

Abstract
Complexes 1-OH and 1-F are related complexes that share similar [X–FeIII–O–FeIV=O]3+ core structures with a total spin S of ½, which arises from antiferromagnetic coupling of an S = 5/2 FeIII-X site and an S = 2 FeIV=O site. EXAFS analysis shows that 1-F has a nearly linear FeIII–O–FeIV core compared to that of 1-OH, which has an Fe–O–Fe angle of ~130° due to the presence of a hydrogen bond between the hydroxo and oxo groups. Both complexes are at least a thousand-fold more reactive at C–H bond cleavage than 2, a related complex with a [OH–FeIV–O–FeIV=O]4+ core having individual S = 1 FeIV units. Interestingly, 1-F is tenfold more reactive than 1-OH. This raises an interesting question about what gives rise to the reactivity difference. DFT calculations comparing 1-OH and 1-F strongly suggest that the H-bond in 1-OH does not significantly change the electrophilicity of the reactive FeIV=O unit and that the lower reactivity of 1-OH arises from the additional activation barrier required to break its H-bond in the course of H-atom transfer by the oxoiron(IV) moiety.
Introduction
Non-heme diiron enzymes catalyze the activation of dioxygen to cleave the C–H bonds of a variety of substrates. This class includes soluble methane monooxygenase (sMMO), related bacterial multi-component monooxygenases, and fatty acid desaturases.1–6 High-valent intermediates are implicated in the oxygen activation mechanisms for these enzymes.3,5–7 For example, intermediate Q of sMMO is a two-electron oxidant that effects the hydroxylation of methane8–14 and has been proposed to have an [FeIV2(μ-O)2] diamond core on the basis of extended x-ray absorption fine structure (EXAFS) studies.15 Related diiron(IV) oxidants may also be involved in the catalytic cycles of fatty acid desaturases and other diiron monooxygenases resulting from cleavage of the O-O bond in observed peroxo intermediates,16–18 but direct evidence for such diiron(IV) species has not yet been obtained. Similar oxygen activation chemistry is utilized by ribonucleotide reductases (RNR) with diiron and iron-manganese centers, which generate an intermediate called X that is used for the one-electron oxidation of a specific Cys residue that is needed to initiate the deoxygenation of ribonucleotides to deoxyribonucleotides.19 For these enzymes, FeIII–O–MIV (M = Fe or Mn) oxidants have been trapped,20–22 and the best structurally characterized is the intermediate for the RNR from Chlamydia tranchomatis, which has been shown to have an [FeIIIMnIV(μ-O)(μ-OH)] diamond core on the basis of Fe and Mn K-edge EXAFS experiments and associated density functional theory (DFT) calculations.22
In our effort to obtain synthetic analogs of such high-valent diiron species, we have characterized the first examples of complexes with [FeIIIFeIV(μ-O)2]23 (1 in Scheme 1) and [FeIV2(μ-O)2]24 core structures, providing synthetic precedents for the [FeIV2(μ-O)2] core proposed for Q.15 More recently, we reported the generation of 1-OH, a complex with an open HO–FeIII–O–FeIV=O core structure (Scheme 2), by the addition of hydroxide to 1 or by the one-electron reduction of its HO–FeIV–O–FeIV=O precursor 2 (Scheme 1).25 As EXAFS characterization of 2 shows an Fe-Fe distance of 3.32 Å and an Fe–O–Fe angle of 130°, it is clear that the Fe–O–Fe unit is bent,26 implicating a hydrogen bonding interaction between the hydroxo proton on one Fe to the oxo on the other Fe. By extension, 1-OH is also proposed to have such an H-bond. Indeed direct spectroscopic evidence for the hydrogen bond in 1-OH has recently been obtained by 1H-ENDOR experiments.27
Scheme 1.
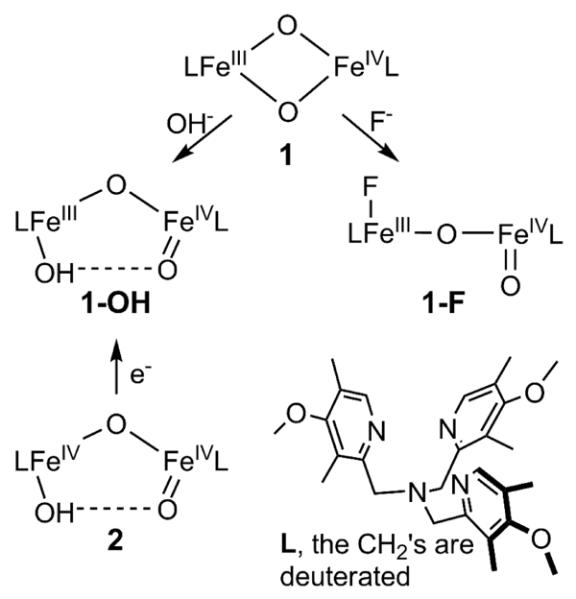
Structures of high-valent diiron complexes in this study.
Scheme 2.
In this paper, we compare the reactivities of 1-OH and 1-F, another open-core complex derived from the addition of F− to 1, and find that 1-F is an order of magnitude more reactive than 1-OH in both H-atom abstraction and oxo-atom transfer. Unlike the hydroxo ligand in 1-OH, the iron(III)-bound fluoride should not be capable of hydrogen bonding to the terminal FeIV=O unit, so the difference in reactivity may be related to the presence of the hydrogen bond. Indeed EXAFS analysis establishes the presence of a linear Fe–O–Fe unit in 1-F. DFT calculations have been carried out on both 1-OH and 1-F to shed further light on this reactivity behavior.
Experimental and Computational Details
Complexes 1 and 2 were prepared according to reported procedures.24,26 9,10-dihydroanthracene (DHA, 97%), Fluorene (>99%) and ferrocene (Fc, 98%) purchased from Aldrich were recrystallized (from EtOH for the former two and MeOH for the latter one) prior to use. Butyronitrile (PrCN, 99%+) purchased from Aldrich was purified and dried according to reported procedures.28 9,9,10,10-d4-DHA was synthesized according to reported procedures.25 Tetrabutylammonium fluoride hydrate (Bu4NF.xH2O) purchased from Aldrich (98%) was dried under vacuum at 40 °C.29 Anhydrous dichloromethane (>99.8%) and acetonitrile (>99.8%) purchased from Aldrich were used without further treatment. Bu4NOCD3 was prepared according to reported procedures.30 UV-vis spectra and kinetic time traces were recorded on a Hewlett-Packard 8453A diode array spectrometer equipped with a cryostat from Unisoku Scientific Instruments, Osaka, Japan. This combination allows kinetic studies to be performed at temperatures down to −85 °C and to record a spectrum (in the range of 190 to 1100 nm) every 0.1 second. For some rapid reactions with a reaction time of 10 seconds, time traces at one wavelength can be obtained with about 100 data points for reliable kinetic fits (See Figures 1). 31P-NMR data were collected on a Varian VXR-300 spectrometer.
Figure 1.
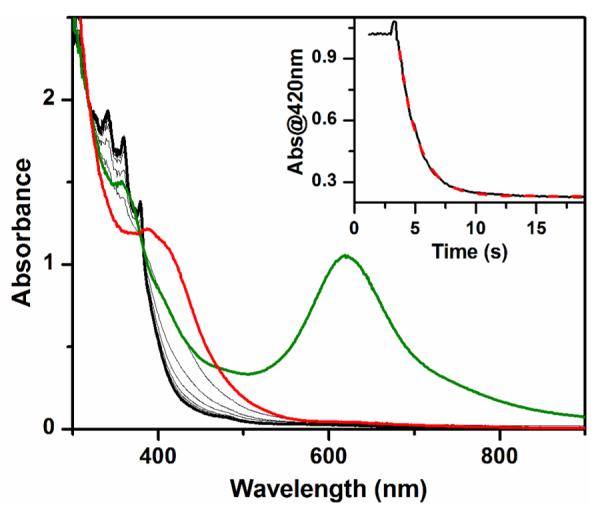
UV-vis spectra of complexes 1 (green line, 0.2 mM) and 1-F (red line) obtained upon addition of Bu4NF to the solution of 1. Additional gray lines show the spectroscopic changes during the reaction of 1-F with 2.0 mM DHA until the reaction is complete (black line). Inset: time trace at 420 nm (black solid line) together with the fit (red dashed line) using a first-order model. Conditions: in 3:1 CH2Cl2-MeCN under Ar at −80 °C.
Reactivity studies
All reactivity measurements were performed in a 3:1 mixture of CH2Cl2-MeCN under Ar to allow measurements to be made at −85 °C. A solution of 1-F was prepared by addition of 1.5 equivalents Bu4NF to 1 (typically 0.2 mM), while a solution of 1-OH was prepared by reduction of 2 (typically 0.2 mM) with 1 equivalent ferrocene. For a typical reactivity experiment, an appropriate amount of substrate (from a stock solution in CH2Cl2) was introduced to the solution of the complex via a micro syringe, and the reaction solution was monitored by UV-vis spectroscopy. The pseudo-first-order rate constants kobs were obtained by fitting the decay time traces and the second-order rate constants k2 were obtained by fitting the kobs versus substrate-concentration plots. The reaction solutions were filtered through silica gel columns to remove iron complexes prior to product analyses. The yield of the anthracene product from DHA oxidation was quantified by the absorbance of the filtrates at 377 nm (ε = 7700 M−1 cm−1).
XAS study
Fe K-edge X-ray absorption spectra (XAS, fluorescence excitation, Ge detector) of a frozen solution of 1-F were recorded at ~10 K at the Stanford Synchrotron Radiation Laboratory (SSRL). The solution contained 3 mM of diiron species in 3:1 PrCN-MeCN, with 75% yield (2.2 mM) of 1-F based on Mössbauer analysis. The energy range was 6.9 to 8.0 keV. The monochromator was calibrated using the K-edge energy of iron foil at 7112.0 eV. The program EXAFSPAK31 was used for evaluation of the data and for EXAFS fitting, the latter in conjunction with FEFF 8.32 SSExafs33,34 was used for fitting of the pre-edge region of XAS spectra.
Computational studies
All calculations were performed with ORCA program package.35 For geometry optimizations, the pure BP8636 and hybrid B3LYP density functionals37,38 in combination with triple-ζquality basis sets (TZVP)39 for key surrounding atoms involved in C-H bond activation and SVP basis sets40 for the remaining atoms were used throughout the study. The resolution of the identity41–43 (RI, for BP86) and RI plus chain of spheres44 (RIJCOSX, for B3LYP) approximations were used to accelerate the calculations using the auxiliary basis set SV/J.42 All the geometries were full optimized without symmetry constraints. Harmonic vibrational frequencies were computed by two-sided numerical differentiation of analytic gradients to verify the nature of the stationary points. The minimum structures reported in this paper have only positive eigenvalues of the Hessian matrix, whereas the transition states (TSs) have only one negative eigenvalue. The zero-point energies, thermal corrections and entropy terms for the optimized geometries were obtained from the frequency calculations.
In order to obtain single-point energies closer to the basis set limit, B3LYP calculations with the much larger def2-TZVPP basis set45 on all elements were carried out. The energies reported in this paper refer to these calculations.
Solvent effects are taken into account via the conductor like screen model (COSMO) for all calculations. Acetonitrile (epsilon = 36.6) was chosen as the solvent. To consider dispersion forces, geometry optimizations and single point calculations were also undertaken included semiempirical van der Waals (VDW) corrections.46–48
Results and Discussion
Comparing the reactivities of 1-F and 1-OH
As shown in Figure 1, addition of 1.5 equiv. Bu4NF to a solution of 1 at −80 °C causes decay of its 620 nm chromophore (green line) and its complete conversion to a new species with λmax at about 400 nm (red line), which is characteristic of 1-F.49 Subsequent addition of 9,10-dihydroanthracene (DHA) to this mixture speeds up the decay of the 400-nm chromophore and forms near-UV absorption features characteristic of the anthracene product at 377 and 357 nm (black line). The anthracene yield is 35% with respect to 1 (and ~50% with respect to 1-F, based on its ~75% yield relative to 1 that has been estimated by Mössbauer analysis.49). The resulting solution is EPR silent, suggesting that a diiron(III) product is formed. Taken together, these results suggest that 1-F effectively acts as a one-electron oxidant in DHA oxidation. There is an isosbestic point at about 380 nm in the course of DHA oxidation, suggesting that this is a simple A-to-B reaction and no intermediate is involved. (In Figure 1, note that the red line, corresponding to the spectrum of 1-F right after Bu4NF addition, does not cross this isosbestic point, because subsequent addition of substrate solution results in sample dilution and a baseline shift. This perturbation is also indicated by the bump at the beginning of the absorption time trace shown in the inset of Figure 1.)
The progress of DHA oxidation can be monitored by following the decay of the absorption at 420 nm, and the time traces can be fit with a pseudo-first-order model to obtain kobs values (Figure 1, inset). As 1-F is a one-electron oxidant, two equivalents of 1-F are required to oxidize one molecule of DHA. Thus pseudo-first order conditions can be achieved even with a substrate/1-F ratio of 5, as less than 10% of the added substrate will have been consumed at the end of the reaction. Indeed excellent fits to a first order decay were obtained for all experiments represented in Figure 2. (We were constrained to use DHA concentrations of 2.0 mM or less, because the oxidation rate was too fast for higher DHA concentrations to be accurately measured by our UV-vis spectrometer.) The second order rate constant (k2) can then be obtained from the slope of the linear kobs-versus-[substrate] plot (Figure 2).
Figure 2.
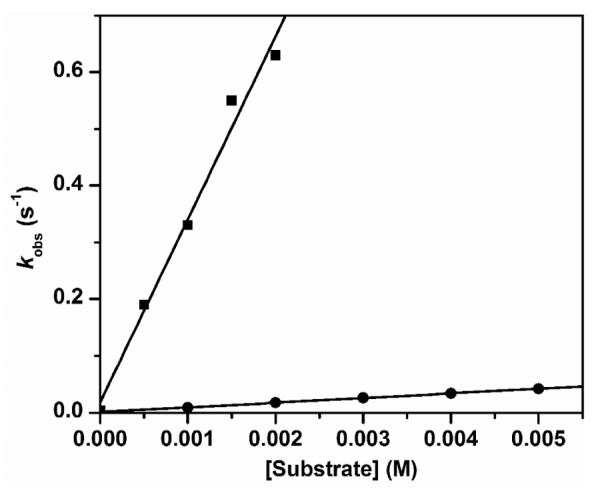
Plots of kobs vs [substrate] for the oxidation of DHA (squares) and DHA-d4 (circles) by 1-F, with k2 of 3.2(2) × 102 M−1 s−1 and 8.2(1) M−1 s−1, respectively. Conditions: in 3:1 CH2Cl2-MeCN under Ar at −80 °C. With one exception, all reactions were carried out with 1-F generated from 0.2 mM 1. For the data point corresponding to 0.5 mM DHA, 0.1 mM 1 was used to generate 1-F to maintain pseudo-first order conditions. Experimental constraints in our ability to measure the high rate of the DHA reaction limited how high a DHA concentration could be used.
The reaction of 1-F with DHA slows down significantly when 9,9,10,10-d4-dihydroanthracene (DHA-d4) is used as the substrate (Figure 2). The kinetic isotope effect (KIE) value of 40 at −80 °C is comparable to that observed for 1-OH (50 under the same conditions)25 and confirms that hydrogen atom transfer (HAT) is the major component of the rate determining step in DHA oxidation by 1-F and 1-OH. However, 1-F oxidizes DHA about 10-fold faster than 1-OH at −80 °C (see Table 1). The same difference in rates was observed at −85 °C (Figure S1). Similar reactivity differences are also observed for fluorene, another hydrocarbon substrate with a stronger C–H bond (BDE = 80 kcal/mol vs 78 kcal/mol for DHA;50 Table 1 and Figure S2). Under the same conditions, the reactivity of 1-F is quite comparable to that of 1-OCD3 (Table 1), a recently reported complex with a [CD3O–FeIII–O–FeIV=O]3+ core structure.30
Table 1.
Comparison of second order rate constants (M−1 s−1) for C–H bond cleavage reactions of high-valent diiron complexes.
Activation parameters for DHA oxidation by 1-OH
Figure 3 shows the Eyring plot for DHA oxidation by 1-OH in the temperature range of −85 °C to −40 °C. The activation parameters calculated from the plot are ΔH‡ = 5.1(4) kcal mol−1 and ΔS‡ = −26(2) cal mol−1 K−1. For comparison, the temperature dependence of the intramolecular oxidation of the OCH3 group in 1-OCH3 afforded Eyring parameters of ΔH‡ = 9.7 kcal mol−1 and ΔS‡ = −15 cal mol−1 K−1.30 The smaller ΔH‡ value for DHA oxidation by 1-OH reflects the lower activation barrier for cleaving the significantly weaker C–H bond of DHA, while its more negative ΔS‡ value is consistent with the intermolecular nature of the reaction. Unfortunately, the activation parameters for the reaction of 1-F with DHA could not be determined for comparison with those of 1-OH, because the reactions at higher temperature were too fast for us to measure.
Figure 3.
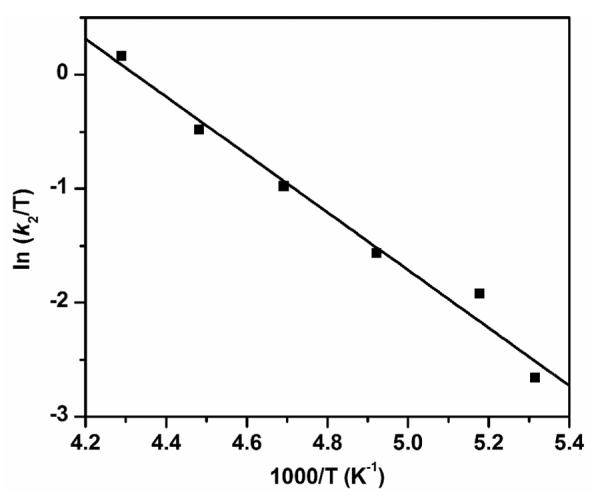
Eyring plot for DHA oxidation by 1-OH in 3:1 CH2Cl2-MeCN under Ar. ΔH‡ = 5.1(4) kcal mol−1, and ΔS‡ = −26(2) cal mol−1 K−1. The k2 values at different temperatures are listed in Table S1.
XAS Characterization of 1-F
The geometric structure of 1-F has been examined with X-ray absorption spectroscopy (XAS). As shown in Figure 4, the first derivative of the XAS intensity reveals two distinct edge energies at E0 = 7124.8 and 7128.6 eV, similar to what is found for its precursor 1 at 7124.8 and 7129.4 eV.24 The pre-edge region can be fit with three discernible features at 7113.8, 7115.5, and 7117.1 eV (Table S2), which are found at energies almost identical to those observed for synthetic mononuclear high-spin oxoiron(IV) complexes51,52 and provide further support for the assignment of an S = 2 spin state for the oxoiron(IV) moiety in 1-F. These pre-edge features have a total area of 24.8 units, which is within the range of values found for the synthetic mononuclear high-spin oxoiron(IV) complexes.51,52 The features associated with the six-coordinate high-spin iron(III) center of 1-F are expected to be much less intense53,54 and would thus be obscured by the more intense bands of the high-spin oxoiron(IV) unit.
Figure 4.
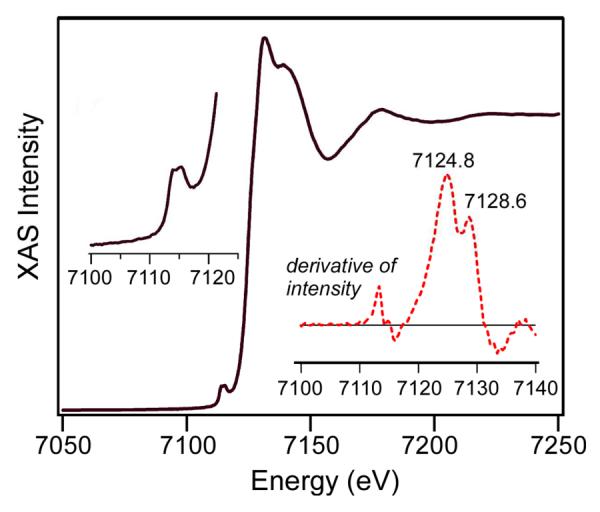
Black line: X-ray absorption near-edge structure (XANES) of 1-F (2.1 mM) in 3:1 PrCN:MeCN. Left inset: pre-edge features observed between 7100 and 7120 eV. Right inset (red dashed line): first derivative of the XAS spectrum in the 7100–7140 eV region. Two distinct edge energies are observed at E0 = 7124.8 and 7128.6 eV.
The Fourier transform (FT, r'-space) of the Fe K-edge EXAFS data collected for 1-F is shown in Figure 5. It displays intense features at r' = 1.7 and 3.2 Å, along with two smaller peaks in between (r' = r – ρ, where r is the actual metal-scatterer distance and ρ is a phase shift of ~0.4 Å). Initial analysis of the EXAFS data assumed that these spectral contributions were derived entirely from single-scattering mechanisms. The best fit obtained with this approach included three first-sphere shells at 1.80, 2.07, and 2.18 Å, each consisting of two O/N atoms. The shells with r > 2 Å correspond to the N atoms of ligand L, while the 1.80 Å shell is a conglomeration of the fluoride, terminal oxo, and bridging oxo ligands. Fitting of the second-sphere features required six C scatterers at 2.99 Å (standard for high-valent complexes with the tris(2-pyridylmethyl)amine-type (TPA) ligands24) and an Fe scatterer at 3.64 Å.
Figure 5.
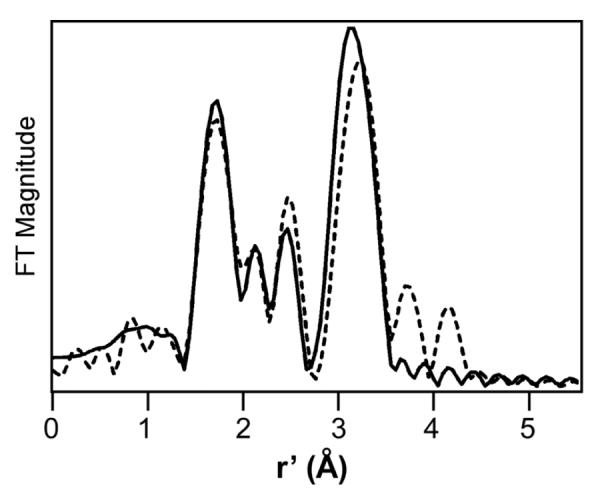
Fourier transform of Fe K-edge data (dashed line) for a 2.1 mM solution of 1-F in 3:1 PrCN:MeCN obtained at 10 K. Fourier-transform range: k = 2.1 – 14.5 Å−1. The best fit (solid line) was obtained with the following parameters: 0.5 O/N at 1.66 Å (Δσ2, 0.0084), 1.5 O/N at 1.83 Å (0.0049), 1 N/O at 2.04 Å (0.0012), 3 N/O at 2.17 (0.0021), 6.5 C at 3.04 Å (0.0085), and 1 Fe at 3.56 Å (0.0037). The fit also included a multiple-scattering feature arising from the nearly linear Fe-O-Fe unit.
While the single-scattering fit is satisfactory, it does not accurately reproduce the unusually high intensity of the Fe scatterer peak at 3.2 Å in the FT (Figure S3). This discrepancy was likely due to neglect of multiple-scattering effects in our first-order analysis, as multiple-scattering effects amplify the intensity of the distant scatterer in a linear triatomic array,55 such as the Fe–O-Fe unit in 1-F. Thus, the FEFF program was used to account for multiple-scattering intensity arising from the Fe–O–Fe unit. Our initial model, depicted in Figure S4, assumed six-coordinate metal centers with iron-ligand distances largely derived from our first-order EXAFS analysis. However, based on insights into the structure of 1-F from other methods, the first shell was split into two components: (i) an O scatterer at 1.65 Å (0.5 occupancy) corresponding to the terminal oxo ligand of the iron(IV) center, and (ii) an O/F scatterer at 1.80 Å (1.5 occupancy) corresponding to the fluoride ligand of the iron(III) ion and the μ-oxo group. As shown in Figure S5, the FEFF-calculated FT from this model nicely matches the experimental data, suggesting that multiple-scattering effects indeed make significant contributions to the EXAFS data. The distances and Debye-Waller factors (Δσ2) of all the scatterers derived from either single scattering or multiple scattering mechanisms were then allowed to vary (with certain constraints) to improve the correspondence between the experimental and computed data. This procedure yielded a high-quality fit that accounts for all salient experimental features, including the intensity of the Fe scatterer peak in the FT (Figure 5; see Figure S6 for the fit of the EXAFS data prior to Fourier transformation). Although most of the first-sphere bonds lengths were relatively unchanged, this second-order approach resulted in a modest shortening of the Fe⋯Fe distance from 3.64 to 3.56 Å. As complexes with linear FeIII–O–FeIII units typically have Fe⋯Fe distances of 3.6 Å,23,56,57 the somewhat shorter Fe⋯Fe distance observed for 1-F probably reflects the expected contraction of the FeIV–μ-O bond length.58 In contrast, the EXAFS analysis of 2, the one-electron oxidized diiron(IV) analog of 1-OH, has revealed an Fe⋯Fe distance of 3.32 Å,26 which is 0.24 Å shorter than that of 1-F and corresponds to an Fe–O–Fe angle of 130°. These geometric differences are proposed to result from the presence of an H-bond between the FeIV–O–H and the FeIV=O units. A similar Fe⋯Fe distance and a comparable Fe–O–Fe angle are observed in the crystal structure of a [H2O–FeIII–O–FeIII–OH] complex with a related 5-ethyl-substituted TPA supporting ligand, where H-bonding is observed between the OH− and H2O ligands.23 The r(Ohydroxo-Ooxo) of 2.46 Å in 2 predicted from DFT geometry optimization26 is also very similar that of the diferric complex (2.464 Å),23 further supporting the presence of H-bond. We speculate that the H-bond remains upon one-electron reduction of 2 to form 1-OH. This speculation is supported by the fact that irradiation of frozen solution of 2 at 77 K with 60Co, conditions under which only electron transfer can occur and no structural change is possible, also generate 1-OH.49
As previously determined by Mössbauer spectroscopy,49 the 1-F sample prepared for spectroscopic studies consists of about 75% 1-F with 22% associated with the diiron(III) decay product. The high fraction of 1-F in the XAS sample is supported by the presence of relatively intense pre-edge features associated with a high-spin oxoiron(IV) unit (see discussion above). While the diiron(III) decay product in the sample could potentially affect the EXAFS results, its presence is unlikely to alter the two major conclusions of our analysis: (i) the presence of an scatterer at 1.66 Å arising from an FeIV=O unit and (ii) the Fe-Fe bond distance of 3.56 Å. Regarding the scatterer at 1.66 Å, the diiron(III) contaminant would not possess such a short Fe-O bond, so we are confident that this feature arises from 1-F. With respect to the Fe-Fe distance, it is likely that the diiron(III) contaminant would also have a linear Fe–O–Fe unit and could thus contribute to the observed Fe scatterer at 3.56 Å. However, given that it only represents 22% of the sample, it is highly unlikely that the decay product would solely be responsible for the intense Fe contribution at 3.56 Å. In the best fit, the Fe scatterer has an N-value of 1.0 and a reasonable Δσ2 value of 0.0037. If we assume that the decay product alone gave rise to the observed Fe scatterer, the N value would have to be decreased to 0.22 and the associated Δσ2 value would become unreasonably small or perhaps even negative in value. Lastly, we emphasize that the EXAFS data can be well simulated using a DFT model of 1-F (Figure S5).
DFT calculations
DFT calculations were performed on 1-OH and 1-F in both syn and anti conformations as shown in Figure 6. For the syn conformer 1-OHsyn, the oxo and hydroxo groups are linked by hydrogen bonding, which enforces an Fe–O–Fe angle about 130°. Documented below are the results obtained with B3LYP functionals; very similar results were obtained with BP86 functionals, which are presented in Table S3. The optimized structure of 1-OHsyn (shown in Figure 6) features an open [HO–FeIII–O–FeIV=O]2+ core structure, similar to that of its one-electron oxidized diiron(IV) analog.26 The calculated O1–H1 bond distance (1.78 Å) and the O1⋯O3 separation (2.73 Å) clearly indicate the presence of a weak hydrogen bond between the terminal oxo and hydroxyl groups, consistent with the van der Waals radii of the H- (1.20 Å) and O-atoms (1.52 Å).59 Formation of the hydrogen bond is facilitated by the proximity of the Fe=O and Fe–OH units. Consequently, 1-OHsyn has a relatively short Fe-Fe distance of 3.34 Å and a bent Fe1–O2–Fe2 angle of 131.9°. For comparison, 1-OHanti is only about 2.4 kcal/mol higher in energy than 1-OHsyn, reflecting the H-bond strength in 1-OHsyn is rather weak. Due to the loss of the H-bond, 1-OHanti adopts a nearly linear Fe1O2Fe2 arrangement with an Fe–O–Fe angle of 174.5° and a longer Fe⋯Fe distance of 3.62 Å. The results of these calculations are fully consistent with the earlier DFT results of De Hont et al.49 (Figure S7) and with recent 1H-ENDOR data that provides direct spectroscopic evidence of the H-bond.27
Figure 6.
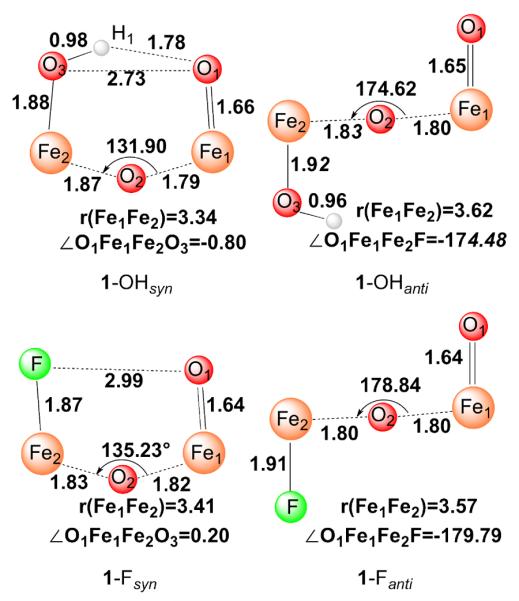
Optimized core structures of 1-OH and 1-F. Bond lengths are in angstroms, angles in degrees. Atom color scheme: H, white; O, red; Fe, orange; F, green.
Like 1-OH, two conformations were taken into account for 1-F computationally, both conformers represent local minima in the calculations. However, the hypothetical syn conformer 1-Fsyn, which most closely resembles 1-OHsyn, is computed to be 5.2 kcal/mol higher in energy than the anti conformer, 1-Fanti. Thus, in agreement with experiment, the calculations confirm that the anti configuration is energetically more favored. Our calculated key geometric parameters for 1-Fanti agree with the results from previous DFT calculations (Figure S7)49 and match the experimental EXAFS data (Figure 5) reasonably well. For both conformers, the calculated FeIV-oxo bond length of 1.64 Å is comparable to those found for 1-OHsyn (1.66 Å) and 1-OHanti (1.65 Å). 1-Fanti features a linear Fe1O2Fe2 arrangement with a long Fe1⋯Fe2 distance of 3.57 Å, similar to that calculated for 1-OHanti. The absence of the hydrogen bond in 1-Fsyn results in a loose “pocket” as evidenced by the rather long F⋯O1 separation (2.99 Å) and the slightly larger Fe1–O2–Fe2 angle (135.2°) compared to that of 1-OHsyn. In analogy to 1-OHsyn, 1-Fsyn possesses a bent Fe1O2Fe2 core and hence displays a shorter Fe1⋯Fe2 distance relative to 1-Fanti (Figure 6).
As shown in Table 1, 1-Fanti is found to be 10-fold faster than 1-OHsyn in cleaving C–H bonds, which in turn is a thousand-fold faster than 2, its one-electron oxidized form.25 To gain insight into the factors that may contribute to this difference, the reactivities of 1-OHsyn and 1-Fanti were theoretically modeled, focusing on the rate-determining H-atom abstraction step. Table 2 lists selected structural parameters from the B3LYP-optimized geometries, and Figure 7 shows the Gibbs free energy profiles for DHA C–H bond activation by 1-OHsyn and 1-Fanti.60 As shown in Figure 7 and Figure S8, the energy barrier for H-atom abstraction by 1-OHsyn is calculated to be 3.3 kcal/mol at the B3LYP + VDW level of theory, 0.8 kcal/mol higher than that calculated for 1-Fanti (2.5 kcal/mol). This barrier difference corresponds to a ratio of 6 between the reaction rates for 1-Fanti and 1-OHsyn, in good agreement with the 10-fold rate enhancement observed experimentally. This factor increases to 7, when the hydrogen-tunneling correction due to Wigner61 is included. The DFT results nicely reproduce the experimental findings, although the calculations without VDW corrections may slightly overestimate the barrier difference. Because there is a large error in the calculated entropy contribution to the free energy (> 10 kcal/mol) for a given combination reaction, especially in solution,62,63 i.e. approach of the substrate towards the reactive center in the present case, we chose the reaction complex (RC), where the substrate weakly bonds to the FeIV=O site, as the reference point to calculate the reaction barrier. With this caveat, the direct comparison of the computed activation barrier with the experimental data requires more caution.
Table 2.
Selected geometric parameters obtained from the calculations including VDW corrections for the key points along the reaction pathways. (Values without VDW corrections are given in parentheses.)
| r(Fe1-O1) | r(Fe1-Fe2) | ∠Fe1O2Fe2(°) | ∠O1Fe1Fe2X (°) | r(O1-O3) | r(O1-H1) | r(O1-H2) | r(C-H2) | |
|---|---|---|---|---|---|---|---|---|
| RC(1-OHsyn) | 1.66(1.66) | 3.34(3.37) | 133.34(134.04) | 2.59(0.02) | 2.73(2.74) | 1.79(1.80) | ~ | 1.10(1.10) |
| TSH(1-OHsyn) | 1.73(1.75) | 3.33(3.38) | 131.90(134.31) | −5.55(−15.53) | 2.77(2.84) | 1.82(1.89) | 1.40(1.40) | 1.20(1.21) |
| IN(1-OHsyn) | 1.88(1.89) | 3.33(3.39) | 132.25(135.71) | 1.42(0.96) | 2.75(2.79) | 1.80(1.84) | 0.96(0.96) | 2.41(2.59) |
| RC(1-Fanti) | 1.64(1.64) | 3.49(3.54) | 152.69 (156.80) | −134.62 (−135.03) | ~ | ~ | 2.31(2.42) | 1.10(1.10) |
| TSH(1-Fanti) | 1.72(1.73) | 3.59(3.63) | 172.79(175.92) | 173.44(172.99) | ~ | ~ | 1.39(1.41) | 1.20(1.20) |
| IN(1-Fanti) | 1.86(1.88) | 3.60(3.62) | 167.86(166.38) | 175.29(170.65) | ~ | ~ | 0.96(0.96) | 2.51(3.12) |
| RC(1-OHanti) | 1.65(1.65) | 3.54(3.60) | 163.41(166.68) | −145.95(−143.79) | ~ | ~ | 2.10(2.50) | 1.10(1.10) |
| TSH(1-OHanti) | 1.72(1.73) | 3.59(3.64) | 176.02(178.25) | −172.54(172.92) | ~ | ~ | 1.38(1.40) | 1.21(1.22) |
| IN(1-OHanti) | 1.88(1.87) | 3.63(3.64) | 174.06(174.13) | −178.27(178.94) | ~ | ~ | 0.96(0.96) | 2.58(3.04) |
| r(Fe1-O1) | r(Fe1-Fe2) | ∠Fe1O2Fe2(°) | ∠O1Fe1Fe2X (°) | r(O1-O3) | r(O1-H1) | r(O1-H2) | r(C-H2) | |
|---|---|---|---|---|---|---|---|---|
| RC(1-OHsyn) | 1.66(1.66) | 3.34(3.37) | 133.34(134.04) | 2.59(0.02) | 2.73(2.74) | 1.79(1.80) | ~ | 1.10(1.10) |
| TSH(1-OHsyn) | 1.73(1.75) | 3.33(3.38) | 131.90(134.31) | −5.55(−15.53) | 2.77(2.84) | 1.82(1.89) | 1.40(1.40) | 1.20(1.21) |
| IN(1-OHsyn) | 1.88(1.89) | 3.33(3.39) | 132.25(135.71) | 1.42(0.96) | 2.75(2.79) | 1.80(1.84) | 0.96(0.96) | 2.41(2.59) |
| RC(1-Fanti) | 1.64(1.64) | 3.49(3.54) | 152.69(156.80) | −134.62 (−135.03) | ~ | ~ | 2.31(2.42) | 1.10(1.10) |
| TSH(1-Fanti) | 1.72(1.73) | 3.59(3.63) | 172.79(175.92) | 173.44(172.99) | ~ | ~ | 1.39(1.41) | 1.20(1.20) |
| IN(1-Fanti) | 1.86(1.88) | 3.60(3.62) | 167.86(166.38) | 175.29(170.65) | ~ | ~ | 0.96(0.96) | 2.51(3.12) |
| RC(1-OHanti) | 1.65(1.65) | 3.54(3.60) | 163.41(166.68) | −145.95(-143.79) | ~ | ~ | 2.10(2.50) | 1.10(1.10) |
| TSH(1-OHanti) | 1.72(1.73) | 3.59(3.64) | 176.02(178.25) | −172.54(172.92) | ~ | ~ | 1.38(1.40) | 1.21(1.22) |
| IN(1-OHanti) | 1.88(1.87) | 3.63(3.64) | 174.06(174.13) | −178.27(178.94) | ~ | ~ | 0.96(0.96) | 2.58(3.04) |
Figure 7.
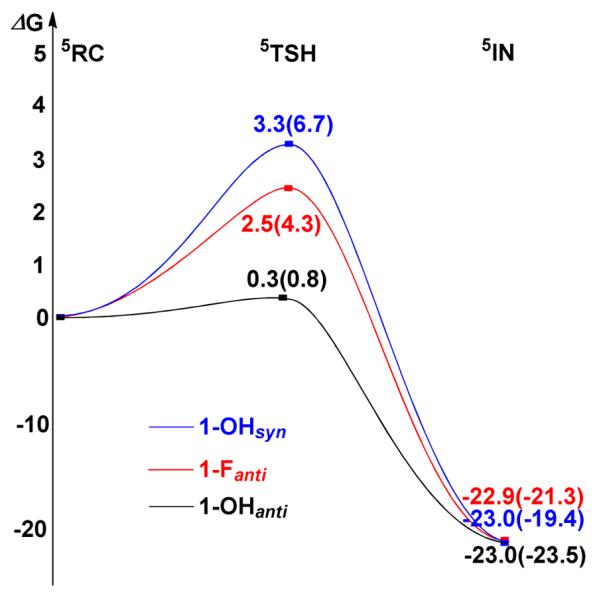
Calculated schematic Gibbs free energy (ΔG) surfaces for the cleavage of the C–H bond of DHA by 1-OHsyn (blue line), 1-Fanti (red line), and 1-OHanti (black line). Energies without VDW corrections are given in parentheses.
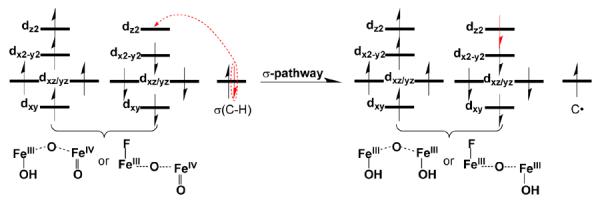
In fact, the process of C–H bond oxidation by the two complexes follows the same reaction mechanism. As expected, the reaction takes place at the FeIV=O unit and proceeds by the σ-mechanism that has been well established for S = 2 mononuclear oxoiron(IV) complexes.64–68 In the σ-pathway of C–H bond activation by a high-spin FeIV=O center, one electron from the substrate that has the same spin as the remaining electrons in the FeIV center is transferred into the σ*(Fe=O) antibonding orbital (Scheme 2). During this process, the oxidation state of the diiron core changes from a mixed valence FeIII-FeIV state in the RCs to an FeIII-FeIII state in the transition states (TSH) and the intermediates (IN). As the two complexes are supported by the same ligand and, more importantly, share similar open core [X–FeIII–O–FeIV=O]3+ structures, the main question is why 1-Fanti exhibits a stronger oxidizing ability than 1-OHsyn.
We considered several factors that could account for the increase in reactivity upon going from 1-OHsyn to 1-Fanti. The first factor might be the changes in the electronic properties of the FeIV=O reactive center that is directly involved in the reaction. As shown in Table 2, the estimated Fe=O bond distance in 1-OHsyn is marginally longer than that in 1-Fanti. Moreover, the calculated Fe=O bond order of 1.7 for 1-OHsyn is slightly lower than that for 1-Fanti (1.8), which is consistent with the computed Fe=O stretching frequencies (834 cm−1 for 1-OHsyn vs. 867 cm−1 for 1-Fanti). (Unfortunately we were unsuccessful in our attempt to obtain resonance Raman data for these complexes that could have experimentally substantiated these calculated changes.) Therefore, the hydrogen bond does not noticeably change the bonding properties of the FeIV=O motif in 1-OHsyn. As such, one may predict that the FeIV=O sites in 1-OHsyn and 1-Fanti would exhibit similar reactivity. In line with this reasoning, nearly identical Fe1–O1, C–H2 and O1–H2 bond distances were found in TSH(1-OHsyn) and TSH(1-Fanti) (Table 2).
The second factor for the increased reactivity of 1-Fanti over 1-OHsyn could be the different steric barriers encountered in the two systems. In fact, as shown in Figure 8, the reaction center (terminal oxo) in 1-Fanti is partially shielded by the pyridine group that is oriented syn with respect to the terminal oxo group. Therefore, it is easier for the substrate to approach the reactive center in 1-OHsyn than in 1-Fanti. Consequently, we would expect higher reactivity of 1-OHsyn compared to that of 1-Fanti from such an analysis. However, experiment demonstrated the opposite trend for the reactivity of the two complexes. Thus, the underlying reason for this intriguing reactivity difference must lie elsewhere.
Figure 8.
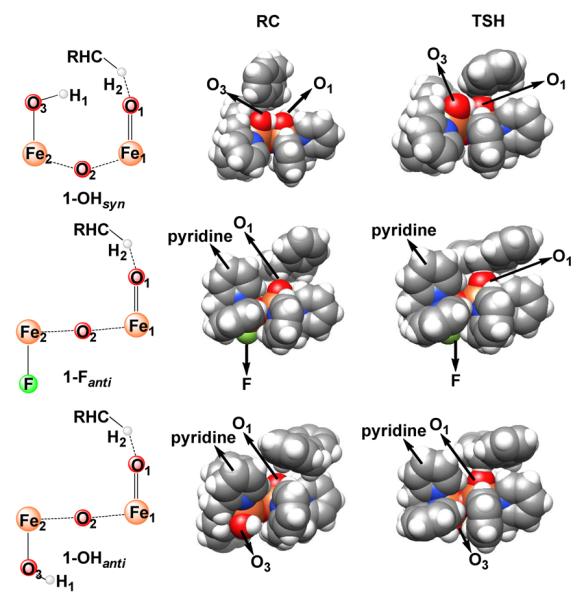
Space-filling models of reaction complexes (RCs) and transition states (TSHs) for 1-Fanti, 1-OHsyn and 1-OHanti. Atom color scheme: C, gray; H, white; N, blue; O, red; Fe, orange; F, green.
In the H-atom abstraction process by mononuclear iron-oxo complexes, the key reaction coordinates are the lengthening of the target C–H bond of the substrate and the FeIV=O bond of the oxidant.67,69,70 Indeed, for the reaction with 1-Fanti, we have not observed any other coordinates that undergo significant changes en route to the transition state. Interestingly, an additional reaction coordinate was identified in the reaction with 1-OHsyn. This motion involves lengthening of the hydrogen bond between the hydroxide and terminal oxo groups. As shown in Table 2, comparison of the structures of RC(1-OHsyn) and TSH(1-OHsyn) clearly demonstrates weakening of the hydrogen bond during the reaction process, especially for the calculations without VDW correction. This is readily ascribed to the changes in the electronic structure of the FeIV=O unit along the reaction coordinate. As discussed elsewhere, as the Fe=O bond lengthens, the FeIV=O intermediate evolves to a species that is best characterized as FeIII–oxyl.64,71 The lengthening of the hydrogen bond (O1–H1) in TSH(1-OHsyn) reflects the fact that the oxyl group has lower electron donating capability relative to the more negatively charged oxo ligand. Thus, the larger geometric distortion resulting from this additional reaction coordinate leads to a higher barrier for the reaction with 1-OHsyn compared to 1-Fanti. To corroborate this notion, we have theoretically investigated the same reaction with the anti conformer 1-OHanti. It turns out that 1-OHanti is more efficient in C–H bond activation than 1-OHsyn and 1-Fanti (Figure 7), although the computed Fe1–O1 and C–H2 distances in TSH(1-OHanti) are almost the same as those found for 1-OHsyn and 1-Fanti (Table 2). This indicates that partially breaking the hydrogen bond indeed slows down the reaction for 1-OHsyn and explains the higher energy barrier encountered by 1-OHsyn than that for 1-Fanti. The difference in reactivity between the two anti conformers mainly originates from the larger geometry reorganization required for the reaction with 1-Fanti relative to 1-OHanti upon going from RC to TSH, as indicated by geometric parameters such as the Fe1-Fe2 distances, the Fe1-O1-Fe2 angles and the O1-Fe1-Fe2-X dihedral angels (Table 2). Our calculations show that the change in the reorganization energies gives ~1 kcal/mol difference in the reaction barriers.
Taken together, our calculations reveal that the hydrogen bond between the oxo and hydroxo group in 1-OHsyn does not significantly change the bonding properties of the FeIV=O unit and hence its reactivity. However, during the reaction of C–H bond oxidation, this hydrogen bond has to be partially broken. This leads to the slightly higher barrier for 1-OHsyn relative to 1-Fanti, which has a similar open-core structure but no hydrogen bond.
Concluding Remarks
Complexes 1-OH and 1-F are related complexes that are supported by the same tetradentate tripodal ligand and, more importantly, share similar [X–FeIII–O–FeIV=O]3+ core structures. They both have a high-spin (S = 2) terminal FeIV=O moiety based on EPR and Mössbauer analysis.30,49 They are also much more reactive at cleaving C–H bonds than 2, the one-electron more oxidized precursor of 1-OH. Complex 2 differs from 1-OH and 1-F in having an S = 1 FeIV=O unit, which supports the DFT-derived hypothesis that a high-spin oxoiron(IV) center is more reactive than an intermediate-spin one due to exchange-enhanced reactivity. H-atom abstraction by an S = 2 FeIV=O unit would introduce an α electron into the empty dz2(σ*) orbital, leading to an increase in the number of exchange interactions, while H-atom abstraction by an S = 1 FeIV=O unit would likely introduce a β electron into a dxz/yz(π*) orbital, leading to a decrease in the number of exchange interactions. This difference leads to a much lower activation barrier for this key step of the reaction in the case of the S = 2 FeIV=O unit.
Interestingly, the C–H bond cleavage reactivity of 1-F is tenfold higher than that of 1-OH. Based on DFT calculations, we attribute this reactivity difference to the distinct core structures of 1-F and 1-OH. In conjunction with an earlier DFT study,27,49 a recent ENDOR study experimentally demonstrated that there is a hydrogen bond between the FeIII–OH and the FeIV=O units of 1-OH, resulting in a bent Fe-O-Fe angle of ~130° and a shorter Fe⋯Fe distance of ~3.3 Å.27,49 In this paper, we demonstrate by EXAFS analysis that 1-F has a Fe⋯Fe distance of 3.56 Å and consequently a nearly linear Fe-O-Fe angle due to the absence of a hydrogen bond. The presence of the H-bond in 1-OH may be responsible for attenuating the H-atom abstracting capability of 1-OH. However the present DFT calculations comparing 1-OH and 1-F strongly suggest that the H-bond in 1-OH does not significantly change the electrophilicity of the reactive FeIV=O unit but instead increases the activation barrier for C–H bond cleavage by requiring the weakening of the H-bond in the course of HAT by the oxoiron(IV) moiety. This study thus sheds light on how Nature might employ hydrogen bonding to modulate the reactivities of oxoiron(IV) intermediates in the active sites of various dioxygen activating iron enzymes.
Supplementary Material
Acknowledgements
The work at Minnesota was supported by US National Institutes of Health via grant GM-38767 to L.Q. and postdoctoral fellowship GM-079839 to A.T.F. C.-Y.G. gratefully acknowledges a grant from China Scholarship Council (CSC). C.-Y.G., S.Y. and F.N. gratefully acknowledge financial support by the German Science Foundation (DFG), the University of Bonn and the Max Planck Society. XAS data were collected on beamline 7-3 at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, and Biomedical Technology Program.
References
- (1).Wallar BJ, Lipscomb JD. Chem. Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- (2).Kurtz DM., Jr. J. Biol. Inorg. Chem. 1997;2:159–167. [Google Scholar]
- (3).Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem. Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- (4).Fox BG, Lyle KS, Rogge CE. Acc. Chem. Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- (5).Murray LJ, Lippard SJ. Acc. Chem. Res. 2007;40:466–474. doi: 10.1021/ar600040e. [DOI] [PubMed] [Google Scholar]
- (6).Tinberg CE, Lippard SJ. Acc. Chem. Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Groves JT. J. Inorg. Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- (8).Lee S-K, Fox BG, Froland WA, Lipscomb JD, Münck E. J. Am. Chem. Soc. 1993;115:6450–6451. [Google Scholar]
- (9).Lee S-K, Nesheim JC, Lipscomb JD. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- (10).Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J. Am. Chem. Soc. 1995;117:10174–10185. [Google Scholar]
- (11).Brazeau BJ, Lipscomb JD. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- (12).Brazeau BJ, Wallar BJ, Lipscomb JD. J. Am. Chem. Soc. 2001;123:10421–10422. doi: 10.1021/ja016632i. [DOI] [PubMed] [Google Scholar]
- (13).Brazeau BJ, Austin RN, Tarr C, Groves JT, Lipscomb JD. J. Am. Chem. Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- (14).Baik M-H, Newcomb M, Friesner RA, Lippard SJ. Chem. Rev. 2003;103:2385–2420. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- (15).Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- (16).Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- (17).Murray LJ, Naik SG, Ortillo DO, García-Serres R, Lee JK, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2007;129:14500–14510. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L., Jr. Proc. Natl. Acad. Sci. USA. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nordlund P, Reichard P. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- (20).Bollinger JM, Jr., Krebs C. J. Inorg. Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- (21).Bollinger JM, Jr., Jiang W, Green MT, Krebs C. Curr. Opin. Struct. Biol. 2008;18:650–657. doi: 10.1016/j.sbi.2008.11.007. [DOI] [PubMed] [Google Scholar]
- (22).Younker JM, Krest CM, Jiang W, Krebs C, Bollinger JM, Jr., Green MT. J. Am. Chem. Soc. 2008;130:15022–15027. doi: 10.1021/ja804365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dong Y, Fujii H, Hendrich MP, Leising RA, Pan G, Randall CR, Wilkinson EC, Zang Y, Que L, Jr., Fox BG, Kauffmann K, Münck E. J. Am. Chem. Soc. 1995;117:2778–2792. [Google Scholar]
- (24).Xue G, Wang D, De Hont R, Fiedler AT, Shan X, Münck E, Que L., Jr. Proc. Natl. Acad. Sci., USA. 2007;104:20713–20718. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Xue G, De Hont R, Münck E, Que L., Jr. Nat. Chem. 2010;2:400–405. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xue G, Fiedler AT, Martinho M, Münck E, Que L., Jr. Proc. Natl. Acad. Sci., USA. 2008;105:20615–20620. [Google Scholar]
- (27).Shanmugam M, Xue G, Que L, Jr., Hoffman BM. Inorg. Chem. 2012;51:10080–10082. doi: 10.1021/ic3015783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. Butterworth-Heinemann; Oxford: 1997. [Google Scholar]
- (29).Cox DP, Terpinski J, Lawrynowicz W. J. Org. Chem. 1984;49:3216–3219. [Google Scholar]
- (30).Xue G, Pokutsa A, Que L., Jr. J. Am. Chem. Soc. 2011;133:16657–16667. doi: 10.1021/ja207131g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).George GN, Pickering IJ. Stanford Synchrotron Radiation Laboratory. Stanford Linear Accelerator Center; Stanford, California: 2000. [Google Scholar]
- (32).Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. J. Am. Chem. Soc. 1991;113:5135–5140. [Google Scholar]
- (33).Scarrow RC, Maroney MJ, Palmer SM, Que L., Jr., Roe AL, Salowe SP, Stubbe J. J. Am. Chem. Soc. 1987;109:7857–7864. [Google Scholar]
- (34).Scarrow RC, Trimitsis MG, Buck CP, Grove GN, Cowling RA, Nelson MJ. Biochemistry. 1994;33:15023–15035. doi: 10.1021/bi00254a011. [DOI] [PubMed] [Google Scholar]
- (35).Neese F. An Ab Initio, Density Functional and Semiempirical Program Package. Version 2.8 ed. Bonn University; Germany: 2010. [Google Scholar]
- (36).Becke AD. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- (37).Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- (38).Lee CT, Yang WT, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- (39).Schafer A, Huber C, Ahlrichs R. J. Chem. Phys. 1994;100:5829–5835. [Google Scholar]
- (40).Schafer A, Horn H, Ahlrichs R. J. Chem. Phys. 1992;97:2571–2577. [Google Scholar]
- (41).Kendall RA, Fruchtl HA. Theor. Chem. Acc. 1997;97:158–163. [Google Scholar]
- (42).Eichkorn K, Treutler O, Ohm H, Haser M, Ahlrichs R. Chem. Phys. Lett. 1995;240:283–289. [Google Scholar]
- (43).Eichkorn K, Weigend F, Treutler O, Ahlrichs R. Theor. Chem. Acc. 1997;97:119–124. [Google Scholar]
- (44).Neese F, Wennmohs F, Hansen A, Becker U. Chem. Phys. 2009;356:98–109. [Google Scholar]
- (45).Weigend F, Haser M, Patzelt H, Ahlrichs R. Chem. Phys. Lett. 1998;294:143–152. [Google Scholar]
- (46).Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132 doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- (47).Grimme S. J. Comput. Chem. 2004;25:1463–1473. doi: 10.1002/jcc.20078. [DOI] [PubMed] [Google Scholar]
- (48).Grimme S. J. Comput. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- (49).De Hont RF, Xue G, Hendrich MP, Que L, Jr., Bominaar EL, Münck E. Inorg. Chem. 2010;49:8310–8322. doi: 10.1021/ic100870v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- (51).England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L., Jr. Angew. Chem. Int. Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).England J, Guo Y, Van Heuvelen KM, Cranswick MA, Rohde GT, Bominaar EL, Münck E, Que L., Jr. J. Am. Chem. Soc. 2011;133:11880–11883. doi: 10.1021/ja2040909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L., Jr. J. Am. Chem. Soc. 1984;106:1676–1681. [Google Scholar]
- (54).Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1997;119:6297–6314. [Google Scholar]
- (55).Que L Jr., editor. Physical Methods in Bioinorganic Chemistry. Spectroscopy and Magnetism. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- (56).Norman RE, Holz RC, Ménage S, O'Connor CJ, Zhang JH, Que J, L. Inorg. Chem. 1990;29:4629–4637. [Google Scholar]
- (57).Kurtz DM., Jr. Chem. Rev. 1990;90:585–606. [Google Scholar]
- (58).Ghosh A, Tiago de Oliveira F, Yano T, Nishioka T, Beach ES, Kinoshita I, Münck E, Ryabov AD, Horwitz CP, Collins TJ. J. Am. Chem. Soc. 2005;127:2505–2513. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]
- (59).Bondi A. J. Phys. Chem. 1964;68:441. [Google Scholar]
- (60).The DHA H-abstraction by 1-Fsyn proved to be problematic, since several hydrogen atoms of the substrate are susceptible to the attraction of the F atom, forming hydrogen bonds between them. Therefore, we cannot directly compare the HAT reactivity of 1-Fsyn with the other systems because these hydrogen bonds may exert an influence on its reactivity.
- (61).Wigner E. Phys. Rev. 1932;40:749–759. [Google Scholar]
- (62).Wirstam M, Lippard SJ, Friesner RA. J. Am. Chem. Soc. 2003;125:3980–3987. doi: 10.1021/ja017692r. [DOI] [PubMed] [Google Scholar]
- (63).Ye S, Riplinger C, Hansen A, Krebs C, Bollinger JM, Jr., Neese F. Chem. Eur. J. 2012;18:6555–6567. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI. Proc. Natl. Acad. Sci. USA. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bernasconi L, Louwerse MJ, Baerends EJ. Eur. J. Inorg. Chem. 2007:3023–3033. [Google Scholar]
- (66).Latifi R, Bagherzadeh M, de Visser SP. Chem. Eur. J. 2009;15:6651–6662. doi: 10.1002/chem.200900211. [DOI] [PubMed] [Google Scholar]
- (67).Geng CY, Ye S, Neese F. Angew. Chem. Int. Ed. 2010;49:5717–5720. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- (68).Janardanan D, Usharani D, Chen H, Shaik S. J. Phys. Chem. Lett. 2011;2:2610–2617. [Google Scholar]
- (69).Hirao H, Kumar D, Que L, Jr., Shaik S. J. Am. Chem. Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- (70).Kumar D, Hirao H, Que L, Jr., Shaik S. J. Am. Chem. Soc. 2005;127:8026–8027. doi: 10.1021/ja0512428. [DOI] [PubMed] [Google Scholar]
- (71).Ye SF, Neese F. Proc. Natl. Acad. Sci. USA. 2011;108:1228–1233. doi: 10.1073/pnas.1008411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.