


Abstract
Our knowledge of prokaryotic defense systems has vastly expanded as the result of comparative genomic analysis, followed by experimental validation. This expansion is both quantitative, including the discovery of diverse new examples of known types of defense systems, such as restriction-modification or toxin-antitoxin systems, and qualitative, including the discovery of fundamentally new defense mechanisms, such as the CRISPR-Cas immunity system. Large-scale statistical analysis reveals that the distribution of different defense systems in bacterial and archaeal taxa is non-uniform, with four groups of organisms distinguishable with respect to the overall abundance and the balance between specific types of defense systems. The genes encoding defense system components in bacterial and archaea typically cluster in defense islands. In addition to genes encoding known defense systems, these islands contain numerous uncharacterized genes, which are candidates for new types of defense systems. The tight association of the genes encoding immunity systems and dormancy- or cell death-inducing defense systems in prokaryotic genomes suggests that these two major types of defense are functionally coupled, providing for effective protection at the population level.
INTRODUCTION
Arms race between viruses and their hosts is arguably the most powerful and relentless driving force in evolution (1–3). As a result, numerous extremely diverse and elaborate antiviral defense systems have evolved and occupy a substantial part of the genome especially in free-living archaea and bacteria (4,5). Although some of these systems have been known for many years and have been thoroughly characterized, recent advances in comparative genomics and experimental study of virus-host interaction have revealed many new antiviral defense mechanisms (5–8).
The defense systems of prokaryotes can be classified into two broad groups that differ in their modes of action. The first group includes those defense systems that function on the self–non-self discrimination principle, with DNA usually being the target of the discriminatory recognition; these defense mechanisms can be viewed as prokaryotic immunity. At least three types of defense systems and their derivatives belong to this group. The best characterized of these are the extremely numerous and diverse restriction-modification (R-M) system that use methylation to label the ‘self’ genomic DNA and recognize and cleave any unmodified ‘non-self’ DNA (9–11). Another defense system in this group is DNA phosphorothioation (known as the DND system), which labels DNA by phosphothiolation and destroys unmodified DNA (8,12,13). The R-M and DND systems represent the prokaryotic version of innate immunity.
Unlike R-M and DND systems, which attack non-self invaders indiscriminately, the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas (CRISPR-associated genes) systems is able to memorize the encounters with infectious agent and attack it specifically afterwards (14–18). Thus, CRISPR-Cas is often viewed as a prokaryotic adaptive immunity system.
The second group of defense systems is generally based on programmed cell death or dormancy induced by infection. Numerous and diverse toxin-antitoxin (TA) systems belong in this category. Depending on the nature of toxins and antitoxins, the TA systems are currently classified into three types: type I with antisense RNA as antitoxin and a protein, usually a small membrane holin-like protein as a toxin; type II, in which both toxin and antitoxin are proteins, and type III, in which with the RNA antitoxin directly inactivates the protein toxin (7,19–28). Two additional types of TA systems (IV and V) have been recently proposed based on distinct mechanisms of action of the respective antitoxins (29,30). In addition to the TA systems, abortive infection (ABI) or phage exclusion systems also often use the mechanism of cell death or dormancy. These systems have not been so far classified in detail, but some of them fit well into the TA systems description (31). The vast majority of toxins in both TA systems and ABI systems interfere with the translation process, mostly via mRNA or tRNA cleavage.
Numerous recent comparative genomic studies not only revealed the high abundance of the known defense system and predicted new ones whose molecular mechanisms of action remain to be characterized but also highlighted several distinct properties of these systems.
The genes encoding different defense systems often cluster in genomic islands of larger than an operon size.
The immunity systems are often encoded within the same genomic loci with systems that cause cell death or dormancy, and, at least in some cases, the two classes of defense systems functionally cooperate.
Different families of toxins and antitoxins often recombine to form (almost) all possible TA pairs.
Defense systems or their components sometimes change their mode of actions. Thus, R-M systems can switch to the functional mode characteristic of TA systems, whereas individual components of TA systems can act solo as ABI systems.
The purpose of this article is to examine these recent observations in some detail and to focus on several recently predicted and still poorly characterized defense systems of bacteria and archaea. The functions and comparative genomics of well-characterized prokaryotic defense systems such as R-M, TA and CRISPR-Cas have been discussed in detail in multiple reviews; therefore, here, we only include brief summaries of the pertinent features of these systems.
DISTRIBUTION OF DEFENSE SYSTEMS IN ARCHAEA AND BACTERIA AND FOUR DISTINCT DEFENSE STRATEGIES INFERRED FROM GENOME ANALYSIS
The fraction of bacterial and archaeal genomes allotted to defense systems varies broadly, from virtual absence to ∼10% (Figure 1A). These distributions reflect the low bound for each type of defense systems because many more instances undoubtedly remain to be discovered as discussed in the rest of this article. The overall abundance of defense systems shows nearly perfect linear scaling with genome size (5). The number of TA genes generally increases faster than linearly (as a power of ∼1.3 of the total number of genes), ABI system genes take an approximately constant fraction of the genome (∼1 per 1000 genes), and R-M genes scale sublinearly with the genome size (power of ∼0.75) (Figure 1B). The CRISPR-Cas system abundance is statistically the same in large and small genomes. The differential scaling with genome size implies that it is most appropriate to analyse the abundance of defense systems genes relative to the expected abundance, given the host genome size.
Figure 1.
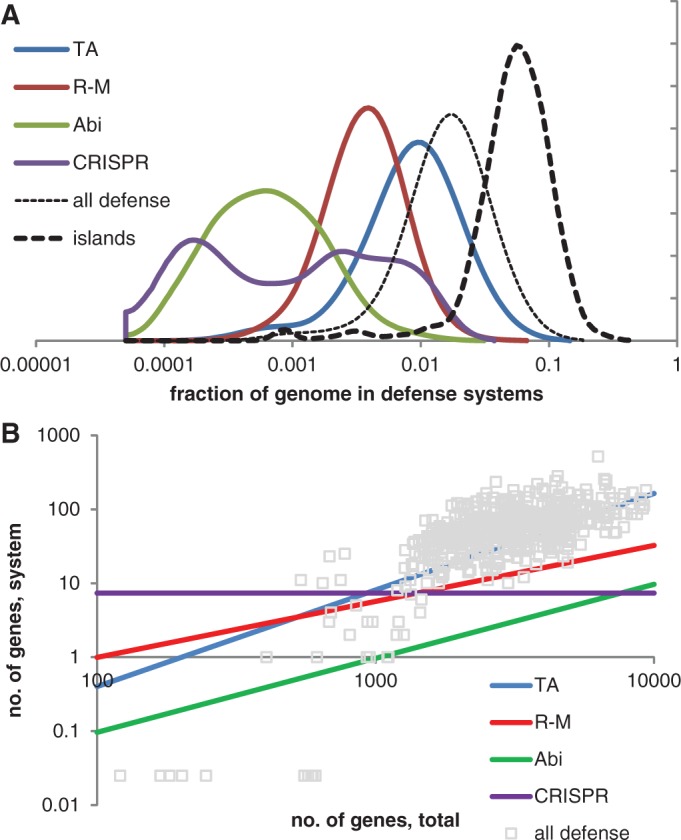
The major types of defense systems in bacterial and archaeal genomes. (A) Distribution (probability density function) of the genome fraction occupied by defense systems in bacteria and archaea. (B) Scaling of the number of genes in defense systems with the total number of genes. A data set of 572 genomes (the largest genome in a genus with addition of E. coli K12 and B. subtilis subsp. subtilis) was selected to represent 1516 genomes that were completely sequenced and available through the NCBI Genome database as of February 2012.
The immediate outcome of the analysis of the distribution of defense genes is their pronounced enrichment in archaea compared with bacteria and in thermophiles (especially hyperthermophiles) compared with mesophiles and psychrophiles (5). The two trends, the dependency on taxonomy and temperature preference, seem to be independent of each other. A deeper analysis of the distribution of the relative abundances of genes belonging to different defense systems reveals four distinct clusters of organisms in the principal component-like space (Figure 2) as indicated by gap function analysis (32). This observation implies the existence of four distinct ‘defense strategies’: (i) all defense systems are under-represented relative to their expected abundance: in the respective organisms, defense is either abandoned altogether or reduced to bare-bones minimum; (ii) the total number of genes dedicated to defense is close to the expected value; prevalence of R-M and ABI over TA and CRISPR; (iii) the total number of genes dedicated to defense is close to the expected value; prevalence of TA and CRISPR over R-M and ABI; and (iv) all defense systems are over-represented, i.e. a greater than average fraction of the genome is dedicated to antivirus defense (Figure 2A).
Figure 2.

Distribution of known and predicted defense systems in archaeal and bacterial genomes. (A) The four ‘defense strategies’. Here, 1–4 refers to the four strategies discussed in the text. The axes show logs of the ratios of the numbers of genes belonging to a given type of defense systems to the number expected from the scaling shown in Figure 1B. The horizontal axis is the sum of the logs for all four types and the vertical axis is (TA + CRISPR) − (R-M + ABI). (B) Defense strategies used by bacterial and archaeal thermophiles and mesophiles. BT, AT, BM and AM stand for bacterial thermophiles, archaeal thermophiles, bacterial mesophiles and archaeal mesophiles, respectively. The axes show logs of the ratios of the numbers of genes belonging to a given type of defense systems to the number expected from the scaling shown in Figure 1B. The horizontal axis is the sum of the logs for all four types and the vertical axis is (TA + CRISPR) − (R-M + ABI). (C) Distribution of the defense strategies among major prokaryotic taxa. Here, 1–4 refers to the four strategies discussed in the text. The number of analysed genomes for each taxon is indicated inside the respective bar. The expected abundance of genes belonging to the defense systems of each type in a given genome was calculated from the genome size using the observed scaling relationships (Figure 1B). Logarithms of the ratios of the observed and expected frequencies of defense system genes in genomes were analysed using Principal Component Analysis; then the data were projected into the space of two orthogonal axes with integer coefficients closest to the first principal components.
An overwhelming majority of bacterial thermophiles, along with the archaea, regardless of the optimal growth temperature, follow strategies (iii) or (iv), including a general over-representation of defense system genes (Figure 2B and C). Bacteria are widely spread across the entire parameter space, with most of the large bacterial groups showing a range of defense strategies among the representative genomes (Figure 2C).
Certainly, one has to keep in mind that the aforementioned partitioning of the archaeal and bacterial defense strategies is conditioned on our ability to identify defense systems by genome analysis. In particular, assignment of an organism to the first strategy (no or little defense) could be somewhat naïve in the sense that some of these organisms might use completely uncharacterized novel defense systems. This concern is minor when it comes to parasitic or symbiotic organisms with very small genomes to which this strategy (or perhaps more precisely, lack of defense strategy) trivially applies. However, extreme paucity of identifiable defense systems has been noted also for some bacteria with large genomes, e.g. Paenibacillus sp, with a genome of more than seven megabases (5). In these cases, the potential unknowns loom large, and it is a question of major interest whether the lifestyle of these organisms renders defense systems superfluous or favours novel defense mechanisms.
DEFENSE ISLANDS
Many cases of clustering of defense genes on the chromosomes have been described (27,33,34) as well as involvement of transposable elements in horizontal transfer of defense genes (35–37), indicating high mobility and preferential attachment of these systems. Thus, unlike other functional groups of bacterial and archaeal genes (such as sugar metabolism, energy metabolism, etc.), defense systems and mobilome-related genes, such as prophages, form clusters the size of which by far exceeds the size of typical operons and that are unlikely to appear by chance. Statistically significant over-clustering of different defense systems has been demonstrated (5). Briefly, many defense operons tend to be in closer physical proximity to each other on the chromosome, compared with the random expectation [see (5) for details]. This finding suggests the possibility of synergistic interactions between different types of defense systems. Although currently there is no unequivocal definition of the defense islands and no clear understanding of the mechanism(s) of their formation, a simple operational definition has been proposed. A defense island is defined as a string of continuous genes, at least one of which belongs to a known defense gene families, which are flanked by house-keeping genes; such islands are significantly enriched by defense and mobilome-related genes, compared with analogous blocks formed by other genomic systems (5). The percentage of genes found in defense islands varies from 0 to 30% across the current collection of prokaryotic genomes (Figure 1A) (5). The greatest fraction of the genome dedicated to antiviral defense was detected in the cyanobacterium Microcystis aeruginosa, the proteobacterium Bartonella tribocorum and the bacteroidetes bacterium Pelodictyon phaeoclathratiforme. The detection of extreme abundance of defense systems in taxonomically scattered bacteria implies that such over-representation is not lineage-specific but is perhaps dictated by the ecology of the respective organisms that might be subject to unusual massive assault by invasive agents (5).
This simple operational definition of defense islands has proved extremely useful for the prediction of new defense systems (5) and understanding the cooperation between them (see later in the text). Figure 3 shows several examples of defense islands that are specifically enriched for genes from different defense systems and include several still experimentally uncharacterized genes that are implicated in antivirus defense.
Figure 3.

Examples of defense islands in archaeal and bacterial genomes. The genes are shown by block arrows with the size roughly proportional to the size of the corresponding gene. The genomic position of each region is indicated given in parentheses after the species name in the form of the range of genes denoted using the systematic names for the respective species. Colour coding is the following: pink are components of TA systems, read, components of CRISPR-Cas systems; dark blue, Pgl system; light blue, regulatory components; green, R-M systems; yellow, ABI system; orange, pAgo; brown, components that are spredicted to be involved in defense; grey, unknown protein. The protein family or domains names are provided above the respective arrows; some of these families were recently introduced and described in the course of comparative genomic analysis of defense islands (5); COG or Pfam families are indicated in parentheses. Pgl, Phage Growth Limitation; HTH, helix-turn-helix; RHH, ribbon-helix-helix; GIY-YIG, conserved motif in a nuclease family.
DEFENSE MECHANISMS IN BACTERIA AND ARCHAEA
Innate immunity: DNA modification systems
The R-M systems are probably the best studied phage defense mechanism in bacteria owing to the extensive application of restriction endonucleases in molecular biology (9–11). Because of this practical importance, as well as the extreme diversity in the genomic organization and protein domain architecture of the R-M systems, detailed rules for restriction enzyme classification and nomenclature have been developed (38). This classification divides the R-M systems into four major types (I–IV), on the basis of subunit composition, ATP(GTP) requirement and cleavage mechanism (39–41). All the R-M systems function on the same principle of self–non-self discrimination, with one enzyme, a methyltransferase (MTase), modifying the self DNA and the other one, restriction endonuclease (REase), cleaving non-methylated foreign DNA (38,42). Type II R-M systems are the simplest and by far the most common and are mostly used for experimental applications owing to the fact that these enzymes cleave the target DNA at highly specific sites. The Type II R-M systems have been further classified into several subtypes, primarily on the basis of cleavage specificity (41). The Type II systems consist solely of the MTase–REase pair that is typically encoded within the same operon, although some cases of apparent disjointed localization of the two genes have been reported (43). The most complex ATP-dependent Type I R-M systems encompass three genes, which encode the R (restriction), M (modification) and S (specificity) subunits of the R-MA complex; the R subunit also contains a distinct ATPase domain that belongs to the helicase Superfamily II (42,44,45). Type III R-M system resemble Type II systems in that they consist of only R and M subunit but, on the other hand, are similar to Type I systems in that the R subunit also contains the helicase domain and the reaction is ATP-dependent (46,47). Type IV R-M systems are distinct two-subunit complex that consist of a AAA + family GTPase and an endonuclease, and cleave the target DNA non-specifically (45,48).
Many genomic loci that encompass R-M systems of all four major types also include variable groups of additional genes that appear to be co-expressed with the genes for R-M system subunits (5) (Figure 3). Although most of these genes have not been experimentally characterized, one such case has been studied in considerable detail and presents a remarkable example of the interplay between different defense mechanisms. The Escherichia coli anticodon nuclease (ACNase) prrC co-localizes with three genes for R-M type Ic system prrI and contributes to the T4 phage exclusion mechanism (49–51). This genomic association that is conserved in diverse bacteria implies also a functional connection, and at least one case has been studied in detail. The PrrC nuclease, normally inactive, can be allosterically activated either by unmodified DNA or by the small anti-restriction peptide encoded by the T4-like enterobacteriophages. The activated PrrC ACNase cleaves the anticodon of tRNALys in a GTP-dependent manner; the GTP hydrolysis is catalysed by the N-terminal ABC NTPase domain of PrrC. The cleavage of tRNALys inhibits the host translation and as a consequence the reproduction of the T4 phage. The RloC enzyme that is homologous to PrrC does not seem to be linked to R-M systems, has similar biochemical properties and is activated under genotoxic stress (52,53). Recent analysis has shown that the ACNase domain of both proteins belongs to the HEPN superfamily that is merging as a major group of ribonucleases that are involved in various forms of defense and stress response (54,55).
Site-specific DNA backbone S-modification and cleavage of unmodified DNA and the dndABCDE genes (after DNA degradation phenotype; alternatively, these genes are designated dpt, i.e. DNA phosphothiolation) involved in this system have been first discovered in Streptomyces lividans 1326. Five additional genes (dndFGHI) that are strongly linked to this system have been found by analysis of the genomic neighbourhoods (12,13). Recently, the genes required for modification (dndABCDE) and restriction (dndFGH) have been identified in the related system from Salmonella enterica serovar Cerro 87 (8). The structures and biochemical activities of the DndA and DndC proteins that are directly involved in S-modification are relatively well-understood (56,57), and the functions of the other genes associated with this system are less clear. Moreover, the neighbourhood around the genes that comprise this system is highly flexible, including cysteine desulfurase dndA, which often is not linked to the other dnd genes (8). Here, we present results of additional sequence and gene context analysis for these genes that show a strong link of several components of the DND systems with ABI and TA systems (Supplementary Table S1). For instance, DndB, the potential negative regulator of restriction (13,58), contains an N-terminal region that belongs to the ABI protein family AbiU1/AIPR/COG1479, which encompasses a ParB superfamily nuclease domain often fused to other nuclease domains from different families and linked to R-M systems (55,59). In DndB, the ParB-like domain is additionally fused to a HEPN domain. A distinct HEPN domain from a different subfamily (DUF4145) is fused to DndF NTPase. Domains of the latter subfamily are often fused to REase components of Type I R-M systems (55).
The third DNA modification system, which is involved in Phage Growth Limitation (Pgl) system, is so far poorly characterized experimentally. The Pgl system is centred around the PglZ protein family in which the only recognizable domain belongs to the alkaline phosphatase superfamily (pfam08665) (60). The scarce experimental evidence indicates that PglZ confers protection against the temperate bacteriophage phiC31 in Streptomyces coelicolor A3(2) (61,62). This system also includes the P-loop ATPase domain-containing protein PglY, the methylase PglW and the serine-threonine kinase PglX (the latter two proteins are encoded in a different locus in S. coelicolor genome). The bacteria that possess the Pgl system support a phage burst on initial infection, but subsequent phage growth cycles are severely restricted (62). Although the molecular mechanism of the Pgl system has not been experimentally elucidated, it has been hypothesized that it methylates the DNA of the phage progeny rather than the host DNA so that on re-infection, the surviving cells in the same Streptomyces colony could activate the system and prevent phage growth (61,62). Thus, the Pgl system might function via a reverse R-M mechanism combining the self–non-self discrimination and virus-induced cell death modes of antivirus defense in a novel defense strategy. The recent comparative analysis of the neighbourhoods of the pglZ gene revealed a substantial complexity of genetic organization of this system that could be possibly compared only with the CRISPR-Cas system (see later in the text) (5). Supplementary Table S2 lists the gene families that are associated with pglZ gene. One of these families is COG1479 (or DUF262 or DGQHR domain) that has been previously identified within the Type I R-M system locus in Campylobacter jejuni (63). The core domain of the COG1479 family belongs to the ParB-like superfamily and is often fused to other nucleases such HNH-type nuclease domain, PD-(D/E)xK-like nuclease and HEPN domain, suggesting that it might be another case of a programmed cell-death system associated with various DNA modification systems (5). Based on the presence of the pglZ gene, this system is found in 174 of 1516 completely sequenced genomes that represent most of the major bacterial lineages and several methanogenic and halophilic archaea. The remarkable complexity of the Pgl system seems to reflect a still poorly understood elaborate molecular mechanism of self–non-self discrimination and fine-tuned regulation.
Adaptive immunity: the CRISPR-Cas system
The CRISPR-Cas system uses a unique defense mechanism that involves incorporation of virus DNA fragments into CRISPR repeat arrays and subsequent utilization of transcripts of these inserts (spacers) as guide RNAs to cleave the cognate virus genome (34,64–67). Thus, the CRISPR-Cas system represents bona fide adaptive immunity that until recently has not been discovered in prokaryotes and, moreover, is the most clear-cut known case of Lamarckian inheritance (68). The role in antiviral defense that initially was predicted for this system on the basis of the detection of spacers identical to fragments of virus and plasmid genomes and comparative analysis of Cas protein sequences has been successfully confirmed experimentally (69). Within the few years since this key breakthrough, the CRISPR research evolved into a distinct, highly dynamic field of microbiology with considerable biotechnology potential (70–73). The recent advances in the study of CRISPR-Cas systems are covered in many reviews (15,74–76); therefore, here we present only a brief outline of the functions and comparative genomics of prokaryotic adaptive immunity and discuss the likely scenarios for the evolution of the different types of CRISPR-Cas.
The CRISPR-Cas systems are classified into three distinct types (I, II and III) (18) and several yet unclassified minor variants (77). This classification was developed through a combination of comparisons of the sequences of the Cas proteins, cas gene repertoires and genomic organization of the CRISPR-Cas loci. For each type and subtype, a specific signature gene has been identified allowing easy classification of the highly variable CRISPR-Cas loci in the course of genome analysis (18). The mechanism of CRISPR-Cas is usually divided into three stages: (i) adaptation, when new spacers homologous to protospacer sequences in viral genomes or other alien DNA molecules are integrated into the CRISPR repeat cassettes; (ii) expression and processing of pre-crRNA into short guide crRNAs; and (iii) interference, when the alien DNA or RNA is targeted by a complex containing a CRISPR RNA (crRNA) guide and a set of Cas proteins [for review, see (15)]. Below, we focus on the basic building blocks of the distinct types of CRISPR-Cas systems and summarize the current considerations on the origin and evolution of this system.
Most of the Cas protein sequences evolve under relaxed purifying selection (78) and/or undergo accelerated evolution resulting from the virus-host arms race [e.g. (79)]. Consequently, most of these sequences are weakly conserved in evolution so that conventional sequence comparison partitions the Cas proteins into >100 families (18). However, advanced sequence analysis combined with structural comparison identifies conserved domains between Cas protein families that were originally considered unrelated and thus enables the identification of the major building blocks that are shared by different CRISPR-Cas types (Figure 4A) (18,34,64,77). The two proteins that are present in the great majority of the CRISPR-Cas systems are Cas1 and Cas2 that together are required and sufficient for spacer integration (the adaptation phase of the CRISPR-Cas response) (80). The only CRISPR-Cas loci that lack Cas1 and Cas2 genes are some Type III systems that co-exist with Type I systems within the same genome and apparently borrow Cas1 and Cas2 proteins from the latter (18). Although both Cas1 and Cas2 are involved in adaptation, Cas1 endonuclease that adopts a unique α-helical fold (81) appears to possess all the required enzymatic activities, whereas Cas2 might perform a distinct function that is not mechanistically related to spacer acquisition (see discussion later in the text).
Figure 4.

General principles of the structure and organization of four CRISPR-Cas types. (A) The building blocks of four distinct CRISPR-Cas system types. The cas genes and domain description for each building block are given. Gene names follow the current nomenclature and classification (18). The symbol ‘#’ indicates the putative small subunit that appears to be fused to the large subunit in several Type I subtypes (77). Asterisk indicates that those COG1517 family proteins that contain a third effector (toxin) domain are implicated in immunity-dormancy/suicide coupling. (B) RRM domain-containing proteins in CRISPR-Cas systems. General organization of operons is shown by arrows with size roughly proportional to the size of respective gene. Homologous genes are shown by the arrows of same colour or hashing. Colour coding is the same as in the (A). Gene and family names are taken from (18,77). Additional designations: LS, large subunit; SS, small subunit; R, RAMPs. RRM domains are shown by pink rectangles, with semitransparent rectangles indicating deteriorated RRM fold. The protein representing families with RRM domains for which structures have been solved are denoted by asterisks. A topology diagram of the RRM fold is shown in the bottom left: beta strands are shown by red arrows; the purple shapes each denotes a single alpha helix in the typical RRM fold that, however, are replaced by more complex secondary structure arrangements in some variants including RAMPs. The structure of Cas6, the typical RAMP superfamily protein with two RRM domains, is shown in the bottom right. The colours of the core RRM elements are the same as in the topology diagram; in addition, the glycine-rich loop, the signature feature of the RAMP superfamily proteins, is shown in blue; amino acids involved in catalysis are rendered in yellow.
With the exception of Cas1, most of the common Cas proteins contain various versions of the RNA Recognition Motif (RRM) domain, a widespread RNA-binding domain that in particular comprises the core of diverse DNA and RNA polymerases (where it is denoted the Palm domain). Among the Cas proteins, different variants of the RRM domain are present in Cas2 (a toxin-like ribonuclease), Cas10 (the so-called CRISPR polymerase, a protein that is homologous to polymerases and cyclases but whose actual biochemical activity remains unknown) and in the largest group of Cas proteins known as the RAMP (Repeat-Associated Mysterious Proteins) superfamily (Figure 4B). In particular, all CRISPR-Cas systems of Type I and most of the systems of Type III include a dedicated ribonuclease for the pre-crRNA processing that typically belongs to the Cas6 family of the RAMPs (82,83). In some cases, e.g. in CRISPR-Cas systems of Type I-C, the function of Cas6 is displaced by a catalytically active RAMP of the Cas5 family (84). In contrast, Type II CRISPR-Cas systems use an unrelated mechanism of pre-crRNA cleavage. This version of pre-crRNA processing requires the involvement of the double-stranded RNA-specific RNase III, a specialized trans-encoded small RNA, which is complementary to a single CRISPR repeat, and still unidentified domains of the Cas9 protein (18,69,85,86).
In Type I-E and I-F CRISPR-Cas systems, the endoribonuclease that catalyses the processing of the pre-crRNA is a subunit of a multisubunit (or multidomain) complex known as CASCADE (CRISPR-associated complex for antiviral defense) (87). The mature crRNA remains associated with the CASCADE complex that scans the target DNA for a match, and once one is found, recruits the Cas3 protein that cleaves the target via its HD endonuclease domain (88). In Type III systems (at least the model system from the archaeon Pyrococcus furiosus), the Cas6 endoribonuclease does not belong to the CASCADE complex that is apparently not directly involved in the processing but instead binds the mature crRNA (89,90). This distinction apart, the architectures of the CASCADE complexes in Type I and Type III CRISPR-Cas are similar and include a large subunit, a small subunit and a pair of RAMPs that belong to the Cas5 and Cas7 families (84,87,90–92) (Figure 4A). Despite the high level of sequence divergence and structural rearrangements that is typical of many Cas proteins, there appears to be a direct homologous relationships between the respective subunits of the Type I and Type III CASCADEs (77). A notable difference is that Type I CRISPR-Cas encompasses a single Cas7 protein that is present in several copies in the CASCADE, whereas in Type III systems, there are several paralogous Cas7-like proteins. In Type II CRISPR-Cas, a single large multidomain protein, Cas9, is responsible for all the functions that in Type I and Type III systems are performed by the CASCADE and the Cas3 protein (93).
The target DNA cleavage in Type I (88) and most likely in Type III systems (77) is catalysed by homologous HD family nucleases. In many Type III systems, the HD domain is fused to the cas10 gene, the large subunit of the CASCADE-like complex, whereas in Type I systems, the most common protein architecture is Cas3 in which the HD domain is fused to a distinct helicase domain that is essential for the interference stage (88,94). Type II systems use an unrelated mechanism that involves two distinct nuclease domains, HNH and RuvC-like, both contained within the Cas9 protein (95). This mechanism involves a unique two-RNA structure that consists of the mature crRNA base-paired, which is base-paired with the trans-encoded small RNA and directs Cas9 to the cognate DNA sequence where this protein introduces double-stranded breaks. During this process, the HNH nuclease domain of Cas9 cleaves the strand of the target DNA that is complementary to the crRNA, whereas the RuvC domain cleaves the second strand (95).
The Cas1 endonuclease, the CASCADE subunits and the Cas3 helicase-nuclease are essential for the immune function of the respective CRISPR-Cas systems. In addition, the CRISPR-Cas loci encompass many other genes that encode proteins whose mechanistic role in adaptive immunity remains unclear but that belong to protein families implicated in other defense systems. These CRISPR-associated gene products include the ribonuclease Cas2, the RecB-like nuclease Cas4 and numerous representatives of the COG1517 superfamily of helix-turn-helix and putative ligand-binding domain containing proteins (34,77). Most of these proteins, in particular Cas2, contain domains that are predicted to be nucleases and toxins, suggesting a secondary role as associated immunity components [see details later in the text and (55)]. Finally, the functions of several Cas proteins remain completely obscure.
Taken together, the results of comparative sequence analysis, structural studies and experimental data suggest that despite the remarkable complexity and diversity, all CRISPR-Cas systems use the same architectural and functional principles and, given the conservation of the principal building blocks, share a common ancestry (Figure 4A). It is notable, however, that some of the essential components of the CRISPR-Cas systems can be replaced either by homologous proteins, such as the substitution of Cas5 for Cas6 in Type I-C CASCADE complexes, or by non-homologous but functionally analogous proteins, such as the substitution of the HNH and RuvC-like domains of Cas9 for the HD nuclease.
Under the recently proposed parsimonious evolutionary scenario, only a few evolutionary events would suffice to explain the emergence of CRISPR-Cas system types and subtypes (55). Furthermore, comparison of the recently solved structures of all major components of the CASCADE complex suggests that the RAMPs and the small subunits might have evolved from the ancestral large subunit resembling the Cas10 protein that contains two RRM domains and an alpha-helical domain resembling the small subunit (96,97). The Cas10 protein (the large subunit of Type III CRISPR-Cas systems) could have evolved from an ancestor RRM (Palm) domain-containing polymerase or cyclase and, combined with the HD domain, might have originally functioned as a CRISPR-independent defense (innate immunity) system (55). The Cas1–Cas2 module originally might have functioned independently as a TA system (see discussion later in the text). Joining this module with the hypothetical ancestral CASCADE-HD system might have led to the emergence of the adaptation stage and accordingly the transformation of an innate immunity mechanism into one for adaptive immunity.
The ancestral Cas10-like protein and the entire ancestral, subtype III-like CRISPR-Cas system most likely evolved in hyperthermophilic archaea and was subsequently horizontally transferred to bacteria. Indeed, in archaeal hyperthermophiles, this variant of the CRISPR-Cas system is (nearly) universal in these organisms, in a sharp contrast to the presence of any form of CRISPR-Cas in <50% of archaeal and bacterial mesophiles (18,77,98). In accord with this scenario, a recent mathematical modelling study has shown that the benefits of adaptive immunity are substantially greater under the conditions of limited virus mutability that seems to be characteristic of hyperthermophilic habitats (99).
Putative defense systems associated with prokaryotic Argonaute homologs
Another putative defense system that remains to be experimentally characterized centres around prokaryotic homologues of the slicer nuclease argonaute (pAgo), the central component of the eukaryotic RNAi system (100). In all, 189 pAgo sequences have been identified in complete or draft genomes that represent most of the major branches of archaea and bacteria. For bacterial pAgos from Aquifex aeolicus and Thermus thermophiles, site-specific DNA-guided endoribonuclease activity has been demonstrated in vitro (101,102), but the natural target and the source of the guide DNA molecule(s) remain to be determined. The pAgos could be classified into two large monophyletic groups: the ‘long’ form that contains a PAZ (oligonucleotide binding) and PIWI (active or inactivated ribonuclease) domains and the ‘short’ form that lacks the PAZ domain (100). Almost all pAgos that lack a PAZ domain appear to be inactivated, and the genes encoding for these proteins are associated with a variety of predicted deoxyribonucleases in putative operons, including those from PD-(D/E)xK, Sir2 and phospholipase D superfamilies. Furthermore, strong association of the pAgo gene with defense islands has been demonstrated (100). Thus, it can be the hypothesized that the PAZ domain-containing pAgos directly destroy virus or plasmid transcripts via their endoribonuclease activity, whereas the apparently inactivated PAZ-lacking pAgos could be structural subunits of protein complexes that contain endonucleases targeting DNA. An alternative possibility is that pAgo represents a distinct ABI system (see later in the text) that targets host nucleic acid and causes death or dormancy of the infected cell. Regardless of the specific mechanisms, it is likely that pAgos are key components of a novel defense system that uses guide DNA or RNA molecules to cleave target nucleic acids (100).
SYSTEMS INDUCING PERSISTENCE AND PROGRAMMED CELL DEATH
Toxins–antitoxins
Both Type I and Type II TA systems originally have been characterized as ‘addictive modules’ that are encoded in plasmids and ensuring their persistence in a host lineage after a cell division (103,104). The toxin component of all TA systems is a protein that kills cells if expressed above a certain level, whereas the antitoxin component reversibly inactivates the toxin and/or regulates its expression, thereby preventing cell killing. Unlike the toxin, the antitoxin is metabolically unstable so that, unless the antitoxin is continuously expressed, the free toxin can be accumulated in amounts sufficient to kill a cell (25,105–108). Once the first genomes have been sequenced, it became clear that numerous TA systems are present not only on plasmids but also on the chromosomes of bacteria and archaea (25,107).
This surprising discovery stimulated a debate on the functions of the chromosomal TA systems and prompted a series of comparative genomic and experimental studies that resulted in the discovery of dozens of new TA systems. These findings and the current ideas on the biological roles of TA systems are summarized in several recent reviews (19,26,109–111). Briefly, it appears that the TA systems provide a mechanism for cell persistence to cope with various stress conditions (23,24,111). The majority of Type II toxins target different components of translation systems, especially mRNA (112,113), whereas Type I toxins affect membrane integrity (114). However, other targets of toxins have been identified as well, such as DNA gyrase (115) and the cell division GTPase FtsZ (116). Because Type I toxins have never been implicated in virus resistance and are not frequently observed in defense islands, we do not consider them here. Instead, we focus on Type II TA systems, particularly poorly characterized variants (Supplementary Table S3), and discuss the results of the recent efforts to identify new TA families using in silico approaches.
The computational approaches for prediction of new TA systems can be classified into three groups: (i) ‘guilt by association’ when a new toxin or antitoxin is predicted by virtue of linkage, in bacterial and archaeal genomes, to genes that belong to known antitoxin or toxin families (27,117); (ii) identification of gene pairs with characteristic features of TA systems such as tight linkage of genes encoding small proteins, propensity for HGT and presence on plasmids or within genomic islands with other defense genes (5,27); and (iii) statistical analysis of whole genome sequencing clones aimed at identification of genes that are unclonable (toxic) in E. coli (118).
The new predicted TA systems usually are validated experimentally in E. coli by a kill/rescue assay in which overexpression of a toxin is expected to inhibit cell growth or kill the cell, whereas co-expression of the toxin and the antitoxin restores growth (117). However, the recent comprehensive study revealed numerous genes that appear to be unclonable in E. coli but do not meet the definition of TA systems, including many metabolic enzymes and informational genes such as ribosomal proteins (118,119). Although not all of these genes form two-gene operons that are typical of TA systems, these findings indicate that dosage imbalance or toxicity of an intermediate substrate can result in toxicity of a gene that can be mitigated by a proper regulation or co-expression by enzyme using a toxic product, mimicking the TA behaviour. Thus, prediction of new TA systems from experimental results obtained with this approach requires caution and should involve assessment of the known and predicted functions and operonic organization of the candidate genes. Several experimentally validated TA systems (e.g. GinA and GinC) do not form evolutionarily conserved two gene operons, suggesting modes of actions distinct from the typical toxin–antitoxin mechanism (120). For example, GinA, a close homologue of the phage Mu host-nuclease inhibitor protein Gam, which inhibits RecBCD binding to dsDNA ends (121), and its ‘antitoxin’ Sak, a single-strand annealing protein (122), are often linked to other enzymes involved in recombination and repair (120). Accordingly, it appears most likely that GinA and GinC are involved in repair-related functions as well. These complications associated with the interpretation of the guilt by association predictions and the standard validation experiments indicate that additional experimental approaches are required to determine whether some recently identified systems are bona fide TA systems.
Additional examples of poorly characterized (predicted) TA systems are given in Supplementary Table S3. One of the most abundant of the predicted TA systems, that is particular common in hyperthermophilic archaea, consists of a HEPN domain-containing protein the minimal nucleotidyltransferase (MNT). Among the two components of this TA system, the HEPN domain protein is likely the toxin (118) that is predicted to function as a RNAse probably targeting an RNA during translation (54,55), whereas the MNT is the antitoxin. Although the HEPN–MNT module shares all the typical characteristics of TA systems (27), the molecular mechanism of this system, and in particular the role of the nucleotidyltransferase activity of the antitoxin, remains unclear. The HEPN proteins in these systems belong to two groups, one of which is over-represented in thermophiles and the other one in mesophiles (27). The HEPN and MNT domains are often fused to each other, which is not typical of other TA pairs. Furthermore, the paRep1/paRep8 (Pyrobaculum aerophilum repetitive family) family of HEPN domains, which is represented almost exclusively in thermophiles and is specifically expanded in crenarchaea, is not associated with MNT; therefore, it remains to be determined whether these proteins are toxins of a distinct family of TA systems using a still unidentified antitoxin.
Another two component system in which one of the proteins is a predicted nucleotidyltransferase is DUF1814-COG5340. More than 700 occurrences of this system were detected in 430 sequenced genomes of most major lineages of archaea and bacteria including several Mycoplasma species with small genomes. Homology of the DUF1814 family with the ABI AbiG (123) and AbiE families (124) has been demonstrated (5). In this case, however, the nucleotidyltransferase (DUF1814) appears to function as the toxin, whereas the COG5340 protein that contains a predicted HTH domain is the antitoxin [(5), see also Supplementary Table S4]. Both ABI systems appear to act at the stage of phage DNA replication, but their molecular mechanisms remain unknown (22).
Yet another putative new toxin is COG2856, a metzincin superfamily protease associated with a potential antitoxin, a HTH-domain protein of the Xre family, often fused to the protease (125). These putative operons are abundant in bacterial and archaeal genomes, phages and plasmids, with lineage-specific expansions in several bacteria. Interestingly, in the bacterium Deinococcus radiodurans, a COG2856 gene (irrE) is a major radiation resistance determinant (126).
Comprehensive comparative genomic analysis of the distribution and co-occurrence of known and predicted families of toxins and antitoxins leads to the following principal conclusions:
The abundance of TA systems in the genomes scales superlinearly with the genome size (5,27).
So far, no TA systems have been detected in most endosymbionts and, among archaea, in Thermoplasmatales, several methanotrophs with small genomes, and the only known symbiotic archaeon, Nanoarchaeum equitans (27,117,127,128).
The distribution of TA systems across phyla is distinctly non-uniform, with many systems significantly over- and under-represented in various taxa (27,117).
Genomic occurrence of TA systems shows exceptional variability even in closely related genomes (27,117).
TA systems are prone to HGT and can be considered a part of the prokaryotic mobilome (27).
The network of associations between different families of toxins and antitoxins contains a giant connected component and only a few isolated systems (Figure 5). The existence of such a strongly connected network is due to the modularity of the TA systems whereby toxins and antitoxins typically can have more than one partner. The principal hubs of the TA systems network are the PIN and RelE toxins and the RHH and Xre antitoxins (Figure 5) (27).
The high prevalence of stand-alone toxin and antitoxin genes (>50% of the genes in the largest families do not belong to TA pairs) suggests potential in trans interaction between toxins and antitoxins that remain to be discovered experimentally (27,117,128).
Figure 5.

A network graph of the relationships between different families of toxins and antitoxins. Known and predicted (magenta) toxins (red circles) and antitoxins (blue circles) and their operon organizations. The edges connect genes with five or more two-component operons identified; the thickness of an edge is proportional to the abundance of the respective operon.
Taken together, all these findings indicate that the TA systems comprise an extremely complex, versatile and certainly not fully investigated network of ‘semi-selfish’ mobile elements that permeates the prokaryotic world. The principal role of the TA systems in bacteria and archaea appears to be induction of dormancy or programmed cell death in response to stress, in particular virus infection. However, it is currently impossible to rule out that the TA systems perform additional cellular functions.
ABI (phage exclusion) systems
The ABI (phage exclusion) systems represent another widespread group of defense mechanisms that abrogate virus infection at different stages, often by causing death of infected cell (21,22). Furthermore, some of the ABI systems are two-component modules with all the properties of TA systems (e.g. the Type III TA systems aforementioned). Numerous ABI systems were identified mostly by genetic methods in lactic acid bacteria and E. coli, but only for a few of them the molecular mechanism is known (21). Supplementary Table S4 briefly summarizes the available information on these systems together with the results of computational analysis that could aid further experimental study. These findings indicate extensive domain sharing between ABI and TA systems and support the observation that most of the systems of both classes act by inducing cell death or dormancy. For example, the two-component AbiG system aforementioned is predicted to function as a TA system (5). Many ABI proteins or domains superfamily including AbiD, AbiF, AbiJ, AbiU2, AbiV and the C-terminal domain of AbiA belong to the HEPN endoribonuclease and are predicted to target the translation system (54). A HEPN domain is also predicted to be responsible for the anticodon tRNase activity of PrrC and RloC [(54), Figure 6]. AbiI, a predicted ribonuclease H superfamily nuclease, has a similar potential. Several membrane ABI systems often cause the membrane leakage similarly to Type I TA systems (129,130). Several ABI systems including AbiU1, AbiL and AbiR are often associated and might interact with R-M systems (5,131). Finally, there is a strong link with mobile elements through the reverse transcriptase domain of AbiA and AbiK proteins (132), although, unlike typical reverse transcriptase, AbiK catalyses non-templated synthesis of random sequence DNA that remains covalently attached to the protein and contributes to ABI (133).
Figure 6.

Examples of genomic loci encoding different immunity systems and containing HEPN and PD-(D/E)xK domains. The genes are depicted as colored block arrows. The HEPN domain is shown by a light green shape with a red outline. The PD-(D/E)xK (RecB-like) domain is shown by a yellow shape with a red outline. HEPN, higher eukaryotes and prokaryotes nucleotide-binding domain, predicted endoribonuclease (54); Sir2, ParB and PD-(D/E)xK, DEDD are nucleases from distinct superfamilies. CRISPR-Cas gene names follow the nomenclature and classification from (18); R-M names follow the nomenclature and classification from (38). (A) HEPN domain associations. (B) PD-(D/E)xK domain associations.
The ∼30 currently known ABI systems come from only two model organisms, suggesting that they represent only a minor fraction of the total diversity of this type of defense modules in bacteria and archaea. Indeed, the analysis of selected defense islands reveals numerous uncharacterized gene families that could be candidates for ABI-like defense systems (5).
IMMUNITY-DORMANCY/SUICIDE COUPLING HYPOTHESIS
As aforementioned, at the deepest level, all archaeal and bacterial defense systems can be classified into two major groups that function on two contrasting principles: (i) immune systems that discriminate self DNA from non-self DNA and specifically destroy the foreign, in particular viral, genomes, whereas the host genome is protected and (ii) systems that induce dormancy or programmed cell suicide in response to infection. Most of the genomic loci that encode immunity systems such as CRISPR-Cas, R-M, DND or Pgl also encompass genes that encode toxins, in particular nucleases implicated in the induction of dormancy or cell death (Figure 6). The most common among these immunity-associated toxins are HEPN domain-containing (predicted) nucleases (Figure 6). In contrast, the immunity loci do not seem to encode antitoxins, at least not those from well-characterized antitoxin families. So far, there is no indication that the toxins are mechanistically involved in the immune functions. Hence, the immunity-dormancy/suicide coupling hypothesis, which posits that antivirus response in prokaryotes involves decision-making steps at which the cell chooses the path to follow by sensing the course of virus infection (55).
According to the coupling hypothesis, the toxins associated with immune systems induce dormancy or cell suicide unless controlled by components of the respective immunity system that act as antitoxins. This type of coupling is illustrated by the activity of the E. coli anticodon nuclease PrrC that interacts with the PrrI R-M system. The coupling of diverse immunity and dormancy/suicide systems in prokaryotes could have evolved under selective pressure to provide robustness to the antivirus response. It can be further proposed that the involvement of dormancy/suicide systems in the coupled antivirus response could take two distinct forms: (i) induction of a dormancy-like state in the infected cell to ‘buy time’ for the activation of adaptive immunity and (ii) dormancy or suicide as the final recourse to prevent viral spread triggered by the failure of immunity.
The first route is likely to realize in the activity of Cas2, a protein that is present in all CRISPR-Cas systems, essential for adaptive immunity and homologous to toxin interferases. Conceivably, this mechanism switches on when the CRISPR-Cas system encounters a new virus so that Cas1 protein has to detect and insert a new spacer. The dormancy-like response through the action of Cas2 and/or a COG1517 protein containing an effector domain, of which the most common are the HEPN and the PD-(D/E)xK (RecB-like) family nuclease, would prevent virus reproduction allowing the host the time required to prime the immunity response, which could be a relatively slow and ineffective process. The same reasoning could apply to other self–non-self discrimination systems if their action is slower than the action of viral phage counter-defenses blocking the immunity response. The second coupling mode is more straightforward. When an immunity system fails and/or the level of genotoxic stress increases, the cell uses the associated toxins for abrogation of key cell processes, typically translation, resulting in persistence or cell death. The cell suicide in such a case can be considered altruistic, i.e. preventing infection of other bacteria or archaea within the same colony or community.
Although multiple associations of (predicted) toxins with prokaryotic immune systems have been observed (Figure 6), it seems likely that many more members of known toxin families as well as novel toxins remain to be identified within immune system loci. Indeed, many of the toxins are highly diverged, small proteins and could be easily overlooked, especially when they are fused to larger proteins as distinct domains (5,27). Finally, in trans interactions between immunity systems and TA modules cannot be ruled out.
The coupling hypothesis might apply not only to antivirus defense systems but more generally to any stress response systems, mimicking the hypothetical functions ascribed to TA systems. For example, recently described bactericidal system (134), polymorphic virulence systems (58) and Ter-dependent chemical stress response system (135) are linked with various nucleases that are likely to possess toxin properties. Finally, it cannot be ruled out that some of the genes associated with immune systems perform functions different from the induction of dormancy or programmed cell death, such as repair of the DNA, RNA or even protein damage that is incurred during the action of the immunity systems.
This immunity-dormancy/suicide coupling hypothesis implies many experimentally testable predictions. In particular, it can be predicted that Cas2 protein present in all CRISPR-Cas operons is an mRNA-cleaving nuclease (interferase) that is activated at an early stage of virus infection to enable incorporation of virus-specific spacers into the CRISPR locus or to trigger cell suicide when the immune function of CRISPR-Cas systems fails. Similarly, toxin-like activity is predicted for components of numerous other defense loci.
CONCLUDING REMARKS
Defense mechanisms in bacteria, in particular R-M systems and TA systems, have been known for decades. However, recent comparative genomic analysis followed by experimental testing of the predictions has vastly expanded the scope of defense systems in prokaryotes. This expansion is both quantitative, including the discovery of diverse R-M and TA systems, and qualitative when fundamentally new defense mechanisms are discovered as was the case with the DND, Pgl and especially CRISPR-Cas. Given that genes encoding components of defense systems often evolve fast, that many of these genes encode small proteins and that the available genomes only represent a small fraction of the actual bacterial and archaeal diversity, there is little doubt that numerous defense systems, probably more than already known, remain to be discovered. Moreover, some of these findings have the potential to reveal new classes of defense mechanisms as suggested, for example, by the prediction of the pAgo-centred defense system(s) that remain to be experimentally characterized.
The prevalence of different defense systems in bacterial and archaeal taxa shows pronounced trends, with four large groups of organisms being readily distinguishable with respect to the overall abundance of defense systems and the prevalence of specific types of defense. Although understanding of some of these trends, such as the over-representation of CRISPR-Cas in hyperthermophiles, is starting to develop, the biological relevance of most aspects of the phyletic distribution of defense systems remains to be discovered.
Statistical analysis of the localization of genes encoding defense system components in bacterial and archaeal genomes shows highly significant clustering in defense islands. Although the evolution of defense islands remains to be investigated in details, in general, they seem to emerge through a preferential attachment mechanism in genome regions characterized by high rate of recombination and relaxed selection for the maintenance of local synteny. Although in itself the formation of defense islands is likely to be a non-adaptive, essentially neutral process, the islands become a ‘playground’ for rapid evolution and shuffling of genes and domains of the defense systems. Furthermore, defense islands, in addition to known defense systems, contain numerous uncharacterized genes that can be considered candidates for the discovery of new defense mechanisms.
The tight genomic association of immunity systems and the defense systems that induce dormancy or cell death suggests that these two major types of defense systems are often functionally coupled. Such coupling could manifest in cell death being triggered when the primary immunity mechanism fails or in the persistence state being forced potentially providing conditions for more effective and less damaging action of the immune systems. Which of these mechanisms is realized under what conditions and how do the defense decisions depend on various factors remains to be studied. All the immune systems that act on the self–non-self discrimination principle possess at least one component (such as RE) that can act as a toxin so that the entire system causes cell death or persistence instead of immunity. One example of such conversion, where a R-M system becomes a TA system, has been experimentally studied (136).
The versatility of the defense systems is to a large extent supported by the combinatorial shuffling of their constituents. The prime case in point is the two-component TA systems that form a strongly connected network owing to the fact that the same toxin family typically combines with more than one antitoxin family and vice versa. Furthermore, the distinction between TA and ABI systems is starting to fade away. A more appropriate view of these systems should focus on toxins that are activated or inactivated by numerous different signals encoded either in cis or in trans. Thus, substantial revisions of the definitions and classification of these defense systems appear inevitable.
Although the approaches for comparative genomic prediction and further experimental analysis of bacterial and archaeal defense systems have substantially advanced during the past few years, the study of viral counter-defense mechanisms is in its embryonic stage, despite the extensive experimental evidence that such systems are numerous and could either be generic or specifically target distinct host defense systems. For example, RNA ligase encoded by phage T4 can repair tRNAs cleaved by PrrC ACNase in E. coli (51), the Dmd protein of bacteriophage T4 functions as an antitoxin against E.coli LsoA and RnlA (137,138) and a short RNA gene from bacteriophage PhiTE functions as antitoxin to ToxIN system (139).
The recent advances in the study of bacterial and archaeal defense systems are uncovering the remarkable complexity of prokaryotic evolution that is in large part shaped by the virus-host arms race. Moreover, the newly discovered defense systems might eventually lead to breakthroughs in biotechnology that could be comparable with that brought about by the discovery of the R-M systems.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4.
FUNDING
Intramural funds of the US Department of Health and Human Services (to National Library of Medicine). Funding for open access charge: Intramural funds of the US Department of Health and Human Services (to National Library of Medicine).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Stern A, Sorek R. The phage-host arms race: shaping the evolution of microbes. Bioessays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koonin EV, Wolf YI. Evolution of microbes and viruses: a paradigm shift in evolutionary biology? Front. Cell. Infect. Microbiol. 2012;2:119. doi: 10.3389/fcimb.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forterre P, Prangishvili D. The great billion-year war between ribosome- and capsid-encoding organisms (cells and viruses) as the major source of evolutionary novelties. Ann. NY Acad. Sci. 2009;1178:65–77. doi: 10.1111/j.1749-6632.2009.04993.x. [DOI] [PubMed] [Google Scholar]
- 4.Haaber J, Samson JE, Labrie SJ, Campanacci V, Cambillau C, Moineau S, Hammer K. Lactococcal abortive infection protein AbiV interacts directly with the phage protein SaV and prevents translation of phage proteins. Appl. Environ. Microbiol. 2010;76:7085–7092. doi: 10.1128/AEM.00093-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS, Wolf YI, Snir S, Koonin EV. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011;193:6039–6056. doi: 10.1128/JB.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng Y, Posfai J, Morgan RD, Vincze T, Roberts RJ. Using shotgun sequence data to find active restriction enzyme genes. Nucleic Acids Res. 2009;37:e1. doi: 10.1093/nar/gkn883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, Luisi BF, Salmond GP. Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012;40:6158–6173. doi: 10.1093/nar/gks231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu T, Yao F, Zhou X, Deng Z, You D. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010;38:7133–7141. doi: 10.1093/nar/gkq610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovall RA, Matthews BW. Type II restriction endonucleases: structural, functional and evolutionary relationships. Curr. Opin. Chem. Biol. 1999;3:578–583. doi: 10.1016/s1367-5931(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 10.Williams RJ. Restriction endonucleases: classification, properties, and applications. Mol. Biotechnol. 2003;23:225–243. doi: 10.1385/mb:23:3:225. [DOI] [PubMed] [Google Scholar]
- 11.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:3552–3569. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He X, Ou HY, Yu Q, Zhou X, Wu J, Liang J, Zhang W, Rajakumar K, Deng Z. Analysis of a genomic island housing genes for DNA S-modification system in Streptomyces lividans 66 and its counterparts in other distantly related bacteria. Mol. Microbiol. 2007;65:1034–1048. doi: 10.1111/j.1365-2958.2007.05846.x. [DOI] [PubMed] [Google Scholar]
- 13.Liang J, Wang Z, He X, Li J, Zhou X, Deng Z. DNA modification by sulfur: analysis of the sequence recognition specificity surrounding the modification sites. Nucleic Acids Res. 2007;35:2944–2954. doi: 10.1093/nar/gkm176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrangou R, Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annu. Rev. Food Sci. Technol. 2012;3:143–162. doi: 10.1146/annurev-food-022811-101134. [DOI] [PubMed] [Google Scholar]
- 15.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 16.Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol. Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Syed MA, Levesque CM. Chromosomal bacterial type II toxin-antitoxin systems. Can. J. Microbiol. 2012;58:553–562. doi: 10.1139/w2012-025. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi Y, Inouye M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011;9:779–790. doi: 10.1038/nrmicro2651. [DOI] [PubMed] [Google Scholar]
- 21.Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 22.Chopin MC, Chopin A, Bidnenko E. Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 2005;8:473–479. doi: 10.1016/j.mib.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. Bacterial persistence by RNA endonucleases. Proc. Natl Acad. Sci. USA. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Tashiro Y, Kawata K, Taniuchi A, Kakinuma K, May T, Okabe S. RelE-mediated dormancy is enhanced at high cell density in Escherichia coli. J. Bacteriol. 2012;194:1169–1176. doi: 10.1128/JB.06628-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 26.Hayes F, Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 27.Makarova KS, Wolf YI, Koonin EV. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009;4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blower TR, Salmond GP, Luisi BF. Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 2011;21:109–118. doi: 10.1016/j.sbi.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012;8:855–861. doi: 10.1038/nchembio.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masuda H, Tan Q, Awano N, Wu KP, Inouye M. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 2012;84:979–989. doi: 10.1111/j.1365-2958.2012.08068.x. [DOI] [PubMed] [Google Scholar]
- 31.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl Acad. Sci. USA. 2009;106:894–899. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a dataset via the gap statistic. J. R. Stat. Soc. B. 2001;63:411–423. [Google Scholar]
- 33.Khan F, Furuta Y, Kawai M, Kaminska KH, Ishikawa K, Bujnicki JM, Kobayashi I. A putative mobile genetic element carrying a novel type IIF restriction-modification system (PluTI) Nucleic Acids Res. 2010;38:3019–3030. doi: 10.1093/nar/gkp1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi N, Ohashi S, Sadykov MR, Mizutani-Ui Y, Kobayashi I. IS-linked movement of a restriction-modification system. PLoS One. 2011;6:e16554. doi: 10.1371/journal.pone.0016554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009;33:376–393. doi: 10.1111/j.1574-6976.2008.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuta Y, Abe K, Kobayashi I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010;38:2428–2443. doi: 10.1093/nar/gkp1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35:D269–D270. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson GG, Murray NE. Restriction and modification systems. Annu. Rev. Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 40.Wilson GG. Organization of restriction-modification systems. Nucleic Acids Res. 1991;19:2539–2566. doi: 10.1093/nar/19.10.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005;8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Ershova AS, Karyagina AS, Vasiliev MO, Lyashchuk AM, Lunin VG, Spirin SA, Alexeevski AV. Solitary restriction endonucleases in prokaryotic genomes. Nucleic Acids Res. 2012;40:10107–10115. doi: 10.1093/nar/gks853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dryden DT, Murray NE, Rao DN. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 2001;29:3728–3741. doi: 10.1093/nar/29.18.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourniquel AA, Bickle TA. Complex restriction enzymes: NTP-driven molecular motors. Biochimie. 2002;84:1047–1059. doi: 10.1016/s0300-9084(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 46.Szczelkun MD. Roles for Helicases as ATP-Dependent Molecular Switches. Adv. Exp. Med. Biol. 2013;767:225–244. doi: 10.1007/978-1-4614-5037-5_11. [DOI] [PubMed] [Google Scholar]
- 47.Raghavendra NK, Bheemanaik S, Rao DN. Mechanistic insights into type III restriction enzymes. Front. Biosci. 2012;17:1094–1107. doi: 10.2741/3975. [DOI] [PubMed] [Google Scholar]
- 48.Raleigh EA. Organization and function of the mcrBC genes of Escherichia coli K-12. Mol. Microbiol. 1992;6:1079–1086. doi: 10.1111/j.1365-2958.1992.tb01546.x. [DOI] [PubMed] [Google Scholar]
- 49.Penner M, Morad I, Snyder L, Kaufmann G. Phage T4-coded Stp: double-edged effector of coupled DNA and tRNA-restriction systems. J. Mol. Biol. 1995;249:857–868. doi: 10.1006/jmbi.1995.0343. [DOI] [PubMed] [Google Scholar]
- 50.Kaufmann G. Anticodon nucleases. Trends Biochem. Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 51.Uzan M, Miller ES. Post-transcriptional control by bacteriophage T4: mRNA decay and inhibition of translation initiation. Virol. J. 2010;7:360. doi: 10.1186/1743-422X-7-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klaiman D, Steinfels-Kohn E, Krutkina E, Davidov E, Kaufmann G. The wobble nucleotide-excising anticodon nuclease RloC is governed by the zinc-hook and DNA-dependent ATPase of its Rad50-like region. Nucleic Acids Res. 2012;40:8568–8578. doi: 10.1093/nar/gks593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidov E, Kaufmann G. RloC: a wobble nucleotide-excising and zinc-responsive bacterial tRNase. Mol. Microbiol. 2008;69:1560–1574. doi: 10.1111/j.1365-2958.2008.06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. HEPN: a major nucleic-acid targeting domain involved in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 2013 doi: 10.1186/1745-6150-8-15. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makarova KS, Anantharaman V, Aravind L, Koonin EV. Live virus-free or die: coupling of antivirus immunity and programmed suicide or dormancy in prokaryotes. Biol. Direct. 2012;7:40. doi: 10.1186/1745-6150-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.You D, Wang L, Yao F, Zhou X, Deng Z. A novel DNA modification by sulfur: DndA is a NifS-like cysteine desulfurase capable of assembling DndC as an iron-sulfur cluster protein in Streptomyces lividans. Biochemistry. 2007;46:6126–6133. doi: 10.1021/bi602615k. [DOI] [PubMed] [Google Scholar]
- 57.Chen F, Zhang Z, Lin K, Qian T, Zhang Y, You D, He X, Wang Z, Liang J, Deng Z, et al. Crystal structure of the cysteine desulfurase DndA from Streptomyces lividans which is involved in DNA phosphorothioation. PLoS One. 2012;7:e36635. doi: 10.1371/journal.pone.0036635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iyer LM, Abhiman S, Aravind L. MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol. Direct. 2008;3:8. doi: 10.1186/1745-6150-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zalatan JG, Fenn TD, Herschlag D. Comparative enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J. Mol. Biol. 2008;384:1174–1189. doi: 10.1016/j.jmb.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chinenova TA, Mkrtumian NM, Lomovskaia ND. Genetic characteristics of a new phage resistance trait in Streptomyces coelicolor A3(2) [in Russian] Genetika. 1982;18:1945–1952. [PubMed] [Google Scholar]
- 62.Sumby P, Smith MC. Genetics of the phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2) Mol. Microbiol. 2002;44:489–500. doi: 10.1046/j.1365-2958.2002.02896.x. [DOI] [PubMed] [Google Scholar]
- 63.Miller WG, Pearson BM, Wells JM, Parker CT, Kapitonov VV, Mandrell RE. Diversity within the Campylobacter jejuni type I restriction-modification loci. Microbiology. 2005;151:337–351. doi: 10.1099/mic.0.27327-0. [DOI] [PubMed] [Google Scholar]
- 64.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 66.Koonin EV, Makarova KS. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol. Rep. 2009;1:95. doi: 10.3410/B1-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 68.Koonin EV, Wolf YI. Is evolution Darwinian or/and Lamarckian? Biol. Direct. 2009;4:42. doi: 10.1186/1745-6150-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 70.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, de Romans S, Lim C, Roux C, Passet V, et al. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One. 2012;7:e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe. 2012;12:177–186. doi: 10.1016/j.chom.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 72.Carroll D. A CRISPR approach to gene targeting. Mol. Ther. 2012;20:1658–1660. doi: 10.1038/mt.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 2012;30:1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 74.Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–209. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 75.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. The CRISPRs, they are a-Changin': how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 76.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 77.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takeuchi N, Wolf YI, Makarova KS, Koonin EV. Nature and Intensity of Selection Pressure on CRISPR-Associated Genes. J. Bacteriol. 2012;194:1216–1225. doi: 10.1128/JB.06521-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wiedenheft B, Zhou K, Jinek M, Coyle SM, Ma W, Doudna JA. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure. 2009;17:904–912. doi: 10.1016/j.str.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 82.Carte J, Pfister NT, Compton MM, Terns RM, Terns MP. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010;16:2181–2188. doi: 10.1261/rna.2230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, Ke A. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure. 2012;20:1574–1584. doi: 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 87.van Duijn E, Barbu IM, Barendregt A, Jore MM, Wiedenheft B, Lundgren M, Westra ER, Brouns SJ, Doudna JA, van der Oost J, et al. Native tandem and ion mobility mass spectrometry highlight structural and modular similarities in clustered-regularly-interspaced shot-palindromic-repeats (CRISPR)-associated protein complexes From Escherichia coli and Pseudomonas aeruginosa. Mol. Cell. Proteomics. 2012;11:1430–1441. doi: 10.1074/mcp.M112.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30:1335–1342. doi: 10.1038/emboj.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deng L, Kenchappa CS, Peng X, She Q, Garrett RA. Modulation of CRISPR locus transcription by the repeat-binding protein Cbp1 in Sulfolobus. Nucleic Acids Res. 2012;40:2470–2480. doi: 10.1093/nar/gkr1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S, Reimann J, Cannone G, Liu H, Albers SV, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol. Cell. 2012;45:303–313. doi: 10.1016/j.molcel.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mulepati S, Bailey S. Structural and biochemical analysis of the nuclease domain of the clustered regularly interspaced short palindromic repeat (CRISPR) associated protein 3(CAS3) J. Biol. Chem. 2011;286:31896–31903. doi: 10.1074/jbc.M111.270017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu X, Ye K. Crystal structure of Cmr2 suggests a nucleotide cyclase-related enzyme in type III CRISPR-Cas systems. FEBS Lett. 2012;586:939–945. doi: 10.1016/j.febslet.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 97.Cocozaki AI, Ramia NF, Shao Y, Hale CR, Terns RM, Terns MP, Li H. Structure of the Cmr2 subunit of the CRISPR-Cas RNA silencing complex. Structure. 2012;20:545–553. doi: 10.1016/j.str.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Makarova KS, Wolf YI, Koonin EV. Potential genomic determinants of hyperthermophily. Trends Genet. 2003;19:172–176. doi: 10.1016/S0168-9525(03)00047-7. [DOI] [PubMed] [Google Scholar]
- 99.Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV. Viral diversity threshold for adaptive immunity in prokaryotes. MBio. 2012;3:e00456–12. doi: 10.1128/mBio.00456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Makarova KS, Wolf YI, van der Oost J, Koonin EV. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol. Direct. 2009;4:29. doi: 10.1186/1745-6150-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008;456:209–213. doi: 10.1038/nature07315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yuan YR, Pei Y, Ma JB, Kuryavyi V, Zhadina M, Meister G, Chen HY, Dauter Z, Tuschl T, Patel DJ. Crystal structure of A. aeolicus argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Mol. Cell. 2005;19:405–419. doi: 10.1016/j.molcel.2005.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl Acad. Sci. USA. 1986;83:3116–3120. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ogura T, Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl Acad. Sci. USA. 1983;80:4784–4788. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buts L, Lah J, Dao-Thi MH, Wyns L, Loris R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005;30:672–679. doi: 10.1016/j.tibs.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 106.Van Melderen L, Saavedra De Bast M. Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet. 2009;5:e1000437. doi: 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gerdes K, Wagner EG. RNA antitoxins. Curr. Opin. Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 108.Fozo EM, Hemm MR, Storz G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008;72:579–589, Table of Contents. doi: 10.1128/MMBR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Magnuson RD. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 2007;189:6089–6092. doi: 10.1128/JB.00958-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 111.Gerdes K, Maisonneuve E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012;66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 112.Christensen-Dalsgaard M, Overgaard M, Winther KS, Gerdes K. RNA decay by messenger RNA interferases. Methods Enzymol. 2008;447:521–535. doi: 10.1016/S0076-6879(08)02225-8. [DOI] [PubMed] [Google Scholar]
- 113.Yamaguchi Y, Inouye M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009;85:467–500. doi: 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- 114.Fozo EM, Makarova KS, Shabalina SA, Yutin N, Koonin EV, Storz G. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010;38:3743–3759. doi: 10.1093/nar/gkq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ogura T, Niki H, Mori H, Morita M, Hasegawa M, Ichinose C, Hiraga S. Identification and characterization of gyrB mutants of Escherichia coli that are defective in partitioning of mini-F plasmids. J. Bacteriol. 1990;172:1562–1568. doi: 10.1128/jb.172.3.1562-1568.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tan Q, Awano N, Inouye M. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol. Microbiol. 2011;79:109–118. doi: 10.1111/j.1365-2958.2010.07433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kimelman A, Levy A, Sberro H, Kidron S, Leavitt A, Amitai G, Yoder-Himes DR, Wurtzel O, Zhu Y, Rubin EM, et al. A vast collection of microbial genes that are toxic to bacteria. Genome Res. 2012;22:802–809. doi: 10.1101/gr.133850.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM. Genome-wide experimental determination of barriers to horizontal gene transfer. Science. 2007;318:1449–1452. doi: 10.1126/science.1147112. [DOI] [PubMed] [Google Scholar]
- 120.Guglielmini J, Van Melderen L. Bacterial toxin-antitoxin systems: translation inhibitors everywhere. Mob. Genet. Elements. 2011;1:283–290. doi: 10.4161/mge.18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murphy KC. The lambda Gam protein inhibits RecBCD binding to dsDNA ends. J. Mol. Biol. 2007;371:19–24. doi: 10.1016/j.jmb.2007.05.085. [DOI] [PubMed] [Google Scholar]
- 122.Scaltriti E, Moineau S, Launay H, Masson JY, Rivetti C, Ramoni R, Campanacci V, Tegoni M, Cambillau C. Deciphering the function of lactococcal phage ul36 Sak domains. J. Struct. Biol. 2010;170:462–469. doi: 10.1016/j.jsb.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 123.O'Connor L, Tangney M, Fitzgerald GF. Expression, regulation, and mode of action of the AbiG abortive infection system of lactococcus lactis subsp. cremoris UC653. Appl. Environ. Microbiol. 1999;65:330–335. doi: 10.1128/aem.65.1.330-335.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garvey P, Fitzgerald GF, Hill C. Cloning and DNA sequence analysis of two abortive infection phage resistance determinants from the lactococcal plasmid pNP40. Appl. Environ. Microbiol. 1995;61:4321–4328. doi: 10.1128/aem.61.12.4321-4328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vujicic-Zagar A, Dulermo R, Le Gorrec M, Vannier F, Servant P, Sommer S, de Groot A, Serre L. Crystal structure of the IrrE protein, a central regulator of DNA damage repair in deinococcaceae. J. Mol. Biol. 2009;386:704–716. doi: 10.1016/j.jmb.2008.12.062. [DOI] [PubMed] [Google Scholar]
- 126.Earl AM, Mohundro MM, Mian IS, Battista JR. The IrrE protein of Deinococcus radiodurans R1 is a novel regulator of recA expression. J. Bacteriol. 2002;184:6216–6224. doi: 10.1128/JB.184.22.6216-6224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sevin EW, Barloy-Hubler F. RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 2007;8:R155. doi: 10.1186/gb-2007-8-8-r155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cheng X, Wang W, Molineux IJ. F exclusion of bacteriophage T7 occurs at the cell membrane. Virology. 2004;326:340–352. doi: 10.1016/j.virol.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 130.Parma DH, Snyder M, Sobolevski S, Nawroz M, Brody E, Gold L. The Rex system of bacteriophage lambda: tolerance and altruistic cell death. Genes Dev. 1992;6:497–510. doi: 10.1101/gad.6.3.497. [DOI] [PubMed] [Google Scholar]
- 131.Aravind L, Anantharaman V, Zhang D, de Souza RF, Iyer LM. Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front. Cell. Infect. Microbiol. 2012;2:89. doi: 10.3389/fcimb.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kojima KK, Kanehisa M. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol. Biol. Evol. 2008;25:1395–1404. doi: 10.1093/molbev/msn081. [DOI] [PubMed] [Google Scholar]
- 133.Wang C, Villion M, Semper C, Coros C, Moineau S, Zimmerly S. A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids Res. 2011;39:7620–7629. doi: 10.1093/nar/gkr397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang D, Iyer LM, Aravind L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011;39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Anantharaman V, Iyer LM, Aravind L. Ter-dependent stress response systems: novel pathways related to metal sensing, production of a nucleoside-like metabolite, and DNA-processing. Mol. Biosyst. 2012;8:3142–3165. doi: 10.1039/c2mb25239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ishikawa K, Fukuda E, Kobayashi I. Conflicts targeting epigenetic systems and their resolution by cell death: novel concepts for methyl-specific and other restriction systems. DNA Res. 2010;17:325–342. doi: 10.1093/dnares/dsq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Short FL, Blower TR, Salmond GP. A promiscuous antitoxin of bacteriophage T4 ensures successful viral replication. Mol. Microbiol. 2012;83:665–668. doi: 10.1111/j.1365-2958.2012.07974.x. [DOI] [PubMed] [Google Scholar]
- 138.Otsuka Y, Yonesaki T. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol. Microbiol. 2012;83:669–681. doi: 10.1111/j.1365-2958.2012.07975.x. [DOI] [PubMed] [Google Scholar]
- 139.Blower TR, Evans TJ, Przybilski R, Fineran PC, Salmond GP. Viral evasion of a bacterial suicide system by RNA-Based molecular mimicry enables infectious altruism. PLoS Genet. 2012;8:e1003023. doi: 10.1371/journal.pgen.1003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.