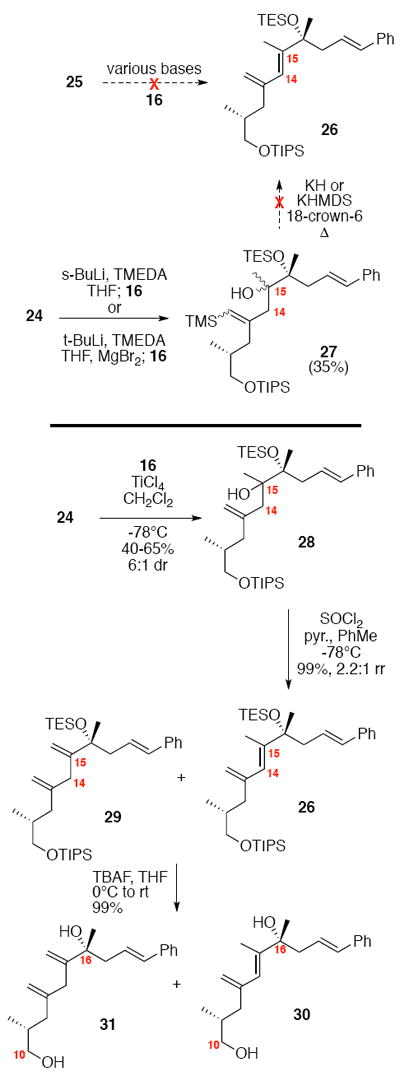
Synthesis of Diene.
The functionalization of the polyene is shown in Scheme 4. Mitsunobu-type conversion of the C10 alcohol into the corresponding nitrile followed by silyl protection yielded 14. Next, we required the selective functionalization of the C18-C19 alkene in the presence of the less sterically hindered 1,1- disubstituted alkene at C13 and the more electron rich alkene at C14-C15. We had hoped that regioselective dihydroxylation at C18-C19 could be possible by exploiting a π-stacking interaction between the phenyl ring and aromatic systems present in Sharpless ligands. While this sort of phenomenon has been used previously to explain enhanced enantioselectivity20 and even diastereoselectivity21 in Sharpless asymmetric dihydroxylation, we were unaware of prior examples to exploit purely a regioselective directing effect. To our delight, the AD Mix β* [the * denotion represents higher percentages of K2OsO4•2H2O and (DHQD)2PHAL as compared to the commercial amounts] provided excellent regioselectivity for the desired location. As hypothesized, use of standard OsO4 / NMO conditions provided a complex mixture of products. Interestingly, AD Mix α* also proved to be a poor reagent of this transformation. While we can rationalize the poor outcome of the ligandless system, a rational for the reactivity differences between the pseudo-enantiomeric ligand systems is less obvious. One possible explanation may be that the C16 stereocenter exerts a pronounced influence on the conformation of the neighboring alkenes – resulting in a mismatched interaction between the (DHQ)2PHAL ligand in AD mix α and the desired alkene. It is also clear that a conformational change occurs between the unconjugated system 33 and the conjugated system 14 – dihydroxylation of polyene 33 results in a regioselective oxidation at C18-C19; however, the reaction does not show any diastereoselectivity. Periodate cleavage under standard conditions yielded the aldehydes 12 and 35.
Scheme 4.
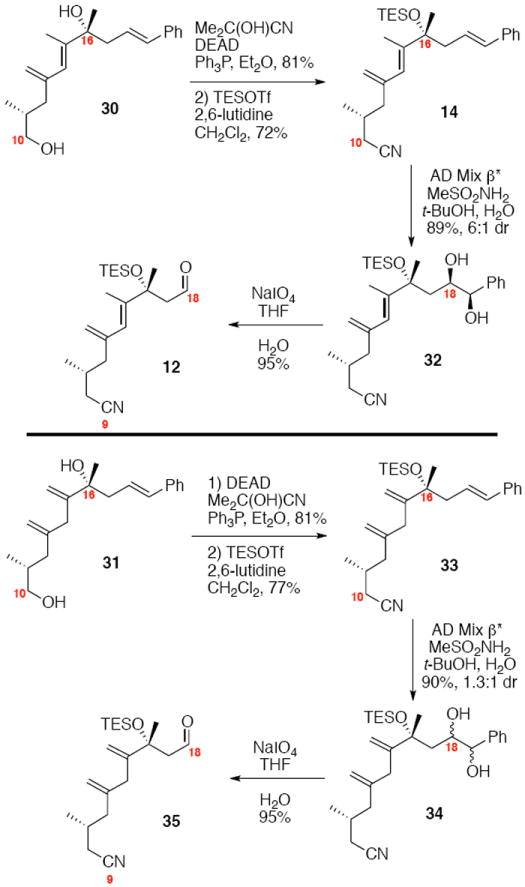
Synthesis of Western Subunit.
Synthesis of the eastern subunit4 and southern fragment are shown in Scheme 5. Evans syn glycolate reaction between oxazolidinone 3622 and readily available aldehyde 378 provided the desired product in reasonable diastereoselectivity (7:1 dr) and good yield. After TES silylation, subsequent conversion to the thioester with lithium thiolate followed by cuprate addition yielded the methyl ketone 13. This strategy for synthesis of the C19-C26 subunit has been subsequently exploited by Früstner and co-workers.3aa,5 The carboxylic acid 11 was available in four straightforward steps from the commercially available ester 38.
Scheme 5.
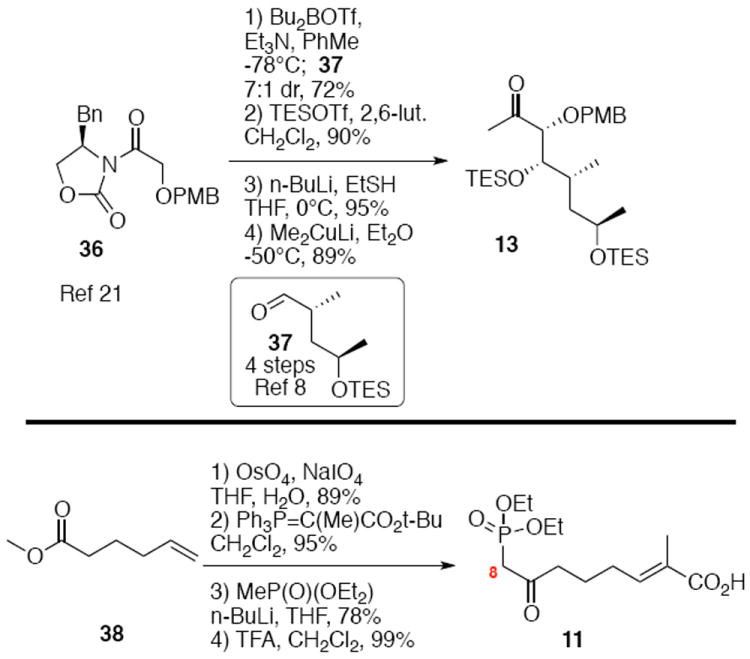
Synthesis of Eastern and Southern Subunits.
With the subunits now constructed, our efforts shifted towards their combination. We were particularly focused initially on addressing the critical diastereoselectivity in the methyl ketone aldol reaction between 12 and 13. Prior to our entry into the field, Pattenden7e and Kobayashi7k had independently explored related coupling strategies with limited success (Scheme 6). In both cases, poor diastereoselectivity was observed (3:2 dr) as well as low chemical yield (50% in Kobayashi’s case, no chemical yield reported in Pattenden’s example). Stereocontrolling models for β-silyloxy aldehydes such as 40 and 44 have been proposed;23 however, it is unclear if the tertiary nature of the C16 silyl ether is compatible with that analysis. In addition, models have been developed for exploiting the stereochemical controlling nature of α-chiral ketones (albeit primarily on ethyl ketone substrates). We had hypothesized the poor stereocontrol in these pioneering examples by Pattenden and Kobayashi could be attributed to the absence of a chelating protecting group at C21, which rigidifies the facial selectivity of the enolate in the aldol transition state. Compelling evidence of the potential for this strategy can be found in Chakraborty’s related work towards amphidinolides G and H (which lack the C16 hydroxyl functionality).24 Pioneering precedent for the stereocontrolling ability of α-oxy enolates had been reported by Paterson,25 Heathcock26 and Masamune;27 however, the majority of the examples are on ethyl ketones. Aldol reactions using methyl ketones have been reported to proceed through both chair and boat transition states – which could lead to an erosion in stereoinduction.28 White,29 Trost30 and Evans31 reported some of the earliest successful examples in this area. Other laboratories have subsequently explored this transformation with varying degrees of stereocontrol.32
Scheme 6.
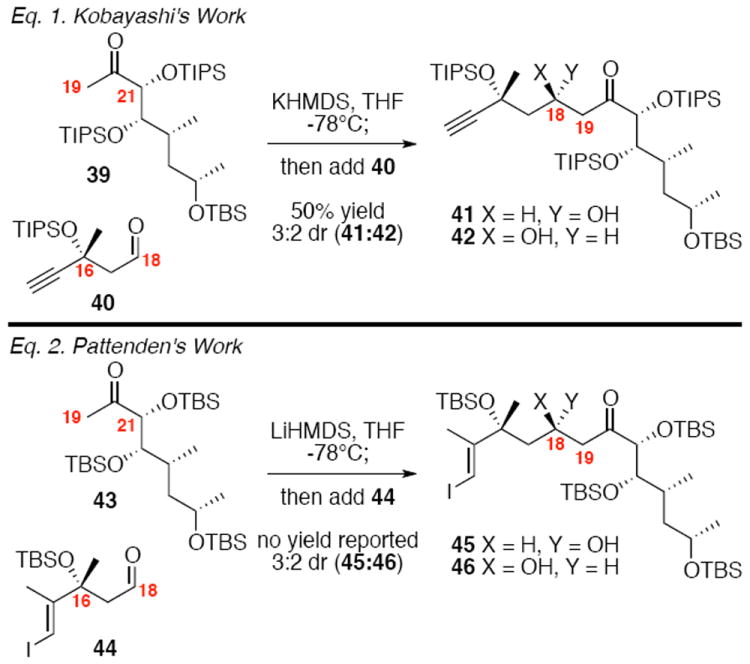
Precedent for C18-C19 Aldol Reaction.
Our exploration into the diastereoselective aldol reaction is detailed in Scheme 7. We initially employed the unconjugated diene 35 as a model for the actual system. To our considerable disappointment, this substrate 35 performed poorly in the transformation – providing low diastereoselectivity (2:1 dr) in modest chemical yield (60%) (Eq. 1). Similar results were observed with the achiral aldehyde 3,3-dimethylbutanal (2-3:1 dr) (Eq. 2). Undeterred by these discouraging model studies, we next studied the conjugated diene system 12 (Eq. 3). We were delighted to see that this transformation proceeded with high levels of stereocontrol – we only observed a single diastereomer in 69% yield. The configuration of the newly established stereocenter was confirmed by advanced Mosher ester method.33 The subtle nature of the controlling influences of this aldol reaction warrants further comment. We are hesitant to put forth a model for the controlling influences of this transformation; however, it is important to note that both the tri-substituted alkene at C14-C15 and the chelating protecting group at C21 appear to work in concert to influence the stereochemical outcome of the aldol reaction. The C16 silyl ether does not appear to exert considerable influence as this chelation strategy has subsequently proven useful in amphidinolide G/H series by both Fürstner3aa,5 and Zhao34 which does not contain the C16 alcohol moiety.
Scheme 7.
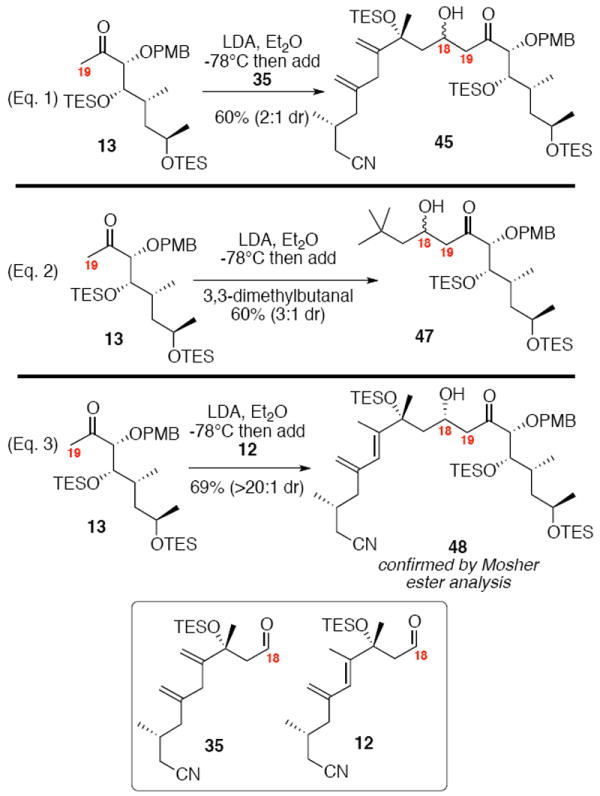
Methyl Ketone Aldol Reaction.
The construction of the macrocyclization precursor is shown in Scheme 8. Silylation of the C18 alcohol proceeded smoothly with TBSOTf and a large excess of triethylamine.35 Interestingly, use of near stoichiometric amounts of an amine base [e.g. 2,6-lutidine (1.2 equiv.)] led to elimination (≈20%) and alkene isomerization (≈10%). Regioselective reduction of the C9 nitrile was accomplished with DIBAL-H. Horner-Wadsworth-Emmons olefination of the resultant aldehyde using Ba(OH)236 yielded the desired α,β-unsaturated ketone in good yield and excellent E/Z selectivity. Alternate conditions on related systems (DBU, LiCl or KHMDS, THF) were less effective for this olefination.37 Subsequent selective cleavage of the C25 TES ether produced the key Mitsunobu macrolactonization12 precursor. Unfortunately, this substrate was resistant to all effort to facilitate the macrocycle formation under these sterochemically invertive conditions.
Scheme 8.
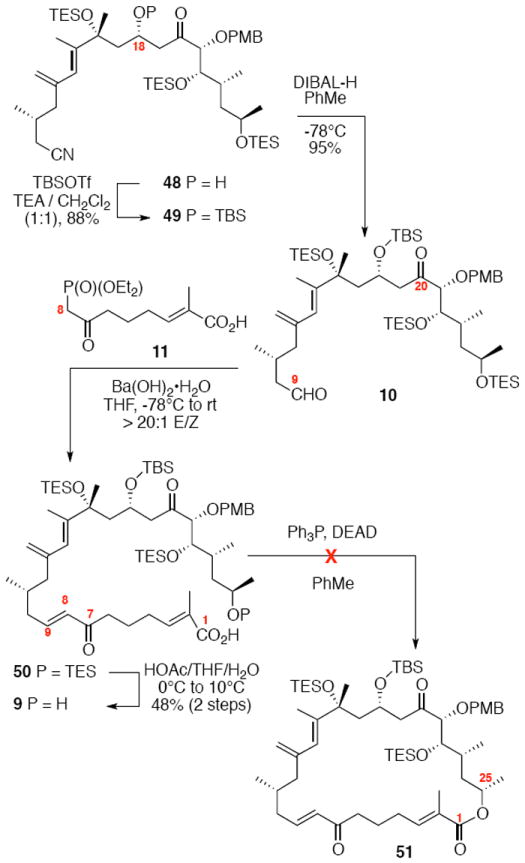
Unsuccessful Macrocyclization Strategy.
Based on our frustrations with facilitating a successful Mitsunobu macrolactonization, we decided a reorganization of our strategy for macrocyclic formation was needed. Specifically, we turned to inverting the C25 stereocenter prior to coupling, forming the C1 ester bond in an intermolecular fashion and constructing the C8-C9 alkene through an intramolecular pathway. Towards this end, synthesis of a modified methyl ketone 54 was necessary (Scheme 9). Selective TES removal could be accomplished under aqueous acidic conditions in excellent yield. Martin’s modified Mitsunobu conditions proceeded smoothly to give the inverted C25 ester. Finally, saponification of the PNB moiety with Ba(OH)2•8H2O and methanol followed by TMS protection revealed the coupling partner 54.
Scheme 9.
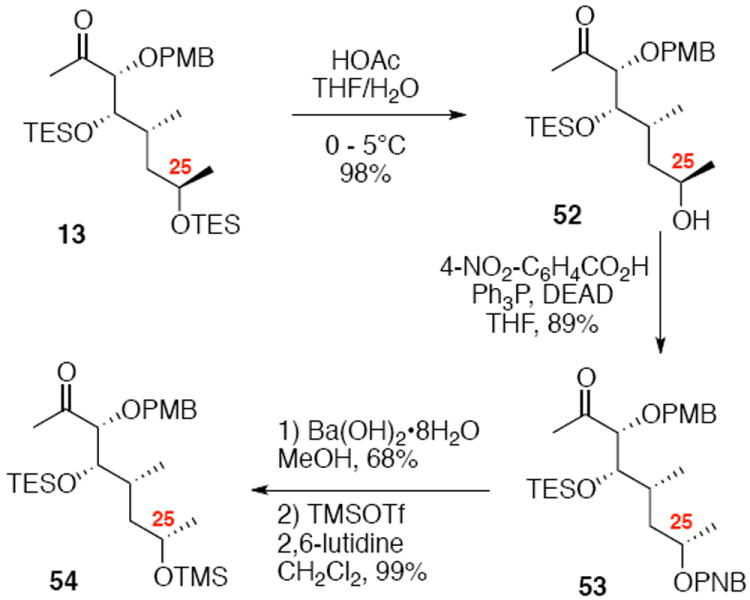
Second Generation Synthesis of Eastern Subunit.
Synthesis of the modified aldehyde coupling partner is illustrated in Scheme 10. Reduction of the C9 nitrile to its corresponding alcohol was accomplished via DIBAL-H reduction to the aldehyde followed by NaBH4 reduction to the alcohol and protection as its acetate. Regioselective functionalization at C18 was again accomplished using Sharpless AD conditions followed by periodate cleavage to reveal the C18 aldehyde.
Scheme 10.
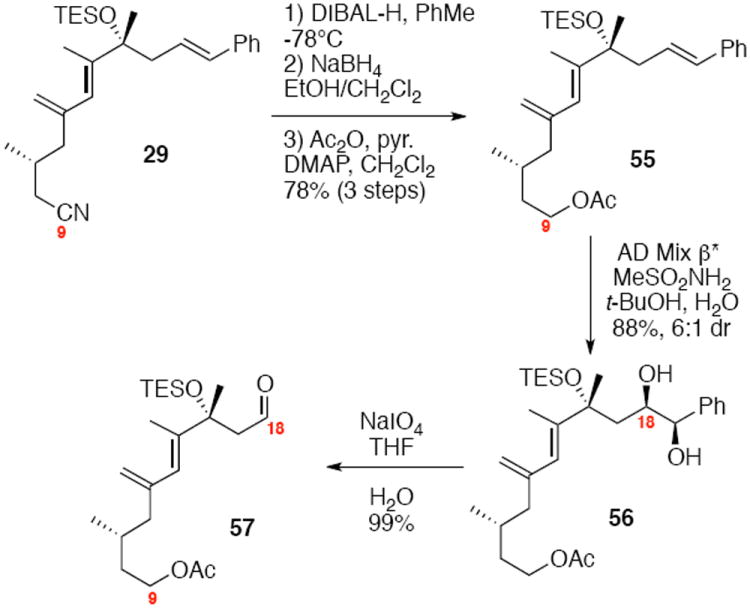
Modification of the Western Subunit.
While the original route to the aldehydes 12 and 57 was effective, it suffered from logistical practicality - notably the titanium-mediated coupling of the allyl silane proved to be a scale dependent reaction and the thionyl chloride elimination step gave a mixture of olefin products. Consequently, we sought to develop a more scalable approach to this subunit (Scheme 11). Starting from the commercially available Oppolzer sultam derivative 59, cuprate addition in accord with conditions developed by Paquette38 cleanly generated the key C11 stereocenter in compound 60. After functional group manipulation to provide aldehyde 61, we were gratified to find that key sequential Wittig / Horner-Wadsworth-Emmons (HWE) reactions cleanly introduced the triene 63 as a single stereoisomer. After DIBAL-H reduction, the resulting trienyl alcohol proved remarkably reactive to Sharpless asymmetric epoxidation conditions (proceeding at -78 °C to -50 °C) to provide epoxide 64. In fact, if this reaction was performed at higher temperatures (e.g. -20 °C), another product was observed that appeared to be the result of a 1,2-alkyl shift to produce an aldehyde.39 Subsequent Red-Al reduction provided the diol 65. Finally, protecting group exchange followed by 1° TES deprotection and oxidation provided the C9-C18 subunit 57. The high efficiency of this route (11.5% overall yield) provided us with access to gram quantities of aldehyde 57.
Scheme 11.
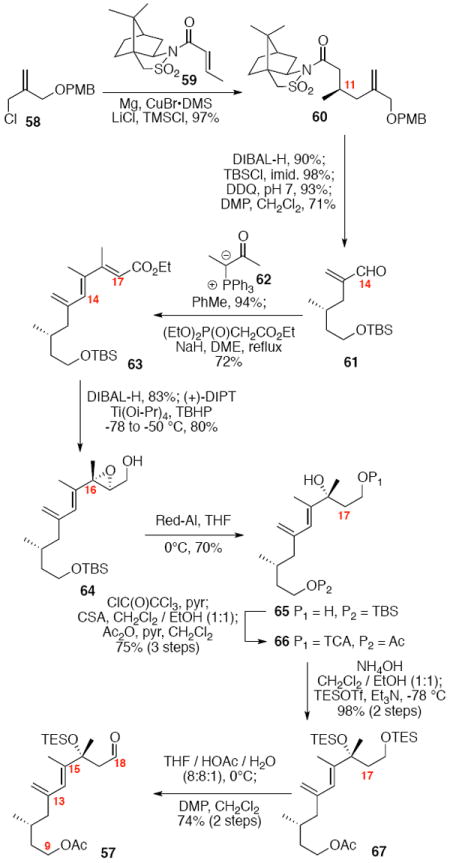
Second Generation Approach to the C9-C18 Western Fragment of Amphidinolide B1.
With these modified coupling partners in hand, we revisited our aldol coupling strategy (Scheme 12). We were pleased to observe that similar levels of diastereoselectivity continued to be observed using the LDA conditions (62% yield, >20:1 dr). Silylation of the C18 alcohol proved significantly more complicated than expected. Use of TBSOTf led to significant exchange of the C25 TMS ether to the corresponding TBS ether. Use of TESOTf minimized this problem – providing a 95% yield of a 5:1 ratio of the C25 TMS to C25 TES. Both of these compounds were productive intermediates and could be converted to the corresponding C25 alcohol using HOAc/THF/H2O conditions. Intermolecular Yamaguchi esterification provided the phosphonate 71. Next, removal of the C9 acetate was cleanly accomplished using Ba(OH)2•8H2O. Subsequent oxidation using TPAP provided the aldehyde cyclization precursor 73. Interestingly, this compound proved to be surprisingly reactive -leading to spontaneous macrocyclization under the TPAP conditions. Ba(OH)2•8H2O was added to drive this transformation to completion with an 85% overall yield from the alcohol 72.
Scheme 12.
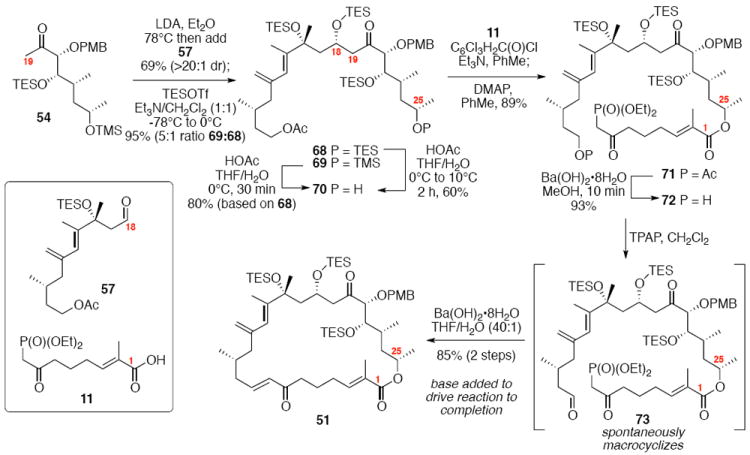
Spontaneous Macrocyclization Strategy.
With the macrocycle in hand, the remaining challenges to be addressed were the successful incorporation of the C6-C9 allylic epoxide and global deprotection (Scheme 13). CBS reduction40 proceeded smoothly to provide the desired allylic alcohol 75 in excellent diastereoselectivity. Subsequent Sharpless asymmetric epoxidation41 provided the desired epoxide 76 in 10:1 dr. Incorporation of the alkene could be best accomplished by inversion at C7 to provide the selenide 7742 followed by TPAP mediated oxidation43 to the selenoxide and syn elimination to yield the allylic epoxide as a single olefin geometry. Despite compelling precedent from Nicolaou and co-workers for a related deprotection in the synthesis of amphidinolide N,2v we were unable to effect PMB removal using DDQ under buffered aqueous or anhydrous conditions. Based on this unfortunate result, it became clear that an alternate C21 protecting group strategy was necessary to complete the total synthesis of amphidinolide B1.
Scheme 13.
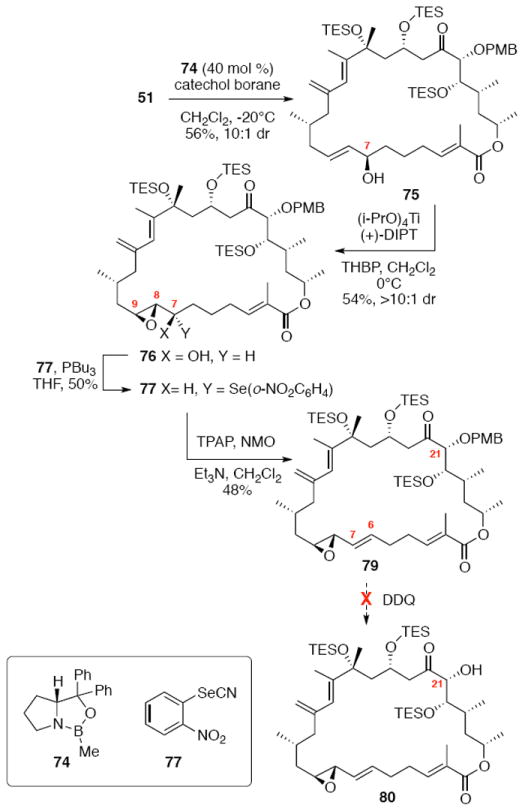
Incorporation of the Allylic Epoxide Moiety.
We initially considered a 3,4-dimethoxybenzyl (DMB) protecting group as a viable alternative to the C21 PMB group as DMB groups are more readily oxidized by DDQ (Scheme 14). We synthesized the necessary C19-C26 subunit via an analogous route as described previously. Interestingly, aldol reaction with the C18 aldehyde 82 proceeded smoothly, but in significantly lower diastereoselectivity (4:1 dr). We are unsure as to the cause of this difference between the PMB and DMB series. Next, we explored the possibility of deprotecting the C21 DMB group using DDQ. Interestingly, treatment of 83 with DDQ under buffer aqueous conditions led to formation of the acetal 86 in modest yield, but as a single diastereomer. This product is likely the result of initial oxidation to the oxonium ion followed by nucleophilic attack by the C22 OTES ether. Subsequent silyl migration to the C18 position would produce the observed product 86. Despite this encouraging early result, we were unable to increase the chemical yield of this transformation through variation of oxidant or reaction conditions. Consequently, this approach was abandoned.
Scheme 14.
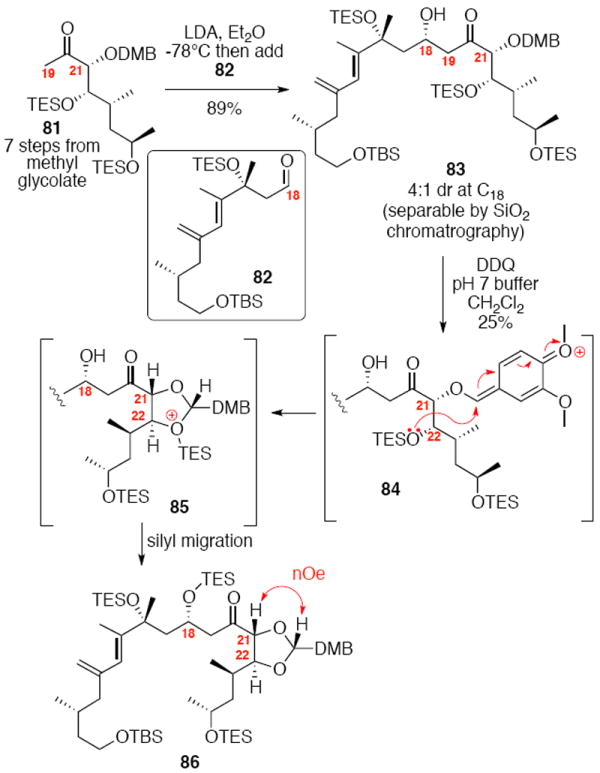
C21 Dimethoxybenzyl Protection Series.
Given our setbacks with benzyl-derived protection at C21, we revisited our central strategy for stereocontrol at C18 through the C21 chelation model described previously (Scheme 15). Traditionally, silyl protecting groups are not viewed as moieties that participate in chelation;44 however, it is important to acknowledge work by Heathcock,45 Eliel and Frye,46 Willard47 and Evans48 which showed that silyl chelation is feasible under certain conditions. We were particularly drawn to Heathcock’s work in the area in which TMEDA was a key additive in their chelation-controlled aldol reaction (Eq. 1). Alternatively, a dipole minimization stereocontrol model produced the alternate facial attack on the aldehyde (Eq. 2). Silyl protection at C21 would simplify the endgame deprotection sequence and circumvent the problematic DDQ deprotection strategy.
Scheme 15.
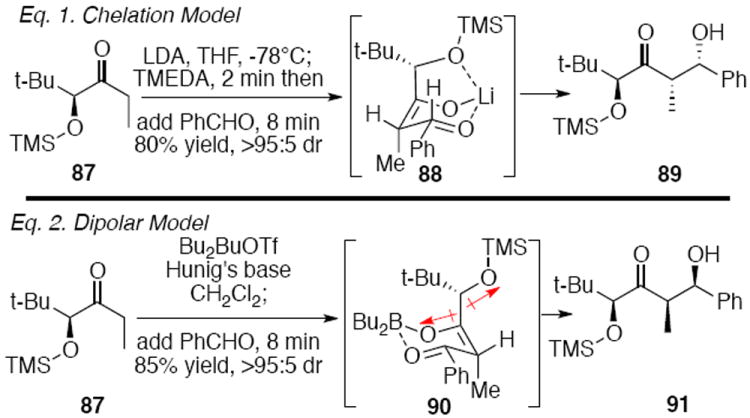
Heathcock’s Pioneering Stereoselective Aldol Work.
The synthesis of the C21 silyl methyl ketone 98 is shown in Scheme 16. One attractive advantage to this strategy is that it greatly simplifies the synthesis of the methyl ketone as differential protection at C21 and C22 is no longer needed. Olefination of aldehyde 37 provided the α,β-unsaturated enone 93. Sharpless dihydroxylation under buffered conditions yielded the diol in good diastereoselectivity. Bis-silyl protection using TESOTf cleanly provided the tris-TES methyl ketone 95. The C25 TES could be cleanly removed using HOAc/THF/H2O conditions followed by inversion and protection as its C25 TMS ether.
Scheme 16.
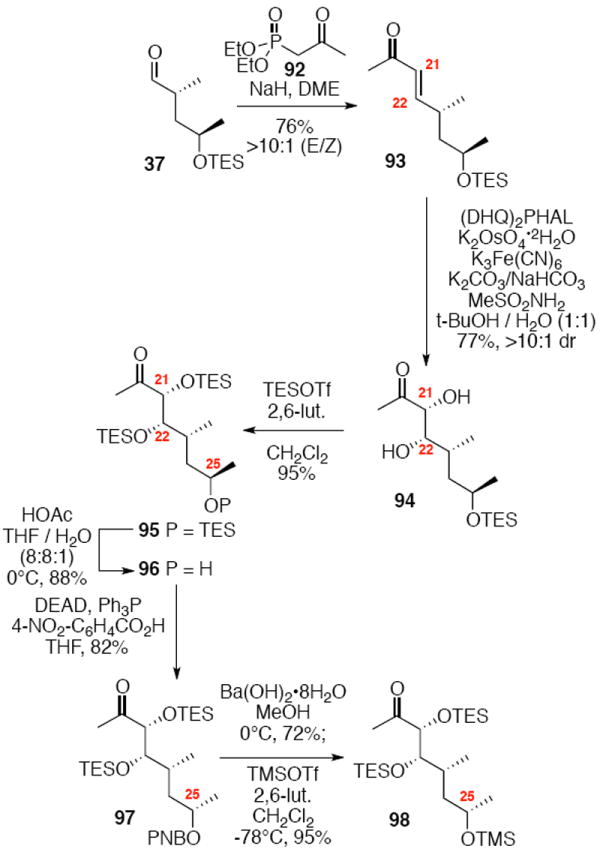
Tris-silyl Methyl Ketone Synthesis.
Exploration of the key aldol reaction on the C21 silyl series generated intriguing results (Table 1). Initial studies with the C25 epimer 95 exhibited poor diastereoselectivity, but good reactivity under the Heathcock’s TMEDA conditions (Entry 1). It should be noted that the replacement of THF with Et2O led to a dramatic decrease in chemical yield (Entry 2). Interestingly, use of the required C25 stereochemistry for amphidinolide B led to a more stereoselective aldol process (Entries 3-6). If the reaction was conducted at -78°C, the transformation appeared to proceed through a dipole minimization model to yield the 18R stereochemistry as the major product (5:1 103:100). If the otherwise identical transformation was conducted at -40°C, the stereoselectivity for the transformation reversed to now favor the 18S stereochemistry. This divergent stereocontrol model could be attributed to reversible nature of aldol reactions.49 A similar phenomenon was observed with the aldehyde 57 (Entries 5 and 6). While unexpected, these results opened the door for the synthesis of both amphidinolide B1 and B2.
Table 1.
Exploration into Aldol Stereoselectivity.
| entry | substrates | conditions | Yield [dr] |
|---|---|---|---|
| 1 | 95, 82 | THF, -78°C | 64% [1.1:1 (99:102)] |
| 2 | 95, 82 | Et2O, -78°C | <10% [1.5:1 (99:102)] |
| 3 | 98, 82 | THF, -78°C | 67% [1:5 (100:103)] |
| 4 | 98, 82 | THF, -40°C | 68% [1.8:1 (100:103)] |
| 5 | 98, 57 | THF, -100°C | 65% [1:8 (101:104)] |
| 6 | 98, 57 | THF, -40°C | 66% [1.2:1 (101:104)] |
Synthesis of the macrocycle using the aldol products was accomplished based on close analogy to our C21 PMB series (Scheme 18). TES protection at C18 using TESCl / DMAP conditions followed by C25 desilylation revealed the C25 alcohol. Yamaguchi esterification followed by saponification of the acetate protecting group produced the C9 alcohol. As employed in our previous macrocyclization, TPAP oxidation produced the reactive aldehyde, which appeared to undergo spontaneous macrocyclization. The cyclization could be driven to completion by addition of Ba(OH)2•8H2O for 18S series and LiCl / Hunig’s base for the 18R series for optimal yields. While the yields for these macrocyclizations were not as high as the C21 PMB series, the ability to successfully close the macrocycle was gratifying. Fortuitously, macrocycle 109 produced crystals suitable for X-ray crystallographic analysis – thereby confirming the stereochemical assignment (Figure 2).
Scheme 18.
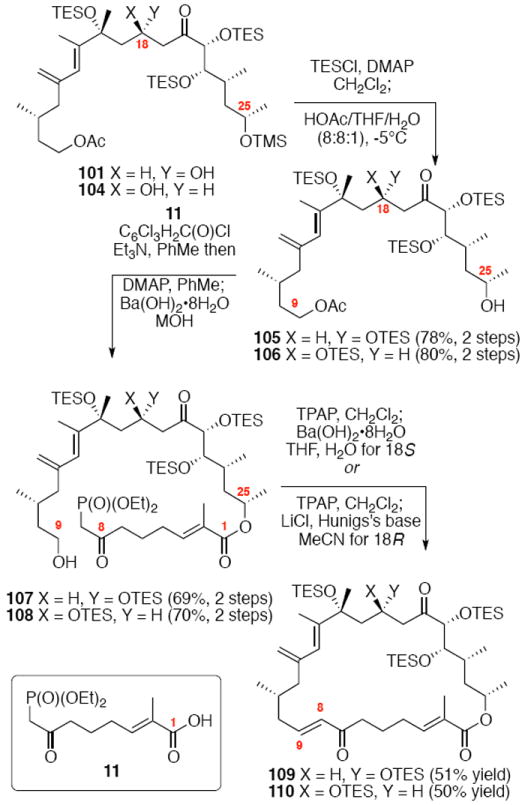
Second Generation Macrocycle Formation.
Figure 2.
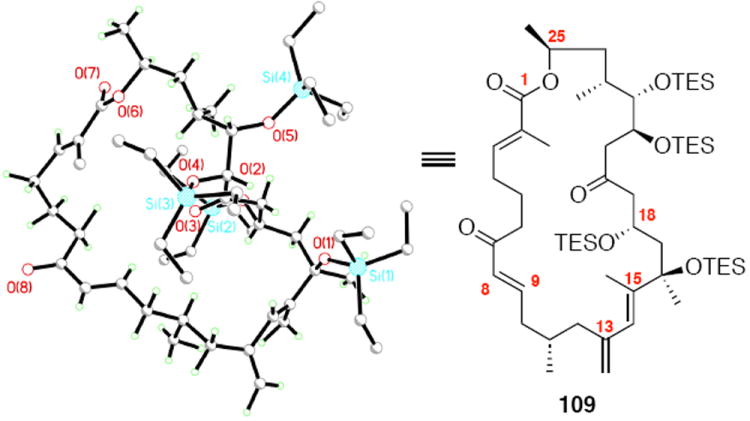
ORTEP Representation of Macrocycle 109.
With the macrocycle in hand and the stereochemistry firmly established, the remaining hurdles for completing the synthesis were incorporation of the allylic epoxide moiety and global deprotection. We first advanced the 18S stereoisomer, which would lead to the synthesis of amphidinolide B2 (Scheme 19). CBS reduction produced the allylic alcohol in good diastereoselectivity. Although epoxidation under tartrate-mediated Sharpless conditions as used previously proved ineffective, titanium-mediated epoxidation (in the absence of tartrate ligand) produced the epoxide alcohols in modest diastereoselectivity (2:1 dr, 74% yield). Given the increased steric demands of the Ti-tartate complex, we attribute the reactivity difference between the C20 OPMB series and the C20 OTES series to a conformational change to the macrocycle, which increases the steric hinderance surrounding the allylic alcohol moiety. While we cannot rigorously assign the stereochemistry of the newly created stereocenters, they were assigned based on literature precedent.40,50 Incorporation of the selenide followed by TPAP-mediated oxidation and syn elimination provided the allyl epoxide 116. Unfortuantely, we were again unable to effect global desilylation under a range of conditions (e.g. TASF or HF•pyr). Fortunately, the ordering of the final events could be altered (global deprotection followed by syn elimination using TMSOOTMS for selenide oxidation) to reveal the proposed structure for amphidinolide B2 (3). It should be noted that the use of TMSOOTMS has not been previously reported for selenide oxidation / elimination. H2O2 was ineffective in this transformation – likely due to unwanted Baeyer-Villiger-type oxidation of the C20 ketone. To our surprise, the spectral data did not match the literature values for amphidinolide B2. We had presumed that the incorrect epoxide had been utilized. Consequently, we repeated the analogous endgame, but this product 114 also did not match the natural product. It became clear that the assignment of amphidinolide B2 was incorrect. Comparison of the synthesized material with the literature data revealed a large chemical shift difference for H14 [Isolation data: 5.93 (bs), Synthetic 3: 6.06 (bs), Synthetic 114: 6.08 (bs)] and H19a,b [Isolation data: 3.09 (dd, J = 2.3, 8.8 Hz, 1H) and 2.69 (dd, J = 8.6, 17.7 Hz, 1H), Synthetic 3: 3.05 (m) and 2.48 (dd, J = 8.0, 17.0 Hz), Synthetic 114: 2.90 (dd, J = 9.9, 17.1 Hz) and 2.45 (m)]. Careful inspection of the structure elucidation paper2c revealed that while amphidinolide B1 was assigned by crystallographic analysis, amphidinolide B2 was assigned based on analogy and comparison of the NMR spectra. We hypothesize that the actual structure of amphidinolide B2 likely contains the opposite stereochemistry at C16 (e.g. 117) – thereby maintaining a syn relationship between the two alcohols at C16 and C18.
Scheme 19.
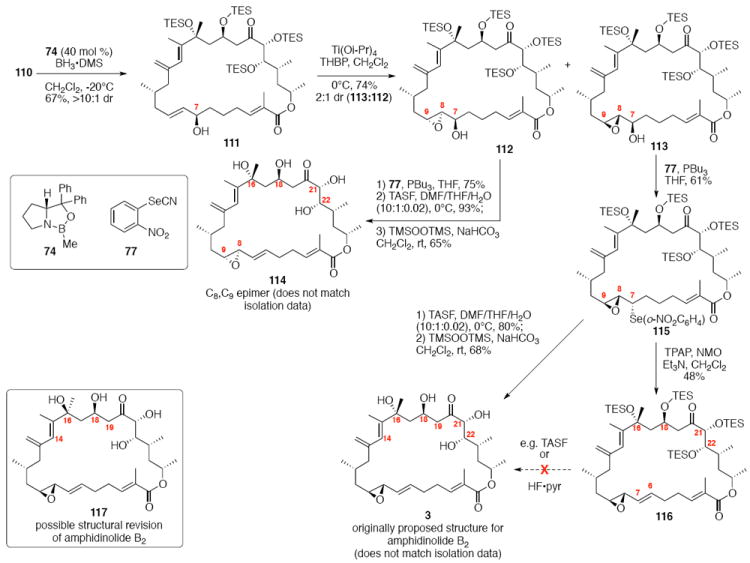
Synthesis of Proposed Structure for Amphidinolide B2.
The successful completion of amphidinolide B1 is shown in Scheme 20. Starting from the macrocycle 109, CBS reduction provided the allylic alcohol 118 in 74% yield (3.5:1 dr). Titanium-mediated epoxidation of the alkene was high yielding, but unselective (1:1.5 dr). Incorporation of the aryl selenide using previously described conditions followed by global deprotection and selenide oxidation / syn elimination yielded amphidinolide B1 (2). Comparison of the synthetic material with literature values showed excellent agreement (1H NMR, 13C NMR, [α]D). For analogue studies, we also carried the epimeric epoxide series onto 8,9-epi-amphidinolide B1 (121).
Scheme 20.
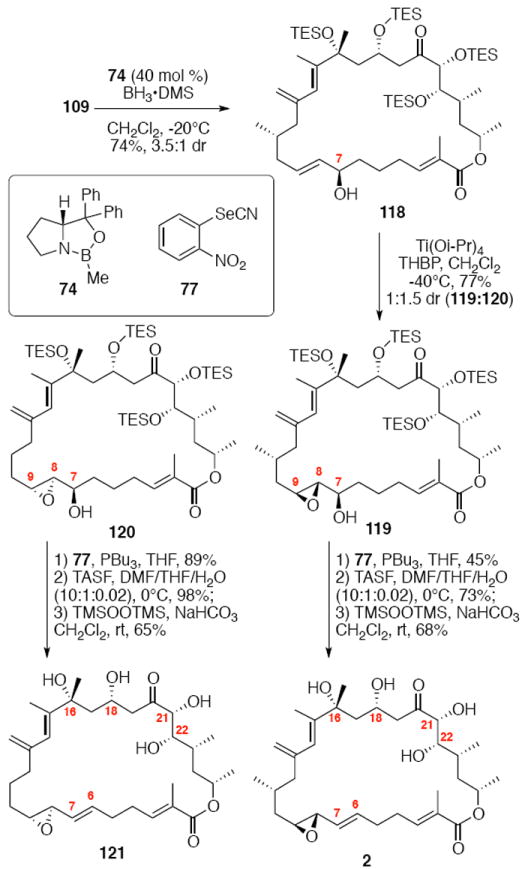
Synthesis of Amphidinolide B1 and 8,9-epi-Amphidinolide B1.
To assess the cytotoxic effects of the proposed structure of amphidinolide B2 on human solid and blood cancer cells, cell viability assays were conducted using nine cancer cell line panel.50 Compound 3 differentially reduced cell viabilities at 0.1 μM in human DU145 prostate cancer, MDA-MB-435 breast cancer, Ly3 lymphoma, K562 CML, MOLT-4 ALL, Reh ALL, U266 myeloma, KG1a AML and HL60 AML cells (Table 2). To further understand these anti-tumor activities of amphidinolide B2, we determined IC50 values = 36.4 ± 2.9 nM, 94.5 ± 8.0 nM, 3.3 ± 0.9 nM and 7.4 ± 0.6 nM against DU145, MDA-MB-435, KG1a and HL60 AML cancer cells, respectively (Table 3). Next, compared the potency of compound 3 to C8,9 epimer 114 and C18 epimer 121 in DU145 prostate cancer cells, compound 3 exhibited over 12-fold increase in biological activity (Figure 3). These findings suggest that the stereochemistry of the epoxide plays an important role in eliciting potent anticancer effects.
Table 2.
Effects of Proposed structure for Compound 3 on Viabilities of Human Cancer Cells (% control).
| DU145 | MDA-MB-435 | OCI-LY3 | K562 | MOLT-4 | Reh | U266 | KG1a | HL60 |
|---|---|---|---|---|---|---|---|---|
| 31 ± 5 | 54 ± 10 | 0 | 67 ± 6 | 47 ± 4 | 82 ± 5 | 76 ± 5 | 10 ± 2 | 0 |
Cell viability assays were carried out as described in Methods. Human DU145 prostate cancer, MDA-MB-435 breast cancer, OCI-LY3 lymphoma, K562 CML, MOLT-4 ALL, Reh ALL, U266 myeloma, KG1a AML and HL60 AML cells were seeded in 96-well plates (5000/well for DU145 and MDA-MB-435, 10000/well for OCI-LY3, K562, MOLT-4, Reh, U266,KG1a and HL60), incubated overnight at 37°C in 5% (v/v) CO2 and exposed to 0.1 μM of compound 3 for 72 h. Absorbance was measured at 490 nm using an automated ELISA plate reader. Each experiment was performed in quadruplicate. Data are mean ± SD.
Table 3.
Determination of IC50 Values of Compound 3 against Human Cancer Cells (nM).
| DU145 | HL60 | KG1a | MDA-MD-435 |
|---|---|---|---|
| 36.4 ± 2.9 | 7.4 ± 0.6 | 3.3 ± 0.9 | 94.5 ± 8.0 |
Cell viability was performed using MTS assays as described in Methods. IC50 values of compound 3 were determined in human DU145 prostate cancer, MDA-MB-435 breast cancer, KG1a AML and HL60 AML cells. Cells were seeded in 96- well plates (5000/well for DU145 and MDA-MB-435, 10000/well for KG1a and HL60), incubated overnight at 37°C in 5% (v/v) CO2 and exposed to compound 3 in a dose-dependent manner for 72 h. Absorbance was measured at 490 nm using an automated ELISA plate reader. Each experiment was performed in quadruplicate. Data are mean ± SD.
Figure 3.
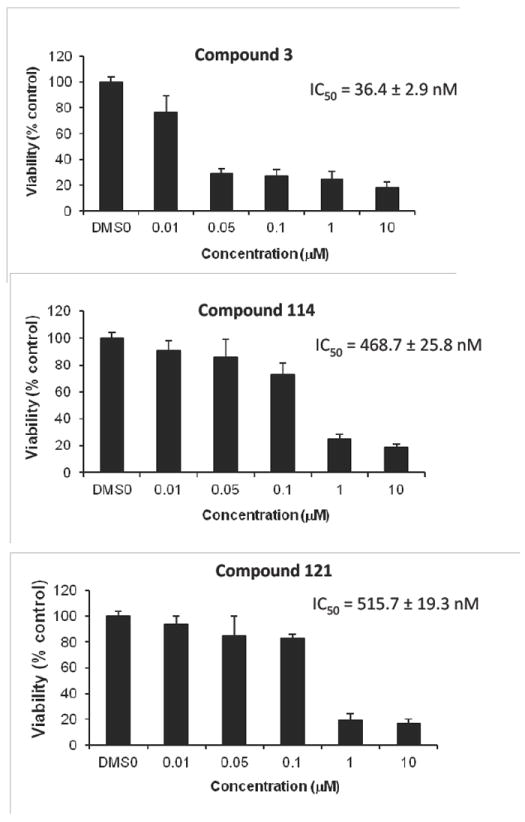
Comparison of Synthetic Analogues for Biological Activity.
Conclusion
In summary, the first total synthesis of amphidinolide B1 and the proposed structure for amphidinolide B2 has been achieved. In our initial approach to this family, we utilized a chelationcontrolled aldol reaction to provide excellent stereoselectivity at C18. Unfortunately, late stage removal of the PMB group proved not feasible in our hands. Consequently, a revised approach utilizing a dipole-minimized aldol provided access to a stereoselective aldol product. The stereoselectivity in this key aldol process could be controlled by variation of the temperature. Other highlights of the synthetic route include two non-metal catalyzed methods to construct the C13-C15 diene, a spontaneous HWE macrocyclization strategy and a novel late stage epoxidation / elimination strategy to incorporate the sensitive C6-C9 allylic epoxide moiety.
The proposed structure for amphidinolide B2 displays potent and stereoselective anti-tumor activities against human solid and blood tumor cells at low nanomolar concentrations that include highly aggressive and metastatic prostate and breast cancer cells. In contrast, C8,9-epimer and C18 epimer exhibit an over 12-fold decrease in anti-tumor activity in comparison. The stereochemistry of the epoxide plays a key role in the anticancer effects.
EXPERIMENTAL SECTION
General
Infrared spectra were recorded neat unless otherwise indicated and are reported in cm-1. 1H NMR spectra were recorded in deuterated solvents and are reported in ppm relative to tetramethylsilane and referenced internally to the residually protonated solvent. 13C NMR spectra were recorded in deuterated solvents and are reported in ppm relative to tetramethylsilane and referenced internally to the residually protonated solvent. HRMS data was collected using a TOF Mass Spectrometer.
Routine monitoring of reactions was performed using EM Science DC-Alufolien silica gel, aluminum-backed TLC plates. Flash chromatography was performed with the indicated eluents on EM Science Gedurian 230-400 mesh silica gel.
Air and/or moisture sensitive reactions were performed under usual inert atmosphere conditions. Reactions requiring anhydrous conditions were performed under a blanket of argon, in glassware dried in an oven at 120°C or by flame, then cooled under argon. Dry THF and DCM were obtained via a solvent purification system. All other solvents and commercially available reagents were either purified via literature procedures or used without further purification
Lactone 20
To a solution of diisopropyl amine (1.67 g, 2.3 mL, 16.5 mmol) in THF (7.6 mL) at −78°C added n-BuLi (6.6 mL, 16.5 mmol, 2.5 M in hexanes) dropwise. After 10 min, the white slurry was warmed to −10°C for 30 min. Then, the solution was cooled back to −78°C and another 58 mL THF was added slowly to the above solution. Next, a solution of Seebach lactone 1851 (2.37 g, 15.0 mmol) in THF (15 mL) was added dropwise to the above solution. After 20 min, a solution of cinnamyl bromide 19 (4.43 g, 3.33 mL, 22.5 mmol) in THF (10 mL) was added dropwise. After 30 min, the reaction was warmed slowly to −10°C over 2 h. After 5 min, the reaction was quenched with sat. aq. NH4Cl (30 mL) and warmed to r.t. After 10 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 30 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2-10% EtOAc / hexanes, to give 20 (3.74 g, 13.7 mmol, 91%) as a white solid: [α]D23 +45.5° (c 1.21, CHCl3); IR (thin film) 3027, 2963, 1796, 1484, 1173, 1138, 970, 692 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.28-7.42 (m, 5H), 6.58 (d, J = 15.6 Hz, 1H), 6.23 (dt, J = 15.6, 6.0 Hz, 1H), 5.24 (s, 1H), 2.72 (dd, J = 6.0, 10.8 Hz, 1H), 2.62 (dd, J = 6.0, 10.8 Hz, 1H), 1.51 (s, 3H), 0.98 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 175.5, 137.1, 135.3, 128.8, 127.9, 126.5, 122.4, 108.9, 80.2, 40.3, 34.8, 23.5, 23.1; HRMS (FAB+) calcd. for C17H23O3 (M+H) 275.1647, found 275.1650.
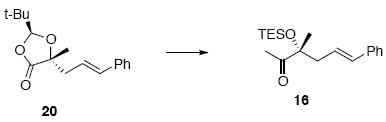
Methyl ketone 16
To a solution of lactone acetal 20 (1.37 g, 5.0 mmol) in THF (100 mL) at −78°C was added MeLi (4.64 mL, 5.6 mmol, 1.2 M in Et2O) via syringe pump. After 15 min, the resulted solution was warmed to r.t. After additional 10 min, the solvent was concentrated in vacuo, diluted with Et2O (50 mL), filtrated through a plug of Celite® and concentrated again in vacuo. The yellow oil was purified by chromatography over silica gel, eluting with 10-30% EtOAc / hexanes, to give a ketone intermediate (0.78 g, 3.85 mmol, 77%) as a colorless oil. To a solution of the above intermediate (340 mg, 1.66 mmol) in THF (8.3 mL) at -78°C was added 2,6-lutidine (0.27 g, 0.29 mL, 2.49 mmol). After 5 min, TESOTf (0.53 g, 0.45 mL, 1.99 mmol) was added. After 30 min, the reaction was warmed to r.t. After 5 min, the reaction was quenched with sat. aq. NH4Cl (10 mL). After 10 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 15 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2-6% EtOAc / hexanes, to give 16 (448 mg, 1.41 mmol, 88%) as a colorless oil: [α]D23 +0.7° (c 0.70, CHCl3); IR (thin film) 3027, 2955, 2916, 2876, 1718, 1457, 1415, 1350, 1239, 1166, 1131, 1016, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.20-7.33 (m, 5H), 6.38 (d, J = 15.9 Hz, 1H), 6.09-6.20 (m, 1H), 2.40-2.63 (m, 2H), 2.21 (s, 3H), 1.38 (s, 3H), 1.01 (t, J = 7.5 Hz, 9H), 0.68 (q, J = 7.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 214.2, 137.8, 133.6, 128.9, 127.6, 126.5, 125.1, 83.0, 45.2, 26.2, 25.4, 7.6, 7.1; HRMS (FAB+) calcd. for C19H31O2Si (M+H) 319.2093, found 319.2094.
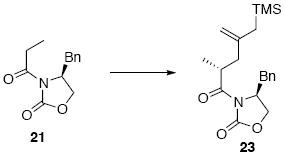
Allylsilane 23
To a solution of NaHMDS (5.6 mL, 5.6 mmol, 1.0 M in THF) at −78°C was added a solution of imide 21 (1.09 g, 4.67 mmol) in THF (4.6 mL). After 20 min, a solution of allyl iodide 2252 (3.6 g, 14 mmol) in THF (3 mL) was added via cannula. After 10 min, the reaction was warmed slowly to 10 °C over 4 h and quenched with sat. aq. NH4Cl (25 mL). After 10 min at r.t., the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 4-15% EtOAc / hexanes, to give 23 (1.46 g, 4.06 mmol, 87%) as a colorless oil: [α]D23 +30.6° (c 0.66, CHCl3); IR (thin film) 2954, 1780, 1699, 1632, 1454, 1385, 1349, 1246, 1207, 1101, 851, 701 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.20-7.35 (m, 5H), 4.66-4.69 (m, 1H), 4.69 (s, 1H), 4.64 (s, 1H), 4.14-4.19 (m, 2H), 4.06 (q, J = 6.9 Hz, 1H), 3.26 (dd, J = 3.3, 13.2 Hz, 1H), 2.71 (dd, J = 3.9, 13.2 Hz, 1H), 2.52 (dd, J = 7.5, 14.4 Hz, 1H), 2.04 (dd, J = 7.5, 14.4 Hz, 1H), 1.59 (d, J = 3.6 Hz, 2H), 1.17 (d, J = 6.9 Hz), 0.04 (s. 9H); 13C NMR (75 MHz, CDCl3) δ 177.4, 153.5, 145.1, 135.8, 129.9, 129.3, 127.7, 109.7, 66.3, 55.7, 42.6, 38.4, 36.2, 26.8, 17.4, -1.0; HRMS (FAB+) calcd. for C20H30NO3Si (M+H) 360.1995, found 360.1989.
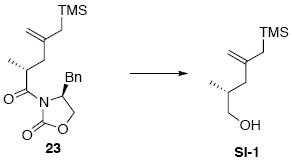
Allylsilane 24
To a solution of 23 (1.88 g, 5.23 mmol) in THF (35 mL) at 0°C was added sequentially MeOH (0.20 g, 0.25 mL, 6.28 mmol) followed by LiBH4 (3.14 mL, 6.28 mmol, 2.0 M in THF). After 2 h at 0°C, the reaction was warmed to r.t. After 2 h, the reaction was quenched with sat. aq. sodium tartrate (25 mL). After stirring for 20 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 25 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15-25% Et2O / pentane, to give free alcohol SI-1 (0.96 g, 5.18 mmol, 99%) as a colorless oil: [α]D23 (+) 9.4° (c 0.84, CHCl3); IR (thin film) 3337 (br), 3072, 2954, 2916, 1631, 1455, 1419, 1248, 1036, 849 cm-1; 1H NMR (300 MHz, CDCl3) δ 5,60 (d, J = 15.6 Hz, 2H), 3.44-3.52 (m, 2H), 2.03-2.10 (m, 1H), 1.77-1.89 (m, 2H), 1.54 (s, 2H), 1.41 (t, OH), 0.91 (d, J = 6.3 Hz, 3H), 0.04 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 146.6, 109.2, 68.7, 43.1, 34.2, 26.7, 17.2, -0.9; HRMS (FAB+) calcd. for C10H22OSi (M) 186.1440, found 186.1447.
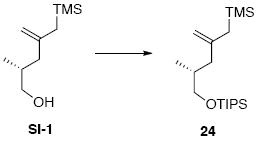
To a solution of alcohol intermediate SI-1 (308 mg, 1.65 mmol) in CH2Cl2 at −78°C was added sequentially Et3N (0.25 g, 0.34 mL, 2.48 mmol) and TIPSOTf (607 mg, 0.53 mL, 1.98 mmol). After 20 min, the reaction was warmed to r.t. After 5 min, the reaction was quenched with sat. aq. NH4Cl (25 mL). After 5 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 1% EtOAc / hexanes, to give 24 (560 mg, 1.64 mmol, 99%) as a colorless oil: [α]D23 -4.9° (c 0.98, CHCl3); IR (thin film) 3072, 2944, 2866, 1631, 1463, 1387, 1248, 1103, 1068, 878, 849, 796, 681 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.57-4.58 (m, 1H), 4.54 (s, 1H), 3.47-3.51 (m, 2H), 2.18 (dd, J = 5.4, 13.8, 1H), 1.78-1.82 (m, 1H), 1.63-1.71 (m, 1H), 1.51 (s, 2H), 1.04-1.08 (m, 21H), 0.87 (d, J = 6.6 Hz, 3H), 0.20 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 146.6, 108.8, 68.9, 42.7, 34.6, 26.7, 18.5, 17.0, 12.4, -0.9; HRMS (FAB+) calcd. for C19H42OSi2 (M+) 342.2774, found 342.2772.
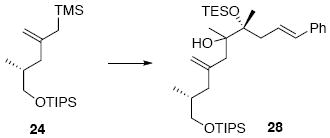
Tertiary alcohol 28
To a solution of allylsilane 24 (214 mg, 0.62 mmol) and ketone 16 (96 mg, 0.3 mmol) in CH2Cl2 (3.0 mL) at −78°C was added freshly distilled TiCl4 (117.6 mg, 70 μL, 0.62 mmol) via micro-syringe dropwise. After 15 min, the dark red solution was quenched with sat. aq. K2CO3 (5 mL) and warmed to r.t. immediately. After 20 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting carefully with 0.5-2% EtOAc / hexanes, to give 28 (382 mg, 0.19 mmol, 65%) as a colorless oil of a mixture of isomers (d.r. = 6:1). The major isomer: 1H NMR (300 MHz, CDCl3) δ 7.24-7.39 (m, 5H), 6.35-6.44 (m, 2H), 4.93 (s, 1H), 4.84 (s, 1H), 3.57-3.61 (m, 1H), 3.43-3.48 (m, 1H), 2.55-2.60 (m, 2H), 2.35-2.40 (m, 1H), 2.31 (dd, J = 5.7, 10.8, 1H), 2.16 (d, J = 9.9 Hz, 1H), 2.03 (dd, J = 5.7, 10.8 Hz, 1H), 1.85-1.88 (m, 1H), 1.33 (s, 3H), 1.20 (s, 3H), 1.08-1.10 (m, 21H), 0.99 (t, J = 6.0 Hz, 9H), 0.94 (d, J = 5.1 Hz, 3 H), 0.70 (q, J = 6.0 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 146.1, 138.0, 132.5, 128.9, 127.9, 127.4, 126.4, 115.4, 81.9, 77.7, 68.6, 43.3, 42.2, 42.1, 34.8, 23.1, 21.3, 18.5, 17.5, 12.4, 7.7, 7.4; HRMS (FAB+) calcd. for C35H64O3Si2 (M+) 588.4394, found 588.4382.
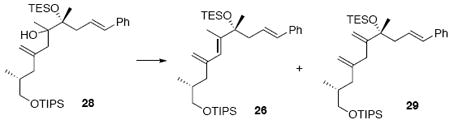
Dienes 26 and 29
To a solution of 28 (118 mg, 0.20 mmol) in PhMe (5.0 mL) at −78°C was added sequentially pyridine (79.0 mg, 0.08 mL, 1.0 mmol) and SOCl2 (71.4 mg, 44 μL, 0.60 mmol) via micro-syringe. After 30 min, the reaction was quenched with sat. aq. NaHCO3 (10 mL) and warmed to r.t. After 20 min, the organic layer was separated and the aqueous layer was extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting carefully with 0.5-1% EtOAc / hexanes, to give mixture of 26 and 29 (114 mg, 0.20 mmol, 99%, 2.2:1 ratio) was separable by HPLC as colorless oil. Conjugate diene 26: [α]D23 -43.2° (c 0.1, CHCl3); IR (thin film) 2954, 2866, 1640, 1459, 1156, 1119, 1013, 882, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.20-7.34 (m, 5H), 6.35 (d, J = 12.0 Hz, 1H), 6.18 (dt, J = 12.0, 5.4 Hz, 1H), 5.89 (s, 1H), 5.01 (s, 1H), 4.82 (s, 1H), 3.44 (d, J = 4.2 Hz, 2H), 2.48-2.50 (m, 2H), 2.35 (dd, J = 3.6, 9.9 Hz, 1H), 1.84 (d, J = 0.9 Hz, 3H), 1.82 (dd, J = 3.6, 9.9 Hz, 1H), 1.66-1.72 (m, 1H), 1.46 (s, 3H), 1.07-1.09 (m, 21H), 1.00 (t, J = 5.7 Hz, 9H), 0.78 (d, J = 5.6 Hz, 3H), 0.64 (q, J = 5.7 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 145.7, 142.8, 138.3, 131.9, 128.8, 127.4, 127.2, 126.3, 125.5, 114.5, 78.7, 68.6, 46.3, 42.6, 34.9, 27.2, 18.5, 16.7, 15.0, 12.4, 7.6, 7.2; HRMS (FAB+) calcd. for C35H62O2Si2 (M+) 570.4288, found 570.4277.
Unconjugated diene 29
[α]D23 -1.3° (c 1.74, CHCl3); IR (thin film) 3026, 2942, 2865, 1643, 1495, 1462, 1381, 1241, 1097, 1012, 882 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.22-7.36 (m, 5H), 6.38 (d, J = 12.0 Hz, 1H), 6.16-6.20 (m, 1H), 5.16 (s, 1H), 4.92 (s, 1H), 4.89 (s, 1H), 4.88 (s, 1H), 3.55-3.59 (m, 1H), 3.44-3.47 (m, 1H), 2.84 (s, 2H), 2.47-2.50 (m, 2H), 2.25 (dd, J = 3.6, 9.9 Hz, 1H), 1.74-1.85 (m, 2H), 1.43 (s, 3H), 1.08-1.10 (m, 21H), 1.00 (t, J = 6.0 Hz, 9H), 0.92 (d, J = 4.8 Hz, 3H), 0.64 (q, J = 6.0 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 152.0, 146.7, 138.3, 132.2, 128.9, 127.5, 127.2, 126.4, 113.9, 111.1, 78.3, 68.8, 46.4, 39.8, 38.8, 34.6, 27.5, 18.5, 17.2, 12.4, 7.6, 7.3.
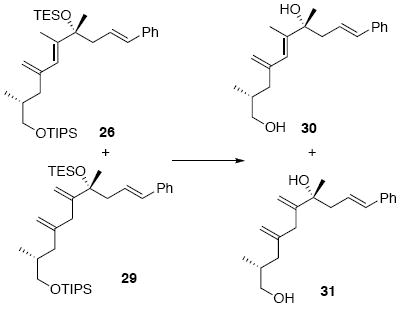
Diols 30 and 31
To a mixture of 26 and 29 (75 mg, 0.13 mmol) in THF (0.1 mL) at 0°C was added TBAF (0.6 mL, 1.0 M in THF). After 10 min, the reaction was warmed to r.t. After 1 h, the reaction was quenched with sat. aq. NH4Cl (3 mL). After 10 min the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20% EtOAc / hexanes, to give diol 30 (20 mg, 0.069 mmol, 61%) as a white foam: [α]D23 -4.9° (c 0.98, CHCl3); IR (thin film) 3373 (br), 3026, 2955, 2925, 2870, 1722, 1627, 1448, 1373, 1032, 966, 897, 741, 692 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.23-7.38 (m, 5H), 6.46 (d, J = 15.6 Hz, 1H), 6.12-6.20 (m, 1H), 5.97 (s, 1H), 5.07 (s, 1H), 4.86 (s, 1H), 3.28-3.40 (m, 2H), 2.66 (dd, J = 7.2, 14.0 Hz, 1H), 2.46 (dd, J = 8.0, 14.0 Hz, 1H), 2.26 (dd, J = 5.6, 13.6 Hz, 1H), 1.89 (s, 3H), 1.84-1.88 (m, 1H), 1.61-1.66 (m, 1H), 1.42 (s, 3H), 0.85 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 145.0, 142.9, 137.7, 133.8, 129.0, 127.8, 126.5, 125.9, 125.1, 115.2, 76.0, 68.5, 44.4, 42.5, 34.9, 28.1, 16.6, 15.4; HRMS (ES+) calcd. for C20H28O2Na (M+Na) 323.1987, found 323.1996.
Diol 31: is colorless oil
[α]D23 +7.6° (c 1.21, CHCl3); IR (thin film) 3373, 3081, 3026, 2957, 2926, 2870, 1724, 1644, 1496, 1449, 1373, 1112, 1034, 967, 900, 739, 692 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.23-7.39 (m, 5H), 6.49 (d, J = 15.6 Hz, 1H), 6.16-6.23 (m, 1H), 5.23 (s, 1H), 4.96 (s, 3H), 3.48-3.52 (m, 2H), 2.85-2.96 (m, 2H), 2.62-2.66 (m, 1H), 2.48-2.52 (m, 1H), 2.18-2.24 (m, 1H), 1.86-1.93 (m, 2H), 1.40 (s, 3H), 0.94 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 151.9, 146.9, 137.7, 134.3, 128.9, 127.7, 126.6, 125.7, 114.3, 111.8, 75.7, 68.6, 45.0, 40.1, 39.2, 34.3, 28.2, 17.1; HRMS (ES+) calcd. for C20H28O2Na (M+Na) 323.1987, found 323.1989.
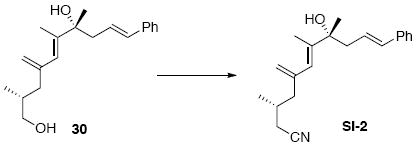
Nitrile 14
To a solution of 30 (98 mg, 0.33 mmol) in Et2O (1.6 mL) at 0°C was added sequentially PPh3 (342 mg, 1.3 mmol) and DEAD (0.24 mL, 1.3 mmol, 40% w/w in PhMe). After 15 min, acetone cyanohydrin (70.0 mg, 74.5 μL, 0.82 mmol) was added dropwise. After 5 min, the reaction was warmed to r.t. After 6 h, the reaction was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / hexanes, to give a corresponding nitrile intermediate (80 mg, 0.26 mmol, 81%) as a colorless oil: [α]D23 +7.8° (c 0.99, CHCl3); IR (neat) 3479 (br), 2962, 2927, 2246, 1733, 1496, 1456, 1420, 1381, 1105, 968, 902, 743, 694 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.26-7.37 (m, 5H), 6.47 (d, J = 16.0 Hz, 1H), 6.12-6.18 (m, 1H), 5.95 (s, 1H), 5.12 (s, 1H), 4.92 (s, 1H), 2.66 (dd, J = 7.2, 14.0 Hz, 1H), 2.48 (dd, J = 7.2, 14.0 Hz, 1H), 2.05-2.20 (m, 4H), 1.88 (s, 3H), 1.76-1.88 (m, 1H), 1.75 (br, 1H), 1.43 (s, 3H), 0.97 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 143.8, 143.5, 137.5, 134.0, 129.0, 127.9, 126.5, 125.5, 124.3, 119.1, 116.4, 76.0, 44.8, 44.5, 29.3, 28.1, 24.2, 19.5, 15.5; HRMS (FAB+) calcd. for C21H28NO (M+1) 310.2171, found 310.2144.
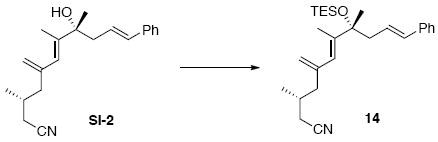
To a solution of the above intermediate (80 mg, 0.26 mmol) in CH2Cl2 (1.0 mL) at −78°C was added 2,6-lutidine (111.0 mg, 0.12 mL, 1.04 mmol) and followed by TESOTf (136.9 mg, 0.12 mL, 0.52 mmol). After 30 min, the reaction was warmed to r.t. and quenched with sat. aq. NH4Cl (3 mL). After 5 min, the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 1% EtOAc / hexanes, to give nitrile 14 (80 mg, 0.19 mmol, 72%) as a colorless oil: [α]D23 -37.6° (c 0.98, CHCl3); IR (neat) 2957, 2933, 2876, 2240, 1733, 1496, 1457, 1420, 1381, 1160, 1122, 1016, 967, 902, 742, 693 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.21-7.33 (m, 5H), 6.33 (d, J = 11.7 Hz, 1H), 6.15-6.19 (m, 1H), 5.87 (s, 1H), 5.07 (s, 1H), 4.87 (s, 1H), 2.50 (d, J = 5.4 Hz, 2H), 2.15 (dd, J = 5.4, 9.9 Hz, 1H), 2.00-2.06 (m, 3H), 1.81 (s, 3H), 1.72-1.79 (m, 1H), 1.47 (s, 3H), 1.02 (t, J = 5.7 Hz, 9H), 0.85 (d, J = 5.1 Hz, 3H), 0.67 (q, J = 5.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 143.9, 143.7, 138.1, 131.9, 129.0, 127.4, 127.3, 126.3, 124.6, 119.0, 116.0, 78.8, 46.0, 44.9, 29.3, 27.6, 24.1, 19.4, 15.1, 7.64, 7.26; HRMS (FAB+) calcd. for C27H42OSiN (M+1) 424.3036, found 424.3044.
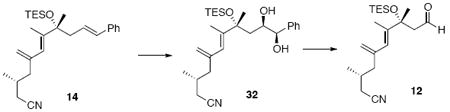
Aldehyde 12
To a solution of 14 (75 mg, 0.18 mmol) in t-BuOH / H2O (3 mL, 1:1) was added MeSO2NH2 (25.6 mg, 0.27 mmol) and AD mix β*53 (250 mg). After 16 h, the reaction was quenched with sat. aq. Na2S2O3 (3 mL). After 20 min, the mixture was extracted with EtOAc (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20% EtOAc / hexanes, to give a mixture of diol isomers (73 mg, d.r. = 6:1, 0.16 mmol, 89%) as a colorless oil. The diol substrate was applied to the next step without further purification.
A solution of the mixture described above in THF/Et2O (2.8 mL, 1:1) was added NaIO4 (304 mg, 1.42 mmol) and H2O (1.4 mL). After 2 h, the reaction was quenched with sat. aq. NaCl (3 mL) and the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give aldehyde 12 (54 mg, 0.15 mmol, 96%) as colorless oil: [α]D23 -12.6° (c 1.22, CHCl3); IR (neat) 3081, 2957, 2917, 2876, 2246, 1722, 1628, 1457, 1417, 1381, 1240, 1163, 1126, 1046, 1017, 904, 802, 743 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.66 (t, J = 3.2 Hz, 1H), 5.99 (s, 1H), 5.10 (s, 1H), 4.89 (s, 1H), 2.61 (dd, J = 3.2, 14.8 Hz, 1H), 2.47 (dd, J = 2.8, 14.8 Hz, 1H), 2.32 (dd, J = 5.2, 16.8 Hz, 1H), 2.15- 2.25 (m, 2H), 2.08 (dd, J = 7.2, 13.6 Hz, 1H), 1.83-1.92 (m, 1H), 1.80 (s, 3H), 1.47 (s, 3H), 1.04 (d, J = 6.4 Hz, 3H), 0.96 (t, J = 8.0 Hz, 9H), 0.63 (q, J = 8.0 Hz, 6 H); 13C NMR (100 MHz, CDCl3) δ 203.2, 143.2, 142.7, 125.2, 118.9, 116.8, 54.5, 44.8, 30.1, 29.6, 28.3, 24.3, 19.8, 15.2, 7.5, 7.2; HRMS (FAB+) calcd. for C20H36O2SiN (M+1) 350.2515, found 350.2508.
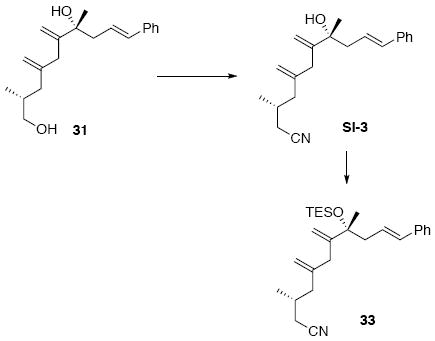
Nitrile 33
To a solution of 31 (30 mg, 0.10 mmol) in Et2O (0.5 mL, 0.2 M) at 0°C was added sequentially PPh3 (104 mg, 0.4 mmol) and DEAD (72 μL, 0.4 mmol, 40% solution in PhMe). After 15 min, acetone cyanohydrin (21.2 mg, 23.0 μL, 0.25 mmol) was added dropwise. After 5 min, the reaction was warmed to r.t. After 6 h, the reaction was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / hexanes, to give a corresponding nitrile intermediate (25 mg, 0.08 mmol, 81%) as a colorless oil.
To a solution of the above nitrile (25 mg, 0.08 mmol) in CH2Cl2 (0.32 mL, 0.25 M) at −78°C added sequentially 2,6-lutidine (34.3 mg, 37 μL, 0.32 mmol) and TESOTf (42.1 mg, 36 μL, 0.16 mmol). After 30 min, the reaction was warmed to r.t. and quenched with sat. aq. NH4Cl (3 mL). After 5 min, the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 1% EtOAc / hexanes, to give 33 (26 mg, 62 μmol, 77%) as a colorless oil: [α]D23 -13.6° (c 0.28, CHCl3); IR (neat) 2957, 2933, 2911, 2875, 2247, 1238, 1157, 1123, 1063, 1005, 967, 905, 742, 693 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.17-7.26 (m, 5H), 6.35 (d, J = 15.9 Hz, 1H), 6.08-6.19 (m, 1H), 5.14 (s, 1H), 4.93 (s, 2H), 4.71 (s, 1H), 2.75-2.85 (m, 2H), 2.46 (dd, J = 0.6, 6.9 Hz, 2H), 2.24-2.32 (m, 1H), 1.97-2.17 (m, 4H), 1.38 (s, 3H), 0.95-1.04 (m, 12H), 0.58-0.68 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 151.7, 144.9, 138.1, 132.3, 128.9, 127.4, 127.1, 126.4, 119.2, 115.8, 111.1, 78.2, 46.4, 42.3, 38.2, 28.9, 27.7, 24.3, 20.0, 7.6, 7.3.
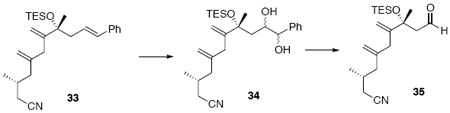
Aldehyde 35
To a solution of 33 (26 mg, 62 μmol) in t-BuOH / H2O (1 mL, 1:1) was added MeSO2NH2 (8.8 mg, 93 μmol) and AD mix β*53 (87 mg). After 16 h, the reaction was quenched with sat. aq. Na2S2O3 (3 mL). After 20 min, the mixture was extracted with EtOAc (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20% EtOAc / hexanes, to give a mixture of the diol isomers (25.5 mg, d.r. = 1.3:1, 56 μmol, 90%) as a colorless oil. The diol substrate was applied to the next step without further purification.
A solution of the mixture described above in THF/Et2O (1.24 mL, 1:1) was added NaIO4 (120 mg, 0.56 mmol) and H2O (0.62 mL). After 2 h, the reaction was quenched with sat. aq. NaCl (3 mL) and the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give aldehyde 35 (18.5 mg, 0.053 mmol, 95%) as colorless oil: [α]D23 -11.4° (c 1.57, CHCl3); IR (neat) 2958, 2915, 2876, 2737, 2246, 1722, 1645, 1457, 1417, 1375, 1239, 1165, 1123, 1045, 1005, 909, 743, 726 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.66 (t, J = 3.0 Hz, 1H), 5.28 (s, 1H), 4.95 (s, 1H), 4.94 (s, 1H), 4.90 (s, 1H), 2.76 (s, 2H), 2.20-2.62 (m, 5H), 2.02-2.06 (m, 2H), 1.46 (s, 3H), 1.08 (d, J = 6.3 Hz, 3H), 0.96 (t, J = 7.8 Hz, 9H), 0.63 (q, J = 7.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 203.3, 150.5, 144.3, 119.0, 116.2, 112.6, 54.5, 42.1, 38.4, 28.9, 28.5, 24.3, 24.0, 19.9, 7.5, 7.2.
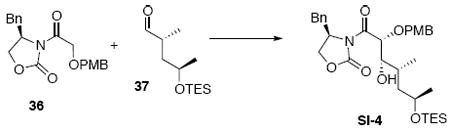
Methyl ketone 13
To a solution of imide 3622 (107 mg, 0.3 mmol) in PhMe (0.7 mL) at −50°C was sequentially added Et3N (34.4 mg, 48.2 μL, 0.34 mmol) and Bu2BOTf (86.0 mg, 80.0 μL, 0.32 mmol). After 1.5 h, a solution of aldehyde 378 (46 mg, 0.20 mmol) in PhMe (0.25 mL) was transferred dropwise into the reaction via cannula. After 40 min, the reaction was warmed to −30°C within 30 min. After 1 h, the reaction was quenched with aq. phosphate buffer solution (0.5 mL, pH 7), MeOH (0.5 mL) and THF (0.5 mL). Next, the mixture was warmed up to r.t. A solution of H2O2 (0.5 mL, 30% H2O2 in H2O) in MeOH (0.5 mL) was added dropwise. After 1 h, the reaction mixture was extracted with EtOAc (3 × 15 mL) and the combined organic extract was diluted with an aqueous phosphate buffer solution (20 mL, pH 7). The mixture was concentrated in vacuo and extracted again with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15-20% EtOAc / hexanes, to give an aldol adduct SI-4 (83 mg, 0.14 mmol, 72%) as a colorless oil: [α]D23 -3.9° (c 0.83, CHCl3); IR (thin film) 3490 (br), 2956, 2875, 1781, 1708, 1612, 1514, 1389, 1248, 1211, 1051, 1012, 744 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.25-7.39 (m, 7H), 6.92 (d, J = 8.8 Hz, 2H), 5.36 (d, J = 2.0 Hz, 1H), 4.64-4.72 (m, 1H), 4.64 (d, J = 10.8 Hz, 1H), 4.49 (d, J = 10.8 Hz, 1H), 4.20-4.29 (m, 2H), 3.84-3.90 (m, 1H), 3.82 (s, 3H), 3.61-3.64 (m, 1H), 3.34 (dd, J = 2.8, 13.2 Hz, 1H), 2.78 (dd, J = 3.6, 13.5 Hz, 1 H), 2.31 (d, J = 10.0 Hz, 1H), 1.74-1.81 (m, 1H), 1.56-1.62 (m, 1H), 1.40-1.48 (m, 1H), 1.15 (d, J = 5.6 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H), 0.99 (t, J = 7.6 Hz, 9H), 0.62 (q, J = 7.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 171.6, 160.0, 153.6, 135.6, 130.7, 129.8, 129.6, 129.4, 127.8, 114.3, 78.4, 76.6, 73.0, 67.4, 67.3, 56.2, 55.7, 43.1, 38.2, 34.6, 23.6, 16.2, 7.3, 5.3; HRMS (FAB+) calcd. for C32H48NO7Si (M+1) 586.3200, found 586.3202.
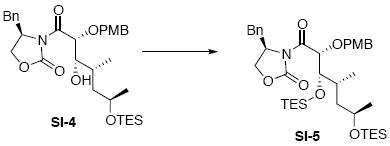
To a solution of the aldol adduct SI-4 (78 mg, 0.13 mmol) in CH2Cl2 (0.3 mL) at 0°C was added sequentially 2,6-lutidine (27.8 mg, 29.8 μL, 0.26 mmol) and TESOTf (68.5 mg, 60.1 μL, 0.26 mmol). After 1 h, the reaction was warmed to r.t. and quenched with sat. aq. NH4Cl (3 mL). After 5 min, the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 4-6% EtOAc / hexanes, to give the corresponding protected alcohol SI-5 (84 mg, 0.12 mmol, 90%) as a colorless oil: [α]D23 -33.8° (c 1.9, CHCl3); IR (thin film) 2956, 2911, 2875, 1785. 1704, 1612, 1513, 1456, 1381, 1248, 1082, 1011, 739 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.34-7.26 (m, 7H), 6.84 (d, J = 8.4 Hz, 2H), 5.25 (d, J = 6.0 Hz, 1H), 4.64 (d, J = 11.7 Hz, 1H), 4.52 (d, J = 11.7 Hz, 1H), 4.46-4.49 (m, 1H), 4.03-4.12 (m, 3H), 3.73- 3.98 (m, 1H), 3.79-3.81 (m, 1H), 3.74 (s, 3H), 3.11 (dd, J = 3.0, 13.2 Hz, 1H), 2.40 (dd, J = 3.0, 13.5 Hz, 1H), 1.47-1.55 (m, 2H), 1.09 (d, J = 6.0 Hz, 3H), 0.87-0.97 (m, 21H), 0.53-0.63 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 172.3, 159.9, 153.3, 135.8, 130.6, 130.3, 129.7, 129.4, 127.8, 114.0, 79.6, 77.8, 73.4, 67.6, 66.8, 56.4, 55.6, 44.7, 37.8, 33.9, 24.1, 15.2, 7.5, 7.4, 5.8, 5.4; HRMS (FAB+) calcd. for C38H60NO7Si2 (M-1) 698.3908, found 698.3890.
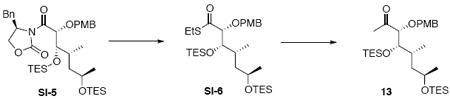
To a solution of EtSH (75 mg, 90.2 μL, 1.21 mmol) in THF (9.0 mL) at 0°C was added n-BuLi (0.43 mL, 1.07 mmol, 2.5 M in hexane). After 20 min, a solution of the above intermediate (500 mg, 0.714 mmol) in THF (3.0 mL) was transferred via cannula dropwise. After 20 min, the reaction was quenched with sat. aq. NH4Cl (10 mL). After 5 min, the mixture was extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by a plug of silica gel, eluting with 10% EtOAc / Hexanes, to give the corresponding thioester intermediate (375 mg, 0.68 mmol, 95%) as a colorless oil.
To a suspension of CuI (796 mg, 4.18 mmol) in Et2O (9.0 mL) at 0°C was added MeLi (5.2 mL, 8.36 mmol, 1.6 M in Et2O). After 15 min, the colorless solution was cooled to −50°C and a solution of the above thioester intermediate (375 mg, 0.68 mmol) in Et2O (2.4 mL) was transferred into the reaction dropwise via cannula. After 2 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) at −50°C, warmed to r.t. and extracted with Et2O (3 × 20 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2-4% EtOAc / hexanes, to give methyl ketone 13 (326 mg, 0.60 mmol, 89%) as a colorless oil: [α]D23 +31.0° (c 0.80, CHCl3); IR 2956, 2912, 2876, 1716, 1613, 1514, 1457, 1416, 1249, 1172, 1082, 1037, 1007, 820, 740 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.23 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 4.40 (d, J = 11.4 Hz, 1H), 4.38 (d, J = 11.4 Hz, 1H), 3.81 (s, 3H), 3.69-3.81 (m, 3H), 2.11 (s, 3H), 1.45-1.48 (m, 3H), 1.09 (d, J = 6.0 Hz, 3H), 0.85-0.97 (m, 21H), 0.53-0.61 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 211.5, 159.8, 130.1, 129.9, 114.1, 88.9, 73.1, 67.4, 55.7, 44.9, 33.5, 27.4, 23.9, 14.3, 7.4, 7.3, 5.7, 5.4; HRMS (FAB+) calcd. for C29H55O5Si2 (M+1) 539.3588, found 539.3559.
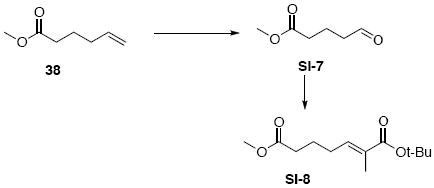
Carboxylic acid 11
To a solution of 38 (320 mg, 0.35 mL, 2.50 mmol) in THF/H2O (35 mL, 1:1) at r.t. was added K2OsO4•H2O (14 mg, 15 μmol) and NaIO4 (2.4 g, 11.25 mmol). After 3 h, the reaction was quenched with sat. aq. Na2S2O3 (5 mL). After 20 min, the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give an aldehyde intermediate (247 mg, 1.90 mmol, 76%) as a colorless oil. The aldehyde intermediate SI-7 was used directly in the next step without further purification.
To a solution of the above aldehyde intermediate SI-7 (247 mg, 1.90 mmol) in CH2Cl2 (5.4 mL) at r.t. was added Wittig reagent Ph3P=C(Me)CO2t-Bu (725 mg, 2.0 mmol). After 30 min, the reaction was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / hexanes, to give Wittig product SI-8 (377 mg, 1.56 mmol, 82%) as a colorless oil: IR (neat) 2977, 1739, 1704, 1652, 1456, 1436, 1367, 1289, 1252, 1159, 1121, 1083, 852, 743 cm-1; 1H NMR (300 MHz, CDCl3) d 6.59 (dt, J = 1.2, 7.5 Hz, 1H), 3.65 (s, 3H), 2.32 (t, J = 7.5 Hz, 2H), 2.17 (q, J = 7.5 Hz, 2H), 1.71-1.83 (m, 5H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) d 174.1, 167.8, 139.9, 130.5, 80.5, 51.9, 33.9, 28.5, 28.3, 24.2, 12.8; HRMS (FAB+) calcd. for C13H23O4 (M+1) 243.1596, found 243.1598.
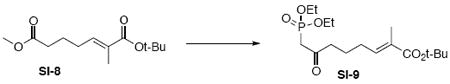
To a solution of methyl diethylphosphonate (318 mg, 0.3 mL, 2.09 mmol) in THF (10 mL) at −78°C was added n-BuLi (0.83 mL, 2.09 mmol, 2.5 M in hexane). After 20 min, this resulted solution was transferred dropwise into a solution of the above Wittig product SI-8 (202 mg, 0.83 mmol) in THF (4 mL) at −78°C via cannula. After 30 min, the reaction was quenched with sat. aq. NH4Cl (3 mL). The mixture was extracted with EtOAc (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 80% EtOAc / Hexanes, to give phosphonate intermediate SI-9 (254 mg, 0.65 mmol, 78%) as a colorless oil: IR (neat) 2978, 2932, 1701, 1652, 1392, 1366, 1254, 1162, 1024, 965, 794 cm-1; 1H NMR (300 MHz, CDCl3) d 6.58 (dt, J = 1.2, 7.5 Hz, 1H), 4.07-4.17 (m, 4H), 3.09 (s, 1H), 3.01 (s, 1H), 2.63 (t, J = 7.2 Hz, 2H), 2.13 (q, J = 7.5 Hz, 2H), 1.75 (s, 3H), 1.67-1.75 (m, 2H), 1.46 (s, 9H), 1.31 (t, J = 7.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) d 202.0, 167.8, 140.2, 130.4, 80.5, 63.0, 43.7, 42.0, 28.5, 28.1, 22.7, 16.7, 12.8; HRMS (FAB+) calcd. for C17H32O6P (M+1) 363.1937, found 363.1942.
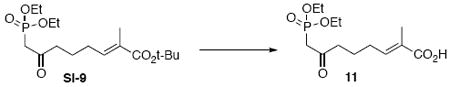
To a solution of the above phosphonate SI-9 (102 mg, 0.28 mmol) in CH2Cl2 (1.5 mL) at 0°C was added TFA (1.15 g, 0.75 mL, 9.7 mmol). After 5 min, the reaction was warmed to r.t. After 2 h, the reaction was concentrated in vacuo and purified by chromatography over silica gel, eluting with 4% MeOH / CH2Cl2, to give 11 (84.8 mg, 0.28 mmol, 99%) as a colorless oil: IR (neat) 3418, 2986, 2934, 1772, 1715, 1395, 1209, 1163, 1023, 973, 799, 698 cm-1; 1H NMR (300 MHz, CDCl3) d 6.86 (t, J = 7.5 Hz, 1H), 6.07 (br, 1H), 4.12-4.23 (m, 4H), 3.18 (s, 1H), 3.10 (s, 1H), 2.64 (t, J = 7.2 Hz, 2H), 2.22 (q, J = 7.5 Hz, 2H), 1.82 (s, 3H), 1.71-1.80 (m, 2H), 1.30 (t, J = 7.5 Hz, 6H); 13C NMR (75 MHz, CDCl3) d 202.2, 170.5, 143.5, 128.5, 63.2, 43.6, 41.9, 28.2, 22.5, 16.7, 12.5; HRMS (FAB+) calcd. for C13H24O6P (M+1) 307.1311, found 307.1322.
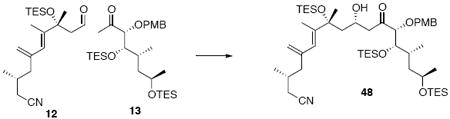
Alcohol 48
To a solution of 13 (73 mg, 0.14 mmol) in Et2O (0.7 mL) at −78°C was added LDA54 (0.15 mL, 1.0 M in THF). After 20 min, a pre-cooled (-78°C) solution of aldehyde 12 (31.5 mg, 0.09 mmol) in Et2O (0.4 mL) was transferred in one portion via cannula at −78°C. After 20 min, the reaction was quenched with sat. aq. NH4Cl (3 mL) and the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / hexanes, to give 48 (55 mg, 0.06 mmol, 69%) as a colorless oil: [α]D23 +21.3° (c 0.60, CHCl3); IR (neat) 3496, 2955, 2911, 2876, 1715, 1614, 1515, 1457, 1418, 1379, 1249, 1086, 1036, 1008, 903, 807, 740 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.87 (s, 1H), 5.09 (s, 1H), 4.89 (s, 1H), 4.47 (d, J = 10.8 Hz, 1H), 4.33-4.36 (m, 2H), 3.80 (s, 3H), 3.76-3.80 (m, 3H), 3.61 (s, 1H), 2.95 (dd, J = 7.2, 18.0 Hz, 1H), 2.05-2.40 (m, 6H), 1.88-1.95 (m, 1H), 1.81 (s, 3H), 1.52 (s, 3H), 1.37-1.52 (m, 5H), 1.07 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.0 Hz, 3H), 0.83-0.98 (m, 30H), 0.51-0.67 (m, 18H); 13C NMR (100 MHz, CDCl3) δ 212.1, 159.7, 144.8, 143.2, 130.1, 124.3, 118.9, 116.7, 114.1, 88.5, 79.7, 72.9, 67.3, 65.1, 55.7, 48.4, 47.2, 45.1, 44.9, 33.5, 29.6, 26.7, 24.3, 23.6, 19.8, 15.2, 13.9, 7.5, 7.4, 7.3, 7.0, 5.7, 5.4; HRMS (FAB+) calcd. for C49H90O7Si3N (M+1) 888.6025, found 888.6052.
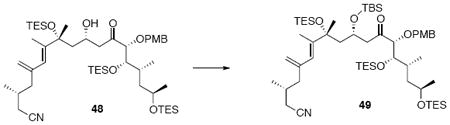
Silylether 49
To a solution of 48 (55 mg, 60 μmol) in CH2Cl2 (0.15 mL) at r.t. was added Et3N (0.15 mL). The solution was then cooled to −78°C. After 5 min, TBSOTf (65.8 mg, 60.4 μL, 0.25 mmol) was added dropwise. After 5 min, the reaction was warmed to r.t. After 20 min, the reaction was quenched with sat. aq. NH4Cl (3 mL) and the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 3% EtOAc / Hexanes, to give 49 (53 mg, 53 μmol, 88%) as a colorless oil: [α]D23 +35.1° (c 0.50, CHCl3); IR (neat) 2954, 2928, 2876, 1714, 1615, 1515, 1458, 1381, 1250, 1167, 1125, 1065, 1006, 987, 835, 776, 740 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.80 (s, 1H), 5.07 (s, 1H), 4.89 (s, 1H), 4.51 (d, J = 10.5 Hz, 1H), 4.21-4.25 (m, 2H), 3.81 (s, 3H), 3.64- 3.81 (m, 3H), 3.17 (dd, J = 9.3, 18.0 Hz, 1H), 2.58 (dd, J = 2.7, 17.7 Hz, 1H), 2.38 (dd, J = 4.5, 16.8 Hz, 1H), 2.19 (dd, J = 6.9, 16.8 Hz, 1H), 2.05-2.10 (m, 2H), 1.86-1.94 (m, 2H), 1.79 (s, 3H), 1.66-1.79 (m, 1H), 1.58-1.62 (m, 1H), 1.40 (s, 3H), 1.28-1.35 (m, 2H), 1.07 (d, J = 6.3 Hz, 3H), 1.05 (d, J = 5.4 Hz, 3H), 0.86-0.97 (m, 30H), 0.78 (s, 9H), 0.50-0.65 (m, 18H), 0.05 (s, 3H), -0.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 212.2, 159.5, 143.6, 143.1, 130.3, 129.9, 124.9, 119.0, 116.6, 113.9, 89.1, 77.3, 72.3, 67.2, 66.1, 55.6, 49.7, 46.4, 45.5, 44.7, 33.7, 29.4, 29.0, 26.3, 24.3, 23.5, 19.7, 18.4, 15.4, 12.9, 7.7, 7.4, 7.3, 5.7, 5.3, -3.7, -4.2; HRMS (FAB+) calcd. for C55H104O7Si4N (M+1) 1002.6890, found 1002.6923.
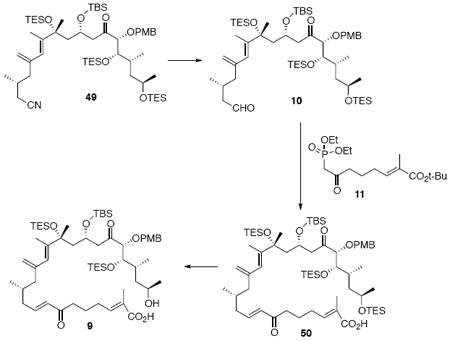
Acid 9
To a solution of 49 (11 mg, 11 μmol) in PhMe (0.25 mL) at −78°C was added DIBAL-H (13.2 μL, 13.2 μmol, 1.0 M in PhMe) dropwise. After 10 min, another portion of DIBAL-H (6.0 μL, 6.0 μmol, 1.0 M in PhMe) was added. the reaction was quenched sequentially with MeOH (0.5 mL), sat. aq. tartaric acid (0.1 mL) and H2O (2 mL). Next, the mixture was warmed to r.t. After 1 h, the mixture was extracted with Et2O (3 × 10 mL) and the dried (MgSO4) extract was concentrated in vacuo to give the unstable aldehyde 10 as a colorless oil. The aldehyde was applied immediately to the next step without further purification.
To a solution of 11 (4.0 mg, 0.012 mmol) in THF/H2O (0.2 mL, 40:1) at r.t. was added Ba(OH)2•8H2O (7.6 mg, 0.024 mmol). After 30 min, a solution of the above aldehyde intermediate 10 in THF (0.15 mL) was added dropwise via cannula. After 16 h, the reaction was quenched with sat. aq. NH4Cl (2 mL) and the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo was applied immediately to the next step without further purification.
To a flask containing crude 50 at 0°C was added a premixed solution of HOAc/THF/H2O (4.0 mL, 8:4:1) dropwise. After 2 h, the solution was allowed to warm to 10°C. After 6 h, the reaction was warmed to rt. After 6 h, the reaction was concentrated in vacuo. Benzene was added to further dry the product 9 (11.3 mg). 1H NMR (300 MHz, CDCl3) d 7.30 (d, J = 8.8 Hz, 2H), 6.84-6.98 (m, 4H), 6.10 (d, J = 15.9 Hz, 2H), 5.83 (s, 1H), 4.99 (s, 1H), 4.83 (s, 1H), 4.52 (d, J = 10.5 Hz, 1H), 4.12-4.27 (m, 2H), 3.81 (s, 3H), 3.68-3.80 (m, 3H), 3.16 (m, 1H), 2.59-2.68 (m, 3H), 2.28-2.38 (m, 1H), 2.21-2.25 (m, 2H), 1.91-2.11 (m, 4H), 1.82 (s, 3H), 1.75 (s, 3H), 1.71-1.82 (m, 4H), 1.64-1.69 (m, 1H), 1.43 (s, 3H), 1.25-1.35 (m, 4H), 1.09 (d, J = 4.0 Hz, 3H), 0.88-1.01 (m, 31H), 0.81 (s, 9H), 0.55-0.68 (m, 18H), 0.09 (s, 3H), 0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) d 212.5, 201.2, 168.6, 159.6, 147.0, 145.0, 144.0, 142.1, 132.1, 130.1, 129.9, 128.9, 125.7, 115.5, 113.9, 88.5, 77.3, 72.2, 67.2, 66.1, 55.6, 49.6, 47.0, 46.0, 43.9, 40.3, 39.3, 33.3, 32.3, 31.5, 29.1, 28.6, 26.3, 23.3, 23.5, 23.0, 19.6, 18.3, 14.7, 14.5, 12.5, 7.7, 7.4, 7.3, 7.0, 6.2, 5.6, -3.8, -4.1.
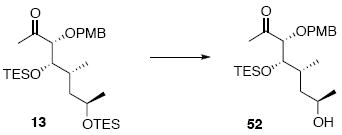
Alcohol 52
To a flask containing 13 (120 mg, 0.22 mmol) at 0°C was added a premixed solution of HOAc/THF/H2O (4.0 mL, 8:8:1) dropwise. After 10 min, the solution was allowed to warm to 5°C. After 2 h, the reaction was quenched with sat. aq. NaHCO3 (10 mL) dropwise. Next, the mixture was extracted with Et2O (3 × 10 mL) and the dried (MgSO4) extract was concentrated in vacuo to give product 52 (90 mg, 0.21 mmol, 95%) as a colorless oil: [α]D23 (+) 17.7° (c 0.34, CHCl3); IR (thin film) 3413, 2958, 2910, 2875, 1712, 1613, 1514, 1457, 1354, 1302, 1249, 1172, 1076, 1055, 1009, 820, 740 cm-1; 1H NMR (300 MHz, CDCl3) d 7.23 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 4.43 (d, J = 11.4 Hz, 1H), 4.38 (d, J = 11.4 Hz, 1H), 3.81-3.91 (m, 2H), 3.80 (s, 3H), 3.74 (d, J = 6.3 Hz, 1H), 2.40 (br, 1H), 2.14 (s, 3H), 1.49-1.65 (m, 2H), 1.32-1.40 (m, 1H), 1.14 (d, J = 6.0 Hz, 3H), 0.85-0.99 (m, 12H), 0.58 (q, J = 7.8 Hz, 6H); 13C NMR (400 MHz, CDCl3) d 210.8, 159.8, 130.2, 129.7, 114.2, 88.4, 75.3, 72.9, 66.2, 55.7, 43.5, 33.5, 27.5, 24.2, 15.1, 7.4, 5.6; HRMS (FAB+) calcd. for C23H41O5Si (M+1) 425.2723, found 425.2730.
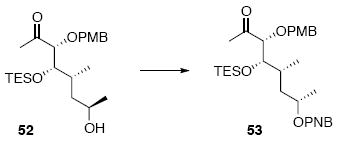
Ester 53
To a solution of 52 (237 mg, 0.56 mmol), p-NO2-benzonic acid (374 mg, 2.24 mmmol) and PPh3 (587 mg, 2.24 mmol) in THF (3.7 mL) at 0 °C was added DEAD (1.0 mL, 1.04 g, 2.24 mmol, 40% w/w in PhMe) dropwise. After 10 min, the reaction was warmed to r.t. After 2 h, the reaction was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-15% EtOAc / Hexanes, to give 53 (46 mg, 0.084mmol, 80%) as a colorless oil: [α]D23 (+) 38.1° (c 0.48, CHCl3); IR (thin film) 2954, 2875, 1719, 1610, 1528, 1514, 1457, 1349, 1275, 1102, 1036, 1013, 823, 720 cm-1; 1H NMR (400 MHz, CDCl3) d 8.28 (d, J = 8.8 Hz, 2H), 8.19 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 5.24-5.29 (m, 1H), 4.53 (d, J = 11.2 Hz, 1H), 4.38 (d, J = 11.2 Hz, 1H), 3.80-3.83 (m, 3H), 2.16 (s, 3H), 2.01-2.08 (m, 1H), 1.67-1.69 (m, 1H), 1.55-1.61 (m, 1H), 1.39 (d, J = 6.0 Hz, 3H), 0.93 (t, J = 8.0 Hz, 9H), 0.89 (d, J = 6.8 Hz, 3H), 0.59 (q, J = 8.0 Hz, 6H); 13C NMR (400 MHz, CDCl3) d 211.3, 164.8, 159.9, 150.8, 136.5, 131.0, 130.3, 129.6, 123.9, 114.2, 87.6, 77.4, 73.1, 71.3, 55.7, 39.9, 33.7, 28.0, 21.4, 14.7, 7.4, 5.5.
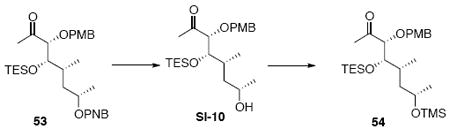
Silylether 54
To a solution of 53 (125 mg, 0.23 mmol) in MeOH (4.7 mL) was added Ba(OH)2.8H2O (36.1 mg, 0.12 mmol). After 1 h, the reaction was quenched with sat. aq. NH4Cl (5 mL). The mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-15% EtOAc / Hexanes, to give an alcohol intermediate SI-10 (63 mg, 0.15 mmol, 65%) as a colorless oil: [α]D23 (+) 38.4° (c 1.87, CHCl3); IR (thin film) 3439, 2958, 2933, 2875, 1713, 1613, 1514, 1458, 1376, 1302, 1249, 1079, 1035, 820, 786, 739 cm-1; 1H NMR (300 MHz, CDCl3) d 7.24 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 4.44 (d, J = 11.4 Hz, 1H), 4.38 (d, J= 11.4 Hz, 1H), 3.75-3.80 (m, 6H), 2.14 (s, 3H), 1.70 (br, 1H), 1.50-1.59 (m, 1H), 1.27-1.35 (m, 2H), 1.16 (d, J = 6.0 Hz, 3H), 0.92 (t, J = 7.8 Hz, 9H), 0.85 (d, J = 6.9 Hz, 3H), 0.58 (q, J = 6.9 Hz, 6H); 13C NMR (400 MHz, CDCl3) d 211.5, 159.8, 130.2, 129.7, 114.2, 88.3, 73.0, 66.4, 55.7, 43.9, 33.8, 27.7, 24.9, 14.7, 7.4, 5.6; HRMS (FAB+) calcd. for C23H40O5SiNa (M+Na) 447.2543, found 447.2561.
To a solution of the alcohol intermediate SI-10 (77 mg, 0.18 mmol) in CH2Cl2 (0.9 mL) at −78°C was added 2,6-lutidine (78 mg, 84.8 μL, 0.73 mmol) and TMSOTf (80 mg, 65.2 μL, 0.36 mmol). After 20 min, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give product 54 (80 mg, 0.16 mmol, 90%) as a colorless oil: [α]D23 (+) 31.8° (c 2.80, CHCl3); IR (thin film) 2955, 2911, 2876, 1715, 1613, 1514, 1458, 1414, 1372, 1352, 1249, 1124, 1088, 1038, 1006, 820, 787, 741 cm-1; 1H NMR (300 MHz, CDCl3) d 7.23 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 4.42 (d, J = 11.4 Hz, 1H), 4.36 (d, J = 11.4 Hz, 1H), 3.74-3.85 (m, 5H), 3.68 (d, J = 6.9 Hz, 1H), 2.09 (s, 3H), 1.55-1.61 (m, 1H), 1.41-1.50 (m, 1H), 1.25-1.35 (m, 1H), 1.12 (d, J = 6.0 Hz, 3H), 0.92 (t, J = 7.8 Hz, 9H), 0.82 (d, J = 6.6 Hz, 3H), 0.58 (q, J = 7.8 Hz, 6H), 0.07 (s, 9H); 13C NMR (75 MHz, CDCl3) d 211.0, 159.7, 130.2, 129.7, 114.1, 89.3, 73.1, 66.2, 55.6, 45.2, 32.4, 26.9, 25.3, 12.9, 7.4, 5.7, 0.7; HRMS (ES+) calcd. for C26H48O5NaSi2 (M+Na) 519.2938, found 519.2914.
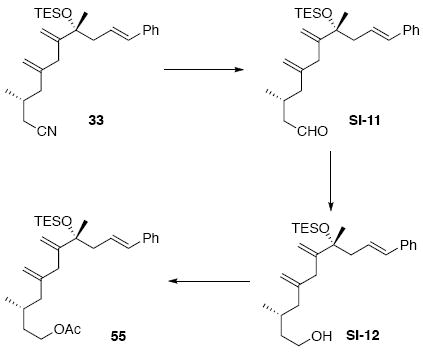
Acetate 55
To a solution of 33 (45 mg, 0.11 mmol) in PhMe (0.53 mL) at -78°C was added DIBAL-H (0.15 mL, 0.15 mmol, 1.0 M in PhMe). After 10 min, the reaction was quenched sequentially with MeOH (0.2 mL), sat. aq. tartaric acid (0.2 mL) and H2O (2.0 mL). After stirring 12 h at r.t., the mixture was extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give the corresponding crude aldehyde (0.11 mmol).
To a mixed solution of the above aldehyde (0.11 mmol) in CH2Cl2 (0.5 mL) and EtOH (0.5 mL) at 0°C was added NaBH4 (8.4 mg, 0.22 mmol). After 15 min, the reaction was quenched with a brine solution (5 mL) and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 6% EtOAc / Hexanes, to give the corresponding alcohol intermediate (38 mg, 0.09 mmol, 85%) as a colorless oil.
To a solution of the above alcohol intermediate (29 mg, 0.07 mmol) in CH2Cl2 at 0°C was added sequentially pyridine (21.4 mg, 22.1 μL, 0.27 mmol), DMAP (8.3 mg, 0.07 mmol) and Ac2O (13.9 mg, 12.8 μL, 0.14 mmol). After 15 min, the reaction was quenched with sat. aq. NH4Cl (5 mL) and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give product 55 (31 mg, 64 μmol, 94%) as a colorless oil: [α]D23 (-) 39.3° (c 0.40, CHCl3); IR (thin film) 3026, 2955, 2913, 2875, 1741, 1496, 1457, 1367, 1236, 1158, 1121, 1053, 1016, 966, 898, 793, 741, 692 cm-1; 1H NMR (300 MHz, CDCl3) d 7.15-7.28 (m, 5H), 6.32 (d, J = 15.9 Hz, 1H), 6.07-6.17 (m, 1H), 5.85 (s, 1H), 4.96 (s, 1H), 4.79 (s, 1H), 3.98-4.07 (m, 2H), 2.41-2.52 (m, 2H), 2.10 (dd, J = 5.7, 13.5 Hz, 1H), 2.00 (s, 3H), 1.85 (dd, J = 7.8, 13.2 Hz, 1H), 1.79 (s, 3H), 1.58-1.67 (m, 2H), 1.43 (s, 3H), 1.28-1.36 (m, 1H), 0.97 (t, J = 7.5 Hz, 9H), 0.75(d, J = 6.6 Hz, 3H), 0.61 (q, J = 7.5 Hz, 6 H); 13C NMR (75 MHz, CDCl3) d 171.6, 145.3, 143.0, 138.3, 132.0, 128.9, 127.4, 127.3, 126.3, 125.3, 114.9, 78.7, 63.3, 46.4, 46.2, 35.6, 29.0, 27.4, 21.4, 19.6, 15.0, 7.6, 7.2; HRMS (FAB+) calcd. for C29H46O3SiNa (M+Na) 493.3114, found 493.3130.
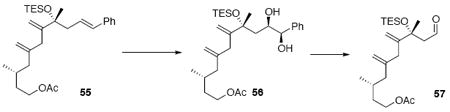
Compound 69
To a solution of 55 (31 mg, 66 μmol) in t-BuOH (0.55 mL) and H2O (0.55 mL) at r.t. was added AD mix β*53 (92 mg) and CH3SO2NH2 (9.5 mg, 0.10 mmol). After 16 h, the reaction was quenched with 10% aq. Na2S2O3 (2 mL). After 30 min, the mixture was extracted with EtOAc (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20% EtOAc / Hexanes, to give the corresponding diol (29 mg, 58 μmol, 88%, 6:1 d.r.).
To a solution of the above diol (26 mg, 52 μmol) in THF/ Et2O/ H2O (1.7 mL, 1:1:1) at r.t. was added NaIO4 (110 mg, 0.52 mmol). After 2 h, the reaction was quenched with a brine solution (2 mL) and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo to give the aldehyde intermediate (20.4 mg, 52 μmol, 99%) as a colorless oil. The aldehyde intermediate was applied to the next step without further purification. [α]D23 = -11.6 (c 1.83, CHCl3); IR (neat) 2957, 2877, 1741, 1724, 1458, 1368, 1239, 1050, 1017, 743 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.66 (t, J = 3.0 Hz, 1H), 6.02 (s, 1H), 5.03 (s, 1H), 4.83 (s, 1H), 4.14-4.04 (m, 2H), 2.65 (dd, J = 15.1, 2.9 Hz, 1H), 2.46 (dd, J = 15.1, 3.1 Hz, 1H), 2.14 (dd, J = 13.6, 5.8 Hz, 1H), 2.04 (s, 3H), 1.92 (dd, J = 13.5, 7.8 Hz, 1H), 1.82 (s, 3H), 1.70 -1.60 (m, 2H), 1.48 (s, 3H), 1.45-1.39 (m, 1H), 1.00-0.94 (m, 9H), 0.87 (d, J = 6.5 Hz, 3H), 0.68-0.56 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 203.0, 171.2, 144.3, 141.2, 125.7, 115.1, 62.8, 54.0, 45.8, 35.3, 28.7, 27.9, 20.9, 19.2, 14.7, 7.1, 6.7, 6.6, 5.8; HRMS (EI+) calcd. for C22H40O4Si (M+) 396.2696, found 396.2678.
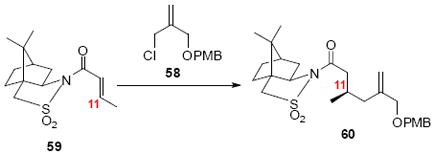
Sultam 60
Following the similar procedure described by Paquette,38a Mg (36.0 g, 1.5 mol) was stirred vigorously at rt in a dry flask under Ar. After 120 h, when black coating formed inside the flask, THF (200 mL) and 1,2-dibromoethane (2.60 g, 1.2 mL, 13.9 mmol) were added sequentially. After 30 min, a solution of allyl chloride 58 (17.0 g, 75.0 mmol) in THF (80 mL) was added slowly to the Mg slurry over 5 h. The resulted mixture was stirred overnight at rt to give 300 mL Grignard reagent (0.12 M, 47%) as gray solution. The concentration of the Grignard reagent was determined by the titration using menthol in the presence of 1,10-phenanthroline.55
Separately, CuBr•SMe2 (7.29 g, 35.5 mmol) and LiCl (1.61 g, 37.9 mmol) were dissolved in THF (80 mL) and added to the solution of Grinard reagent (263 mL, 31.5 mmol) at -78°C via syringe. TMSCl (3.96 g, 4.5 mL, 36.5 mmol) was then added followed by a solution of known sultam 5956 (6.9 g, 24.3 mmol) in THF (60 mL). After another 90 min, the reaction was quenched with NH4Cl-NH4OH (9:1, pH 9, 60 mL), warmed to rt. The aqueous layer was extracted with EtOAc (3 × 200 mL). The organic phase was washed with sat. aq. NaCl (100 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 8-15% EtOAc / Hexanes, to give the product 60 (11.2 g, 34.4 mmol, 97%) as a colorless oil: [α]D23 = -68.0 (c 0.51, CHCl3); IR (neat) 2959, 2927, 2851, 1693, 1512, 1454, 1328, 1246, 1032 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.28 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 5.12 (s, 1H), 4.96 (s, 1H), 4.44 (t, J = 11.8 Hz, 2H), 3.94 (t, J = 11.8 Hz, 2H), 4.00 (dd, J = 14.0 Hz, 2H), 3.88 (t, J = 6.2 Hz, 1H), 3.82 (s, 3H), 3.46 (dd, J = 23.0, 13.8 Hz, 2H), 2.78 (dd, J = 16.1, 5.7 Hz, 1H), 2.51 (dd, J = 16.1, 7.6 Hz, 1H), 2.30-2.40 (m, 1H), 2.02-2.15 (m, 4H), 1.82-1.96 (m, 3H), 1.28-1.45 (m, 3H), 1.15 (s, 3H), 0.99 (d, J = 6.4 Hz, 3H), 0.98 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.4, 159.1, 144.1, 130.6, 129.3, 113.7, 113.6, 72.4, 71.7, 65.2, 55.3, 53.0, 48.3, 47.7, 44.7, 42.5, 40.8, 38.6, 32.9, 28.0, 26.5, 20.8, 19.9; HRMS (ES+) calcd. for C26H37NO5SNa (M+Na) 498.2290, found 498.2271.
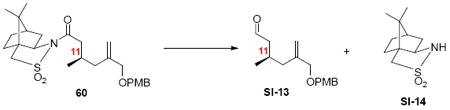
Aldehyde SI-13
To a stirred solution of sultam 60 (11.0 g, 23.1 mmol) in CH2Cl2 (115 mL) at -78°C was added DIBAL-H (50.8 mL, 50.8 mmol, 1.0 M in CH2Cl2). After 2 h, the reaction was carefully quenched with methanol (2.0 mL) and poured into aq. sodium potassium tartrate (250 mL, 10% aq.) at rt. The reaction flask was rinsed with an additional portion of CH2Cl2 (150 mL). After 3 h, the aqueous layer was extracted with CH2Cl2 (3 × 150 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give the aldehyde SI-13 (5.9 g, 22.6 mmol, 98%) as colorless oil. Further elution with 5% MeOH / EtOAc gave recovered auxiliary SI-14 (4.9 g, 22.4 mmol, 97%). SI-13: [α]D23 = +5.93 (c 0.91, CHCl3); IR (neat) 2956, 2929, 2837, 1723, 1612, 1513, 1247, 1077, 1034 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.75 (t, J = 1.5 Hz, 1H), 7.29 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 5.14 (d, J = 1.2 Hz, 1H), 4.95 (s, 1H), 4.44 (s, 2H), 3.93 (s, 2H), 3.83 (s, 3H), 2.47 (ddd, J = 14.0, 4.0 and 1.3 Hz, 1H), 2.17-2.34 (m, 2H), 2.01-2.11 (m, 2H), 0.98 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 202.6, 159.2, 143.9, 130.3, 129.3, 114.1, 113.8, 72.4, 71.7, 55.3, 50.6, 41.0, 26.3, 20.1; HRMS (ES+) calcd. for C16H22O3Na (M+Na) 285.1467, found 285.1494.
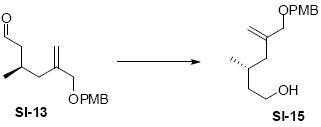
Aldohol SI-15
To a stirred solution of aldehyde SI-13 (5.6 g, 21.4 mmol) in CH2Cl2 (110 mL) at -78°C was added DIBAL-H (28.3 mL, 28.3 mmol, 1.0 M in CH2Cl2). After 1 h, the reaction was carefully quenched with methanol (2.0 mL) and poured into aq. sodium potassium tartrate (250 mL, 10% aq.) at rt. The reaction flask was rinsed with an additional portion of CH2Cl2 (150 mL). After 3 h, the aqueous layer was extracted with CH2Cl2 (3 × 150 mL). The dried extract (MgSO4) was concentrated in vacuo to give the alcohol SI-15 (5.6 g, 20.8 mmol, 97%) as a colorless oil: [α]D23 = -2.94 (c 0.51, CHCl3); IR (neat) 3407, 2926, 2868, 1612, 1513, 1461, 1248, 1059, 1036 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 5.10 (s, 1H), 4.94 (s, 1H), 4.44 (t, J = 12.2 Hz, 2H), 3.91 (t, J = 13.4 Hz, 2H), 3.82 (s, 3H), 3.61-3.78 (m, 2H), 2.15 (dd, J = 13.8, 6.0 Hz, 1H), 1.89-1.96 (m, 1H), 1.89-1.96 (m, 1H), 1.72-1.86 (m, 1H), 1.57-1.69 (m, 1H), 1.23-1.45 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 159.2, 144.6, 130.4, 129.3, 113.8, 113.4, 72.6, 71.6, 61.0, 55.3, 41.3, 39.7, 27.5, 19.7, 18.8; HRMS (ES+) calcd. for C16H24O3Na (M+Na) 287.1623, found 287.1649.
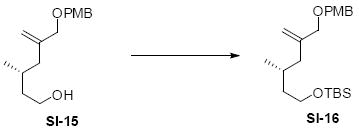
TBS ether SI-16
To a stirred solution of alcohol SI-15 (5.5 g, 20.8 mmol) in DMF (50 mL) at rt was sequentially added imidazole (3.4 g, 50.0 mmol) and TBSCl (3.8 g, 25.2 mmol). After 1 h, the reaction was quenched with sat. aq. NH4Cl (50 mL) and extracted with Et2O (3 × 150 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give TBS ether SI-16 (7.7 g, 20.3 mmol, 97%) as a colorless oil: [α]D23 = -3.27 (c 1.31, CHCl3); IR (neat) 2954, 2928, 2856, 1513, 1249, 1094, 835 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 5.11 (s, 1H), 4.93 (s, 1H), 4.44 (s, 2H), 3.92 (s, 2H), 3.83 (s, 3H), 3.63-3.70 (m, 2H), 2.13 (dd, J = 13.8, 6.3 Hz, 1H), 1.88-1.96 (m, 1H), 1.76-1.86 (m, 1H), 1.57-1.69 (m, 1H), 1.28-1.35 (m, 1H), 0.91 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.06 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 159.1, 144.7, 130.6, 129.3, 113.8, 112.8, 72.6, 71.6, 61.3, 55.3, 41.4, 39.8, 27.5, 26.0, 19.6, 18.3, -5.3; HRMS (ES+) calcd. for C22H38O3NaSi (M+Na) 401.2488, found 401.2489.

Alcohol SI-17
To a stirred solution of TBS ether SI-16 (3.85 g, 10.2 mmol) in CH2Cl2/pH 7 buffer (10 : 1, 110 mL) was added DDQ (2.77 g, 12.2 mmol) at rt. After 1 h, the reaction was quenched with sat. aq. NaHCO3 (50 mL) and extracted with Et2O (3 × 100 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 8% EtOAc / Hexanes, to give a mixture of product SI-17 and 4-methoxybenzaldehyde (3.90 g, 1:1 mole/mole, 9.9 mmol, 97%) as colorless oil. An analytically pure sample was prepared by chromatography over silica gel, eluting with 3%-5% EtOAc / Hexanes, for characterization, but the product mixture was used in the subsequent step without complete removal of 4-methoxybenzaldehyde. SI-17: [α]D23 = -6.09 (c 1.21, CHCl3); IR (neat) 3338, 2955, 2929, 2858, 1471, 1463, 1255, 1098, 835 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.09 (d, J = 1.5 Hz, 1H), 4.89 (s, 1H), 4.07 (d, J = 6.3 Hz, 2H), 3.61-3.75 (m, 2H), 2.13 (dd, J = 13.5, 6.0 Hz, 1H), 1.75-1.95 (m, 2H), 1.55-1.66 (m, 1H), 1.41-1.50 (m, 1H), 1.27-1.38 (m, 1H), 0.91 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 147.6, 110.7, 65.8, 61.2, 41.2, 39.6, 27.6, 26.0, 19.7, 18.3, -5.3; HRMS (EI+) calcd. for C14H31O2Si (M+H) 259.2093, found 259.2091.
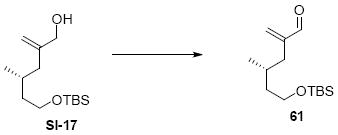
Aldehyde 61
To a stirred solution of alcohol SI-17 and 4-methoxybenzaldehyde (7.8 g, 1:1 mole/mole, 19.7 mmol) in CH2Cl2 (200 mL) was sequentially added NaHCO3 (3.0 g, 35.7 mmol) and DMP (10.0 g, 23.7 mmol) at rt. After 1 h, the reaction was quenched with sat. aq. NaHCO3 (50 mL) and extracted with Et2O (3 × 150 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 3% EtOAc / Hexanes, to give aldehyde 61 (4.6 g, 17.7 mmol, 90%) as a colorless oil: [α]D23 = -8.20 (c 1.21, CHCl3); IR (neat) 2956, 2929, 2857, 1698, 1255, 1099, 835 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.56 (s, 1H), 6.28 (s, 1H), 6.07 (s, 1H), 3.60-3.73 (m, 2H), 2.28 (dd, J = 14.1, 6.9 Hz, 1H), 2.12 (dd, J = 13.8, 8.1 Hz, 1H),1.77-1.86 (m, 1H), 1.53-1.64 (m, 1H), 1.29-1.39 (m, 1H), 0.91 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 194.7, 149.0, 135.2, 61.1, 39.5, 35.2, 28.4, 25.9, 25.5, 19.4, -5.4; HRMS (EI+) calcd. for C14H28O2Si (M) 256.1859, found 256.1861.
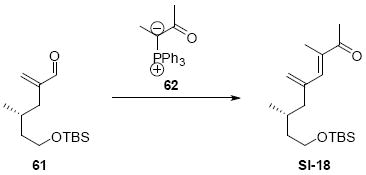
Methyl ketone SI-18
A solution of aldehyde 61 (4.5 g, 17.5 mmol) and ylide 6257 (10.2 g, 30.7 mmol) in toluene (60 mL) was refluxed in a sealed tube (oil bath 112°C). After 16 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes (1% Et3N added), to give diene SI-18 (5.0 g, 16.1 mmol, 92%) as a slightly yellow oil: [α]D23 = -41.1 (c 0.53, CHCl3); IR (neat) 2955, 2928, 2857, 1671, 1255, 1100, 836, 775 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.90 (s, 1H), 5.29 (s, 1H), 5.14 (s, 1H), 3.62-3.73 (m, 2H), 2.37 (s, 3H), 2.30 (dd, J = 10.2, 4.8 Hz, 1H), 2.05 (dd, J = 10.5, 6.3 Hz, 1H), 1.97 (d, J = 10.2 Hz, 3H), 1.71-1.78 (m, 1H), 1.59- 1.64 (m, 1H), 1.34-1.38 (m, 1H), 0.91 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.07 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 200.3, 143.7, 140.9, 137.8, 118.9, 61.0, 45.1, 39.6, 28.4, 25.9, 25.7, 19.3, 18.3, 13.1, - 5.3; HRMS (FAB+) calcd. for C18H35O2Si (M+H) 311.2406, found 311.2400.
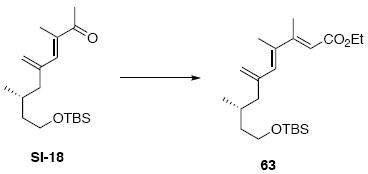
Triene ester 63
To a stirred slurry of NaH (1.29 g, 32.2 mmol) in DME (50 mL) was added ethyl 2-(diethoxyphosphoryl)acetate (6.48 g, 5.74 mL, 28.9 mmol) at rt. After 1 h, a solution of methyl ketone SI-18 (5.00 g, 16.1 mmol) in DME (25 mL) was added. The resulted solution was refluxed for 3 h then quenched with H2O (15 mL) and extracted with Et2O (3 × 150 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2% EtOAc / Hexanes (1% Et3N added), to give triene ester 63 (4.42 g, 11.6 mmol, 70%) as a colorless oil: [α]D23 = - 34.7 (c 1.66, CHCl3); IR (neat) 2955, 2928, 2857, 1716, 1610, 1255, 1163, 1098, 836, 775 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.24 (s, 1H), 5.92 (s, 1H), 5.13 (s, 1H), 4.93 (s, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.61-3.68 (m, 2H), 2.36 (s, 3H), 2.20 (dd, J = 13.3, 5.9 Hz, 1H), 1.93-2.00 (m, 4H), 1.53-1.71 (m, 2H), 1.31 (t, J = 7.2 Hz, 3H), 0.89 (s, 9H), 0.87 (d, J = 6.9 Hz, 3H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 167.4, 156.4, 144.7, 137.8, 132.4, 116.4, 115.9, 61.2, 59.7, 45.7, 39.7, 28.3, 25.9, 19.5, 18.3, 15.8, 15.5, 14.4, -5.3; HRMS (EI+) calcd. for C22H40O3Si (M+H) 380.2747, found 380.2732.
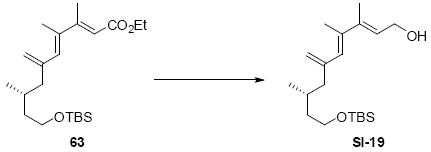
Allyl alcohol SI-19
To a stirred solution of triene ester 63 (8.61 g, 22.6 mmol) in THF (200 mL) was added DIBAL-H (46 mL, 46.0 mmol, 1 M in toluene) at -78°C. After 1.5 h, the reaction was quenched with MeOH (1.0 mL) and poured into aq. sodium potassium tartrate (350 mL, 10% aq.) at rt. The reaction flask was rinsed with an additional portion of Et2O (50 mL). After 3 h, the aqueous layer was extracted with Et2O (3 × 200 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 8% EtOAc / Hexanes, to give allyl alcohol SI-19 (6.20 g, 18.3 mmol, 81%) as a colorless oil: [α]D23 = -36.6 (c 1.66, CHCl3); IR (neat) 3327, 2954, 2928, 2857, 1471, 1462, 1376, 1255, 1098, 1006, 835, 775 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.97 (s, 1H), 5.80 (t, J = 6.3 Hz, 1H), 5.08 (s, 1H), 4.87 (s, 1H), 4.34 (d, J = 6.4 Hz, 2H), 3.46-3.72 (m, 2H), 2.18 (dd, J = 13.6, 6.0 Hz, 1H), 1.99 (d, J = 0.8 Hz, 3H), 1.93 (dd, J = 13.5, 5.2 Hz, 1H), 1.86 (s, 3H), 1.54-1.73 (m, 2H), 1.28-1.37 (m, 1H), 0.90 (s, 9H), 0.87 (d, J = 6.6 Hz, 3H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 145.2, 139.3, 137.6, 128.1, 125.9, 115.4, 61.3, 60.1, 46.0, 39.7, 28.3, 25.9, 19.5, 18.3, 15.6, 14.2, -5.3; HRMS (EI+) calcd. for C20H38O2Si (M) 338.2641, found 338.2612.

Epoxide 64
To a stirred solution of (+)-DIPT (41.5 mg, 0.18 mmol) and 4 Å mol sieves (about 200 mg) in CH2Cl2 (4.0 mL) was sequentially added Ti(O-iPr)4 (34 mg, 34.6 μL, 0.12 mmol) and TBHP (236 μL, 1.30 mmol, 5.0-6.0 M in decane) at -20°C. After 20 min, the reaction mixture was cooled to -78°C and a pre-cooled solution (-78°C) of allyl alcohol SI-19 (200 mg, 0.59 mmol) in CH2Cl2 (4.0 mL) was added via cannula. The resulted solution was warmed up to -50°C. After another 60 min, the reaction was quenched with pH7 phosphate buffer (0.5 mL), filtered over Celite®, and extracted with Et2O (3 × 20 mL). The dried organic layers (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give epoxide 64 (167 mg, 0.47 mmol, 80%) as colorless oil: [α]D23 = -17.3 (c 1.66, CHCl3); IR (neat) 3430, 2954, 2927, 2856, 1471, 1463, 1378, 1255, 1097, 836, 775 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.89 (s, 1H), 5.03 (s, 1H), 4.86 (s, 1H), 3.86-3.92 (m, 1H), 3.73-3.81 (m, 1H), 3.62-3.71 (m, 2H), 3.02 (dd, J = 10.5, 6.3 Hz, 1H), 2.14 (dd, J = 13.5, 5.6 Hz, 1H), 1.91 (dd, J = 13.5, 8.4 Hz, 1H), 1.84 (d, J = 1.1 Hz, 3H), 1.54-1.66 (m, 2H), 1.45 (s, 3H), 1.27-1.34 (m, 1H), 0.91 (s, 9H), 0.83 (d, J = 6.4 Hz, 3H), 0.06 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 144.0, 137.5, 126.2, 115.2, 63.7, 61.3, 45.6, 39.8, 28.2, 26.0, 19.3, 18.3, 16.7, 14.8, -5.3; HRMS (CI+) calcd. for C20H39O3Si (M+H) 355.2669, found 355.2666.
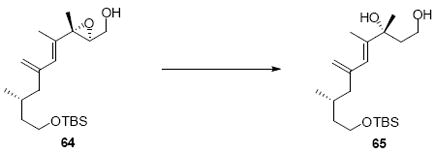
Diol 65
To a stirred solution of epoxide 64 (1.5 g, 4.23 mmol) in THF (30 mL) was added Red-Al (1.5 mL, 9.91 mmol, 65% W/V in toluene) at 0°C. After 1 h, another portion of Red-Al (1.5 mL, 9.91 mmol, 65% W/V in toluene) was added. After another 1.5 h, the reaction was quenched with H2O (0.10 mL), and extracted with CH2Cl2 (3 × 150 mL). The dried organic layers (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 6-12% EtOAc / Hexanes (1% Et3N added), to give diol 65 (1.05 g, 2.96 mmol, 70%) as a colorless oil: [α]D23 = -29.4 (c 0.81, CHCl3); IR (neat) 3389, 2955, 2928, 2858, 1471, 1462, 1382, 1255, 1097, 836, 775 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.10 (s, 1H), 5.04 (d, J = 0.9 Hz, 1H), 4.84 (s, 1H), 3.62-3.77 (m, 4H), 3.04 (s, br, 1H), 2.60 (s, br, 1H), 2.16 (dd, J = 13.2, 5.1 Hz, 1H), 1.88-1.96 (m, 3H), 1.79 (d, J = 0.9 Hz, 3H), 1.55-1.70 (m, 2H), 1.37 (s, 1H), 1.27-1.34 (m, 1H), 0.91 (s, 9H), 0.86 (d, J = 6.5 Hz, 3H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 145.1, 141.7, 125.1, 114.4, 77.1, 61.6, 60.4, 46.1, 40.1, 39.7, 28.6, 28.2, 26.0, 19.6, 18.4, 15.1, -5.2; HRMS (ES+) calcd. for C20H40O3SiNa (M+Na) 379.2644, found 379.2643
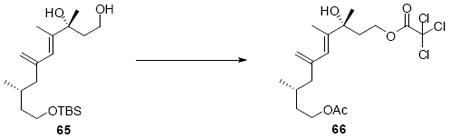
Ester 66
To a stirred solution of diol 65 (2.10 g, 5.89 mmol) in CH2Cl2 (50 mL) was sequentially added pyridine (1.37 g, 1.40 mL, 17.7 mmol) and trichloroacetyl chloride (1.29 g, 0.79 mL, 7.09 mmol). After 3 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with ether (3 × 30 mL). The dried extract (MgSO4) was concentrated in vacuo to give crude ester (3.30 g) as a colorless oil, which was used in the next step without further purification.
To a stirred solution of crude ester (3.30 g) in CH2Cl2 / EtOH (1:1, 50 mL) was added CSA (2.31 g, 9.88 mmol) at 0°C. After 1.5 h, the reaction was quenched with sat. aq. NaHCO3 (10 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo to give crude diol (2.02 g), which was used in the next step without further purification.
To a stirred solution of crude diol (2.02 g) in CH2Cl2 (50 mL) was sequentially added pyridine (1.53 g, 1.57 mL, 19.5 mmol) and Ac2O (0.99 g, 0.92 mL, 9.73 mmol). After 1.5 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give 66 (1.90 g, 4.42 mmol, 75% over 3 steps) as a colorless oil: [α]D 23 = -14.1 (c 1.16, CHCl3); IR (neat) 3481, 2962, 2928, 1766, 1739, 1720, 1458, 1368, 1247, 828, 682 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.01 (s, 1H), 5.04 (s, 1H), 4.85 (s, 1H), 4.47-4.36 (m, 2H), 4.15-4.06 (m, 2H), 2.12-2.02 (m, 3H), 2.03 (s, 3H), 1.97-1.89 (m, 3H), 1.83 (s, 3H), 1.70-1.60 (m, 2H), 1.50-1.38 (m, 1H), 1.41 (s, 3H), 0.89 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.2, 161.9, 144.1, 141.4, 124.9, 115.3, 74.6, 66.5, 62.3, 45.6, 37.8, 35.2, 28.7, 28.3, 21.0, 19.4, 14.9; HRMS (EI+) calcd. for C18H27O5Cl3 (M+) 428.0924, found 428.0932.
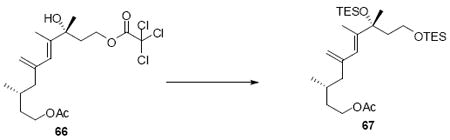
TES ether 67
To a stirred solution of alcohol 66 (1.90 g, 4.4 mmol) in CH2Cl2 / EtOH (1:1, 50 mL) was added NH3•H2O (15 mL) at rt. After 1 h, the reaction was quenched with sat. aq. NH4Cl (15 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo to afford crude diol (1.55 g), which which was used in the next step without further purification.
To a stirred solution of crude diol (1.55 g) in CH2Cl2 / Et3N (1:1, 30 mL) was added freshly distilled TESOTf (3.52 g, 3.01 mL, 13.1 mmol) at -78°C. After 20 min, the reaction was quenched with sat. aq. NaHCO3 (10 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo andpurified by chromatography over silica gel, eluting with 2% EtOAc / Hexanes, to give TES ether 67 (2.12 g, 4.09 mmol, 93% over 2 steps) as a colorless oil: [α]D23 = -18.4 (c 1.11, CHCl3); IR (neat) 29554, 2912, 2876, 1748, 1458, 1238, 1086, 1016, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.89 (s, 1H), 5.00 (d, J = 1.2 Hz, 1H), 4.81 (d, J = 1.2 Hz, 1H), 4.18-4.05 (m, 2H), 3.72-3.63 (m, 1H), 3.54-3.41 (m, 1H), 2.15 (dd, J = 13.5, 5.4 Hz, 1H), 2.05 (s, 3H), 1.95-1.89 (m, 2H), 1.87-1.78 (m, 1H), 1.77 (d, J = 1.2 Hz, 3H), 1.71-1.58 (m, 2H), 1.49-1.39 (m, 1H), 1.42 (s, 3H), 1.12-0.91 (m, 18H), 0.87 (d, J = 6.6 Hz, 3H), 0.74-0.50 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 171.1, 144.8, 142.3, 124.3, 114.4, 77.2, 62.3, 59.5, 46.0, 44.4, 35.3, 28.8, 27.8, 21.0, 19.3, 14.6, 7.2, 6.9, 6.8, 6.4, 5.8, 4.4; HRMS (EI+) calcd. for C28H56O4Si2Na (M+Na) 535.3615, found 535.3637.

Aldehyde 57
TES ether 67 (2.1 mg, 4.09 mmol) was dissolved in a stirred solution of HOAc / THF / H2O (34 mL, 8:8:1) at 0°C. After 1.5 h, the reaction was then quenched with solid NaHCO3 and extracted with ether (4 × 50 mL). The dried extract (MgSO4) was concentrated in vacuo to afford crude alcohol (2.0 g), which was used in the next step without further purification.
To a stirred solution of crude alcohol (2.0 g) in CH2Cl2 (20 mL) was added sequentially solid NaHCO3 (1.0 g, 11.9 mmol) and DMP (2.16 g, 5.09 mmol) at rt. After 1 h, the reaction was quenched with sat. aq. NaHCO3 (15 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo andpurified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give aldehyde 57 (1.36 g, 3.43 mmol, 74% over 2 steps) as a colorless oil.

Aldol Product SI-20
To a solution of methyl ketone 54 (38 mg, 76 μmol) in Et2O (0.4 mL) at −78°C was added LDA54 (85.0 μL, 1.0 M in THF). After 20 min, a pre-cooled (-78°C) solution of aldehyde intermediate (20 mg, 50 μmol) in Et2O (0.25 mL) was transferred in one portion via cannula at −78°C. After 20 min, the reaction was quenched with sat. aq. NH4Cl (3 mL) and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give the alcohol intermediate SI-20 (28 mg, 31 μmol, 62%) as a colorless oil: [α]D23 (+) 15.7° (c 1.55, CHCl3); IR (neat) 3515, 2955, 2912, 2876, 1741, 1717, 1615, 1515, 1417, 1368, 1247, 1117, 1037, 1008, 899, 806, 741 cm-1; 1H NMR (300 MHz, CDCl3) d 7.28 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 5.87 (s, 1H), 5.01 (s, 1H), 4.83 (s, 1H), 4.50 (d, J = 11.1 Hz, 1H), 4.34-4.51 (m, 1H), 4.33 (d, J = 11.1 Hz, 1H), 4.04-4.14 (m, 2H), 3.80 (s, 3H), 3.71-3.78 (m, 3H), 3.03 (dd, J = 7.5, 17.4 Hz, 1H), 2.29 (dd, J = 4.5, 17.4 Hz, 1H), 2.12 (dd, J = 5.7, 8.7 Hz, 1H), 2.03 (s, 3H), 1.83-1.96 (m, 2H), 1.82 (s, 3H), 1.59-1.73 (m, 3H), 1.52 (s, 3H), 1.42-1.49 (m, 4H), 1.11 (d, J = 6.0 Hz, 3H), 0.86-0.98 (m, 21H), 0.82 (d, J = 6.6 Hz, 3H), 0.51-0.67 (m, 12H), 0.06 (s, 9H); 13C NMR (75 MHz, CDCl3) d 211.7, 171.6, 159.6, 144.6, 143.8, 130.3, 130.1, 125.2, 115.6, 114.0, 88.4, 79.8, 77.6, 72.8, 66.2, 65.1, 63.3, 55.6, 48.5, 46.7, 46.2, 44.8, 35.7, 32.5, 29.1, 26.5, 25.2, 21.4, 19.7, 15.1, 13.3, 7.5, 7.4, 7.0, 5.7, 0.7; HRMS (ES+) calcd. for C48H88O9Si3Na (M+Na) 915.5634, found 915.5564.
TES Ethers 68 and 69
To a solution of the above alcohol intermediate (40 mg, 45 μmol) in CH2Cl2 (0.15 mL) and Et3N (0.15 mL) at −78°C was added TESOTf (32.0 mg, 28.3 μL, 0.12 mmol). After 20 min, the reaction was quenched with sat. aq. NH4Cl (3 mL). The mixture was extracted with Et2O (3 × 10 mL) and the dried (MgSO4) extract was concentrated in vacuo to give 69 (36 mg, 36 μmol, 82%) as a colorless oil: [α]D23 (+) 18.6° (c 1.58, CHCl3); IR (neat) 2955, 2926, 2876, 2855, 1742, 1716, 1615, 1515, 1458, 1366, 1249, 1127, 1084, 1006, 899, 835, 776, 741 cm-1; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 5.84 (s, 1H), 5.04 (s, 1H), 4.89 (s, 1H), 4.59 (d, J = 10.8 Hz, 1H), 4.29-4.35 (m, 1H), 4.27 (d, J = 10.8 Hz, 1H), 4.10-4.17 (m, 2H), 3.84 (s, 4H), 3.76-3.78 (m, 1H), 3.71 (d, J = 7.2 Hz, 1H), 3.19 (dd, J = 8.4, 17.6 Hz, 1H), 2.54 (dd, J = 3.3, 17.6 Hz, 1H), 2.11-2.15 (m, 1H), 2.06 (s, 3H), 1.86-1.98 (m, 2H), 1.85 (s, 3H), 1.63-1.77 (m, 4H), 1.52-1.56 (m, 1H), 1.45 (s, 3H), 1.30-1.38 (m, 2H), 1.13 (d, J = 6.4 Hz, 3H), 0.83-0.98 (m, 30H), 0.86 (d, J = 6.4 Hz, 3H), 0.52-0.64 (m, 18H), 0.15 (s, 9H); 13C NMR (75 MHz, CDCl3) d 211.3, 171.2, 159.1, 144.4, 142.6, 130.1, 129.7, 125.1, 115.0, 113.5, 88.2, 77.4, 76.4, 72.0, 65.9, 63.0, 55.3, 50.0, 46.5, 46.0, 44.0, 35.3, 32.3, 29.7, 28.7, 28.0, 24.7, 21.0, 19.3, 14.9, 13.3, 7.3, 7.1, 7.0, 6.8, 6.6, 6.4, 5.3, 5.1, 0.4; HRMS (ES+) calcd. for C54H102O9Si4Na (M+Na) 1029.6499, found 1029.6470.
Alcohol 70
To a flask with 69 (180 mg, 0.18 mmol) at 0°C was added a freshly prepared stock solution of HOAc/THF/H2O (3.0 mL, 8:8:1) dropwise. After 40 min, the reaction was quenched with sat. aq. NaHCO3 (20 mL) carefully and extracted with Et2O (3 × 10 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give 70 (102 mg, 0.11 mmol, 61%) as a colorless oil: [α]D23 (+) 23.7° (c 0.60, CHCl3); IR (neat) 3546, 2955, 2926, 2876, 2855, 1742, 1716, 1615, 1515, 1458, 1366, 1249, 1127, 1084, 1006, 899, 835, 776, 741 cm-1; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 5.87 (s, 1H), 5.02 (s, 1H), 4.86 (s, 1H), 4.55 (d, J = 10.8 Hz, 1H), 4.23-4.29 (m, 1H), 4.23 (d, J = 10.8 Hz, 1H), 4.12-4.17 (m, 2H), 3.85 (s, 4H), 3.75-3.85 (m, 3H), 3.18 (dd, J = 9.2, 17.6 Hz, 1H), 2.54 (dd, J = 2.8, 17.6 Hz, 1H), 2.13 (dd, J = 5.6, 13.6 Hz, 1H), 2.06 (s, 3H), 1.86-1.94 (m, 2H), 1.83 (s, 3H), 1.63-1.80 (m, 4H), 1.52-1.56 (m, 1H), 1.45 (s, 3H), 1.38-1.45 (m, 2H), 1.19 (d, J = 6.0 Hz, 3H), 0.88-1.01 (m, 33H), 0.55-0.68 (m, 18H); 13C NMR (100 MHz, CDCl3) d 211.3, 171.2, 159.1, 144.9, 141.7, 130.0, 129.8, 125.5, 114.9, 113.5, 88.5, 77.4, 77.2, 76.4, 71.8, 65.8, 63.2, 55.3, 49.5, 46.2, 45.8, 44.1, 35.4, 33.2, 29.7, 28.7, 28.5, 24.5, 21.0, 19.3, 14.9, 13.6, 7.3, 7.0, 6.95, 6.87, 5.2, 5.1; HRMS (ES+) calcd. for C51H94O9Si3Na (M+Na) 957.6103, found 957.6157.
Ketophosphonate 71
To a solution of phosphonate acid 11 (74 mg, 0.24 mmol) in PhMe (0.5 mL) at r.t. was added triethylamine (26.2 mg, 36.0 μl, 0.24 mmol) and 2,4,6-trichlorobenzoyl chloride (56 mg, 36.0 μL, 0,24 mmol). After 6 h, solvent of the reaction was removed in vacuo. A solution of the alcohol 70 (60 mg, 64 μmol) in PhMe (0.5 mL) was transferred into the above flask via cannula and DMAP (29.6 mg, 0.24 mmol) was sequentially added. After 16 h, the reaction was purified by chromatography over silica gel, eluting with 70% EtOAc / Hexanes, to give 71 (50 mg, 41 μmol, 64%) as a colorless oil: [α]D23 (+) 25.3° (c 1.50, CHCl3); IR (neat) 2954, 2933, 2876, 1739, 1712, 1613, 1515, 1461, 1366, 1251, 1165, 1124, 1026, 968, 901, 835, 770, 741 cm-1; 1H NMR (400 MHz, CDCl3) d 7.31 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.69 (t, J = 7.2 Hz, 1H), 5.85 (s, 1H), 5.03 (br, 2H), 4.88 (s, 1H), 4.62 (d, J = 10.8 Hz, 1H), 4.25-4.31 (m, 1H), 4.25 (d, J = 10.8 Hz, 1H), 4.05-4.21 (m, 6H), 3.84 (s, 3H), 3.81-3.83 (m, 1H), 3.72 (d, J = 6.0 Hz, 1H), 3.16 (dd, J = 8.8, 17.6 Hz, 1H), 3.12 (s, 1H), 3.07 (s, 1H), 2.67 (t, J = 7.2 Hz, 2H), 2.60 (dd, J = 2.8, 17.6 Hz, 1H), 2.16 (dd, J = 7.6, 14.8 Hz, 2H), 2.08-2.12 (m, 1H), 2.06 (s, 3H), 1.83-1.94 (m, 4H), 1.83 (s, 3H), 1.81 (s, 3H), 1.61-1.77 (m, 5H), 1.52-1.55 (m, 1H), 1.43 (s, 3H), 1.40-1.43 (m, 1H), 1.36 (t, J = 6.8 Hz, 6H), 1.22 (d, J = 6.0 Hz, 3H), 0.90-1.00 (m, 30H), 0.86 (d, J = 6.4 Hz, 3H), 0.54-0.66 (m, 18H); 13C NMR (100 MHz, CDCl3) d 211.7, 201.5, 201.4, 171.2, 167.7, 159.2, 144.5, 142.3, 140.6, 129.9, 129.7, 129.0, 125.2, 115.0, 113.6, 87.4, 76.0, 72.0, 68.9, 66.0, 63.1, 62.6, 62.5, 55.3, 49.9, 46.8, 45.9, 43.4, 43.1, 41.8, 39.3, 35.3, 33.0, 28.7, 28.2, 27.7, 22.3, 21.0, 20.8, 19.3, 16.4, 16.3, 14.9, 14.1, 12.5, 7.3, 7.0, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd. for C64H115O14PSi3Na (M+Na) 1245.7230, found 1245.7181.
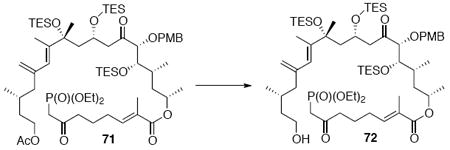
Alcohol 72
To a flask containing 71 (50.0 mg, 41 μmol) at r.t. was added a premixed solution of Ba(OH)2•8H2O (1.5 mL, 0.1 M in MeOH). After 10 min, the mixture was purified by chromatography over silica gel, eluting with 50% EtOAc / Hexanes, to give 72 (42 mg, 36 μmol, 88%) as a colorless oil: [α]D23 (+) 19.8° (c 0.78, CHCl3); IR (neat) 3444, 2954, 2929, 2876, 1713, 1614, 1515, 1458, 1379, 1251, 1165, 1125, 1059, 1024, 970, 900, 835, 809, 741 cm-1; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.69 (t, J = 7.6 Hz, 1H), 5.85 (s, 1H), 5.03-5.06 (m, 2H), 4.85 (s, 1H), 4.62 (d, J = 10.4 Hz, 1H), 4.12-4.27 (m, 7H), 3.84 (s, 3H), 3.81 (dd, J = 3.2, 6.8 Hz, 1H) 3.70-3.76 (m, 3H), 3.19 (dd, J = 9.2, 17.2 Hz, 1H), 3.13 (s, 1H), 3.07 (s, 1H), 2.68 (t, J = 7.2 Hz, 2H), 2.60 (dd, J = 2.8, 17.6 Hz, 1H), 2.18 (q, J = 7.6 Hz, 2H), 2.10 (dd, J = 5.2, 18.4 Hz, 1H), 1.83-1.91 (m, 3H), 1.83 (s, 3H), 1.81 (s, 3H), 1.51-1.81 (m, 7H), 1.43 (s, 3H), 1.38-1.43 (m, 1H), 1.36 (t, J = 7.2 Hz, 6H), 1.23 (d, J = 6.0 Hz, 3H), 0.87-1.00 (m, 33H), 0.54-0.66 (m, 18H); 13C NMR (100 MHz, CDCl3) d 212.3, 201.5, 201.4, 167.7, 159.2, 144.9, 141.7, 140.7, 130.0, 129.7, 129.0, 125.6, 114.9, 113.5, 87.6, 77.5, 75.9, 72.0, 68.9, 66.2, 62.6, 62.5, 61.0, 55.3, 49.5, 46.6, 46.2, 43.4, 43.1, 41.8, 40.1, 39.5, 32.9, 29.7, 28.4, 28.3, 27.7, 22.3, 20.8, 19.2, 16.4, 16.3, 15.1, 14.1, 13.9, 12.5, 7.3, 7.0, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd. for C62H113O13PSi3Na (M+Na) 1203.7124, found 1203.7054.

Macrolactone 51
To a solution of 72 (42 mg, 36 μmol) in CH2Cl2 (1.2 mL) at r.t. was added TPAP (15 mg, 43 μmol). After 1 h, THF (3.3 mL), H2O (82.8 μL) and Ba(OH)2•8H2O (43 mg, 0.13 mmol) were sequentially added to the reaction. After 3 h, the reaction was directly purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give 51 (30.6 mg, 30 μmol, 85%) as a colorless oil: [α]D23 (+) 1.6° (c 0.45, CHCl3); IR (neat) 2954, 2925, 2875, 2854, 1710, 1673, 1614, 1515, 1461, 1377, 1251, 1171, 1120, 1068, 1018, 985, 835, 808, 742 cm-1 ; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.6 Hz, 2H), 6.94-7.01 (m, 1H), 6.88 (d, J = 8.6 Hz, 2H), 6.71 (t, J = 7.6 Hz, 1H), 6.09 (d, J = 16.0 Hz, 1H), 5.77 (s, 1H), 5.05 (s, 1H), 4.96-5.04 (m, 1H), 4.87 (s, 1H), 4.53 (d, J = 10.8 Hz, 1H), 4.21-4.27 (m, 1H), 4.21 (d, J = 10.8 Hz, 1H), 3.84 (s, 3H), 3.67-3.74 (m, 2H), 3.15 (dd, J = 9.2, 17.6 Hz, 1H), 2.56-2.67 (m, 3H), 2.08-2.27 (m, 3H), 1.90 (s, 3H), 1.78 (s, 3H), 1.47-1.78 (m, 9H), 1.43 (s, 3H), 1.26-1.42 (m, 2H), 1.25 (d, J = 6.0 Hz, 3H), 0.90-1.00 (m, 30H), 0.83 (d, J = 6.4 Hz, 3H), 0.54-0.66 (m, 18H); 13C NMR (100 MHz, CDCl3) d 209.8, 200.8, 167.6, 159.2, 147.0, 144.5, 142.7, 140.8, 132.3, 130.0, 129.5, 129.2, 125.3, 115.5, 113.6, 87.9, 71.7, 68.9, 65.4, 55.3, 49.2, 47.1, 45.8, 41.8, 40.6, 37.4, 32.0, 31.5, 31.4, 27.4, 22.9, 21.0, 19.5, 15.2, 14.2, 12.5, 7.3, 7.0, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd. for C58H100O9Si3Na (M+Na) 1047.6573, found 1047.6494.
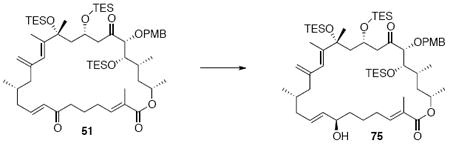
Allylic alcohol 75
To a solution of (S)-2-methyl-CBS-oxazaborolidine (10.1 μL, 10 μmol, 1.0 M in PhMe) in CH2Cl2 (0.3 mL) at r.t. was added catechol borane (0.20 mL, 0.20 mmol, 1.0 M CH2Cl2). After 10 min, the resulted solution was cooled to -20°C. A solution of macrolactone 51 (26 mg, 25 μmol) in CH2Cl2 (0.65 mL) was added into the above solution dropwise via cannula. After 2 h, the reaction was quenched with MeOH (2.0 mL) and concentrated in vacuo. The resultant mixture was purified by preparative TLC, eluting with 25% EtOAc/Hexane, to give 75 (14 mg, 14 μmol, 56%) as a colorless oil: [α]D23 (+) 7.6° (c 0.88, CHCl3); IR (neat) 3459, 2954, 2917, 2875, 2849, 1709, 1614, 1515, 1461, 1377, 1251, 1173, 1125, 1070, 1018, 835, 776, 743 cm-1 ; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 6.72 (t, J = 7.6 Hz, 1H), 5.79 (s, 1H), 5.68-5.72 (m, 1H), 5.49 (dd, J = 6.8, 15.2 Hz, 1H), 5.05 (s, 1H), 4.97-5.04 (m, 1H), 4.87 (s, 1H), 4.57 (d, J = 10.8 Hz, 1H), 4.28-4.32 (m, 1H), 4.22 (d, J = 10.8 Hz, 1H), 4.13-4.17 (m, 1H), 3.85 (s, 3H), 3.65-3.73 (m, 2H), 3.16 (dd, J = 9.2, 18.0 Hz, 1H), 2.53 (dd, J = 3.2, 18.0 Hz, 1H), 2.21-2.24 (m, 4H), 1.98-2.04 (m, 4H), 1.88 (s, 3H), 1.82 (s, 3H), 1.75-1.84 (m, 2H), 1.53-1.70 (m, 7H), 1.44 (s, 3H), 1.30-1.36 (m, 1H), 1.24 (d, J = 5.6 Hz, 3H), 0.89-1.00 (m, 30H), 0.82 (d, J = 6.0 Hz, 3H), 0.54-0.66 (m, 18H); 13C NMR (100 MHz, CDCl3) d 210.6, 167.8, 159.2, 145.1, 142.6, 141.7, 134.5, 130.4, 129.9, 129.8, 128.5, 125.6, 115.1, 113.5, 88.1, 77.2, 76.6, 72.4, 71.7, 68.6, 65.5, 55.3, 53.5, 49.5, 46.4, 44.9, 41.1, 39.4, 36.6, 32.2, 31.4, 28.6, 27.5, 23.7, 21.1, 19.8, 15.0, 12.6, 11.9, 7.3, 7.0, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd. for C58H102O9Si3Na (M+Na) 1049.6729, found 1049.6678.
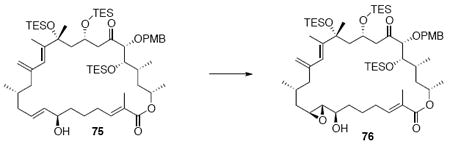
Hydroxyl epoxide 76
To a solution of (+)-DIPT (0.11 mL, 89 μmol, 0.8 M in CH2Cl2) was added CH2Cl2 (0.30 mL) and 4 Å MS (30 mg). The resulted mixture was cooled to -20°C and Ti(OiPr)4 (21.6 mg, 20.0 μL, 76 μmol) was added. After 30 min, TBHP (14.2 μL, 76 μmol, 5.5 M in decane) was added dropwise. After another 30 min, a solution of alcohol 75 (13 mg, 13 μmol) in CH2Cl2 (0.30 mL) was added dropwise via cannula and the reaction was allowed to warm to 0°C. After 16 h, the reaction was cooled back to -20°C and a brine solution of NaOH (0.5 mL, 1.0 M) was added. After 5 min, the reaction was diluted with brine (0.5 mL) and CH2Cl2 (0.5 mL) and warmed up to r.t. Next, the mixture was extracted with CH2Cl2 (3 × 15 mL) and the dried (MgSO4) extract concentrated in vacuo. The crude mixture was purified by preparative TLC, eluting with 25% EtOAc/Hexane, to give vinyl epoxide 76 (7.0 mg, 7.0 μmol, 54%) as a colorless oil: [α]D23 (+) 12.2° (c 0.36, CHCl3); IR (neat) 3467, 3017, 2954, 2917, 2875, 2849, 2107, 1709, 1515, 1462, 1378, 1251, 1173, 1125, 1070, 1018, 835, 756, 667 cm-1; 1H NMR (400 MHz, CDCl3) d 7.33 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.77 (t, J = 7.6 Hz, 1H), 5.80 (s, 1H), 5.06 (s, 1H), 4.95-5.00 (m, 1H), 4.87 (s, 1H), 4.60 (d, J = 10.8 Hz, 1H), 4.22-4.30 (m, 1H), 4.21 (d, J = 10.8 Hz, 1H), 3.85 (s, 3H), 3.75-3.84 (m, 1H), 3.67-3.74 (m, 2H), 3.22 (dd, J = 9.2, 18.0 Hz, 1H), 3.05-3.10 (m, 1H), 2.78-2.81 (m, 1H), 2.54 (dd, J = 3.2, 18.0 Hz, 1H), 2.38 (br, 1H), 2.21-2.30 (m, 4H), 1.89-1.92 (m, 1H), 1.89 (s, 3H), 1.84 (s, 3H), 1.75-1.86 (m, 3H), 1.53-1.70 (m, 7H), 1.45 (s, 3H), 1.30-1.36 (m, 1H), 1.26 (d, J = 5.6 Hz, 3H), 0.91-1.01 (m, 30H), 0.56-0.67 (m, 18H); 13C NMR (100 MHz, CDCl3) d 210.8, 167.8, 159.2, 144.7, 142.4, 141.2, 129.9, 129.8, 128.9, 125.6, 115.4, 113.6, 88.1, 77.1, 71.8, 68.7, 68.2, 65.4, 61.3, 55.3, 54.2, 49.8, 46.5, 45.4, 41.8, 39.5, 33.3, 31.1, 30.8, 28.5, 27.7, 24.1, 21.1, 20.1, 15.1, 12.5, 11.8, 7.3, 7.0, 6.9, 5.3, 5.1; HRMS (ES+) calcd. for C58H102O10Si3Na (M+Na) 1065.6679, found 1065.6774.
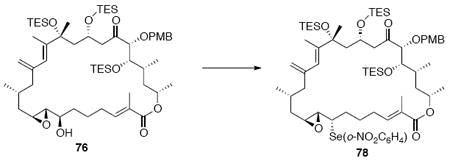
Selenide 78
To a solution of vinyl epoxide 76 (5.0 mg, 5 μmol) in THF (0.15 mL) at r.t. was added 77 (33 mg, 0.15 mmol) and PBu3 (29 mg, 36.2 μL, 0.15 mmol). After 1 h, the reaction mixture was directly purified by preparative TLC, eluting with 25% EtOAc/Hexane, to give selenide 78 (3.0 mg, 2.5 μmol, 50%) as a colorless oil: [α]D23 (-) 9.9° (c 0.22, CHCl3); IR (neat) 2956, 2932, 2874, 1701, 1515, 1457, 1250, 1074, 1007, 806, 730 cm-1 ; 1H NMR (300 MHz, CDCl3) d 8.22 (d = 6.9 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.48-7.54 (m, 1H), 7.48-7.55 (m, 3H), 6.87 (d, J = 8.4 Hz, 2H), 6.69 (t, J = 7.6 Hz, 1H), 5.84 (s, 1H), 5.05 (s, 1H), 4.96-5.00 (m, 1H), 4.87 (s, 1H), 4.56 (d, J = 10.5 Hz, 1H), 4.25-4.30 (m, 1H), 4.21 (d, J = 10.5 Hz, 1H), 3.84 (s, 3H), 3.70 (s, 2H), 3.26 (dd, J = 9.6, 17.4 Hz, 1H), 3.20 (d, J = 8.1, 14.4 Hz, 1H), 2.99-3.04 (m, 1H), 2.96 (dd, J = 8.1, 15.0 Hz, 1H), 2.57 (dd, J = 2.8, 17.4 Hz, 1H), 2.21-2.30 (m, 4H), 1.89-1.92 (m, 1H), 1.84 (s, 3H), 1.79 (s, 3H), 1.45-1.78 (m, 10H), 1.42 (s, 3H), 1.30-1.36 (m, 1H), 1.25 (d, J = 5.6 Hz, 3H), 0.91-1.01 (m, 30H), 0.56-0.67 (m, 18H); 13C NMR (75 MHz, CDCl3) d 210.9, 167.5, 159.1, 147.7, 144.5, 142.4, 140.3, 133.4, 131.3, 129.9, 129.7, 129.2, 126.1, 126.0, 125.5, 115.4, 113.5, 88.5, 77.2, 71.7, 68.9, 65.6, 62.1, 59.6, 55.3, 49.6, 45.9, 45.7, 44.8, 41.1, 39.6, 31.7, 31.6, 30.9, 28.3, 26.2, 21.0, 19.9, 15.2, 12.6, 12.1, 7.3, 7.0, 6.9, 5.3, 5.1; HRMS (ES+) calcd. for C64H106NO11Si3Se (M+1) 1228.6239, found 1228.6216.
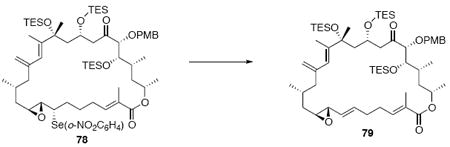
Allyl epoxide 79
To a solution of selenide 78 (5.7 mg, 5.0 μmol) in CH2Cl2 (0.25 mL) at r.t. was sequentially added triethylamine (19 mg, 26.0 μL, 0.18 mmol), TPAP (16 mg, 45 μmol) and NMO (35 mg, 0.3 mmol). After 30 min, the reaction mixture was purified directly by preparative TLC, eluting with 25% EtOAc/Hexanes, to give vinyl epoxide 79 (2.5 mg, 2.4 μmol, 48%) as a colorless oil: [α]D23 -1.3° (c 0.15, CHCl3); IR (neat) 3467, 3017, 2954, 2917, 2875, 2849, 2107, 1709, 1515, 1462, 1378, 1251, 1173, 1125, 1070, 1018, 835, 756, 667 cm-1 ; 1H NMR (400 MHz, CDCl3) d 7.34 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.73 (t, J = 7.6 Hz, 1H), 5.85-5.92 (m, 1H), 5.78 (s, 1H), 5.27 (dd, J = 8.0, 15.2 Hz, 1H), 5.01-5.04 (m, 2H), 4.85 (s, 1H), 4.57 (d, J = 10.4 Hz, 1H), 4.28-4.32 (m, 1H), 4.21 (d, J = 10.4 Hz, 1H), 3.85 (s, 3H), 3.70-3.74 (m, 2H), 3.25 (dd, J = 9.6, 17.2 Hz, 1H), 3.09 (dd, J = 2.0, 6.4 Hz, 1H), 2.90-2.94 (m, 1H), 2.54 (dd, J = 2.8, 17.2 Hz, 1H), 2.06-2.28 (m, 4H), 1.94 (d, J = 2.4, 10.8 Hz, 1H), 1.86 (s, 3H), 1.80-1.86 (m, 3H), 1.79 (s, 3H), 1.53-1.70 (m, 7H), 1.46 (s, 3H), 1.30-1.36 (m, 1H), 1.25 (d, J = 6.0 Hz, 3H), 0.90-1.01 (m, 27H), 0.86 (d, J = 6.0 Hz, 3H), 0.54-0.68 (m, 18H); 13C NMR (100 MHz, CDCl3) d 211.0, 167.6, 159.1, 144.6, 142.4, 140.5, 134.3, 130.1, 129.8, 129.5, 128.7, 125.2, 115.3, 113.5, 88.4, 76.4, 71.7, 68.9, 65.7, 59.6, 58.5, 55.3, 49.4, 46.1, 45.6, 40.6, 40.2, 32.1, 30.9, 30.5, 29.7, 28.0, 27.8, 20.4, 19.8, 15.2, 13.0, 12.5, 7.3, 7.0, 6.9, 6.8, 5.2, 5.1; HRMS (ES+) calcd. for C58H100O9Si3Na (M+Na) 1047.6573, found 1047.6664.
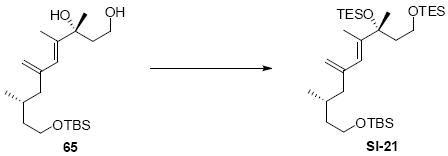
TES ether SI-21
To a stirred solution of diol 65 (470 mg, 1.32 mmol) in DCM / TEA (6 mL, 1:1) was added freshly distilled TESOTf (1.05 g, 0.89 mL, 3.96 mmol) at -78°C. After 30 min, the reaction was quenched with sat. aq. NaHCO3 (1 mL) and extracted with ether (3 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2-5% EtOAc / Hexanes, to give SI-21 (732 mg, 1.25 mmol, 95%) as a colorless oil: [α]D23 = -21.3° (c 1.37, CHCl3); IR (neat) 2954, 2929, 2876, 1460, 1254, 1093, 1007, 835, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.88 (s, 1H), 4.99 (s, 1H), 4.80 (s, 1H), 3.61-3.71 (m, 3H), 3.47 (dt, J = 15.6, 5.4 Hz, 1H), 2.15 (dd, J = 13.5, 5.4 Hz, 1H), 1.80-1.99 (m, 5H), 1.77 (d, J = 0.9 Hz, 3H), 1.54-1.66 (m, 1H), 0.88-1.00 (m, 27H), 0.84 (d, J = 6.3 Hz, 3H), 0.55-0.66 (m, 12H), 0.063 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 145.2, 142.0, 124.6, 114.1, 77.2, 61.7, 59.3, 46.2, 44.5, 40.0, 28.7, 26, 19.6, 18.4, 14.7, 7.2, 7.0, 6.9, 6.8, 4.4, - 5.3; HRMS (EI+) calcd. for C32H68O3Si3 (M+) 584.4476, found 584.4500.
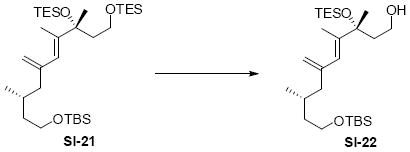
Alcohol SI-22
TES ether SI-21 (300 mg, 0.51 mmol) was dissolved in a stirred solution of HOAc / THF / H2O (8 mL, 8:8:1) at 0°C. After 1.5 h, the reaction was then quenched with solid NaHCO3 and extracted with ether (3 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give alcohol SI-22 (241 mg, 0.51 mmol, 99%) as a colorless oil: [α]D23 = -15.5° (c 0.64, CHCl3); IR (neat) 3437, 2954, 2928, 2876, 1461, 1254, 1099, 1008, 835 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.02 (s, 1H), 5.04 (s, 1H), 4.85 (s, 1H), 3.63-3.73(m, 4H), 2.79 (t, J = 5.6 Hz, 1H), 2.17 (dd, J = 13.6, 5.6 Hz, 1H), 1.88-1.97 (m, 3H), 1.74-1.83 (m, 1H), 1.79 (d, J = 1.2 Hz, 3H), 1.59-1.68 (m, 1H), 1.47 (s, 3H), 1.28-1.40 (m, 1H), 0.99-1.03 (t, J = 7.6 Hz, 9H), 0.92 (s, 9H), 0.88 (d, J = 6.4 Hz, 3H), 0.69 (q, J = 7.2 Hz, 6H), 0.078 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 145.0, 141.4, 125.6, 114.6, 80.0, 61.6, 60.1, 46.1, 42.8, 39.9, 29.7, 28.6, 27.6, 26.0, 19.5, 18.4, 14.9, 7.2, 6.9, 6.8, -5.3; HRMS (EI+) calcd. for C26H54O3Si2 (M+) 470.3611, found 470.3604.
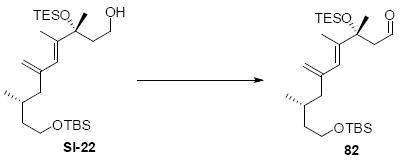
Aldehyde 82
To a stirred solution of alcohol SI-22 (1.29 g, 2.73 mmol) in DCM (40 mL, 1:1) was sequentially added DMP (2.17 g, 5.12 mmol) and NaHCO3 (1.68 g, 20.0 mmol) at rt. After 30 min, the reaction was quenched with sat. aq. NaHCO3 (10 mL) and extracted with ether (3 × 40 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give aldehyde 82 (1.17 g, 2.49 mmol, 91%) as a colorless oil: [α]D23 = -12.5° (c 0.56, CHCl3); IR (neat) 2955, 2929, 2877, 1725, 1255, 1099, 1007, 836, 726 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.69 (t, J = 3.3 Hz, 1H), 6.03 (s, 1H), 5.03 (s, 1H), 4.83 (s, 1H), 3.61-3.68(m, 2H), 2.64 (dd, J = 15.0, 3.0 Hz, 1H), 2.45 (dd, J = 15.0, 3.0 Hz, 1H), 2.15 (dd, J = 13.5, 6.0 Hz, 1H), 1.85-1.92 (m, 1H), 1.83 (d, J = 1.2 Hz, 3H), 1.54-1.64 (m, 1H), 1.49 (s, 3H), 1.25-1.35 (m, 2H), 0.95-1.00 (t, J = 7.7 Hz, 9H), 0.91 (s, 9H), 0.85 (d, J = 6.5 Hz, 3H), 0.65 (q, J = 7.8 Hz, 6H), 0.061 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 203.1, 144.7, 141.0, 126.0, 114.9, 76.9, 61.5, 54.2, 46.0, 39.9, 28.5, 27.8, 26.0, 19.5, 18.3, 14.7, 7.1, 6.7, -5.3; HRMS (EI+) calcd. for C26H52O3Si2 (M+) 468.3455, found 468.3448
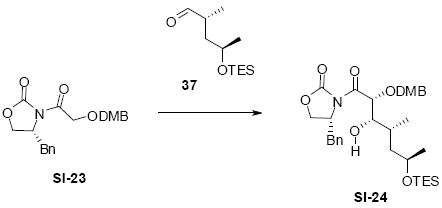
Aldol adduct SI-24
To a stirred solution of SI-23 (1.60 g, 0.15 mmol) in CH2Cl2 (11.2 mL) at -60°C was sequentially added Et3N (0.44 g, 0.60 mL, 4.33 mmol) and Bu2BOTf (1.19 g, 1.08 mL, 4.33 mmol). After 3 h, the resulted solution was warmed up 0°C for 30 min and then cooled back to -60°C. A solution of aldehyde 378 (1.12 g, 4.86 mmol) in DCM (4.8 mL) was transferred to the reaction mixture via cannula. After 2 h, the reaction was allowed to warm up to 0°C. After another 20 min, the reaction was quenched by adding pH 7 phosphate buffer (20 mL) followed by MeOH (15 mL) and 30% H2O2 (4 mL). After 1 h, the reaction mixture was extracted with EtOAc (4 × 35 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15-20% EtOAc / Hexanes, to give SI-24 (1.58 g, 2.57 mmol, 62%) as a colorless oil: [α]D23 = -9.2° (c 0.77, CHCl3); IR (neat) 3493, 2957, 2876, 1781, 1709, 1593, 1517, 1455, 1390, 1265, 1240, 1159, 1052, 1028, 746 cm-1; 1H NMR (400 MHz) δ 7.23-7.38 (m, 5H), 6.83-7.03 (m, 3H), 5.36 (d, J = 2.0 Hz, 1H), 4.63-4.71 (m, 2H), 4.51 (d, J = 10.8 Hz, 1H), 4.21-4.29 (m, 2H), 3.93 (s, 3H), 3.84-3.90 (m, 1H), 3.88 (s, 3H), 3.62-3.66 (m, 1H), 3.32 (dd, J = 13.6, 3.6 Hz, 1H), 2.77 (dd, J = 13.6, 10.0 Hz, 1H), 2.32 (d, J = 10.0 Hz, 1H), 1.78-1.80 (m, 1H), 1.58-1.64 (m, 1H), 1.15 (d, J = 6.0 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H), 0.98 (t, J = 8.0 Hz, 9H), 0.62 (q, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 177.2, 153.2, 149.1, 149.0, 135.2, 129.5, 129.4, 129.0, 127.4, 121.4, 112.1, 110.9, 78.1, 76.1, 66.9, 55.9, 55.7, 42.8, 37.7, 34.1, 23.2, 15.7, 6.9, 4.9; HRMS (ES+) calcd. for C33H49NO8SiNa (M+Na) 638.3125, found 638.3155.

TES ether SI-25
To a stirred solution of adol adduct SI-24 (500 mg, 0.81 mmol) in DCM (3.32 mL) at 0°C was sequentially added 2,6-lutidine (184 mg, 0.20 mL, 1.72 mmol) and TESOTf (287 mg, 0.25 mL, 1.09 mmol). After 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extract with Et2O (3 × 30 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15% EtOAc / Hexanes, to give SI-25 (570 mg, 0.78 mmol, 96%) as a colorless oil: [α]D23 = -40.7° (c 0.42, CHCl3); IR (neat) 2955, 2911, 2876, 1784, 1702, 1517, 1456, 1239, 1084, 740 cm-1; 1H NMR (300 MHz) δ 7.17-7.32 (m,H), 6.80-7.00 (m, 3H), 5.28 (d, J = 6.0 Hz, 1H), 4.71 (d, J = 11.7 Hz, 1H), 4.54 (d, J = 11.7 Hz, 2H), 4.47-4.49 (m, 1H), 4.00-4.12 (m, 3H), 3.90 (s, 3H), 3.84 (s, 3H), 3.78-3.82 (m, 1H), 3.14 (dd, J = 3.0, 13.2 Hz, 1H), 2.40 (dd, J = 10.5, 13.5 Hz, 1H), 1.42-1.56 (m, 3H), 1.11 (d, J = 5.7 Hz, 3H), 0.89-0.97 (m, 21H), 0.55-0.63 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 171.9, 152.9, 148.7, 148.8, 135.3, 130.3, 129.3, 129.0, 127.3, 121.1, 111.9, 110.7, 73.4, 67.2, 66.4, 56.0, 55.9, 55.8, 44.4, 37.4, 33.5, 23.7, 14.8, 7.1, 6.9, 5.4, 5.3, 5.2, 5.0; HRMS (ES+) calcd. for C39H63NO8Si2Na (M+Na) 752.3990, found 752.3992.
Thiol ester SI-26
To a stirred solution of EtSH (90 mg, 0.107 mL, 1.45 mmol) in THF (12.6 mL) at 0°C was added n-BuLi (0.51 mL, 1.27 mmol, 2.5 M in Hexanes). After 1 h, a solution of SI-25 (610 mg, 0.83 mmol) in THF (2.7 mL) was added dropwise via cannula. After another 1 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extract with Et2O (3 × 30 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5-8% EtOAc / Hexanes, to give SI-26 (470 mg, 0.76 mmol, 92%) as a colorless oil: [α]D23 = +42.0° (c 0.39, CHCl3); IR (neat) 2955, 2911, 2876, 1683, 1517, 1458, 1419, 1378, 1266, 1240, 1161, 1079, 1032, 811, 740 cm-1; 1H NMR (400 MHz) δ 7.04 (dd, J = 1.6 Hz, 1H), 6.94 (dd, J = 1.6, 8.0 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 4.63 (d, J = 10.8 Hz, 1H), 4.43 (d, J = 10.8 Hz, 1H), 3.94 (s, 3H), 3.92-3.93 (m, 4H), 3.81-3.86 (m, 2H), 2.91 (q, J = 7.6 Hz, 2H), 1.45-1.57 (m, 3H), 1.30 (t, J = 7.2 Hz, 3H), 1.14 (d, J = 6.0 Hz, 3H), 0.90-0.98 (m, 21H), 0.53-0.62 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 202.2, 148.8, 148.6, 129.9, 120.6, 110.6, 110.7, 88.2, 72.9, 66.9, 55.9, 55.8, 44.9, 32.7, 23.4, 22.5, 14.6, 13.6, 7.0, 6.9, 5.3, 5.2, 5.0; HRMS (ES+) calcd. for C31H58O6Si2SNa (M+Na) 637.3390, found 637.3407.

Methyl ketone 81
To a stirred slurry of CuI (859 mg, 4.51 mmol) in Et2O (8.3 mL) at 0°C was added MeLi (5.6 mL, 9.6 mmol, 1.6 M in Et2O). After 15 min, the colorless solution was cooled to -50°C and a solution of SI-26 (450 mg, 0.75 mmol) in Et2O (4.2 mL) was transferred into the reaction mixture dropwise via cannula. After 2 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) at -50°C, warmed to rt and extracted with Et2O (3 × 30 mL). The dried (MgSO4) extract was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2-4% EtOAc / Hexanes, to give 81 (296 mg, 0.52 mmol, 71%) as a colorless oil: [α]D23 = +22.6 (c 0.23, CHCl3); IR (neat) 2955, 2911, 2876, 1716, 1517, 1457, 1267, 1240, 1082, 1031, 1007, 741 cm-1; 1H NMR (400 MHz) δ 6.82-6.90 (m, 3H), 4.50 (dd, J = 11.4, 15.0 Hz, 2H), 3.90 (s, 6H), 3.80-3.85 (m, 2H), 3.76 (d, J = 6.3 Hz, 1H), 2.13 (s, 3H), 1.50-1.52 (m, 3H), 1.13 (d, J = 6.0 Hz, 3H), 0.88-1.00 (m, 21H), 0.57-0.64 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 210.6, 148.8, 129.9, 120.7, 111.4, 110.8, 88.5, 72.9, 67.0, 55.9, 55.8, 44.5, 33.1, 27.0, 23.5, 14.0, 7.0, 6.9, 5.3, 5.0; HRMS (ES+) calcd. for C30H56O6Si2Na (M+Na) 591.3513, found 591.3527.
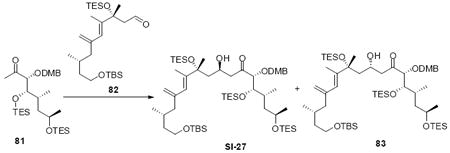
Aldol adduct 83
To a stirred solution of methyl ketone 81 (30 mg, 0.0527 mmol) in Et2O (0.5 mL) at -78°C was added LDA54 (64 μL, 0.064 mmol, 1 M in THF). After 15 min, a pre-cooled (-78°C) solution of aldehyde 82 (50 mg, 0.105 mmol) in THF (0.5 mL) was added via cannula in one portion. After another 0.5 h, the reaction was quenched with sat. aq. NH4Cl (2 mL) at -78°C, warmed up to rt and extracted with ether (3 × 10 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 6-8% EtOAc / Hexanes, to sequentially give aldol adduct SI-27 (10 mg, 0.0096 mmol, 18%) and 83 (39 mg, 0.0375 mmol, 71%) and as colorless oils. 83: [α]D23 = +9.1° (c 0.58, CHCl3); IR (neat) 3503, 2955, 2934, 2876, 1715, 1517, 1463, 1265, 1240, 1095, 1007, 741 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.83-6.97 (m, 3H), 5.90 (s, 1H), 5.05 (s, 1H), 4.85 (s, 1H), 4.53 (d, J = 11.2 Hz, 1H), 4.44-4.50 (m, 1H), 4.39(d, J = 10.8 Hz, 1H), 3.91 (s, 6H), 3.76-3.83 (m, 3H), 3.66-3.70 (m, 2H), 3.05 (dd, J = 17.6, 6.8 Hz, 1H), 2.38 (dd, J = 17.6, 5.6 Hz, 1H), 2.19 (dd, J = 13.5, 4.8 Hz, 1H), 1.80-1.91 (m, 1H), 1.84 (s, 3H), 1.48-1.68 (m, 4H), 1.57 (s, 3H), 1.20-1.42 (m, 4H), 1.10 (d, J=6.0 Hz, 3H), 0.86-1.00 (m, 42H), 0.57-0.66 (m, 18H), 0.08 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 211.0, 148.8, 148.6, 144.6, 142.9, 130.1, 125.2, 120.7, 114.9, 111.5, 110.7, 88.3, 79.5, 72.6, 66.9, 64.8, 61.4, 55.9, 55.8, 48.0, 46.7, 46.0, 44.8, 39.9, 33.1, 28.4, 23.3, 19.5, 14.7, 13.5, 7.1, 7.0, 6.9, 6.5, 5.3, 5.0, -5.2; HRMS (ES+) calcd. for C56H107O9Si4 (M+H) 1035.6992, found 1035.7047.
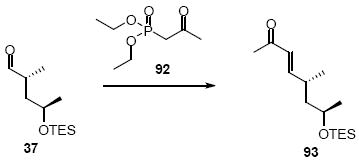
Enone 93
To a stirred slurry of NaH (36 mg, 0.90 mmol, 60% W / W in mineral oil) in DME (2 mL) was added phosphonate 92 (138 mg, 0.83 mmol) at rt. After 1 h, a solution of aldehyde 37 (160 mg, 0.69 mmol) in DME (2 mL) was added via cannula. After another 6 h, the reaction was quenched with sat. aq. NH4Cl (2 mL) and extracted with Et2O (4 × 10 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give enone 93 (142 mg, 0.52 mmol, 76%) as a colorless oil: [α]D23 = -46.0° (c 1.47, CHCl3); IR (neat) 2958, 2877, 1700, 1678, 1627, 1458, 1360, 1252, 1139, 1055, 984, 745 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.76 (dd, J = 15.9, 7.8 Hz, 1H), 6.09 (d, J = 15.9 Hz, 1H), 3.86-3.79 (m, 1H), 2.58-2.53 (m, 1H), 2.26 (s, 3H), 1.41-1.55 (m, 2H), 1.18 (d, J = 6.0 Hz, 3H), 1.09 (d, J = 6.9 Hz, 3H), 0.97 (t, J = 7.8 Hz, 9H), 0.64 (q, J = 7.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 198.8, 153.5, 129.6, 66.4, 46.3, 33.4, 27.0, 24.3, 20.3, 6.9, 5.2, 5.0; HRMS (EI+) calcd. for C15H30O2Si (M+) 270.2015, found 270.2008.
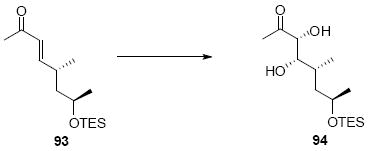
Diol 94
To a stirred solution of enone 93 (142 mg, 0.52 mmol) in tBuOH/H2O (5 mL, 1:1) at 0°C was sequentially added AD-mix-α (0.735 g), NaHCO3 (132 mg, 1.57mmol), MeSO2NH2 (50.6 mg, 0.53 mmol), and K2OsO2(OH)4 (1.9 mg, 0.005 mmol). After 8 h, the reaction was quenched with sat. aq. Na2SO3 (8 mL) and extracted with EtOAc (4 × 10 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15-40% EtOAc / Hexanes, to give diol 94 (122 mg, 0.40 mmol, 77%) as a colorless oil: [α]D23 = -29.2° (c 1.5, CHCl3); IR (neat) 3456, 2957, 2877, 1717, 1380, 1238, 1132, 1048, 1011, 744 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.28 (d, J = 3.6 Hz, 1H), 4.04-3.99 (m, 1H), 3.72-3.78 (m, 1H & OH), 2.41 (d, J = 10.4 Hz, 1H), 2.30 (s, 3H), 1.89-1.96 (m, 1H), 1.71-1.77 (m, 1H), 1.40-1.47 (m, 1H), 1.22 (d, J = 6.0 Hz, 3H), 1.10 (d, J = 6.8 Hz, 3H), 1.00 (t, J = 7.8 Hz, 9H), 0.68 (q, J = 7.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 208.9, 77.7, 75.2, 66.9, 42.8, 34.0, 25.3, 23.1, 16.4, 6.9, 4.9; HRMS (ES+) calcd. for C15H32O4SiNa (M+Na) 327.1968, found 327.1950.
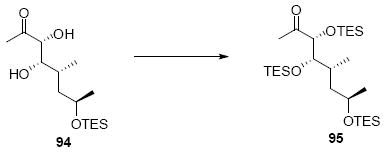
TES ether 95
To a stirred solution of diol 94 (800 mg, 2.63 mmol) in CH2Cl2 (10 mL) at -78°C was sequentially added 2,6-lutidine (1.41 g, 1.53 mL, 13.1 mmol) and TESOTf (1.74 g, 1.49 mL, 6.58 mmol). After 30 min, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (4 × 25 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give TES ether 95 (1.29 g, 2.42 mmol, 92%) as a colorless oil: [α]D23 = -0.83° (c 1.2, CHCl3); IR (neat) 2957, 2878, 1716, 1458, 1238, 1005 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.07 (d, J = 6.0 Hz, 1H), 3.78-3.85 (m, 1H), 3.70 (dd, J = 5.9, 2.6 Hz, 1H), 2.20 (s, 3H), 1.60-1.64 (m, 1H), 1.45-1.51 (m, 2H), 1.13 (d, J = 6.0 Hz, 3H), 0.94-1.00 (m, 27H), 0.83 (d, J = 6.6 Hz, 3H), 0.55-0.70 (m, 18H); 13C NMR (75 MHz, CDCl3) δ 210.0, 81.5, 78.6, 67.1, 45.4, 32.2, 27.3, 23.1, 14.0, 7.0, 6.84, 6.79, 5.2, 4.9, 4.8; HRMS (ES+) calcd. for C27H60O4Si3Na (M+Na) 555.3697, found 555.3683.
Alcohol 96
TES ether 95 (5.60 g, 10.5 mmol) was dissolved in a stirred solution of HOAc / THF / H2O (107 mL, 8:8:1) at 0°C. After 12 h, the reaction was quenched with solid NaHCO3, filtered over Celite® and extracted with ether (4 × 100 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give alcohol 96 (3.90 g, 9.31 mmol, 89%) as a colorless oil: [α]D23 = -30.5 (c 1.45, CHCl3); IR (neat) 3446, 2958, 2878, 1716, 1458, 1239, 1005, 739 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.18 (d, J = 5.6 Hz, 1H), 3.86-3.88 (m, 2H), 2.24 (s, 3H), 1.80-1.90 (m, 1H), 1.41-1.55 (m, 2H), 1.20 (d, J = 6.0 Hz, 3H), 0.97-1.05 (m, 18H), 0.87 (d, J = 6.8 Hz, 3H), 0.61-0.73 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 209.3, 81.4, 76.4, 65.6, 43.7, 31.6, 27.8, 23.6, 15.4, 7.0, 6.8, 5.2, 4.8, 4.7; HRMS (ES+) calcd. for C21H46O4Si2Na (M+Na) 441.2832, found 441.2836

PNB ester 97
To a stirred solution of alcohol 96 (600 mg, 1.43 mmol) in THF (15 mL) at 0°C was sequentially added PPh3 (1.50 g, 5.72 mmol), 4-nitrobenzoic acid (0.96 g, 5.74 mmol), and DEAD (0.99 g, 0.90 mL, 5.70 mmol). After 1 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 1-3% EtOAc / Hexanes, to give ester 97 (670 mg, 1.18 mmol, 82%) as a colorless oil: [α]D23 = +13.6° (c 1.08, CHCl3); IR (neat) 2956, 2878, 1723, 1530, 1319, 1275, 1014, 721 cm-1; 1H NMR (300 MHz, CDCl3) δ 8.31 (d, J=9.0 Hz, 2H), 8.23 (d, J=9.0 Hz, 2H), 5.26 (m, 1H), 4.17 (d, J = 4.8 Hz, 1H), 3.69 (t, J = 4.8 Hz, 1H), 2.18 (s, 3H), 2.02-2.12 (m, 1H), 1.76 (m, 1H), 1.45-1.49 (m, 1H), 1.38 (d, J = 6.1 Hz, 3H), 0.93-1.02 (m, 18H), 0.90 (d, J = 6.6 Hz, 3H), 0.56-0.70 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 209.3, 164.3, 150.4, 136.1, 130.7, 123.4, 81.5, 78.2, 70.8, 40.2, 32.4, 27.9, 20.9, 14.8, 7.0, 6.8, 5.1, 4.8; HRMS (ES+) calcd. for C28H49NO7Si2Na (M+Na) 590.2945, found 590.2926.
Alcohol SI-28
To a stirred solution of ester 97 (700 mg, 1.23 mmol) in MeOH (20 mL) at 0°C was added Ba(OH)2•8H2O (390 mg, 1.24 mmol). After 4 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with EtOAc (4 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 15% EtOAc / Hexanes, to give alcohol SI-28 (369 mg, 0.88 mmol, 72%) as a colorless oil: [α]D23 = -7.6° (c 1.2, CHCl3); IR (neat) 3434, 2957, 2878, 1716, 1459, 1415, 1239, 1005, 739 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.17 (d, J = 5.4 Hz, 1H), 3.81 (m, 1H), 3.69 (dd, J = 5.4, 3.9 Hz, 1H), 2.22 (s, 3H), 1.83 (m, 1H), 1.61-1.69 (m, 1H), 1.24-1.30 (m, 1H), 1.20 (d, J = 6.0 Hz, 3H), 0.94-1.04 (m, 18H), 0.87 (d, J = 6.6 Hz, 3H), 0.58-0.71 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 210.1, 81.5, 78.1, 66.2, 43.8, 32.9, 27.6, 24.4, 15.2, 7.0, 6.8, 5.2, 4.8; HRMS (ES+) calcd. for C21H47O4Si2 (M+H) 419.3013, found 419.2993.

TMS ether 98
To a stirred solution of alcohol SI-28 (1.90 g, 4.54 mmol) in CH2Cl2 (25 mL) at -78°C was sequentially added 2,6-lutidine (1.45 g, 1.58 mL, 13.5 mmol) and TMSOTf (1.51 g, 1.23 mL, 6.81 mmol). After 30 min, the reaction was quenched with sat. aq. NH4Cl (10 mL) and extracted with Et2O (4 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10% EtOAc / Hexanes, to give TMS ether 98 (2.12 g, 4.32 mmol, 95%) as a colorless oil: [α]D23 = +14.4° (c 2.2, CHCl3); IR (neat) 2957, 2878, 1716, 1459, 1415, 1124, 1006, 841, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 4.03 (d, J = 6.3 Hz, 1H), 3.79-3.85 (m, 1H), 3.71 (dd, J = 6.3, 2.4 Hz, 1H), 2.19 (s, 3H), 1.73-1.79 (m, 1H), 1.45-1.54 (m, 1H), 1.23-1.32 (m, 1H), 1.16 (d, J = 6.0 Hz, 3H), 0.95-1.03 (m, 18H), 0.83 (d, J = 6.6 Hz, 3H), 0.58-0.70 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 210.2, 81.8, 78.6, 65.9, 45.3, 31.3, 26.8, 24.7, 13.2, 7.0, 6.8, 5.3, 4.8, 0.3; HRMS (ES+) calcd. for C24H54O4Si3Na (M+Na) 513.3224, found 513.3204.
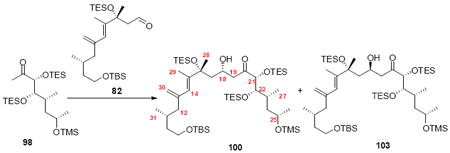
Aldol adducts 100&103
To a stirred solution of methyl ketone 98 (312 mg, 0.64 mmol) in THF (5 mL) at -78°C was added LDA2 (0.765 mL, 1 M in THF). After 15 min, TMEDA (133 mg, 0.172 mL, 1.14 mmol) was added. After 5 min, the reaction was warmed up to -40°C, followed by the addition of a pre-cooled (-40°C) solution of aldehyde 82 (200 mg, 0.43 mmol) in THF (5 mL) via cannula in one portion. After another 0.5 h, the reaction was quenched with sat. aq. NH4Cl (10 mL) -78°C, warmed up to rt and extracted with ether (4 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 1-1.5% EtOAc / Hexanes, to give aldol adduct 100 (142 mg, 0.15 mmol, 35%) and 103 (114 mg, 0.12 mmol, 28%) as colorless oil. 100: [α]D23 = -12.0° (c 1.3, CHCl3); IR (neat) 3511, 2955, 2929, 2877, 1715, 1460, 1413, 1250, 1092, 1006, 838, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.89 (s, 1H), 5.02 (s, 1H), 4.83 (s, 1H), 4.33 (m, 1H), 4.10 (d, J = 5.7 Hz, 1H), 3.82 (m, 1H), 3.74 (s, 1H), 3.64-3.72 (m, 3H), 2.96 (dd, J = 17.7, 6.2 Hz, 1H), 2.60 (dd, J = 18.1, 6.4 Hz, 1H), 2.18 (dd, J = 12.8, 4.0 Hz, 1H), 1.81-1.90 (m, 2H), 1.84 (s, 3H), 1.47-1.67 (m, 4H), 1.56 (s, 3H), 1.22-1.38 (m, 3H), 1.15 (d, J=6.0 Hz, 3H), 0.92-1.03 (m, 36H), 0.87 (d, J=6.4 Hz, 3H), 0.80 (d, J=6.7 Hz, 3H), 0.58-0.70 (m, 18H), 0.10 (s, 9H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 210.7, 144.7, 125.1, 114.7, 81.1, 79.4, 78.4, 65.9, 65.1, 61.5, 48.2, 47.2, 46.0, 45.0, 39.9, 31.1, 28.5,26.2, 26.0, 24.7, 19.5, 18.3, 14.7, 13.8, 7.2, 7.1, 6.9, 6.6, 5.3, 4.9, 0.4, -5.2; HRMS (ES+) calcd. for C50H106O7Si5Na (M+Na) 981.6683, found 981.6646.
MTPA esters
To a solution of 100 (5 mg, 0.005 mmol) in CH2Cl2 (0.5 mL) was sequentially added DMAP (6.4 mg, 0.052 mmol) and (R) or (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride (6.6 mg, 4.9 μL, 0.026 mmol). After 10 min, the solution was evaporated and the residue was loaded directly onto silica gel and purified by chromatography, eluting with 2 - 10% EtOAc / Hexanes, to give product (S)- or (R)- MTPA esters (52-61%) as colorless oils. 1H NMR Difference in ppm [(S)-Mosher Ester – (R)-Mosher ester, CDCl3, CDCl3, 300 MHz NMR] H19 = 2.847 – 2.834 = +0.013, H21: 3.996 – 3.961 = +0.035, H22: 3.686 – 3.678 = +0.008, H25: 3.848 – 3.818 = +0.030, H31’: 0.881 – 0.876 = +0.005, H29: 1.888 – 1.895 = -0.007, H14: 5.723 – 5.824 = -0.101, H28: 4.905 – 4.925 = -0.020, H12: 2.319 – 2.334 = - 0.015, H27: 1.130 – 1.137 = -0.007.
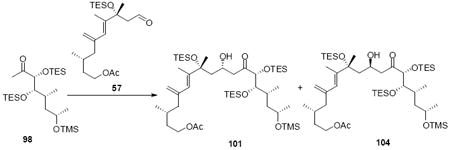
Aldol adducts 101 & 104
Method A (-100°C Conditions)
To a stirred solution of methyl ketone 98 (574 mg, 1.17 mmol) in THF (6 mL) at -78°C was added LDA2 (1.38 mL, 1 M in THF). After 15 min, TMEDA (400 mg, 0.310 mL, 3.44 mmol) was added. After 5 min, the reaction was cooled to -100°C, followed by the addition of a pre-cooled (-100°C) solution of aldehyde 57 (310 mg, 0.78 mmol) in THF (6 mL) via cannula in one portion. After another 0.5 h, the reaction was quenched with 1 M AcOH in THF (1.5 mL) at -100°C. The reaction mixture was then warmed up to rt, diluted with sat. aq. NH4Cl (10 mL) and extracted with ether (4 × 25 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 50% CH2Cl2 / Hexanes -2% EtOAc / Hexanes, to give aldol adduct 104 (405 mg, 0.45 mmol, 58%) and 101 (50 mg, 0.056 mmol, 7%) as colorless oils.
Method B (-40°C Conditions)
To a stirred solution of methyl ketone 98 (37.2 mg, 0.0758 mmol) in THF (0.4 mL) at -78°C was added LDA2 (90 μL, 0.09 mmol, 1 M in THF). After 15 min, TMEDA (15.5 mg, 20 μL, 0.133 mmol) was added. After 5 min, the reaction was warmed up to -40°C, followed by the addition of a pre-cooled (-40°C) solution of aldehyde 57 (20 mg, 0.0504 mmol) in THF (0.4 mL) via cannula in one portion. After another 0.5 h, the reaction was quenched with sat. aq. NH4Cl (2 mL) and extracted with ether (4 × 5 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 50% CH2Cl2 / Hexanes -2% EtOAc / Hexanes, to give aldol adduct 101 (16.1 mg, 0.0181 mmol, 36%) and 104 (13.4 mg, 0.0151 mmol, 30%) as colorless oils. 101: [α]D23 = -9.42 ° (c 1.21, CHCl3); IR (neat) 3516, 2956, 2913, 2877, 1743, 1719, 1458, 1414, 1370, 1249, 1116, 1088, 1008, 841, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.89 (s, 1H), 5.02 (s, 1H), 4.83 (s, 1H), 4.38-4.27 (m, 1H), 4.06-4.10 (m, 3H), 3.86-3.75 (m, 1H), 3.72-3.68 (m, 1H), 3.66 (s, 1H, OH), 2.96 (dd, J = 17.9, 6.2 Hz, 1H), 2.59 (dd, J = 18.0, 6.1 Hz, 1H), 2.15 (dd, J = 13.2, 5.7 Hz, 1H), 2.04 (s, 3H), 1.94-1.76 (m, 4H), 1.81 (s, 3H), 1.70-1.63 (m, 2H), 1.54 (s, 3H), 1.48-1.38 (m, 2H), 1.27-1.22 (m, 1H), 1.13 (d, J = 5.9 Hz, 3H), 1.02-0.93 (m, 27H), 0.89 (d, J = 6.3 Hz, 3H), 0.79 (d, J = 6.6 Hz, 3H), 0.59- 0.69 (m, 18H), 0.09 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 210.8, 171.1, 144.3, 143.3, 124.8, 115.0, 81.0, 79.3, 78.4, 65.9, 65.0, 62.9, 48.2, 47.2, 45.8, 45.0, 35.3, 31.0, 28.7, 26.3, 24.6, 21.0, 19.3, 14.7, 13.8, 7.1, 7.0, 6.8, 6.6, 5.2, 4.9, 0.3; HRMS (ES+) calcd. for C46H94O8Si4Na (M+Na) 909.5924, found 909.5895. 104: [α]D23 = +1.76 ° (c 1.25, CHCl3); IR (neat) 3511, 2956, 2913, 2877, 1743, 1718, 1458, 1369, 1249, 1119, 1088, 1011, 841, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.06 (s, 1H), 5.03 (s, 1H), 4.88 (s, 1H), 4.17-4.06 (m, 4H), 3.88 (s, 1H, OH), 3.88-3.81 (m, 1H), 3.73-3.69 (m, 1H), 2.66-2.78 (m, 2H), 2.17 (dd, J = 13.5, 6.0 Hz, 1H), 2.05 (s, 3H), 1.97-1.93 (m, 1H), 1.85-1.78 (m, 1H), 1.82(s, 3H), 1.74-1.70 (m, 1H), 1.62-1.14 (m, 4H), 1.44 (s, 3H), 1.28-1.20 (m, 2H), 1.14 (d, J = 5.8 Hz, 3H), 1.02- 0.94 (m, 27H), 0.91 (d, J = 6.2 Hz, 3H), 0.78 (d, J = 6.6 Hz, 3H), 0.72-0.57 (m, 18H); 13C NMR (100 MHz, CDCl3) δ 210.5, 171.1, 144.5, 140.8, 125.4, 114.8, 81.3, 80.7, 78.4, 66.0, 65.7, 63.0, 47.1, 45.9, 45.0, 35.2, 31.0, 29.7, 28.7, 28.2, 24.6, 21.0, 19.4, 14.9, 13.6, 7.1, 7.0, 6.9, 6.8, 6.6, 5.2, 4.8, 0.3; HRMS (ES+) calcd. for C46H94O8Si4Na (M+Na) 909.5924, found 909.5948.
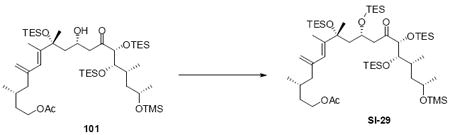
TES ether SI-29
To a stirred solution of aldol adduct 101 (295 mg, 0.332 mmol) in CH2Cl2 (10 mL) at rt was sequentially added DMAP (608 mg, 4.98 mmol) and TESCl (375 mg, 0.418 mL, 2.49 mmol). After 3 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2% EtOAc / Hexanes, to give TES ether SI-29 (290 mg, 0.289 mmol, 87%) as a colorless oil: [α]D23 = -31.4° (c 0.85, CHCl3); IR (neat) 2955, 2913, 2877, 1745, 1718, 1459, 1368, 1249, 1127, 1086, 1007, 841, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.78 (s, 1H), 5.03 (s, 1H), 4.92 (s, 1H), 4.20-4.08 (m, 3H), 4.04 (d, J = 5.7 Hz, 1H), 3.84-3.81 (m, 1H), 3.72-3.68 (m, 1H), 2.86 (dd, J = 18.0, 5.8 Hz, 1H), 2.72 (dd, J = 17.4, 7.2 Hz, 1H), 2.17 (dd, J = 13.8, 6.0 Hz, 1H), 2.05 (s, 3H), 1.93-1.64 (m, 5H), 1.84 (s, 3H), 1.54-1.39 (m, 3H), 1.45 (s, 3H), 1.27-1.21 (m 1H), 1.14 (d, J = 6.0 Hz, 3H), 1.03-0.87 (m, 39H), 0.78 (d, J = 6.7 Hz, 3H), 0.71-0.55 (m, 24H), 0.11 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 208.6, 171.1, 144.3, 143.0, 125.0, 115.1, 81.0, 78.3, 77.7, 66.1, 65.7, 63.0, 49.8, 48.5, 46.0, 44.9, 35.4, 31.0, 28.6, 27.9, 24.6, 21.0, 19.2, 14.5, 14.2, 7.2, 7.0, 6.9, 6.8, 5.2, 4.9, 0.4; HRMS (ES+) calcd. for C52H108O8Si5Na (M+Na) 1023.6788, found 1023.6785.
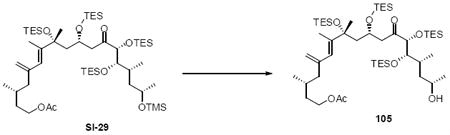
Alcohol 105
To a stirred solution of TMS ether SI-29 (290 mg, 0.289 mmol) in THF / H2O (6.52 mL, 8:1) at -20°C was added HOAc (4 × 1.45 mL) in 4 portions every 60 min. After 5 h, the reaction was quenched with solid NaHCO3, filtered over Celite® and extracted with ether (4 × 15 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5-10% EtOAc / Hexanes, to give alcohol 105 (220 mg, 0.237 mmol, 82%) as a colorless oil: [α]D23 = -31.6° (c 1.01, CHCl3); IR (neat) 3510, 2956, 2912, 2877, 1744, 1720, 1458, 1414, 1368, 1239, 1062, 1006, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.78 (s, 1H), 5.02 (s, 1H), 4.90 (s, 1H), 4.22-4.08 (m, 4H), 3.81-3.77 (m, 1H), 3.69-3.66(m, 1H), 2.94 (dd, J = 18.3, 6.1 Hz, 1H), 2.78 (dd, J = 18.3, 5.7 Hz, 1H), 2.15 (dd, J = 13.1, 5.7 Hz, 1H), 2.04 (s, 3H), 1.93-1.60 (m, 6H), 1.83 (s, 3H), 1.45-1.34 (m, 2H), 1.44 (s, 3H), 1.30-1.20 (m, 1H), 1.17 (d, J = 6.0 Hz, 3H), 1.04-0.87 (m, 39H), 0.82 (d, J = 6.0 Hz, 3H), 0.72-0.56 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 208.8, 171.2, 144.4, 142.6, 125.2, 115.1, 81.4, 78.1, 77.7, 66.2, 65.4, 63.1, 49.7, 49.0, 45.9, 44.2, 35.3, 32.5, 28.6, 28.0, 24.3, 21.0, 19.2, 15.5, 14.6, 7.2, 7.0, 6.8, 5.3, 5.2, 4.8; HRMS (ES+) calcd. for C49H100O8Si4Na (M+Na) 951.6393, found 951.6398.
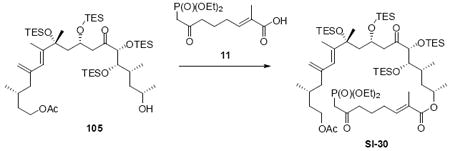
Phosphonate SI-30
To a stirred solution of acid 11 (450 mg, 1.47 mmol) in PhMe (3.2 mL) at rt was sequentially added Et3N (149 mg, 0.204 mL, 1.47 mmol) and 2, 4, 6-trichlorobenzoyl chloride (346 mg, 0.222 mL, 1.47 mmol). After 12 h, the resulted solution was concentrated in vacuo. DMAP (180 mg, 1.47 mmol) was added, followed by the addition of a solution of alcohol 105 (240 m mg, 0.258 mmol) in PhMe (5.7mL). After another 19 h, the reaction was quenched with sat. aq. NH4Cl (8 mL) and extracted with EtOAc (4 × 50 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20-60% EtOAc / Hexanes, to give phosponate SI-30 (235 mg, 0.193 mmol, 78%) as a colorless oil: [α]D23 = -23.6° (c 0.83, CHCl3); IR (neat) 2955, 2912, 2877, 1734, 1716, 1458, 1369, 1241, 1056, 1019, 970, 741 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.69 (t, J = 6.3 Hz, 1H), 5.79 (s, 1H), 5.10-5.00 (m, 1H), 5.03 (s, 1H), 4.92 (s, 1H), 4.22-4.08 (m, 8H), 3.72 (dd, J = 5.1, 3.6 Hz, 1H), 3.13 (d, J = 27.8 Hz, 2H), 2.90 (dd, J = 18.0, 5.9 Hz, 1H), 2.77 (dd, J = 17.8, 6.0 Hz, 1H), 2.68 (t, J = 7.2 Hz, 2H), 2.18-2.07 (m, 3H), 2.06 (s, 3H), 1.94-1.68 (m, 9H), 1.842 (s, 3H), 1.82 (s, 3H), 1.46 (s, 3H), 1.43-1.39 (m, 2H), 1.36 (t, J = 7.0 Hz, 6H), 1.25 (d, J = 6.0 Hz, 3H), 1.03-0.92 (m, 36H), 0.90 (d, J = 6.4 Hz, 3H), 0.80 (d, J = 6.7 Hz, 3H), 0.70-0.55 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 208.4, 201.3, 171.1, 167.6, 144.3, 142.7, 140.5, 127.9, 125.1, 115.1, 80.9, 77.8, 77.6, 68.8, 65.6, 63.0, 62.9, 62.8, 48.7, 49.0, 45.9, 43.4, 41.7, 40.6, 35.3, 31.4, 28.6, 28.0, 27.7, 22.3, 21.0, 20.7, 19.2, 16.3, 16.2, 14.7, 14.5, 12.4, 7.2, 7.0, 6.9, 6.8, 5.3, 5.2, 4.9; HRMS (ES+) calcd. for C62H121O13Si4PNa (M+Na) 1239.7520, found 1239.7458.
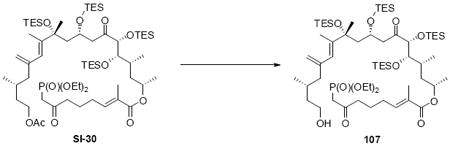
Alcohol 107
To a stirred solution of ester SI-30 (230 mg, 0.189 mmol) in MeOH (10 mL) at rt was added Ba(OH)2•8H2O (66.4 mg, 0.189 mmol). After 1 h, the reaction mixture was purified directly by chromatography over silica gel, eluting with 20-60% EtOAc / Hexanes, to give alcohol 107 (204 mg, 0.168 mmol, 89%) as a colorless oil: [α]D23 = -27.0° (c 0.80, CHCl3); IR (neat) 3434, 2955, 2877, 1716, 1458, 1376, 1242, 1019, 742 cm-1; 1H NMR (300 MHz, CDCl3): δ 6.68 (t, J = 6.8 Hz, 1H), 5.78 (s, 1H), 5.11-5.00 (m, 1H), 5.00 (s, 1H), 4.86 (s, 1H), 4.19-4.05 (m, 6H), 3.73-3.66 (m, 3H), 3.07 (d, J = 27.8 Hz, 2H), 2.93 (dd, J = 18.0, 6.1 Hz, 1H), 2.78 (dd, J = 18.3, 5.7 Hz, 1H), 2.65 (t, J = 7.1 Hz, 2H), 2.22- 2.11 (m, 3H), 1.92-1.59 (m, 8H), 1.81 (s, 6H), 1.43 (s, 3H), 1.42-1.39 (m, 3H), 1.33 (t, J = 7.0 Hz, 6H), 1.23 (d, J = 6.3 Hz, 3H), 1.02-0.85 (m, 39H), 0.78 (d, J = 6.5 Hz, 3H), 0.69-0.52 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 209.0, 201.4, 167.6, 144.8, 142.3, 140.5, 129.0, 125.5, 114.9, 80.8, 77.8, 77.7, 68.8, 65.6, 62.6, 62.5, 61.0, 49.5, 49.0, 46.2, 43.3, 41.6, 40.6, 40.0, 31.4, 28.3, 28.1, 27.7, 22.3, 20.7, 19.3, 16.3, 16.2, 14.7 (2C), 12.4, 7.2, 7.0, 6.9, 6.8, 5.2, 5.1, 4.9; HRMS (ES+) calcd. for C60H119O12Si4PNa(M+Na) 1197.7414, found 1197.7422.
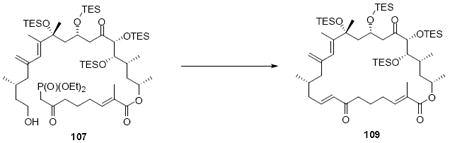
Macrocycle 109
To a stirred solution of alcohol 107 (125 mg, 0.106 mmol) in CH2Cl2 (4 mL) at rt was added TPAP (45 mg, 0.127 mmol). After 0.5 h, the reaction mixture was diluted with THF (6.5 mL) / H2O (16 μL) and Ba(OH)2•8H2O (3 × 74 mg, 0.636 mmol) was added in 3 portions every 30 min. After another 2 h, the reaction mixture was purified directly by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give macrocycle 109 (54 mg, 0.053 mmol, 50% over 2 steps) as colorless crystals: [α]D23 = -27.0° (c 0.40, CHCl3); IR (neat) 2955, 2925, 2876, 1727, 1708, 1675, 1458, 1417, 1260, 1127, 1064, 1009, 741 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.15-7.03 (m, 1H), 6.73 (t, J = 6.9 Hz, 1H), 6.19 (d, J = 16.3 Hz, 1H), 5.78 (s, 1H), 5.00 (s, 1H), 5.05-4.97 (m, 1H), 4.82 (s, 1H), 4.18-4.09 (m, 1H), 4.11 (d, J = 5.1 Hz, 1H), 3.62-3.58 (m, 1H), 3.00-2.93 (m, 2H), 2.85-2.70 (m, 1H), 2.62-2.50 (m, 1H), 2.37-2.21 (m, 3H), 2.15-2.00 (m, 2H), 1.91-1.63 (m, 7H), 1.80 (s, 6H), 1.45-1.35 (m, 2H), 1.41 (s, 3H), 1.25 (d, J = 6.1 Hz, 3H), 1.08-0.83 (m, 39H), 0.75-0.47 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 206.2, 201.0, 167.6, 147.2, 144.7, 141.8, 140.6, 132.4, 129.2, 125.8, 115.4, 80.5, 79.6, 77.9, 68.3, 64.9, 49.7, 48.6, 46.7, 43.2, 41.1, 37.5, 31.0, 29.2, 28.9, 27.8, 22.6, 21.0, 18.8, 15.6, 13.3, 12.5, 7.3, 7.1, 7.0, 5.2, 4.8; HRMS (ES+) calcd. for C56H106O8Si4Na (M+Na) 1041.6863, found 1041.6812.
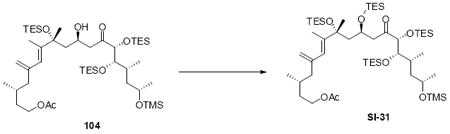
TES ether SI-31
To a stirred solution of aldol adduct 104 (440 mg, 0.496 mmol) in CH2Cl2 (20 mL) at rt was sequentially added DMAP (910 mg, 7.44 mmol) and TESCl (557 mg, 0.620 mL, 3.72 mmol). After 3 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 2% EtOAc / Hexanes, to give TES ether SI-31 (445 mg, 0.444 mmol, 90%) as a colorless oil: [α]D23 = -20.0° (c 0.24, CHCl3); IR (neat) 2955, 2912, 2877, 1744, 1717, 1458, 1249, 1127, 1069, 1008, 840, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.81 (s, 1H), 5.00 (s, 1H), 4.85 (s, 1H), 4.43-4.30 (m, 1H), 4.16-4.03 (m, 3H), 3.88-3.78 (m, 1H), 3.69-3.72 (m, 1H), 2.88 (dd, J = 17.4, 4.8 Hz, 1H), 2.73 (dd, J = 17.4, 7.2 Hz, 1H), 2.13 (dd, J = 13.8, 6.0 Hz, 1H), 2.04 (s, 3H), 1.82-1.94 (m, 3H), 1.79 (s, 3H), 1.64-1.75 (m, 3H), 1.46 (s, 3H), 1.39-1.51 (m, 2H), 1.20-1.28 (m, 1H), 1.14 (d, J = 6.0 Hz, 3H), 0.84-1.03 (m, 39H), 0.77 (d, J = 6.6 Hz, 3H), 0.56-0.71 (m, 24H), 0.11 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 208.4, 171.1, 144.3, 143.8, 124.3, 114.8, 81.1, 78.4, 77.3, 66.1 (2C), 62.9, 50.3, 48.7, 45.9, 45.2, 35.2, 30.7, 28.7, 26.9, 24.7, 21.0, 19.3, 14.8, 13.8, 7.2, 7.0, 6.9, 6.7, 5.4, 5.3, 5.0, 0.3; HRMS (ES+) calcd. for C52H108O8Si5Na (M+Na) 1023.6788, found 1023.6737.

Alcohol 106
To a stirred solution of TMS ether SI-31 (432 mg, 0.43 mmol) in THF / H2O (9 mL, 8:1) at -20°C was added HOAc (8 mL). After 5 h, the reaction was quenched with solid NaHCO3, filtered over Celite® and extracted with ether (4 × 20 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 5-10-20% EtOAc / Hexanes, to give alcohol 106 (338 mg, 0.36 mmol, 85%) as a colorless oil: [α]D23 = -20.8° (c 1.01, CHCl3); IR (neat) 3503, 2955, 2912, 2877, 1744, 1720, 1458, 1414, 1367, 1239, 1007, 741 cm-1; 1H NMR (400 MHz, CDCl3) δ 5.85 (s, 1H), 5.02 (s, 1H), 4.86 (s, 1H), 4.50-4.40 (m, 1H), 4.18-4.03 (m, 3H), 3.85-3.77 (m, 1H), 3.67 (t, J = 5.0 Hz, 1H), 2.74-2.86 (m, 2H), 2.19 (br, OH), 2.14 (dd, J = 13.6, 6.3 Hz, 1H), 2.06 (s, 3H), 1.89-1.96 (m, 2H), 1.80 (s, 3H), 1.66-1.85 (m, 5H), 1.45 (s, 3H), 1.39-1.42 (m, 1H), 1.19 (d, J = 6.0 Hz, 3H), 1.10-1.22 (m, 1H), 0.95-1.05 (m, 36H), 0.90 (d, J = 6.4 Hz, 3H), 0.85 (d, J = 6.4 Hz, 3H), 0.59-0.72 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 208.4, 171.2, 144.3, 143.5, 124.4, 115.0, 82.1, 78.5, 77.3, 66.0, 65.9, 63.0, 50.0, 48.0, 45.9, 44.4, 35.2, 32.3, 28.7, 27.3, 24.2, 21.0, 19.3, 15.4, 15.0, 7.3, 7.1, 7.0, 6.9, 6.8, 5.3, 5.2, 4.9; HRMS (ES+) calcd. for C49H100O8Si4Na (M+Na) 951.6393, found 951.6418.
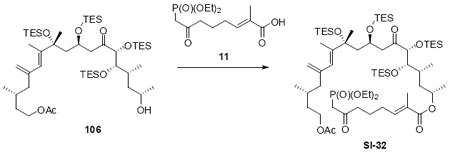
Phosphonate SI-32
To a stirred solution of acid 11 (837 mg, 2.73 mmol) in PhMe (6 mL) at rt was sequentially added Et3N (276 mg, 0.379 mL, 2.73 mmol) and 2, 4, 6-trichlorobenzoyl chloride (641 mg, 0.411 mL, 2.73 mmol). After 12 h, the resulted solution was concentrated in vacuo. DMAP (333 mg, 2.73 mmol) was added, followed by the addition of a solution of alcohol 106 (445 mg, 0.479 mmol) in PhMe (10.5 mL). After another 19 h, the reaction was quenched with sat. aq. NH4Cl (15 mL) and extracted with EtOAc (4 × 50 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 20-60% EtOAc / Hexanes, to give phosphonate SI-32 (450 mg, 0.100 mmol, 77%) as a colorless oil: [α]D23 = -20.3° (c 1.23, CHCl3); IR (neat) 2955, 2913, 2877, 1740, 1716, 1458, 1368, 1243, 1056, 1019, 968, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.69 (t, J = 6.4 Hz, 1H), 5.82 (s, 1H), 5.10-5.02 (m, 1H), 5.01 (s, 1H), 4.84 (s, 1H), 4.42-4.32 (m, 1H), 4.21- 4.02 (m, 7H), 3.76-3.68 (m, 1H), 3.12 (d, J = 22.8 Hz, 2H), 2.85 (dd, J = 17.4, 4.1 Hz, 1H), 2.65-2.76 (m, 3H), 2.12-2.22 (m, 2H), 2.10-2.04 (m, 1H), 2.05 (s, 3H), 1.58-1.93 (m, 10H), 1.82 (s, 3H), 1.79 (s, 3H), 1.45 (s, 3H), 1.43-1.39 (m, 1H), 1.35 (t, J = 7.1 Hz, 6H), 1.24 (d, J = 5.9 Hz, 3H), 0.87-1.03 (m, 39H), 0.79 (d, J = 6.6 Hz, 3H), 0.55-0.68 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 208.3, 201.4, 171.1, 167.6, 144.3, 143.7, 140.5, 129.0, 124.4, 114.9, 81.1, 77.9, 77.34, 68.8, 66.2, 62.9, 62.6, 62.5, 50.3, 49.0, 45.9, 43.3, 41.6, 40.8, 35.2, 31.1, 28.7, 27.7, 26.9, 22.3, 21.0, 20.7, 19.3, 16.3, 16.2, 14.8, 14.3, 12.4, 7.2, 7.1, 7.0, 6.9, 6.7, 5.3, 5.2, 5.0; HRMS (ES+) calcd. for C62H121O13Si4PNa (M+Na) 1239.7520, found 1239.7563.
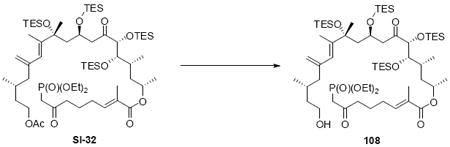
Alcohol 108
To a stirred solution of ester SI-32 (170 mg, 0.140 mmol) in MeOH (0.5 mL) at rt was added a saturated solution of Ba(OH)2•8H2O in MeOH (6.0 mL). After 20 min, the reaction mixture was purified directly by chromatography over silica gel, eluting with 20-60% EtOAc / Hexanes, to give alcohol 108 (150 mg, 0.127 mmol, 91%) as a colorless oil: [α]D23 = -20.3° (c 0.60, CHCl3); IR (neat) 3440, 2955, 2877, 1716, 1458, 1242, 1019, 969, 742 cm-1; 1H NMR (300 MHz, CDCl3): δ 6.69 (t, J = 6.2 Hz, 1H), 5.82 (s, 1H), 5.10-5.03 (m, 1H), 5.00 (s, 1H), 4.83 (s, 1H), 4.42-4.31 (m, 1H), 4.20-4.08 (m, 5H), 3.63-3.74 (m, 3H), 3.12 (d, J = 22.8 Hz, 2H), 2.78-2.72 (m, 2H), 2.66 (t, J = 7.1 Hz, 2H), 2.04-2.19 (m, 3H), 1.89-1.61 (m, 10H), 1.81 (s, 3H), 1.78 (s, 3H), 1.44 (s, 3H), 1.43-1.38 (m, 1H), 1.35 (t, J = 7.1 Hz, 6H), 1.24 (d, J = 5.9 Hz, 3H), 1.03-0.92 (m, 36H), 0.87 (d, J = 6.4 Hz, 3H), 0.79 (d, J = 6.6 Hz, 3H), 0.55-0.71 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 208.5, 201.5, 167.7, 144.6, 143.5, 140.6, 129.0, 124.7, 114.7, 81.2, 78.0, 68.8, 66.1, 62.6, 62.5, 61.1, 50.3, 49.1, 46.1, 43.5, 43.4, 41.8, 40.9, 39.8, 31.1, 28.5, 27.8, 27.1, 22.3, 20.8, 19.5, 16.4, 16.3, 14.9, 14.2, 12.4, 7.3, 7.1, 7.0, 6.9, 6.8, 5.3, 5.2, 5.0; HRMS (ES+) calcd. for C60H119O12Si4PNa (M+Na) 1197.7414, found 1197.7423.

Macrocycle 110
To a stirred solution of alcohol 108 (170 mg, 0.144 mmol) in CH2Cl2 (4 mL) at rt was added TPAP (61 mg, 0.173 mmol). After 0.5 h, the reaction mixture was diluted with CH2Cl2 (3 mL) / CH3CN (7 mL) and Hunig’s base (297 mg, 0.4 mL, 2.29 mmol) was added, followed by the addition of LiCl (20 mg, 0.476 mmol). After 24 h, the reaction mixture was purified directly by chromatography over silica gel, eluting with 5% EtOAc / Hexanes, to give macrocycle 110 (75 mg, 0.073 mmol, 51% over 2 steps) as a colorless oil: [α]D23 = -10.0° (c 0.62, CHCl3); IR (neat) 2955, 2913, 2876, 1708, 1674, 1457, 1417, 1375, 1240, 1124, 1072, 1008, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.88 (dt, J = 16.2, 7.4 Hz, 1H), 6.72 (t, J = 8.0 Hz, 1H), 6.10 (d, J = 16.0 Hz, 1H), 5.82 (s, 1H), 5.08-4.99 (m, 1H), 5.02 (s, 1H), 4.87 (s, 1H), 4.30-4.22 (m, 1H), 4.01 (d, J = 6.4 Hz, 1H), 3.58 (dd, J = 6.4, 2.7 Hz, 1H), 2.86 (dd, J = 17.4, 8.6 Hz, 1H), 2.77 (dd, J = 15.4, 6.0 Hz, 1H), 2.62-2.54 (m, 2H), 2.36-2.19 (m, 4H), 2.14 (dd, J = 17.4, 8.6 Hz, 1H), 2.05-1.76 (m, 7H), 1.80 (s, 3H), 1.79 (s, 3H), 1.70-1.60 (m, 1H), 1.57-1.51 (m, 1H), 1.41 (s, 3H), 1.25 (d, J = 6.2 Hz, 3H), 1.05-0.93 (m, 36H), 0.84 (d, J = 6.6 Hz, 3H), 0.76 (d, J = 6.6 Hz, 3H), 0.73-0.58 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 208.1. 201.0, 167.6, 147.3, 147.1, 144.0, 142.8, 140.8, 132.4, 129.3, 124.7, 115.1, 80.6, 77.4 (2C), 68.5, 65.1, 50.6, 49.0, 46.1, 41.7, 40.2, 37.2, 31.3, 30.8, 27.8, 27.4, 23.1, 21.0, 19.7, 15.3, 12.8, 12.5, 7.3, 7.1, 7.0, 6.9, 5.3, 5.2, 4.9; HRMS (ES+) calcd. for C56H106O8Si4Na (M+Na) 1041.6863, found 1041.6824.
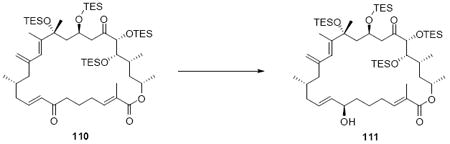
Allylic alcohol 111
To a stirred solution of macrocycle 110 (107 mg, 0.105 mmol) in CH2Cl2 (5.4 mL) at -20°C was sequentially added (S)-CBS (0.42 mL, 0.42 mmol, 1 M in PhMe) and BH3•DMS (0.84 mL, 0.84 mmol, 1 M in THF). After 45 min, the reaction was quenched with MeOH (0.3 mL), diluted with aq. NaHCO3 (5 mL) and extracted with Et2O (3 × 8 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 3-6% EtOAc / Hexanes, to give allylic alcohol 111 (72 mg, 0.0704 mmol, 67%) as a colorless oil: [α]D23 = -16.3° (c 0.30, CHCl3); IR (neat) 3431, 2954, 2913, 2876, 1708, 1674, 1458, 1414, 1376, 1241, 1128, 1073, 1009, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.80 (t, J = 8.0 Hz, 1H), 5.89 (s, 1H), 5.63-5.52 (m, 1H), 5.45 (dd, J = 15.4, 7.5 Hz, 1H), 5.10-5.00 (m, 1H), 4.98 (s, 1H), 4.82 (s, 1H), 4.32-4.20 (m, 1H), 4.19- 4.09 (m, 1H), 4.04 (d, J = 6.2 Hz, 1H), 3.57 (dd, J = 6.2, 2.5 Hz, 1H), 2.87 (dd, J = 17.0, 9.0 Hz, 1H), 2.49 (d, J = 16.8 Hz, 1H), 2.28-2.20 (m, 2H), 2.13-1.98 (m, 3H), 1.90-1.61 (m, 9H), 1.86 (s, 3H), 1.75 (s, 3H), 1.55-1.46 (m, 2H), 1.39 (s, 3H), 1.26 (d, J = 6.0 Hz, 3H), 1.06-0.94 (m, 36H), 0.82 (d, J = 6.4 Hz, 3H), 0.73-0.60 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 208.5, 167.8, 144.6, 142.0, 141.3, 134.6, 130.8, 128.2, 125.4, 114.4, 80.6, 78.0, 72.9, 68.4, 65.2, 50.3, 49.2, 45.4, 41.8, 39.7, 36.8, 31.3, 30.4, 28.8, 28.2, 24.0, 20.9, 19.6, 15.4, 12.5, 12.4, 7.3, 7.1, 7.0, 5.3, 5.2, 4.9; HRMS (ES+) calcd. for C56H108O8Si4Na (M+Na) 1043.7019, found 1043.7052
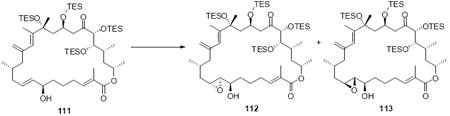
Epoxide 113 & 112
To a stirred solution of allylic alcohol 111 (70 mg, 0.0685 mmol) in CH2Cl2 (6 mL) at -20°C was sequentially added 4Å MS (50 mg), TBHP (37 μL, 0.206 mmol, 5.5 M in decane) and Ti(O-iPr)4 (23.3 mg, 24 μL, 0.082 mmol). After 5 h, the reaction was quenched with aq. NaHCO3 (3 mL) and extracted with Et2O (3 × 7 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 6-10% EtOAc / Hexanes, to give epoxide 113 (35 mg, 0.0342 mmol, 50%) and epoxide 112 (17 mg, 0.0166 mmol, 24%) as colorless oils. 113: [α]D23 = -29.0° (c 0.42, CHCl3); IR (neat) 3431, 2954, 2923, 2876, 1708, 1647, 1458, 1414, 1377, 1242, 1128, 1073, 1009, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.78 (t, J = 8.0 Hz, 1H), 5.88 (s, 1H), 5.05-4.98 (m, 1H), 5.01 (s, 1H), 4.86 (s, 1H), 4.30-4.20 (m, 1H), 4.05 (d, J = 6.1 Hz, 1H), 3.59 (dd, J = 6.1, 2.0 Hz, 1H), 3.52-3.43 (m, 1H), 3.22-3.15 (m, 1H), 2.91 (dd, J =16.3, 9.1 Hz, 1H), 2.70 (dd, J =6.0, 2.0 Hz, 1H), 2.44-2.25 (m, 3H), 2.18 (dd, J =13.0, 5.6 Hz, 1H), 2.10-1.63 (m, 12H), 1.87 (s, 3H), 1.78 (s, 3H), 1.41 (s, 3H), 1.26 (d, J = 6.0 Hz, 3H), 1.15-1.09 (m, 1H), 1.07-0.89 (m, 36H), 0.80 (d, J = 6.4 Hz, 3H), 0.76-0.59 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 208.4, 167.7, 144.0, 142.1, 141.4, 128.6, 125.0, 114.9, 80.8, 78.2, 77.5, 71.8, 68.3, 65.6, 62.8, 56.4, 50.6, 49.1, 46.6, 42.1, 38.9, 33.1, 30.1, 29.4, 28.8, 28.3, 23.7, 21.0, 19.8, 15.5, 12.5, 12.4, 7.3, 7.1, 7.0, 5.3, 5.2, 5.0; HRMS (ES+) calcd. for C56H108O9Si4Na (M+Na) 1059.6968, found 1059.7009. 112: [α]D = -26.2 (c 0.60, CHCl3); IR (neat) 3482, 2954, 2923, 2876, 1708, 1458, 1414, 1377, 1240, 1128, 1008, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.79 (t, J = 7.7 Hz, 1H), 5.87 (s, 1H), 5.05-4.98 (m, 1H), 4.99 (s, 1H), 4.84 (s, 1H), 4.30-4.20 (m, 1H), 4.09 (d, J = 5.9 Hz, 1H), 3.75-3.69 (m, 1H), 3.57 (dd, J = 5.6, 3.2 Hz, 1H), 2.91-2.78 (m, 3H), 2.68-2.60 (m, 2H), 2.38-2.20 (m, 3H), 2.07 (dd, J =12.9, 6.2 Hz, 1H), 1.93-1.62 (m, 10H), 1.85 (s, 3H), 1.76 (s, 3H), 1.50-1.44 (m, 2H), 1.40 (s, 3H), 1.25 (d, J = 6.0 Hz, 3H), 1.05-0.93 (m, 39H), 0.74-0.59 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 208.6, 167.8, 144.1, 142.5, 141.5, 128.4, 124.9, 114.9, 81.0, 78.2, 77.4, 69.0, 68.4, 65.5, 60.7, 54.9, 50.3, 49.2, 46.3, 41.7, 38.4, 33.3, 30.7, 30.3, 29.0, 27.9, 23.7, 21.0, 20.0, 15.3, 13.2, 12.4, 7.3, 7.1, 7.0, 6.9, 5.3, 5.2, 4.9; HRMS (ES+) calcd. for C56H108O9Si4Na (M+Na) 1059.6968, found 1059.7009.
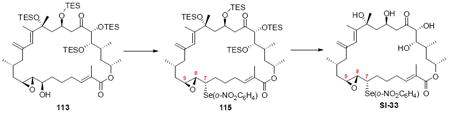
Selenide 115
To a stirred solution of epoxide 113 (42 mg, 0.0405 mmol) in THF (2 mL) at rt was sequentially added o-NO2C6H4SeCN (184 mg, 0.809 mmol) and PBu3 (164 mg, 202 μL, 0.809 mmol). After 5 h, the reaction mixture was concentrated in vacuo purified by chromatography over silica gel, eluting with Hexanes then with 4% EtOAc / Hexanes, to give crude selenide 115 (30 mg) as yellow oils which was used directly in next step without further purification.
Polyol SI-33
To a stirred solution of selenide 115 (30 mg) in THF /DMF / H2O (10:1:0.02, 1.8 mL / 180 μL / 3.6 μL) at 0°C was added TAS-F (33.7 mg, 0.123 mmol). The reaction mixture was then warmed up to rt. After 2 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-65% EtOAc / Hexanes, to give polyol SI-33 (15 mg, 0.0196 mmol, 48% over 2 steps) as a yellow solid: [α]D23 = -20.1° (c 0.12, CHCl3); IR (neat) 3447, 2925, 2854, 1701, 1520, 1456, 1334, 1273, 759, 732 cm-1; 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 8.3 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.56 (t, J = 8.1 Hz, 1H), 7.38 (t, J = 8.1 Hz, 1H), 6.81 (t, J = 6.4 Hz, 1H), 6.05 (s, 1H), 5.10-5.00 (m, 1H), 5.02 (s, 1H), 4.80 (s, 1H), 4.38 (d, J = 3.6 Hz, 1H), 4.18 (s, 1H), 4.17-4.11 (m, 1H), 4.07 (t, J = 9.6 Hz, 1H), 3.58 (t, J = 8.6 Hz, 1H), 3.18-3.10 (m, 1H), 2.97 (d, J = 8.1 Hz, 1H), 2.79 (dd, J = 12.8, 10.1 Hz, 1H), 2.37-2.28 (m, 1H), 2.40-2.30 (m, 1H), 2.22-2.04 (m, 3H), 1.94-1.76 (m, 7H), 1.83 (s, 3H), 1.79 (s, 3H), 1.72-1.62 (m, 3H), 1.38 (s, 3H), 1.31 (d, J = 6.1 Hz, 3H), 1.25-1.19 (m, 2H), 1.07 (d, J = 6.4 Hz, 3H), 0.83 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.1, 167.6, 149.6, 144.4, 141.0 (2C), 133.1, 132.5, 128.9, 128.3, 127.0, 126.0, 125.1, 115.0, 78.3, 77.5, 75.3, 68.7, 68.3, 62.3, 59.3, 46.7, 45.7, 45.5, 43.8, 40.4, 40.0, 32.8, 31.6, 29.1, 29.0, 28.1, 26.1, 21.2, 17.7, 16.2, 15.3, 12.5; HRMS (ES+) calcd. for C38H55NO10NaSe (M+Na) 788.2889, found 788.2859.

Proposed structure of Amphidinolide B2 (3)
To a stirred solution of selenide SI-33 (6.0 mg, 0.00784 mmol) in CH2Cl2 (1.2 mL) at rt was sequentially added NaHCO3 (60 mg, 0.714 mmol) and TMSOOTMS (41.7 mg, 50 μL, 0.233 mmol). After 1.5 h, the yellow color vanished and the reaction mixture was purified by preparative TLC, eluting with 60% EtOAc / Hexanes, to give allylic epoxide 3 (3.0 mg, 0.00533 mmol, 68%): [α]D23 = -52.3° (c 0.21, CHCl3); IR (neat) 3446, 2923, 2853, 1701, 1457, 1273, 1120 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.74 (t, J = 6.4 Hz, 1H), 6.06 (s, 1H), 5.92 (ddd, J = 15.0, 8.9, 4.4 Hz, 1H), 5.20 (dd, J = 15.5, 8.8 Hz, 1H), 5.11-5.07 (m, 1H), 5.07 (s, 1H), 4.86 (s, 1H), 4.28 (d, J =5.4, 1H), 4.14 (s, OH), 4.14-4.09 (m, 1H), 3.73 (d, J = 5.6 Hz, OH), 3.69 (t, J = 9.5 Hz, 1H), 3.50 (d, J = 6.8 Hz, 1H), 3.23 (dd, J = 8.7, 2.2 Hz, 1H), 3.08-3.03 (m, 2H), 2.95 (d, J = 9.2 Hz, OH), 2.53-2.45 (m, 1H), 2.45-2.36 (m, 1H), 2.28 (dd, J = 13.2, 1.8 Hz, 1H), 2.17-2.12 (m, 3H), 1.97-1.93 (m, 4H), 1.85 (s, 3H), 1.82-1.79 (m, 1H), 1.80 (s, 3H), 1.78-1.75 (m, 1H), 1.64-1.60 (m, 1H), 1.34 (s, 3H), 1.31 (d, J = 6.1 Hz, 3H), 1.17-1.12 (m, 1H), 1.03 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 5.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.5, 167.7, 144.7, 141.6, 139.5, 136.3, 128.4, 128.3, 124.9, 114.6, 78.1, 75.6, 69.28, 68.23, 61.45, 59.5, 47.1, 46.4, 44.1, 40.0, 39.4, 33.3, 31.0, 29.3, 28.3, 26.7, 21.2, 17.5, 15.9, 15.2, 12.6; HRMS (ES+) calcd. for C32H50O8Na (M+Na) 585.3403, found 585.3390.
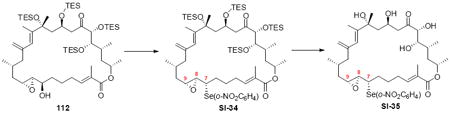
Selenide SI-34
To a stirred solution of epoxide 112 (12 mg, 0.0116 mmol) in THF (0.7 mL) at rt was sequentially added o-NO2C6H4SeCN (53 mg, 0.232 mmol) and PBu3 (47 mg, 58 μL, 0.232 mmol). After 0.5 h, the reaction mixture was concentrated in vacuo purified by chromatography over silica gel, eluting with Hexanes then with 4% EtOAc / Hexanes, to give crude selenide SI-34 (10.6 mg) as a yellow oil which was used directly in next step without further purification.
Polyol SI-35
To a stirred solution of selenide SI-34 (10.6 mg) in THF / DMF / H2O (10:1:0.02, 1.0 mL / 100 μL / 2.0 μL) at 0°C was added TAS-F (12 mg, 0.0434 mmol). The reaction mixture was then warmed up to rt. After 2 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-65% EtOAc / Hexanes, to give polyol SI-35 (6.2 mg, 0.00811 mmol, 70% over 2 steps) as a yellow oil: [α]D = -47.0° (c 0.30, CHCl3); IR (neat) 3446, 2925, 2854, 1701, 1515, 1456, 1332, 1271, 757, 731 cm-1; 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 6.85 (t, J = 6.3 Hz, 1H), 6.02 (s, 1H), 5.11-5.05 (m, 1H), 5.04 (s, 1H), 4.81 (s, 1H), 4.38 (d, J = 3.6 Hz, 1H), 4.32-4.25 (m, 1H), 4.17-4.12 (m, 1H), 3.89 (d, J = 4.6 Hz, 1H), 3.72-3.65 (m, 1H), 3.62-3.55 (m, 1H), 3.10-3.05 (m, 2H), 2.84 (dd, J = 14.8, 9.2 Hz, 1H), 2.52 (dd, J = 14.8, 2.5 Hz, 1H), 2.38 (d, J = 9.5 Hz, 1H), 2.40-2.20 (m, 3h), 1.97-1.74 (m, 8H), 1.83 (s, 3H), 1.76 (s, 3H), 1.70-1.60 (m, 3H), 1.36 (s, 3H), 1.33 (d, J = 6.1 Hz, 3H), 1.06 (d, J = 6.7 Hz, 3H), 0.87 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.0, 167.7, 147.7, 144.6, 141.3, 140.9, 133.7, 132.3, 130.1, 128.6, 126.5, 126.1, 125.6, 115.1, 78.0, 77.0, 75.5, 69.1, 67.7, 60.6, 58.4, 45.7, 45.6, 43.7, 43.1, 40.4, 39.2, 33.8, 30.3, 29.5, 28.9, 28.0, 27.0, 21.2, 19.7, 16.3, 15.2, 12.5; HRMS (ES+) calcd. for C38H55NO10NaSe(M+Na) 788.2889, found 788.2897.
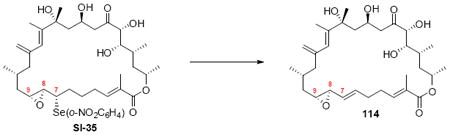
Allylic epoxide 114
To a stirred solution of selenide SI-35 (2.5 mg, 0.00327 mmol) in CH2Cl2 (0.5 mL) at rt was sequentially added NaHCO3 (20 mg, 0.238 mmol) and TMSO-OTMS (19.1 mg, 23 μL, 0.107 mmol). After 1.5 h, the yellow color vanished and the reaction mixture was purified by preparative TLC, eluting with 60% EtOAc / Hexanes, to give allylic epoxide 114 (1.2 mg, 0.00213 mmol, 65%): [α]D = -27.5° (c 0.12, CHCl3); IR (neat) 3443, 2924, 2852, 1703, 1457, 1379, 1272, 1118 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.71-6.63 (m, 1H), 6.08 (s, 1H), 5.88-5.78 (m, 1H), 5.26 (dd, J = 15.1, 8.4 Hz, 1H), 5.12-5.05 (m, 1H), 5.05 (s, 1H), 4.84 (s, 1H), 4.29 (dd, J =5.7, 1.5 Hz, 1H), 4.30-4.20 (m, 1H), 3.71 (t, J = 7.9 Hz, 1H), 3.66 (d, J = 5.7 Hz, 1H), 3.19 (d, J = 9.6 Hz, 1H), 3.12 (dd, J = 8.6, 2.2 Hz, 1H), 3.01-2.87 (m, 2H), 2.46 (dd, J = 14.4, 2.1 Hz, 1H), 2.36-2.20 (m, 5H), 2.00-1.74 (m, 6H), 1.83 (s, 3H), 1.77 (s, 3H), 1.60-1.55 (m, 1H), 1.35 (s, 3H), 1.33-1.28 (m, 1H), 1.30 (d, J = 6.1 Hz, 3H), 1.18-1.11 (m, 1H), 1.03 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.9, 167.5, 144.9, 141.6, 140.3, 136.4, 128.7, 128.6, 125.7, 115.0, 78.2, 76.6, 75.9, 68.5, 68.2, 60.2, 60.0, 45.8, 45.1, 43.8, 39.4, 39.3, 33.6, 31.0, 30.3, 28.5, 27.0, 21.2, 20.0, 15.8, 15.2, 12.7; HRMS (ES+) calcd. for C32H50O8Na (M+Na) 585.3403, found 585.3394.
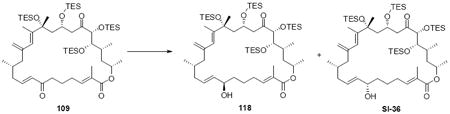
Allylic alcohol 118
To a stirred solution of macrocycle 109 (50 mg, 0.049 mmol) in CH2Cl2 (2.3 mL) at -30°C was sequentially added (S)-CBS (0.196 mL, 0.196 mmol, 1 M in PhMe) and BH3•DMS (0.3934 mL, 0.393 mmol, 1 M in THF). After 45 min, the reaction was quenched with MeOH (0.3 mL), diluted with aq. NaHCO3 (3 mL) and extracted with Et2O (4 × 6 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 3-6% EtOAc / Hexanes, to give allylic alcohol 118 (29 mg, 0.028 mmol, 58%) and SI-36 (8 mg, 0.0078 mmol, 16%) as colorless oils. 118: [α]D23 = -23.6° (c 0.25, CHCl3); IR (neat) 3503, 2954, 2911, 2876, 1707, 1458, 1376, 1240, 1128, 1007, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.81 (t, J = 6.9 Hz, 1H), 5.76 (s, 1H), 5.70-5.62 (m, 1H), 5.53 (dd, J = 13.2, 6.3 Hz, 1H), 5.00-4.95 (m, 1H), 4.98 (s, 1H), 4.84 (s, 1H), 4.15- 4.04 (m, 3H), 3.58 (dd, J = 5.7, 2.4 Hz, 1H), 2.93 (dd, J = 18.4, 6.2 Hz, 1H), 2.78 (dd, J = 18.4, 6.0 Hz, 1H), 2.28-2.22 (m, 2H), 2.18-2.10 (m, 2H), 2.08-1.99 (m, 1H), 1.89-1.57 (m, 9H), 1.84 (s, 3H), 1.81 (s, 3H), 1.48-1.40 (m, 2H), 1.45 (s, 3H), 1.26 (d, J = 6.1 Hz, 3H), 1.07-0.91 (m, 36H), 0.88 (d, J = 6.6 Hz, 3H), 0.74-0.53 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 207.8, 167.8, 145.5, 142.5 (2C), 134.9, 129.7, 127.9, 125.7, 114.9, 80.6, 79.2, 77.9, 71.9, 68.2, 65.3, 49.6, 49.2, 45.6, 42.2, 39.4, 37.0, 31.8, 29.8, 29.0, 28.1, 23.6, 21.0, 19.9, 14.7, 12.7, 12.4, 7.2, 7.1, 7.0, 6.9, 6.8, 5.3, 5.2, 4.7; HRMS (ES+) calcd. for C56H108O8Si4Na (M+Na) 1043.7019, found 1043.7072. SI-36: [α]D = -29.1 (c 0.80, CHCl3); IR (neat) 3481, 2954, 2876, 1707, 1458, 1241, 1130, 1008, 742 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.74 (t, J = 7.4 Hz, 1H), 5.77 (s, 1H), 5.75-5.70 (m, 1H), 5.55 (dd, J = 15.5, 7.0 Hz, 1H), 5.00-4.96 (m, 1H), 4.99 (s, 1H), 4.83 (s, 1H), 4.22-4.15 (m, 1H), 4.11-4.06 (m, 1H), 4.07 (d, J = 5.8 Hz, 1H), 3.57 (dd, J = 5.6, 2.3 Hz, 1H), 2.96 (dd, J = 18.4, 7.4 Hz, 1H), 2.78 (dd, J = 18.3, 4.2 Hz, 1H), 2.23-2.08 (m, 4H), 1.95-1.52 (m, 11H), 1.834 (s, 3H), 1.80 (s, 3H), 1.48-1.40 (m, 1H), 1.43 (s, 3H), 1.25 (d, J = 6.1 Hz, 3H), 1.06- 0.87 (m, 39H), 0.73-0.55 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 207.4, 167.9, 145.3, 142.2, 141.7, 134.2, 130.9, 128.3, 125.9, 114.9, 80.9, 79.0, 77.7, 73.2, 68.4, 65.1, 49.2, 48.8, 45.5, 42.1, 39.8, 36.8, 31.7, 30.1, 28.5, 28.3, 24.5, 21.0, 19.4, 15.0, 13.1, 12.4, 7.3, 7.1, 7.0, 6.9, 6.8, 5.2, 5.1, 4.8; HRMS (ES+) calcd. for C56H108O8Si4Na (M+Na) 1043.7019, found 1043.6984.
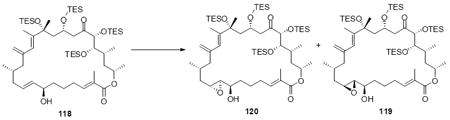
Epoxide 119 & 120
To a stirred solution of allylic alcohol 118 (29 mg, 0.0284 mmol) in CH2Cl2 (2.2 mL) at -40°C was sequentially added 4Å MS (20 mg), TBHP (15.5 μL, 0.0852 mmol, 5.5 M in decane) and Ti(O-iPr)4 (16.1 mg, 16.6 μL, 0.0567 mmol). After 5 h, the reaction was quenched with aq. NaHCO3 (3 mL) and extracted with Et2O (4 × 4 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 6-10% EtOAc / Hexanes, to give epoxide 120 (13.6 mg, 0.0131 mmol, 46%) and epoxide 119 (9.1 mg, 0.00867 mmol, 31%) as colorless oils. 120: [α]D23 = -32.3° (c 0.73, CHCl3); IR (neat) 3482, 2954, 2911, 2876, 1706, 1458, 1380, 1239, 1131, 1073, 1009, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.83 (t, J = 7.0 Hz, 1H), 5.76 (s, 1H), 5.00-4.92 (m, 1H),4.98 (s, 1H), 4.83 (s, 1H), 4.20-4.10 (m, 1H), 4.10(d, J = 5.5 Hz, 1H), 3.58 (dd, J = 5.4, 1.7 Hz, 1H), 3.60-3.50 (m, 1H), 3.12-3.05 (m, 1H), 2.98-2.92 (m, 2H), 2.90-2.82 (m, 1H), 2.77 (dd, J = 5.0, 2.0 Hz, 1H), 2.40-2.30 (m, 2H), 2.17 (dd, J = 12.7, 4.3 Hz, 1H), 2.12-2.00 (m, 1H), 1.91-1.61 (m, 8H), 1.82 (s, 3H), 1.79 (s, 3H), 1.50-1.40 (m, 3H), 1.42 (s, 3H), 1.30-1.20 (m, 1H), 1.26 (d, J = 6.0 Hz, 3H), 1.08-0.87 (m, 39H), 0.75-0.50 (m, 27H); 13C NMR (75 MHz, CDCl3) δ 208.1, 167.8, 144.7, 141.8, 141.7, 128.2, 125.9, 115.0, 80.5, 79.4, 77.9, 70.4, 68.2, 65.1, 60.8, 55.8, 49.8, 48.9, 45.9, 42.9, 39.3, 33.6, 30.2, 29.2, 28.6, 28.5 24.0, 21.1, 19.5, 15.1, 12.9, 12.4, 7.3, 7.1, 6.9, 5.2, 4.7; HRMS (ES+) calcd. for C56H108O9Si4Na (M+Na) 1059.6968, found 1059.7001. 119: [α]D23 = -25.7 (c 0.42, CHCl3); IR (neat) 3482, 2954, 2876, 1708, 1458, 1378, 1240, 1130, 1008, 742 cm-1; 1H NMR (300 MHz, CDCl3) δ 6.76 (t, J = 6.1 Hz, 1H), 5.83 (s, 1H), 5.05-4.90 (m, 1H), 5.01 (s, 1H), 4.86 (s, 1H), 4.15-4.10 (m, 1H), 4.08 (d, J = 5.8 Hz, 1H), 3.58 (dd, J = 5.5, 2.6 Hz, 1H), 3.59-3.49 (m, 1H), 3.11-3.03 (m, 1H), 2.95-2.91 (m, 2H), 2.77 (dd, J = 5.5, 2.0 Hz, 1H), 2.35-2.20 (m, 3H), 2.02-1.59 (m, 10H), 1.83 (s, 3H), 1.81 (s, 3H), 1.44 (s, 3H), 1.50-1.40 (m, 3H), 1.25 (d, J = 6.0 Hz, 3H), 1.06-0.89 (m, 39H), 0.74-0.52 (m, 27H); 13C NMR (75 MHz, CDCl3) δ 207.7, 167.8, 144.8, 142.3, 141.5, 128.4, 125.4, 115.3, 80.8, 78.9, 77.8, 71.5, 68.4, 65.3, 61.8, 55.5, 49.2 (2C), 45.4, 41.7, 38.8, 33.3, 30.4, 29.6, 28.5, 28.3, 23.9, 21.0, 20.0, 15.0, 13.2, 12.5, 7.2, 7.1, 7.0, 6.9, 5.2, 4.8; HRMS (ES+) calcd. for C56H108O9Si4Na (M+Na) 1059.6968, found 1059.6982.
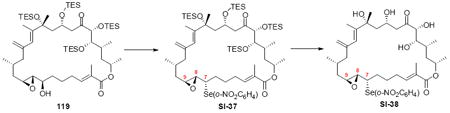
Selenide SI-37
To a stirred solution of epoxide 119 (8.5 mg, 0.00819 mmol) in THF (0.5 mL) at rt was sequentially added o-NO2C6H4SeCN (37 mg, 0.164 mmol) and PBu3 (33.2 mg, 41μL, 0.164 mmol). After 5 h, the reaction mixture was concentrated in vacuo purified by chromatography over silica gel, eluting with Hexanes then with 4% EtOAc / Hexanes, to give crude selenide SI-37 (4.5 mg) as a yellow oil which was used directly in next step without further purification.
Polyol SI-38
To a stirred solution of selenide SI-37 (4.5 mg) in THF / DMF / H2O (10:1:0.02, 0.50 mL / 50 μL / 1 μL) at 0°C was added TAS-F (5 mg, 0.0180 mmol). The reaction mixture was then warmed up to rt. After 2 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-65% EtOAc / Hexanes, to give polyol SI-38 (2.0 mg, 0.00261 mmol, 32% over 2 steps) as a yellow oil: [α]D23 = -41.7° (c 0.12, CHCl3); IR (neat) 3446, 2924, 2854, 1701, 1519, 1457, 1378, 1334, 1121, 759, 732 cm-1; 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 6.88 (t, J = 6.3 Hz, 1H), 5.87 (s, 1H), 5.15-5.08 (m, 1H), 5.03 (s, 1H), 4.82 (s, 1H), 4.50 (s, 1H), 4.20-4.10 (m, 2H), 3.78-3.69 (m, 1H), 3.20-3.10 (m, 1H), 2.94-2.77 (m, 3H), 2.39-2.33 (m, 1H), 2.30-2.13 (m, 2H), 2.07-1.50 (m, 11H), 1.84 (s, 6H), 1.41 (s, 3H), 1.40-1.28 (m, 5H), 1.07 (d, J = 6.5 Hz, 3H), 0.77 (t, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.4, 167.7, 149.7, 144.0, 143.0, 141.1, 133.1, 132.6, 128.8, 128.4, 127.1, 125.9, 123.9, 115.2, 77.6, 76.0, 75.3, 68.3, 66.0, 62.1, 59.0, 46.8, 46.1, 45.3, 44.7, 40.3, 40.2, 32.7, 31.9, 29.2, 28.9, 28.0, 26.3, 21.2, 17.8, 15.9, 15.4, 12.5; HRMS (ES+) calcd. for C38H55NO10NaSe(M+Na) 788.2889, found 788.2891.

Amphidinolide B1 (2)
To a stirred solution of selenide SI-38 (2.0 mg, 0.00261 mmol) in CH2Cl2 (0.4 mL) at rt was sequentially added NaHCO3 (20 mg, 0.238 mmol) and TMSO-OTMS (16.6 mg, 20 μL, 0.0929 mmol). After 1.5 h, the yellow color vanished and the reaction mixture was purified by preparative TLC, eluting with 60% EtOAc / Hexanes, to give amphidinilide B1 (2) (1.0 mg, 0.00178 mmol, 68%): [α]D23 = -63.7° (c 0.08, CHCl3), Literature Value:2c -62.5° (c 0.39, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.78 (t, J = 7.8 Hz, 1H), 5.99 (s, 1H), 5.93 (ddd, J = 15.2, 8.5, 4.8 Hz, 1H), 5.18 (dd, J = 15.8, 8.6 Hz, 1H), 5.06 (m, 1H), 5.05(s, 1H), 4.84 (s, 1H), 4.34 (dd, J =4.8, 1.4 Hz, 1H), 4.20 (m, 1H), 3.92 (d, J = 3.3 Hz, 1H), 3.88 (d, J = 5.0 Hz, 1H), 3.73 (ddd, J = 10.3, 8.8, 1.5 Hz, 1H), 3.19 (d, J = 10.0 Hz, 1H), 3.16 (dd, J = 8.3, 2.0 Hz, 1H), 2.94 (ddd, J = 8.9, 2.6, 2.2 Hz, 1H), 2.86 (d, J = 7.3 Hz, 1H), 2.80 (dd, J = 15.9, 3.2 Hz, 1H), 2.43 (m, 1H), 2.38 (m, 1H), 2.25 (m, 1H), 2.20 (m, 1H), 2.17 (m, 1H), 2.16 (s, 1H), 1.98-1.91 (m, 4H), 1.80 (s, 6H), 1.76 (dd, J =14.5, 5.2 Hz, 1H), 1.64 (m, 1H), 1.49 (ddd, J = 13.6, 10.9, 3.0 Hz, 1H), 1.44 (s, 3H), 1.31 (m, 1H), 1.30 (d, J = 6.1 Hz, 3H), 1.26 (m, 1H), 1.03 (d, J = 6.6 Hz, 3H), 0.90 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.5, 167.8, 144.5, 143.2, 140.0, 135.5, 128.6, 128.5, 124.4, 114.9, 77.9, 76.1, 75.7, 68.4, 66.7, 60.2, 47.0, 46.0, 45.4, 39.5, 39.4, 33.3, 31.0, 29.4, 28.4, 26.9, 21.1, 18.3, 15.7, 15.2, 12.5; HRMS (ES+) calcd. for C32H50O8Na (M+Na) 585.3403, found 585.3411.
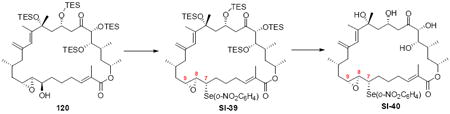
Selenide SI-39
To a stirred solution of epoxide 120 (14.5 mg, 0.0139 mmol) in THF (0.8 mL) at rt was sequentially added o-NO2C6H4SeCN (63 mg, 0.279 mmol) and PBu3 (56.7 mg, 70 μL, 0.279 mmol). After 1 h, the reaction mixture was concentrated in vacuo purified by chromatography over silica gel, eluting with Hexanes then with 4% EtOAc / Hexanes, to give crude selenide SI-39 (15.2 mg) as a yellow oil which was used directly in next step without further purification.
Polyol SI-40
To a stirred solution of selenide SI-39 (15.2 mg, 0.0122 mmol) in THF / DMF / H2O (10:1:0.02, 1.6 mL / 0.16 mL / 3.2 μL) at 0°C was added TAS-F (16.8 mg, 0.0610 mmol). The reaction mixture was then warmed up to rt. After 2 h, the reaction mixture was concentrated in vacuo and purified by chromatography over silica gel, eluting with 10-65% EtOAc / Hexanes, to give polyol SI-40 (9.1 mg, 0.0119 mmol, 86% over 2 steps) as yellow oils: [α]D23 = -51.8 ° (c 0.44, CHCl3); IR (neat) 3447, 2926, 2855, 1701, 1514, 1456, 1332, 1271, 1037, 902, 756, 731 cm-1; 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 7.3 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 6.81 (t, J = 7.0 Hz, 1H), 5.97 (s, 1H), 5.10-5.03 (m, 1H), 5.03 (s, 1H), 4.83 (s, 1H), 4.47 (s, OH), 4.40-4.32 (m, 1H), 4.14 (dd, J = 14.2, 7.2 Hz, 1H), 3.88-3.78 (m, 1H), 3.42-3.35 (m, 1H), 3.05-2.97 (m, 1H & OH), 2.85 (d, J = 4.8 Hz, 2H), 2.50-2.38 (br, 1H), 2.30-2.20 (m, 2H), 2.07 (s, OH), 2.11-2.02 (m, 1H), 1.96-1.65 (m, 11H), 1.83 (s, 6H), 1.44 (s, 3H), 1.40-1.32 (m, 2H), 1.33 (d, J = 5.8 Hz, 3H), 1.08 (d, J = 6.3 Hz, 3H), 0.90 (d, J = 4.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.6, 167.9, 147.6, 144.0, 143.5, 141.2, 133.6, 132.3, 130.6, 128.7, 126.4, 126.0, 124.0, 115.5, 78.0, 76.1, 75.0, 69.5, 66.3, 60.7, 58.7, 46.1, 45.8, 44.8, 43.8, 40.5, 39.5, 33.5, 30.4 (2C), 28.2, 27.7, 27.1, 20.8, 19.6, 16.5, 15.2, 12.5; HRMS (ES+) calcd. for C38H55NO10NaSe (M+Na) 788.2889, found 788.2934.
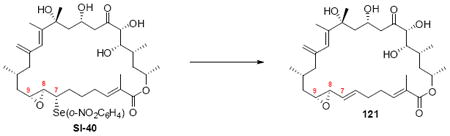
Allylic epoxide 121
To a stirred solution of selenide SI-40 (2.7 mg, 0.00353 mmol) in CH2Cl2 (0.5 mL) at rt was sequentially added NaHCO3 (30 mg, 0.357 mmol) and TMSO-OTMS (22.5 mg, 27 μL, 0.126 mmol). After 1.5 h, the yellow color vanished and the reaction mixture was purified by preparative TLC, eluting with 60% EtOAc / Hexanes, to give allylic epoxide 121 (1.3 mg, 0.00231 mmol, 65%): [α]D23 = +10.0° (c 0.13, CHCl3); IR (neat) 3422, 2923, 2853, 1701, 1457, 1377, 1261, 1103; 1H NMR (400 MHz, CDCl3) δ 6.72 (t, J = 4.9 Hz, 1H), 6.04 (s, 1H), 5.88-5.80 (m, 1H), 5.28 (dd, J = 15.4, 8.4 Hz, 1H), 5.10-5.05 (m, 1H), 5.06 (s, 1H), 4.84 (s, 1H), 4.35 (dd, J = 4.2, 1.5 Hz, 1H), 4.30-4.20 (m, 1H), 4.13 (s, 1H), 3.79 (t, J = 9.8 Hz, 1H), 3.77 (d, J = 4.6 Hz, 1H), 3.40 (d, J = 10.2 Hz, 1H), 3.09 (dd, J = 8.5, 2.1 Hz, 1H), 2.98-2.93 (m, 2H), 2.76 (dd, J = 15.4, 2.4 Hz, 1H), 2.38-2.23 (m, 5H), 2.35 (s, 1H), 1.98 (m, 1H), 1.95 (m, 1H), 1.88 (m, 1H), 1.85 (m, 1H), 1.84 (s, 6H), 1.78 (dd, J =14.6, 4.7 Hz, 1H), 1.70 (m, 1H), 1.64 (m, 1H), 1.32 (m, 1H), 1.30 (d, J = 6.1 Hz, 3H), 1.13 (m, 1H), 1.03 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.4 Hz, 3H) 13C NMR (100 MHz, CDCl3) δ 212.6, 167.5, 144.6, 143.2, 140.8, 135.9, 128.9, 128.7, 124.3, 114.9, 78.3, 76.1, 75.5, 68.3, 66.8, 60.3, 59.2, 46.5, 46.1, 45.6, 39.6, 33.3, 31.2, 30.5, 29.7, 28.6, 27.1, 21.0, 19.9, 15.7, 15.4, 12.7; HRMS (ES+) calcd. for C32H50O8Na (M+Na) 585.3403, found 585.3409.
Cell viability assays
MTS assays were conducted for cell viability as described by the supplier (Promega).58 Human DU145 prostate cancer, OCI-LY3 lymphoma, K562 CML, MOLT-4 ALL, Reh ALL, U266 myeloma, KG1a AML, HL60 AML and MDA-MB-435 breast cancer cells were seeded in 96-well plates, incubated overnight at 37°C in 5% (v/v) CO2 and exposed to 3, 114 or 121 at 0.1 mM or in a dose-dependent manner for 72 h. DMSO was used as the vehicle control. Cell viability was determined by tetrazolium conversion to its formazan dye and absorbance was measured at 490 nm using an automated ELISA plate reader.
Supplementary Material
Scheme 17.
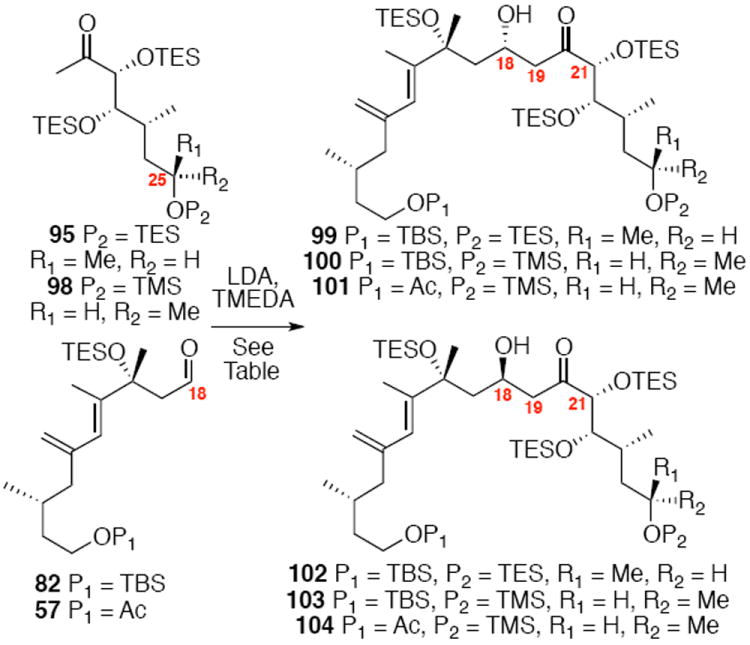
Exploration into Aldol Stereoselectivity.
Acknowledgments
Financial support was provided by the National Institutes of Health (NIH) (GM63723) and Oregon State University). The authors are grateful to Professor Claudia Maier & Jeff Morré (OSU) for mass spectra data and Dr. Lev Zakharov (OSU) for x-ray crystallographic analysis of 109. Finally, the authors thank Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for his helpful discussions.
Footnotes
SUPPORTING INFORMATION. Copies of 1H and 13C spectra for all new compounds are provided. X-ray crystallographic data (CIF) for compound 109 is also provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews of the amphidinolides: Ishibashi M, Kobayashi J. Heterocycles. 1997;44:543–572.. Chakraborty TK, Das S. Curr Med Chem Anti-cancer Agents. 2001;1:131–49. doi: 10.2174/1568011013354660.. Kobayashi J, Shimbo K, Kubota T, Tsuda M. Pur App Chem. 2003;75:337–342.. Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77–93. doi: 10.1039/b310427n.. Colby EA, Jamison TF. Org Biomol Chem. 2005;3:2675–2684. doi: 10.1039/b507315b.. Kobayashi J, Kubota T. J Nat Prod. 2007;70:451–460. doi: 10.1021/np0605844.. Hiersemann N, Kobayashi J. J Antibiot. 2008;61:271–284. doi: 10.1038/ja.2008.39.. Fürstner A. Israel J Chem. 2011;51:329–345.
- 2.Amphidinolide A: Kobayashi J, Ishibashi M, Nakamura H, Ohizumi Y, Yamasu T, Sasaki T, Hirata Y. Tetrahedron Lett. 1986;27:5755–5758.. Amphidinolide B: Ishibashi M, Ohizumi Y, Hamashima M, Nakamura H, Hirata Y, Sasaki T, Kobayashi J. J Chem Soc Chem Commun. 1987:1127–29.. Bauer I, Maranda L, Shimizu Y, Peterson RW, Cornell L, Steiner JR, Clardy J. J Am Chem Soc. 1994;116:2657–58.. Ishibashi M, Ishiyama H, Kobayashi J. Tetrahedron Lett. 1994;35:8241–42. Tsuda M, Kariya Y, Iwamoto R, Fukushi E, Kawabata J, Kobayashi J. Mar Drugs. 2005;3:1–8.. Oguchi K, Tsuda M, Iwamoto R, Okamoto Y, Endo T, Kobayashi J, Ozawa T, Masuda A. J Nat Prod. 2007;70:1676–1679. doi: 10.1021/np0703085.. Amphidnolide C / F: Kobayashi J, Ishibashi M, Wälchli NR, Nakamura H, Yamasu T, Hirata Y, Sasaki T, Ohizumi Y. J Am Chem Soc. 1988;110:490–94.. Kubota T, Tsuda M, Kobayashi J. Org Lett. 2001;3:1363–66. doi: 10.1021/ol015741z.. Kubota T, Sakuma Y, Tsuda M, Kobayashi J. Mar Drugs. 2004;2:83–87.. Tsuda M, Kariya Y, Iwamoto R, Fukushi E, Kawabata J, Kobayashi J. Mar Drugs. 2005;3:1–8.. Kubota T, Suzuki A, Yamada M, Baba S, Kobayashi J. Heterocycles. 2010;82:333–338.. Amphidinolide D: Kobayashi J, Ishibashi M, Nakamura H, Ohizumi Y, Yamasu T, Hirata Y, Sasaki T, Ohta T, Nozoe S. J Nat Prod. 1989;52:1036–1041. doi: 10.1021/np50065a021.. Amphidinolide E: Kobayashi J, Ishibashi M, Murayama T, Takamatsu M, Iwamura M, Ohizumi Y, Sasaki T. J Org Chem. 1990;55:3421–3423.. Kubota T, Tsuda M, Kobayashi J. J Org Chem. 2002;67:1651–1656. doi: 10.1021/jo016326n.. Amphidinolide F: Kobayashi J, Tsuda M, Ishibashi M, Shigemori H, Hirota H, Sasaki T. J Antibiot. 1991;44:1259–1261. doi: 10.7164/antibiotics.44.1259.. Amphidinolides G/H: Kobayashi J, Shigemori H, Ishibashi M, Yamasu T, Hirota H, Sasaki T. J Org Chem. 1991;56:5221–5224.. Kobayashi J, Shimbo K, Sato M, Shiro M, Tsuda M. Org Lett. 2000;2:2805–2807. doi: 10.1021/ol006223b.. Kobayashi J, Shimbo K, Sato M, Tsuda MJ. Org Chem. 2002;67:6565–6592. doi: 10.1021/jo016222c.. Amphidinolide J: Kobayashi J, Sato M, Ishibashi M. J Org Chem. 1993;58:2645–2646.. Amphidinolide K: Ishibashi M, Sato M, Kobayashi J. J Org Chem. 1993;58:6928–6929.. Amphidinolide L: Tsuda M, Sasaki T, Kobayashi J. J Org Chem. 1994;59:3734–3737.. Amphidinolide M: Kobayashi J, Yamaguchi N, Ishibashi M. J Org Chem. 1994;59:3698–4700.. Amphidinolide N: Ishibashi M, Yamaguchi N, Sasaki T, Kobayashi J. J Chem Soc Chem Commun. 1994:1455–1456.. Amphidinolide O/P: Ishibashi M, Takahashi M, Kobayashi J. J Org Chem. 1995;60:6062–6066.. Amphidinolide Q: Kobayashi J, Takahashi M, Ishibashi M. Tetrahedron Lett. 1996;37:1449–1450.. Takahashi Y, Kubota T, Fukushi E, Kawabata J, Kobayashi J. Org Lett. 2008;10:3709–3711. doi: 10.1021/ol8013123.. Amphidinolides R and S: Ishibashi M, Takahashi M, Kobayashi J. Tetrahedron. 1997;53:7827–7832.. Amphidinolide T: Kobayashi J, Kubota T, Endo T, Tsuda M. J Org Chem. 2001;66:134–142. doi: 10.1021/jo005607c.. Kobayashi J, Kubota T, Endo T, Tsuda M. Tetrahedron. 2001;57:6175–6179.. Amphidinolide U: Tsuda M, Endo T, Kobayashi J. Tetrahedron. 1999;55:1465–14570.. Amphidnolide W: Shimbo K, Tsuda M, Izui N, Kobayashi J. J Org Chem. 2002;67:1020–1023. doi: 10.1021/jo016089o.. Amphidinolide X: Tsuda M, Izui N, Shimbo K, Sato M, Fukushi E, Kawabata J, Katsumata K, Horiguchi T, Kobayashi J. J Org Chem. 2003;68:5339–4345. doi: 10.1021/jo0343634.. Amphidinolide Y: Tsuda M, Izui N, Shimbo K, Sato M, Fukushi E, Kawabata J, Kobayashi J. J Org Chem. 2003;68:9109–9112. doi: 10.1021/jo035278z.
- 3.Syntheses of amphidinolide natural products (excluding amphidinolide B): Williams D, Kissel WS. J Am Chem Soc. 1988;120:11198–11199.. Williams DR, Myers BJ, Mi L. Org Lett. 2000;2:945–948. doi: 10.1021/ol0000197.. Williams DR, Meyer KG. J Am Chem Soc. 2001;123:765–766. doi: 10.1021/ja005644l.. Lam HW, Pattenden G. Angew Chem Int Ed. 2002;41:508–511. doi: 10.1002/1521-3773(20020201)41:3<508::aid-anie508>3.0.co;2-7.. Maleczka RE, Jr, Terrell LR, Geng F, Ward JS., III Org Lett. 2002;4:2841–2844. doi: 10.1021/ol0262284.. Fürstner A, Aissa C, Riveiros R, Ragot J. Angew Chem Int Ed. 2002;41:4763–4766. doi: 10.1002/anie.200290042.. Trost BM, Chisholm JD, Wrobleski SJ, Jung M. J Am Chem Soc. 2002;124:12420–12421. doi: 10.1021/ja027883+.. Aiessa C, Riveiros R, Ragot J, Fürstner A. J Am Chem Soc. 2003;125:15512–15520. doi: 10.1021/ja038216z.. Ghosh AK, Liu C. J Am Chem Soc. 2003;125:2374–2375. doi: 10.1021/ja021385j.. Lepage O, Kattnig E, Fürstner A. J Am Chem Soc. 2004;126:15970–15971. doi: 10.1021/ja044130+.. Ghosh AK, Gong G. J Am Chem Soc. 2004;126:3704–3705. doi: 10.1021/ja049754u.. Trost BM, Harrington PE. J Am Chem Soc. 2004;126:5028–5029. doi: 10.1021/ja049292k.. Trost BM, Papillion JPN. J Am Chem Soc. 2004;126:13618–13619. doi: 10.1021/ja045449x.. Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589–13597. doi: 10.1021/ja0533646.. Harrington PE, Chisholm JD, Wrobleski ST. J Am Chem Soc. 2005;127:13598–13610. doi: 10.1021/ja053365y.. Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297–4307. doi: 10.1021/ja042733f.. Trost BM, Papillon JPN, Nussbaumer T. J Am Chem Soc. 2005;127:17921–17937. doi: 10.1021/ja055967n.. Va P, Roush WR. J Am Chem Soc. 2006;128:15960–15961. doi: 10.1021/ja066663j.. Kim CH, An HJ, Shin WK, Yu W, Woo SK, Jung SK, Lee E. Angew Chem Int Ed. 2006;45:8019–8021. doi: 10.1002/anie.200603363.. Ghosh AK, Gong G. J Org Chem. 2006;71:1085–1093. doi: 10.1021/jo052181z.. Fürstner A, Kattnig E, Lepage O. J Am Chem Soc. 2006;128:9194–9204. doi: 10.1021/ja061918e.. Nicoloau KC, Brenzovich WE, Bulger PG, Francis TM. Org Biomol Chem. 2006;4:2119–2157. doi: 10.1039/b602020h.. Nicolaou KC, Bulger PG, Brenzovich WE. Org Biomol Chem. 2006;4:2158–2183. doi: 10.1039/b602021f.. Deng L-S, Huang X-P, Zhao G. J Org Chem. 2006;71:4625–4635. doi: 10.1021/jo0605086.. Jin J, Chen Y, Li Y, Wu J, Dai W-M. Org Lett. 2007;9:2585–2588. doi: 10.1021/ol0710360.. Va P, Roush WR. Tetrahedron. 2007;63:5768–5796. doi: 10.1016/j.tet.2007.02.058.. Fürstner A, Bouchez LC, Funel J-A, Liepins V, Poree F-H, Gilmour R, Beaufils F, Laurich D, Tamiya M. Angew Chem Int Ed. 2007;46:9265–9270. doi: 10.1002/anie.200704024.. Dai W-M, Chen Y, Jin J, Wu J, Lou J, He Q. Synlett. 2008:1737–1741.. Barbazanges M, Meyer C, Cossy J. Org Lett. 2008;10:4489–4492. doi: 10.1021/ol801708x.. Kim CH, An HJ, Shin WK, Yu W, Woo SK, Jung SK, Lee E. Chem Asian J. 2008;3:1523–1534. doi: 10.1002/asia.200800062.. Rodriquez-Escrich C, Urpi F, Vilarrasa J. Org Lett. 2008;10:5191–5194. doi: 10.1021/ol8021676.. Hangyou M, Ishiyama H, Takahashi Y, Kobayashi J. Org Lett. 2009;11:5046–5049. doi: 10.1021/ol902026f.. Ko HM, Lee CW, Kwon HK, Chung HS, Choi SY, Chung YK, Lee E. Angew Chem Int Ed. 2009;47:2364–2366. doi: 10.1002/anie.200805266.. Fürstner A, Flügge S, Larionov O, Takahashi Y, Kubota T, Kobayashi J. Chem Eur J. 2009;15:4011–4029. doi: 10.1002/chem.200802068.. Yadav JS, Reddy CS. Org Lett. 2009;11:1705–1708. doi: 10.1021/ol9002724.. Li H, Wu J, Luo J, Dai W-M. Chem Eur J. 2010;16:11530–11534. doi: 10.1002/chem.201001794.. Wu D, Li H, Jin J, Wu J, Dai W-M. Synlett. 2011:895–898.. Sun L, Wu D, Wu J, Dai W-M. Synlett. 2011:3036–3040.. Mahapatra S, Carter RG. Angew Chem Int Ed. 2012;51:7948–7851. doi: 10.1002/anie.201203935.
- 4.(a) Lu L, Zhang W, Carter RG. J Am Chem Soc. 2008;130:7253–7255. doi: 10.1021/ja803012n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu L, Zhang W, Carter RG. J Am Chem Soc. 2008;130:11834. doi: 10.1021/ja803012n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fürstner A, Bouchez LC, Morency L, Funel J-A, Liepins V, Poree F-H, Gilmour R, Laurich D, Beaufils F, Tamiya M. Chem Eur J. 2009;15:3983–4010. doi: 10.1002/chem.200802067. [DOI] [PubMed] [Google Scholar]
- 6.Biology and Biosynthesis of the Amphidinolides: Matsunaga K, Nakatani K, Ishibashi J, Kobayashi J, Ohizumi Y. Biochem Biophys Acta. 1999;1427:23–32. doi: 10.1016/s0304-4165(98)00175-5.. Sato M, Shimbo K, Tsuday M, Kobayashi J. Tetrahedron Lett. 2000;41:503–506.. Kubota T, Tsuda M, Kobayashi J. Tetrahedron. 2001;57:5975–5977.. Tsuda M, Kubota T, Sakuma Y, Kobayashi J. Chem Pharm Bull. 2001;49:1366–1367. doi: 10.1248/cpb.49.1366.. Tsuda M, Izui N, Sato M, Kobayashi J. Chem Pharm Bull. 2002;50:976–977. doi: 10.1248/cpb.50.976.. Saito S-Y, Feng J, Kira A, Kobayashi J, Ohizumi Y. Biocehm Biophys Res Commun. 2004;320:961–965. doi: 10.1016/j.bbrc.2004.06.050.. Kubota T, Iinuma Y, Kobayashi J. Biolog Pharm Bull. 2006;29:1314–1318. doi: 10.1248/bpb.29.1314.. Fürstner A, Kattnig E, Kelter G, Fiebig H-H. Chem Eur J. 2009;15:4030–4043. doi: 10.1002/chem.200802069.. Trigili C, Pera B, Barbazanges M, Cossy J, Meyer C, Pineda O, Rodriguez-Escrich C, Urpi F, Vilarrasa J, Diaz JF, Barasoain I. ChemBioChem. 2011;12:1027–1030. doi: 10.1002/cbic.201100042.
- 7.(a) Chakraborty TK, Thippeswamy D, Suresh VR, Jayaprakash S. Chem Lett. 1997:563–64. [Google Scholar]; (b) Chakraborty TK, Suresh VR. Chem Lett. 1997:565–66. [Google Scholar]; (c) Lee DH, Lee S-W. Tetrahedron Lett. 1997;38:7909–10. [Google Scholar]; (d) Ohi K, Shima K, Hamada K, Saito Y, Yamada N, Ohba S, Nishiyama S. Bull Chem Soc Jpn. 1998;71:2433–40. [Google Scholar]; (e) Cid MB, Pattenden G. Synlett. 1998:540–542. [Google Scholar]; (f) Ohi K, Nishiyama S. Synlett. 1999:571–72. [Google Scholar]; (g) Ohi K, Nishiyama S. Synlett. 1999:573–75. [Google Scholar]; (h) Eng HM, Myles DC. Tetrahedron Lett. 1999;40:2275–78. [Google Scholar]; (i) Eng HM, Myles DC. Tetrahedron Lett. 1999;40:2279–82. [Google Scholar]; (j) Chakraborty TK, Thippeswamy D. Synlett. 1999:150–152. [Google Scholar]; (k) Ishiyama H, Takemura T, Tsuda M, Kobayashi J. J Chem Soc Perkin Trans. 1999;1:1163–1166. [Google Scholar]; (l) Lee D-H, Rho MD. Tetrahedron Lett. 2000;41:2573–76. [Google Scholar]; (m) Ndubaku CO, Jamison TF. 227th ACS National Meeting; Anaheim. ORGN-392. [Google Scholar]; (n) Mandal AK, Schneekloth JS, Jr, Crews CM. Org Lett. 2005;7:3645–3648. doi: 10.1021/ol051175m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Mandal AK, Schneekloth JS, Jr, Crews CM. Org Lett. 2005;7:5347. doi: 10.1021/ol051175m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Gopalaratham A, Nelson SG. Org Lett. 2006;8:7–10. doi: 10.1021/ol051861l. [DOI] [PubMed] [Google Scholar]; (q) Mandal AK, Schneekloth JS, Jr, Kuramochi K, Crews CM. Org Lett. 2006;8:427–430. doi: 10.1021/ol052620g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Sidera M, Costa AM, Vilarrasa J. Org Lett. 2011;13:4934–4937. doi: 10.1021/ol2020187. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Carter RG, Yokochi AFT. J Org Chem. 2004;69:2569–72. doi: 10.1021/jo035829l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Zhang W, Carter RG. Org Lett. 2005;7:4209–12. doi: 10.1021/ol051544e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang W, Carter RG. 227th American Chemical Society National Meeting; Anheim, CA. ORG-398. [Google Scholar]
- 10.Zhang W. Ph D Dissertation. Oregon State University; 2006. [Google Scholar]
- 11.Hara A, Morimoto R, Iwasaki Y, Saitoh T, Ishikawa Y, Nishiyama S. Angew Chem Int Ed. 2012;51:9877–9880. doi: 10.1002/anie.201204992. [DOI] [PubMed] [Google Scholar]
- 12.(a) Mitsunobu O. Synthesis. 1981;1981:1–28. [Google Scholar]; (b) Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 13.Seebach D, Naef R, Calderari G. Tetrahedron. 1984;40:1313–1324. [Google Scholar]
- 14.Kang K-T, U JS, Park DK, Kim JG, Kim WJ. Bull Kor Chem Soc. 1995;16:464–466. [Google Scholar]
- 15.(a) Ager DJ. Synthesis. 1984:384–398. [Google Scholar]; (b) Van Staden LF, Gravestock D, Ager DJ. Chem Soc Rev. 2002;31:195–200. doi: 10.1039/a908402i. [DOI] [PubMed] [Google Scholar]
- 16.Ehlinger Ed, Magnus P. J Am Chem Soc. 1980;102:5004–5011. [Google Scholar]
- 17.Lau PWK, Chan TH. Tetrahedron Lett. 1978;19:2383–2386. [Google Scholar]
- 18.Evans DA, Golob A-M. J Am Chem Soc. 1975;97:4765–4766. [Google Scholar]
- 19.Hosomi A, Sakurai H. Tetrahedron Lett. 1976;17:1295–1298. [Google Scholar]
- 20.(a) Kolb HC, Andersson PG, Sharpless KB. J Am Chem Soc. 1994;116:1278–91. [Google Scholar]; (b) Corey EJ, Guzman-Perez A, Noe MC. J Am Chem Soc. 1995;117:10805–16. [Google Scholar]
- 21.(a) Mander LN, Morris JC. J Org Chem. 1997;62:7497–99. doi: 10.1021/jo970873k. [DOI] [PubMed] [Google Scholar]; (b) Allen PA, Brimble MA, Prabaharan H. Synlett. 1999:295–298. [Google Scholar]; (c) Carter RG, Weldon DJ. Org Lett. 2000;2:3913–16. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]
- 22.Evans DA, Gage JR, Leighton JL, Kim AS. J Org Chem. 1992;57:1961–1963. [Google Scholar]
- 23.(a) Evans DA, Dart MJ, Duffy JL, Yang MG. J Am Chem Soc. 1996;118:4322–4343. [Google Scholar]; (b) Evans DA, Coleman PJ, Coté B. J Org Chem. 1997;62:788–789. [Google Scholar]; (c) Evans DA, Coté B, Coleman PJ, Connell B. T J Am Chem Soc. 2003;125:10893–10898. doi: 10.1021/ja027640h. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty TK, Suresh VR. Tetrahedron Lett. 1998;39:7775–7778. [Google Scholar]
- 25.Paterson I, Lister MA, Norcross R. D Tetrahedron Lett. 1992;33:1767–1770. [Google Scholar]
- 26.(a) Heathcock CH, White CT. J Am Chem Soc. 1979;101:7076–7077. [Google Scholar]; (b) Heathcock CH, White CT, Morrison JJ, VanDerveer D. J Org Chem. 1981;46:1296–1309. [Google Scholar]
- 27.Masamune S, Ali SA, Snitman DL, Garvey DS. Angew Chem Int Ed. 1980;19:557–558. [Google Scholar]
- 28.(a) Braun M. Angew Chem Int Ed Engl. 1987;26:24–37. [Google Scholar]; (b) Li Y, Padden-Row MN, Houk KN. J Org Chem. 1990;55:484–493. [Google Scholar]; (c) Bernardi A, Capelli AM, Gcnnari C, Goodman JM, Paterson I. J Org Chem. 1990;55:3576–3581. [Google Scholar]; (d) Paterson I, Goodman JM, Lister MA, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663–4684. [Google Scholar]; (e) Gustin DJ, VanNieuwenhze MS, Roush WR. Tetrahedron Lett. 1995;36:3443–3446. [Google Scholar]
- 29.White JD, Bolton GL. J Am Chem Soc. 1990;112:1626–1628. [Google Scholar]
- 30.(a) Trost BM, Urabe H. J Org Chem. 1990;55:3982–3984. [Google Scholar]; (b) Trost BM, Rodriguez MS. Tetrahedron Lett. 1992;33:4675–4678. [Google Scholar]
- 31.Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew Chem Int Ed. 1998;37:2354–2359. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.(a) Wallace GA, Scott RW, Heathcock CH. J Org Chem. 2000;65:4145–4152. doi: 10.1021/jo0002801. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Stavenger RA. J Am Chem Soc. 2000;122:8837–8847. [Google Scholar]; (c) Hosaka M, Hayakawa H, Miyashita M. J Chem Soc Perkin Trans. 2000;1:4227–4230. [Google Scholar]; (d) Crimmins MT, Katz JD, McAtee LC, Tabet EA, Kirincich SJ. Org Lett. 2001;3:949–952. doi: 10.1021/ol015652m. [DOI] [PubMed] [Google Scholar]; (e) Nakamura S, Inagaki J, Sugimoto T, Kudo M, Nakajima M, Hashimoto S. Org Lett. 2001;3:4075–4078. doi: 10.1021/ol0168364. [DOI] [PubMed] [Google Scholar]; (f) Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661–5663. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]; (g) Nakamura S, Inagaki J, Kudo M, Sugimoto T, Obara K, Nakajima M, Hashimoto S. Tetrahedron. 2002;58:10353–10374. [Google Scholar]; (h) Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]; (i) Smith AB, III, Fox RJ, Vanecko JA. Org Lett. 2005;7:3099–3102. doi: 10.1021/ol051119l. [DOI] [PubMed] [Google Scholar]; (j) Paterson I, Coster MJ, Chen DY-K, Acena JL, Bach J, Keown LE, Trieselmann T. Org Biomol Chem. 2005;3:2420–2430. doi: 10.1039/b504149j. [DOI] [PubMed] [Google Scholar]; (k) Kawahara S, Gaunt MJ, Scolaro A, Yamanoi S, Ley SV. Synlett. 2005:2031–2034. [Google Scholar]; (l) Pellicena M, Solsona JG, Romea P, Urpí F. Tetrahedron Lett. 2008;49:5265–5267. [Google Scholar]; (m) Lorenz M, Kalesse M. Org Lett. 2008;10:4271–4374. doi: 10.1021/ol8018105. [DOI] [PubMed] [Google Scholar]; (n) Paterson I, Mühlthau FA, Cordier CJ, Housden MP, Burton PM, Loiseleur O. Org Lett. 2009;11:353–356. doi: 10.1021/ol802562b. [DOI] [PubMed] [Google Scholar]; (o) Paterson I, Findlay AD, Noti C. Chem Asian J. 2009;4:594–611. doi: 10.1002/asia.200800445. [DOI] [PubMed] [Google Scholar]; (p) Lorenz M, Bluhm N, Kalesse M. Synthesis. 2009:3061–3066. [Google Scholar]; (q) Lorente A, Pellicena M, Romea P, Urpí F. Tetrahedron Lett. 2010;51:942–945. [Google Scholar]; (r) Guérinot A, Lepesqueux G, Sablé S, Reymond S, Cossy J. J Org Chem. 2010;75:5151–5163. doi: 10.1021/jo100871m. [DOI] [PubMed] [Google Scholar]; (s) Smith AB, III, Dong S, Fox RJ, Brenneman JB, Vanecko JA, Maegawa T. Tetrahedron. 2011;67:9809–9828. doi: 10.1016/j.tet.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–96. [Google Scholar]; (b) Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–19. [Google Scholar]; (c) Sullivan GR, Dale JA, Mosher HS. J Org Chem. 1973;38:2143–47. [Google Scholar]
- 34.Deng L, Ma Z, Zhang Y, Zhao G. Synlett. 2007:87–80. [Google Scholar]
- 35.Magnus P, Carter R, Davies M, Elliott J, Pitterna T. Tetrahedron. 1996;52:6283–6306. [Google Scholar]
- 36.Paterson I, Yeung K. Tetrahedron Lett. 1993;34:5347–5350. [Google Scholar]
- 37.Blanchette MA, Choy W, Davis T, Essenfeld AP, Masamune S, Roush WR, Sakai T. Tetrahedron Lett. 1984;25:2183–2186. [Google Scholar]
- 38.(a) Boulet SL, Paquette LA. Synthesis. 2002:895–900. [Google Scholar]; (b) Lipshultz BH, Hackmann CJ. J Org Chem. 1994;59:7437–7444. [Google Scholar]; (c) Zhou XT, Liang L, Furkert DK, Wells CE, Carter RG. Angew Chem Int Ed. 2006;45:7622–7626. doi: 10.1002/anie.200603353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Maruoka K, Ooi T, Yamamoto H. J Am Chem Soc. 1989;111:6431–6432. [Google Scholar]; (b) Maruoka K, Ooi T, Nagahara S, Yamamoto H. Tetrahedron. 1991;47:6983–6998. [Google Scholar]; (c) Suda K, Kikkawa T, Nakajima S-I, Takanami T. J Am Chem Soc. 2004;126:9554–9555. doi: 10.1021/ja047104k. [DOI] [PubMed] [Google Scholar]
- 40.(a) Hirao A, Itsuno S, Nakahama S, Yamazaki N. J Chem Soc Chem Commun. 1981:315–317. [Google Scholar]; (b) Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]; (c) Corey EJ, Bakshi K, Shibata S, Chen C, Singh VK. J Am Chem Soc. 1987;109:7925–7926. [Google Scholar]; (d) Mathre DJ, Jones TK, Xavier LC, Blacklock TJ, Reamer RA, Mohan JJ, Jones ETT, Hoogsteen K, Baum MW, Grabowski EJJ. J Org Chem. 1991;56:751–762. [Google Scholar]
- 41.(a) Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]; (b) Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]; (c) Ahmed A, Hoegenar EK, Enev SV, Hanbeur M, Kaehlig H, Ohler E, Mulzer J. J Org Chem. 2003;68:3026–3042. doi: 10.1021/jo026743f. [DOI] [PubMed] [Google Scholar]
- 42.Grieco PA, Gilman S, Nishizawa M. J Org Chem. 1976;41:1485–1486. [Google Scholar]
- 43.Zhou X-T, Carter RG. Angew Chem Int Ed. 2006;45:1787–1790. doi: 10.1002/anie.200503733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.(a) Frye SV, Eliel E. Tetrahedron Lett. 1986;27:3223–3226. [Google Scholar]; (b) Kahn SD, Keck GE, Hehre WJ. Tetrahedron Lett. 1987;28:279–280. [Google Scholar]; (c) Keck GE, Castellino S. Tetrahedron Lett. 1987;28:281–284. [Google Scholar]; (d) Shambiayati S, Blake JF, Wierschke SG, Jorgensen WJ, Schreiber SL. J Am Chem Soc. 1990;112:697–703. [Google Scholar]
- 45.Van Draanen NA, Arseniyadis S, Crimmins MT, Heathcock CH. J Org Chem. 1991;56:2499–2506. [Google Scholar]
- 46.Chen X-N, Hortelano ER, Eliel EL, Frye VS. J Am Chem Soc. 1992;114:1778–1784. [Google Scholar]
- 47.Willard PG, Hintze MJ. J Am Chem Soc. 1987;109:5539–5541. [Google Scholar]
- 48.(a) Evans DA, Allison BD, Yang MG. Tetrahedron Lett. 1999;40:4457–4460. [Google Scholar]; (b) Evans DA, Halstead DP, Allison BD. Tetrahedron Lett. 1999;40:4461–4464. [Google Scholar]
- 49.Heathcock CH, Lampe J. J Org Chem. 1983;48:4330–4337. [Google Scholar]
- 50.Lurain AE, Carroll PJ, Walsh PJ. J Am Chem Soc. 2005;70:1262–1268. doi: 10.1021/jo048345d. [DOI] [PubMed] [Google Scholar]
- 51.Seebach D, Naef R, Calderari G. Tetrahedron. 1984;40:1313–1324. [Google Scholar]
- 52.The iodide was prepared from commercially available 2-(chloromethyl)allyl-trimethylsiliane [NaI (2.5 equiv), acetone (0.5 M), 16 h, flask covered in aluminum foil, r.t., 99%]. 1H NMR (300 MHz, CDCl3) δ 5.16 (s, 1H), 4.70 (s, 1H), 3.88 (s, 2H), 1.72 (s, 2H), 0.04 (s, 9H).
- 53.AD Mix β* = (DHQD)2PHAL (15.2 mg), K2OsO4•2H2O (2.55 mg), K2CO3 (293.6 mg), K3Fe(CN)6 (699.6 mg). Commercially available AD mix β proved to be slow and inefficient.
- 54.Preparation of LDA Solution: To a solution of diisopropylamine (101.9 mg, 0.14 mL, 1.0 mmol) in THF (0.46 mL) at −78°C was added n-BuLi (0.4 mL, 1.0 mmol, 2.5 M in THF). After 5 min, the white slurry was warmed to −10°C and stirred for an additional 15 min.
- 55.Titration of Grignard reagent: To a stirred solution of menthol (15.6 mg, 0.1 mmol) and 1,10-phenanthroline (2 mg) in THF (0.5 mL) was added prepared Grignard reagent solution until a burgundy color persists. The concentration was calculated using the formula: [RMgX] = 0.1 mmol / volume of added RMgX in mL. For the references, see: Lin H-S, Paquette LA. Synth Comm. 1994;24:2503. Watson SC, Eastham JF. J Organomet Chem. 1967;9:165.
- 56.Vanderwalle M, Van der Eychen J, Oppolzer W, Vullioud C. Tetrahedron. 1986;42:4035. [Google Scholar]
- 57.Aitken RA, Atherton JI. J Chem Soc Perkin Trans. 1994;1:1281. [Google Scholar]
- 58.Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, Lee FY, Jove R. Mol Cancer Ther. 2007;6:1400–1405. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.