

Abstract
HIV enters the brain during the early stages of initial infection and can result in a complicated array of diverse neurological dysfunctions. While neuronal injury and loss are at the heart of neurological decline and HIV-associated neuropathology, HIV does not productively infect neurons and the effects of HIV on neurons may be described as largely indirect. Viral proteins released from infected cells in the CNS are a well-characterized source of neuronal toxicity. Likewise, host-derived inflammatory cytokines and chemokines released from infected and/or activated glial cells can damage neurons, as well. Newly identified host–virus interactions and the current state of our knowledge regarding HIV-associated neuronal toxicity will be addressed in this review. Aspects of HIV-associated neurotoxic mechanisms, patterns of neuronal damage, viral effects on neurotrophic signaling, clade variations and comorbid substance abuse will be discussed. Recent advances in our understanding of the impact of HIV infection of the CNS on neuronal dysfunction and cell death will also be highlighted.
Keywords: blood–brain barrier, cART, CNS, excitotoxicity, HIV-associated neurocognitive disorders, inflammation, neurons, synaptodendritic
HIV infection of the CNS
HIV infects a subset of circulating immune cells, including T cells and macrophages. HIV infection is dependent primarily on host cell expression of the CD4 receptor and expression of the coreceptors CXCR4 and CCR5 [1,2]. Although other coreceptors and CD4-independent mechanisms are reported to play roles in HIV infection [3,4], their potential contributions are beyond the scope of this review. Shifts in population dynamics (preferential use of CXCR4 or CCR5) in circulating virus throughout the course of disease are important because the major route by which HIV enters the CNS is via HIV-infected macrophages trafficking from the bloodstream across the blood–brain barrier [5]. HIV is believed to enter the CNS during the earliest stages of infection, seeding the brain with virus [6]. Once inside the CNS, HIV replicates in permissive cells and maturing virions are released into the brain parenchyma. During the course of disease, plasma viral loads often fluctuate, and during times of high viremia, the virus may enter the brain again, thereby reseeding the CNS. It is well established that in the brain, microglia and macrophages are the main cellular targets for HIV infection and productive viral replication. Very early studies as well as more recent reports conclude that HIV-associated CNS dysfunction is due in a large part to indirect mechanisms that damage neurons [7]. The effects of HIV infection on neuronal fitness and functioning are a main focus of this review, and the current understanding of how HIV infection impacts neurons will be discussed.
Viral replication in the host cell
The complete picture of HIV infection involves key steps in the viral lifecycle rather than simply the presence of virus inside a cell. Infection and productive replication depend in part on the expression of specific receptors and coreceptors by permissive host cells [2]. In brief, entry of HIV into a target host cell begins with viral envelope glycoprotein gp160-mediated interaction of the virus with the host receptor. gp160 is comprised of two components, gp120 and gp41, which interact with the CD4 receptor on the surface of the host cell. Upon the binding of gp120 to CD4, a conformational change in gp120 unmasks the coreceptor binding site. Upon interaction of gp120 with the coreceptors CXCR4 or CCR5, gp41 inserts into the host cell membrane to orient the cell and virus close together so that membrane fusion can occur. Upon entry, the viral particle releases two viral RNA strands and three viral enzymes: protease, reverse transcriptase and integrase. Viral RNA is transcribed by reverse transcriptase into proviral dsDNA. The dsDNA is processed, transported into the nucleus and inserted into the host cell genome by integrase. Upon host cell DNA transcription, the viral DNA is transcribed, as well. Following a series of splicing events of the single 9.2-kb transcript, translation into viral proteins, encapsidation and viral particle assembly, the virion can bud from the host cell. Maturation of the virus into an infectious virion is completed after the particle is released from the host cell. Thus, the presence of viral DNA sequences in the host cell genome provides evidence for a key step constituting infection. Production and release of replication-competent viral particles from the target is also important in the concept of infection and is considered below in the context of HIV infection of neurons.
HIV infection of neurons
One of the most controversial aspects of HIV neurobiology is whether or not HIV can infect neurons, even at low levels, and numerous studies dating back to the 1980s and 90s address the ability of HIV to infect neurons in the brain in vivo [8–10]. Studies utilizing in situ PCR and immunohistochemistry reported the presence of HIV genetic material and antigens in neurons, respectively [11,12]. Further studies isolating neurons from post-mortem brain tissues of HIV-infected patients using laser capture microdissection (LCM) reported the presence of HIV proviral DNA in neurons by PCR [13,14]. Another study, published in 2008, employed hyperbranched multidisplacement methods for whole-gene amplification via PCR and reported the presence of HIV DNA in single neurons collected by LCM from autopsy brain tissue [15]. In vitro studies reported that human neuronal cell lines could be infected with HIV [16,17]. However, confirmation of pathologically significant infection of adult human neurons in vivo is still lacking. In addition to its presence in the CNS of adult patients, HIV is also detected in the CNS of infected pediatric patients, as well as in the developing fetal brain [18,19]. As with adult brain tissue studies, findings in pediatric HIV patients report conflicting results regarding whether HIV infects neurons [20–23].
Evidence for infection of neural stem cells and/or neural progenitor cells may be more substantial than that for infection of adult neurons, and this area of study is currently a topic of investigation by several groups. In fact, evidence for HIV-1 infection of nestin-positive neural progenitor cells was detected by multiple approaches, including in situ hybridization, LCM, DNA extraction and PCR in brain tissues from four of seven pediatric AIDS patients, suggesting in vivo infection of neural progenitor cells [24]. As early as 1995, Ensoli et al. reported that primary neural cells derived from the human fetal olfactory system representative of the developing CNS were susceptible to HIV infection in vitro, although the cells did not express CD4 [25]. Later, studies by Lawrence et al. tested whether HIV-1 NL4-3 or IIIB, X4 T-tropic virus strains, could infect human fetal progenitor cells or progenitor-derived astrocytes in vitro. Their findings suggested that human neural progenitor cells constitute a reservoir for HIV-1 in the brain, since peak replication occurred between 3 and 6 days but was consistently undetectable after 10 days [26].
In a seminal review by Bell in 1998, productive HIV infection in the adult CNS was suggested to be restricted to cells of the microglial/macrophage lineage, with limited, nonproductive infection of astrocytes [27]. Based on these and other studies, it may be concluded that productive infection of neurons by HIV is not likely to be responsible for the neurotoxicity observed in HIV patients’ brains. As described above, mechanisms involved in neuronal degeneration in HIV patients are complex, and most evidence points to chemokines, cytokines and viral proteins released from infected and/or activated microglia/macrophages as causative agents of neuronal damage observed in HIV infection [28].
Direct & indirect effects of HIV on neuronal fitness
Direct
Direct HIV-mediated neurotoxicity will be considered in the context of viral protein interactions with neurons that result in neuronal damage or death. As described in a 2002 review in the Journal of Infectious Diseases, there are several mechanisms by which HIV proteins can be released into the extracellular space, where they will encounter neurons [29]. During the uncoating process of viral fusion with the host cell, the viral envelope containing gp120 and gp41 is shed and may injure neurons in close proximity. Release of viral proteins also occurs during a cytopathic infection when the rate of viral replication is high and viral particles and proteins are released from infected cells. During nonproductive or restricted infection of astrocytes, certain viral regulatory genes are expressed that result in the production and release of Tat, Nef and Rev proteins (Figure 1). The production and release of replication-defective viral particles or secreted proteins may impact neuronal cell fitness, as well. The neurotoxic effects of some HIV proteins have been studied and described in detail, and most elicit their damage by both direct and indirect mechanisms. Figure 1 illustrates some of the well-described mechanisms by which gp120, gp41, Nef, Rev, Tat and Vpr can damage neurons [30]. As shown, some HIV proteins can disrupt ion homeostasis and depolarize neurons. Others can localize to neurons to disrupt neuronal membranes or induce apoptosis. Perhaps the most well-characterized HIV proteins that contribute to neurotoxicity are gp120 and Tat, and these will be discussed in detail below.
Figure 1. Simplified gene map of HIV-1 indicates reported effects of viral gene proteins on cells of the CNS.

HIV genes encode proteins that can be divided into three classes: structural: Gag, Pol (polymerase) and Env (envelope gp120, gp41); regulatory: Tat (transactivator of transcription) and Rev (regulatory for expression of viral proteins); and accessory: Vif (viral infectivity factor), Vpu (viral protein U), Vpr (viral protein R) and Nef (negative factor).
gp: Glycoprotein; iNOS: Inducible nitric oxide synthase; LTR: Long terminal repeat.
Reproduced with permission from [30].
gp120
As described in previous sections, the gp120 envelope protein is required for viral interaction with the host cell. In addition to its infectivity properties, gp120 has been shown to be toxic to neurons by numerous studies. Some of the first studies to report gp120-mediated neuronal toxicity were published in 1986–1987 and suggested that gp120 may be interfering with the neurotrophic factor neuroleukin [31]. Since then, an entire literature of in vitro and in vivo systems has developed to address gp120’s impact on neurons, which is too extensive to address completely. Most recently, Louboutin et al. describe anti-oxidant-mediated inhibition of gp120’s induction of caspases as a potential neuroprotective strategy [32]. Likewise, Zhang et al. report the neuroprotective effects of a Sigma-1 receptor agonist that blocks the pro-apoptotic properties of gp120 [33].
Tat
Tat, the trans-activator for transcription of HIV, has been referred to as a jack-of-all-trades, in that it can promote transcription, enhance angiogenesis, mediate the delivery of genes and peptides into cells, and mimic host cell growth factors and cytokines. As with gp120, the literature devoted to the neurotoxic effects of Tat is expansive and cannot be covered in the context of this review. In brief, with regard to neuronal toxicity, one of the first studies to use synthetic Tat characterized regions of the protein that were necessary to cause neuronal death [34]. Among the deleterious effects of Tat on neurons is neuronal depolarization by direct excitation independent of synaptic interactions, NMDA receptor dysregulation and disruption of calcium homeostasis [35,36]. For example, a study from 2008 described a mechanism by which Tat induced ryanodine receptor-mediated loss of calcium from the ER followed by the unfolded protein response and leading to pathologic dilatation of the ER in cortical neurons in vitro [37]. A more recent study described the protective effects of treating neurons with ketone bodies to block Tat-mediated mitochondrial dysfunction and changes in cellular calcium levels, and suggested this as a potential therapy, since ketones can readily cross the blood–brain barrier [38]. Recently, studies have shown that Tat-mediated neuronal dysfunction is dependent in part on small noncoding miRNA molecules. Specifically, Sawaya’s group showed that miR-34a was induced in neurons exposed to HIV-1 Tat and in Tat-transgenic mice [39], adding another level at which Tat may regulate host cell machinery.
Indirect
To consider the neurotoxic effects of HIV proteins on neurons, the effects on glial cells must be considered as well, since toxic factors derived from astrocytes and microglia significantly impact neuronal fitness [29]. Human primary neurons cultured in vitro are damaged not only by exposure to viral proteins including gp120 and Tat, but also by inflammatory factors [40–42]. Indirect effects of HIV on neurons involve the activation of glial cells (both infected and uninfected), resulting in the production and release of neurotoxic cytokines such as TNF-α. Supernatant collected from peripheral blood mononuclear cells infected with HIV contains not only viral proteins but also a variety of cytokines, and is toxic to human primary neurons in vitro (Figure 2). Moreover, recent studies from the Gelbard laboratory show that Tat-mediated interactions between central and peripheral myeloid cells can induce synaptic damage and disrupt neuroimmune homeostasis [43]. Other pathways through which HIV can damage neurons involve complement activation via increased expression of mannose-binding lectin in the axons of HIV encephalitis (HIVE) patients [44].
Figure 2. HIV proteins and host-derived inflammatory factors damage human neurons.
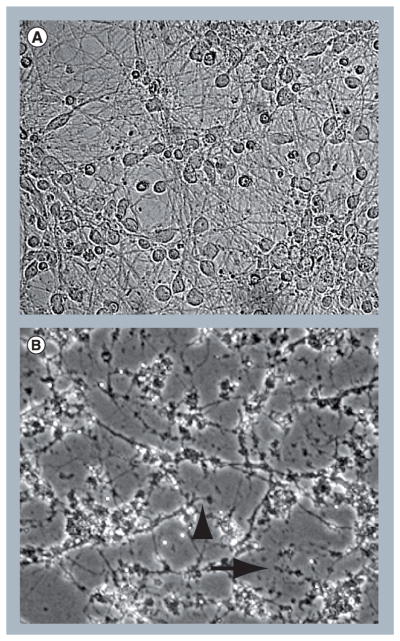
(A) Human primary neurons exposed to media from peripheral blood mononuclear cells not infected with HIV show normal morphology with intact process branching and normal cell bodies. (B) Neurons exposed to the supernatant from peripheral blood mononuclear cells infected with HIV show abnormal branching with beading (arrowhead), dystrophic process extensions (arrow) and degenerating cell bodies. Magnification = ×40.
Neuropathology of HIV-associated infection
The possible manifestations of CNS pathology resulting from HIV-associated infection are complex and include a broad array of alterations. The clinical features of HIV-associated neurocognitive disorders (HAND) range from mild neurocognitive alterations to the most severe form, HIV-associated dementia (HAD). In some HIV patients, however, there are no detectable neurocognitive alterations. The efficacy of combination antiretroviral therapy (cART) to control peripheral viral loads and allow for immune system recovery has impacted significantly on the prevalence of HAND. With the use of cART, the incidence of HAD has decreased, but the prevalence of milder forms of HAND has increased [30,45]. Thus, widespread implementation of cART has resulted in the partial recovery from or reversal of HAD in some patients, but milder forms of HAND persist in HIV patients before progression to AIDS. This may be due in part to the ability of cART regimens with higher CNS-penetrating efficiency (CPE) to suppress viral replication in the brain [46,47]. In this context, HIVE has been described as a neuropathological correlate of HAND. Hallmarks of HIVE include multinucleated giant cells, microglial nodules, perivascular cuffing by inflammatory cells and gliosis [48]. Apoptotic death of both reactive astrocytes and neurons has been detected in the brains of HIVE patients, as well [49]. With improved cART and the development of regimens with higher CPE, HIVE has shifted from a subacute to a chronic condition, while HAND remains common among many patients.
Patterns of neuronal damage
Neuronal populations vary in susceptibility to HIV-mediated damage based in part upon their expression levels of cytokine and chemokine receptors [50]. One specific population in the neocortex that is damaged by HIV is pyramidal neurons expressing MAP2, neurofilament, glutamate and cytokine/chemokine receptors, and low levels of calcium-binding proteins. Interneurons expressing high levels of cytokine/chemokine receptors and the calcium-binding proteins calbindin (CB) and parvalbumin (PV) are also particularly vulnerable to HIV-induced toxicity [51]. Likewise, neurons that express high levels of chemokine receptors, such as CXCR4, and low levels of trophic factor receptors, including fibroblast growth factor receptor (FGFR) or neurotrophin factor receptors for nerve growth factor (NGF), or brain-derived neurotrophic factor (BDNF), are also more vulnerable to HIV-mediated neurotoxicity [52–54]. Regional differences in patterns of neuronal damage have been reported from autopsy studies, as well. For example, hippocampal pyramidal neurons from HIVE patients are relatively spared, but significant loss of PV-immunoreactive interneurons in the CA3 region expressing cytokine receptors for IL-1β, IL-6 and TNF-α has been observed [50,51,55]. In the basal ganglia of HIVE patients, a significant loss of MAP2- and glutamate receptor-immunoreactive large spiny neurons is observed, but CB-immunoreactive neurons show less damage [56,57]. Neuronal loss of specific populations within the basal ganglia likely contributes to dysfunction of the dopaminergic system, which is proposed to play a role in HAD [58–60]. Additionally, somatostatin-immunoreactivity is decreased in interneurons in the frontal cortex, in hippocampal pyramidal and nonpyramidal neurons, and in neurons in the globus pallidus [61,62] and may be related to virus-induced alterations in expression of glutamate receptors. Data from autopsy cases are consistent with results from gp120 transgenic mice that show significant damage to MAP2-immunoreactive pyramidal neurons in the neocortex, while PV-immunoreactive interneurons are spared [63–65]. Taken together, these findings suggest that neocortical neurons may be more vulnerable to direct damage mediated by HIV-derived proteins (such as gp120), while neurons in the hippocampus and basal ganglia may be more vulnerable to indirect damage from cytokines and chemokines.
Early studies characterizing the neuropathological profiles of HIVE reported neuronal damage and neurodegeneration in striato–cortical, cortico–cortical and limbic intrinsic/inhibitory circuitries [55]. Specifically, large pyramidal neurons in the neocortex [66–68], spiny neurons in the putamen, medium-sized neurons in the globus pallidus, and interneurons in the hippocampus were most impacted by HIV infection of the CNS [51,56,62,69]. A statistically significant decrease in the number of large neurons, a reduction in neocortical width and astrogliosis have been observed, as well [51,55,56,66,68,70,71].
Morphologically, dendritic simplification of pyramidal neurons and selective loss of inter-neuron populations have been observed [55]. In addition to neuronal loss, aberrant sprouting and dystrophic synaptodendritic connections are common in HIVE patients [30], with decreased expression of MAP2 and neurofilament, markers for synaptodendritic connectivity (Figure 3). Damage is proposed to begin in synapses and dendrites and then spread to the rest of the neuron, leading to apoptosis [72,73]. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays in HIVE brains indicate DNA fragmentation in neurons, glia and endothelial cells [70,74–76]. Additionally, differential expression of miRNAs involved in the regulation of apoptosis has been reported in HIVE [77]. For a detailed review of the vulnerability of distinct neuronal populations to HIV see [69].
Figure 3. Immunofluorescence labeling of human brain tissue showing loss of synaptodendritic markers in HIV encephalitis.

(A) Frontal cortex from an HIV patient with no CNS alterations, showing normal distribution of dendrites (microtubular-associated protein 2: green) and axons (neurofilament: red). (B) Frontal cortex from an HIV encephalitic patient showing loss of immunolabeling for dendrites (microtubular-associated protein 2: green) and axons (neurofilament: red). Magnification = ×40.
HIV & neurotrophic signaling
As noted above, host-derived inflammatory factors such as TNF-α generated in response to HIV infection can damage neurons. Other factors produced by host cells in response to challenge may be able to provide protection from neurotoxins. NGF, BDNF, platelet-derived growth factor (PDGF) and NT3 are among the neurotrophic factors produced by neurons and glia to promote neuronal growth and survival [78]. Neurotrophic factors and components of the neuroinflammatory response interact with one another during HIV infection of cells in the CNS. Viral proteins such as Tat and gp120 can dysregulate growth factor signaling cascades in a number of ways. For example, through molecular mimicry, viral proteins can exploit the host cell’s machinery by binding to receptors and disrupting normal host cell signaling [79,80]. For instance, molecular mimicry between gp41 and the astrocytic protein α-actinin, originally revealed by antibody cross-reactivity, allows gp41 to bind to astrocyte cell membranes and induce aberrant signaling [81]. As astrocytes play a supportive role in neuronal health, gp41-induced glial signaling dysfunction has important implications for neuronal survival. Additionally, Tat can mimic signaling properties of vascular endothelial cell growth factor (VEGF) by binding to the VEGF receptor on cerebral endothelial cells thereby promoting angiogenesis [82]. Studies also indicate that Tat mimicry of VEGF correlates with increased microvessel density in AIDS-related diffuse large B-cell and Burkitt lymphomas [83]. Molecular mimicry by Tat and gp120 exploit chemokine receptors by mimicking their ligands to promote the neuroinflammatory response and viral infection of host cells. Ultimately, mimicry of host cytokines, chemokines, neurotrophic growth factors or their receptors by viral proteins contributes to the dysregulation of host-mediated immune responses [84–86].
Progression of CNS disease associated with HIV infection is also affected by the interactions of neurotrophic factors with the virus or with neuroinflammatory molecules. For example, neurotrophic factors including FGF 1 and 2, BDNF and NGF may protect neurons from toxic inflammatory factors produced in response to macrophage/microglial HIV infection. Specifically, NGF and BDNF play important roles in neuronal survival in HIV infection of the CNS by activating the NF-κB pathway, thereby inducing expression of the anti-apoptotic Bcl-2 gene that protects neurons from the pro-apoptotic effects of HIV Tat [61]. Interaction of gp120 with BDNF has been shown to result in increased somatostatin neurotransmission [61,87]. gp120 has also been shown to affect the expression of both NGF and nitric oxide synthase in rodent models of HIV disease [88,89]. Furthermore, Tat has been shown to interact with p35 signaling in neurons to dysregulate the NGF pathway, thereby disrupting neuronal survival and differentiation [90]. Levels of some of these neurotrophic factors have been shown to correlate with the degree of cognitive dysfunction and with severity of neuropathology at autopsy [91–93].
PDGF-mediated neuroprotection against both gp120 and Tat is reported in studies with SH-SY5Y neuroblastoma cells [94–96]. Pathways through which PDGF protects differentiated neurons from gp120 and Tat involve blocking apoptosis; however, the signaling cascades differ. PDGF protects neurons from gp120-mediated toxicity via the PI3K/AKT/GSK3β pathway [95,96]. On the other hand, neurons are protected from Tat by PDGF regulation of extracellular glutamate and intracellular calcium levels [46,47,94]. Further studies in mice showed that intrastriatal delivery of PDGF protected dopaminergic neurons in the substantia nigra from Tat toxicity via transient receptor potential canonical channel signaling [97]. Therefore, the balance between toxic and protective factor production contributes to neuronal survival and ultimately influences the degree of HAND and disease progression.
Clade specificity in neuronal damage
Since neuronal damage in HIV is caused by inflammatory factors and viral proteins released from infected and/or activated microglia/macrophages, sequence differences among different clades or subtypes of viruses may result in varying degrees of neurotoxicity. The HIV-1 group M consists of at least nine clades or subtypes: A–D, F–H, J and K, including multiple common circulating recombinant forms that are believed to result from the recombination of different viruses coexisting in an individual. Clade B is responsible for approximately 10% of HIV infections worldwide and is predominant in the Americas, Australia and Europe. Clade C is more common in sub-Saharan Africa and parts of India, and is responsible for nearly 50% of infections worldwide. Several studies have addressed the potential differences in clades with regard to immune response, neurovirulence, disease progression and response to anti-retroviral therapy. For example, studies in Uganda where clades A and D predominate suggest that antiretroviral-naive individuals infected with clade D have a higher prevalence for HAND than those infected with clade A [98]. Studies from different regions in India and in Ethiopia report different degrees of HIV-associated cognitive impairment in antiretroviral-naive patient populations [99–104]. In vitro studies suggest that differences in the genomic sequence of Tat may contribute to clade-specific neurotoxic effects. Studies comparing Tat from HIV-1 B with C suggest that the presence of the dicysteine motif, C30C31, in B confers increased neurotoxicity in human neuronal cultures [105]. Other studies in rat neurons explored the NMDA-mediated neurotoxic effects of HIV-1 B Tat compared with C and found that in zinc-dependent signaling, clade C Tat was less neurotoxic than clade B Tat [106]. Thus, among the numerous factors that impact the degree of neurotoxicity and HAND observed in HIV patients, differences in viral protein sequences among clades may contribute, as well.
HIV & substance abuse
The degree to which neurons are damaged in HIV-infected patients is significantly affected by numerous comorbid conditions, including substance abuse. Both opiate and stimulant use and abuse are reported to contribute to the development of HAND in HIV patients [107,108]. Since stimulants and opioids disrupt normal dopaminergic signaling, the already-compromised dopamine system in HIV patients is likely to be impacted even more significantly in substance abuse patients. In HIV-infected drug-user populations, other comorbid conditions including hepatitis C infection or poly-drug use may impact neuronal functioning, as well. Thus, understanding the potentially unique (additive or synergistic) effects of HIV with opioids or stimulants in patient populations becomes problematic. In this context, many in vitro and in vivo studies have shown that methamphetamine, cocaine and morphine exacerbate the neurotoxic effects of viral proteins, gp120 and Tat while enhancing viral replication.
Stimulants
Studies in HIVE patients who were methamphetamine abusers in life report extensive loss of CB and PV-immunoreactive interneuron populations and significant loss of markers for synapses [109]. Moreover, these patients display significantly greater cognitive deficits than HIV patients that do not use methamphetamine [110].
In vitro and in vivo studies with methamphetamine and HIV proteins show altered mitochondrial membrane potential and cell death, due in part to dopaminergic signaling dysregulation [111–113]. Other in vitro studies report activation of matrix-degrading proteinases, induction of inflammatory factor production, autophagy and apoptosis [114–117].
Opioids
Dysregulation of cytokine signaling, increased oxidative damage, dendritic spine loss and apoptosis are among the many contributors proposed to cause increased neurotoxicity of HIV when combined with morphine [118–122]. A common element in many of these and other studies (both in vivo and in vitro) is the critical role of glia in mediating the neurotoxic effects of HIV proteins and opioids [123,124]. Importantly, increased neurotoxicity from the use of opioids in HIV patients is compounded by the expression of opioid receptors on not only neurons, but also on astrocytes and microglia, making glial cells key mediators of indirect neuronal damage in both infection and morphine-induced aspects of disease. One study reported, however, that release of CCL5 from astrocytes protected neurons from M-tropic gp120 [125]. Recently, findings report that sequence differences in gp120 from different HIV clades determines HIV/opioid-mediated patterns of neurotoxicity [126] and may explain, to some extent, the protective effects such as those reported for CCL5.
Future perspective
As described in a recent review, improved CPE rankings of antiretrovirals have led to lower viral burden in the cerebrospinal fluid and improved neurocognitive functioning in HIV patients [47]. These findings may be indicative of neuronal sparing or recovery during HIV infection [30]. Lower viral counts in the cerebrospinal fluid may reflect lower levels in the brain, and neuronal damage, whether direct or indirect, may be lessened, as well, in these cases. Enhanced contributions of host-derived neurotrophic factors could be possible as well, given that some HIV proteins have been shown to dysregulate normal trophic factor signaling. In combination with antiretrovirals with high CPE, adjuvant therapies aimed at improving host response to toxic factors may be potentially important, as well. However, as pointed out by Tan and McArthur, antiretrovirals have shown benefit for HAND where many adjuvant therapies have not shown significant benefit [127]. Thus, gaps still exist in our capacity to improve HAND beyond controlling viral burdens. Due to the fluctuating state of HAND in some patients, it is clear that numerous factors are involved in CNS disease progression. Along with comorbid conditions, other factors such as aging, cART neurotoxicity and the emergence of more aggressive and resistant HIV strains likely play roles in the increased susceptibility of distinct neuronal populations to damage.
In conclusion, HIV does not directly injure neurons by productive infection, but rather by inducing the production of inflammatory factors from infected or activated macrophages and microglia. Likewise, HIV perturbs the expression of proteins in synaptic compartments, thereby disrupting neuronal communication [128]. These indirect mechanisms of toxicity lead to the damage of selected neuronal populations and white matter tracts, and in many cases result in neurocognitive impairment. A better understanding of the molecular mechanisms underlying neuronal injury and dysfunction during the progression of neuro-AIDS will allow for the evaluation of potential recovery and more targeted and efficacious treatment strategies. As cART CPE improves, maintaining viral latency in the CNS becomes a promising approach to protect neurons and alleviate HAND.
Executive summary.
HIV infection of the CNS
HIV infects a subset of circulating immune cells, including T cells and macrophages.
HIV-infected macrophages traffic from the bloodstream across the blood–brain barrier into the CNS early after infection.
Microglia and macrophages are the main cellular targets for HIV infection and productive viral replication in the CNS.
Viral replication in the host cell
Viral infection of a cell depends largely on the expression of specific host cell surface receptors, CD4, CCR5 and CXCR4.
Once inside the host cell, viral replication is regulated by virus- and host-derived factors.
HIV infection of neurons
Productive infection of neurons by HIV is not likely to be responsible for the neurotoxicity observed in HIV patients’ brains.
Chemokines, cytokines and viral proteins released from infected and/or activated microglia/macrophages and astrocytes are likely to be responsible for neuronal damage.
Direct & indirect effects of HIV on neuronal fitness
Viral proteins including gp120, gp41, Tat, Nef, Rev and Vpr released from infected cells can damage neurons.
Indirect effects of HIV on neurons involve the activation of glial cells (both infected and uninfected) resulting in the production and release of neurotoxic cytokines.
Neuropathology of HIV-associated infection
HIV-associated neurocognitive disorders (HAND) range from mild to severe (dementia).
Combination antiretroviral therapy with increased CNS-penetrating efficiency has resulted in increased prevalence of milder forms of HAND and fewer cases of HIV-associated dementia.
With cART, HIV encephalitis, the neuropathological correlate of HAND, has shifted from a subacute to a chronic condition.
Patterns of neuronal damage
Regional differences in neuronal damage in the brains of HIV-infected patients are reported.
Different neuronal populations vary in susceptibility to HIV-mediated damage based in part on expression of specific cytokine, chemokine, neurotransmitter and growth factor receptors.
Autopsy studies report dendritic simplification, aberrant sprouting, pruning and dystrophic synaptodendritic connectivity.
HIV & neurotrophic signaling
Host-derived trophic factors may provide protection from HIV-associated neurotoxic factors.
Some HIV proteins can mimic neurotrophic factors to dysregulate host cell signaling.
Clade specificity in neuronal damage
Potential differences in immune response, neurovirulence, disease progression and response to antiretroviral therapy by different HIV-1 clades are proposed.
Differences in the prevalence of HAND among patients infected with different clades of HIV-1 are suggested.
Sequence variation in the HIV proteins, including Tat and gp120, may account for some of the differences reported.
HIV & substance abuse
Substance abuse is major risk factor for becoming HIV infected.
Like HIV, opioids and stimulants also disrupt dopaminergic signaling and contribute to HAND.
Future perspective
It is likely that neuronal injury in HIV-infected patients derives from multiple sources, including HIV proteins, inflammatory factors, co-morbid conditions such as substance abuse, host genetics, clade diversity and combination antiretroviral therapy components.
Maintaining viral latency in the CNS is a promising approach to protect neurons.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
- 1.Stevenson M. HIV-1 pathogenesis. Nat Med. 2003;9(7):853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- 2.Gorry PR, Ancuta P. Coreceptors and HIV-1 pathogenesis. Curr HIV/AIDS Rep. 2011;8(1):45–53. doi: 10.1007/s11904-010-0069-x. [DOI] [PubMed] [Google Scholar]
- 3.Fauci AS. Host factors and the pathogenesis of HIV-induced disease. Nature. 1996;384(6609):529–534. doi: 10.1038/384529a0. [DOI] [PubMed] [Google Scholar]
- 4.Lavi E, Kolson DL, Ulrich AM, Fu L, Gonzalez-Scarano F. Chemokine receptors in the human brain and their relationship to HIV infection. J Neurovirol. 1998;4(3):301–311. doi: 10.3109/13550289809114531. [DOI] [PubMed] [Google Scholar]
- 5.Strazza M, Pirrone V, Wigdahl B, Nonnemacher MR. Breaking down the barrier: the effects of HIV-1 on the blood-brain barrier. Brain Res. 2011;1399:96–115. doi: 10.1016/j.brainres.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38(1):9–14. doi: 10.1212/wnl.38.1.9. [DOI] [PubMed] [Google Scholar]
- 7.Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986;83(18):7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Funke I, Hahn A, Rieber EP, Weiss E, Riethmuller G. The cellular receptor (CD4) of the human immunodeficiency virus is expressed on neurons and glial cells in human brain. J Exp Med. 1987;165(4):1230–1235. doi: 10.1084/jem.165.4.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavi E, Strizki JM, Ulrich AM, et al. CXCR-4 (Fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons. Am J Pathol. 1997;151(4):1035–1042. [PMC free article] [PubMed] [Google Scholar]
- 10.Peudenier S, Hery C, Ng KH, Tardieu M. HIV receptors within the brain: a study of CD4 and MHC-II on human neurons, astrocytes and microglial cells. Res Virol. 1991;142(2–3):145–149. doi: 10.1016/0923-2516(91)90051-4. [DOI] [PubMed] [Google Scholar]
- 11.Bagasra O, Lavi E, Bobroski L, et al. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS (London, England) 1996;10(6):573–585. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Nuovo Gj, Gallery F, Macconnell P, Braun A. In situ detection of polymerase chain reaction-amplified HIV-1 nucleic acids and tumor necrosis factor-alpha RNA in the central nervous system. Am J Pathol. 1994;144(4):659–666. [PMC free article] [PubMed] [Google Scholar]
- 13.Torres-Munoz J, Stockton P, Tacoronte N, Roberts B, Maronpot RR, Petito CK. Detection of HIV-1 gene sequences in hippocampal neurons isolated from postmortem AIDS brains by laser capture microdissection. J Neuropathol Exp Neurol. 2001;60(9):885–892. doi: 10.1093/jnen/60.9.885. [DOI] [PubMed] [Google Scholar]
- 14.Trillo-Pazos G, Diamanturos A, Rislove L, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol (Zurich, Switzerland) 2003;13(2):144–154. doi: 10.1111/j.1750-3639.2003.tb00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torres-Munoz JE, Nunez M, Petito CK. Successful application of hyperbranched multidisplacement genomic amplification to detect HIV-1 sequences in single neurons removed from autopsy brain sections by laser capture microdissection. J Mol Diagn. 2008;10(4):317–324. doi: 10.2353/jmoldx.2008.070074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhtar M, Acheampong E, Khan MA, Bouhamdan M, Pomerantz RJ. Down-modulation of the CXCR4 co-receptor by intracellular expression of a single chain variable fragment (SFv) inhibits HIV-1 entry into primary human brain microvascular endothelial cells and post-mitotic neurons. Brain Res Mol Brain Res. 2005;135(1–2):48–57. doi: 10.1016/j.molbrainres.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Alvarez-Losada S, Canto-Nogues C, Munoz-Fernandez MA. A new possible mechanism of human immunodeficiency virus type 1 infection of neural cells. Neurobiol Dis. 2002;11(3):469–478. doi: 10.1006/nbdi.2002.0566. [DOI] [PubMed] [Google Scholar]
- 18.Kure K, Llena JF, Lyman WD, et al. Human immunodeficiency virus-1 infection of the nervous system: an autopsy study of 268 adult, pediatric, and fetal brains. Hum Pathol. 1991;22(7):700–710. doi: 10.1016/0046-8177(91)90293-x. [DOI] [PubMed] [Google Scholar]
- 19.Lyman WD, Kress Y, Kure K, Rashbaum WK, Rubinstein A, Soeiro R. Detection of HIV in fetal central nervous system tissue. AIDS (London, England) 1990;4(9):917–920. doi: 10.1097/00002030-199009000-00014. [DOI] [PubMed] [Google Scholar]
- 20.Canto-Nogues C, Sanchez-Ramon S, Alvarez S, Lacruz C, Munoz-Fernandez MA. HIV-1 infection of neurons might account for progressive HIV-1-associated encephalopathy in children. J Mol Neurosci. 2005;27(1):79–89. doi: 10.1385/JMN:27:1:079. [DOI] [PubMed] [Google Scholar]
- 21.Vallat AV, De Girolami U, He J, et al. Localization of HIV-1 co-receptors CCR5 and CXCR4 in the brain of children with AIDS. Am J Pathol. 1998;152(1):167–178. [PMC free article] [PubMed] [Google Scholar]
- 22.Sharer LR, Saito Y, Da Cunha A, et al. In situ amplification and detection of HIV-1 DNA in fixed pediatric AIDS brain tissue. Hum Pathol. 1996;27(6):614–617. doi: 10.1016/s0046-8177(96)90172-0. [DOI] [PubMed] [Google Scholar]
- 23.Sharer LR, Saito Y, Epstein LG, Blumberg BM. Detection of HIV-1 DNA in pediatric AIDS brain tissue by two-step ISPCR. Adv Neuroimmunol. 1994;4(3):283–285. doi: 10.1016/s0960-5428(06)80268-8. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz L, Civitello L, Dunn-Pirio A, et al. Evidence of human immunodeficiency virus type 1 infection of nestin-positive neural progenitors in archival pediatric brain tissue. J Neurovirol. 2007;13(3):274–283. doi: 10.1080/13550280701344975. [DOI] [PubMed] [Google Scholar]
- 25.Ensoli F, Cafaro A, Fiorelli V, Vannelli B, Ensoli B, Thiele CJ. HIV-1 infection of primary human neuroblasts. Virology. 1995;210(1):221–225. doi: 10.1006/viro.1995.1336. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence DM, Durham LC, Schwartz L, Seth P, Maric D, Major EO. Human immunodeficiency virus type 1 infection of human brain-derived progenitor cells. J Virol. 2004;78(14):7319–7328. doi: 10.1128/JVI.78.14.7319-7328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27▪.Bell JE. The neuropathology of adult HIV infection. Rev Neurol. 1998;154(12):816–829. Describes results from numerous studies that point to cells of the macrophage/microglial lineage as being responsible for productive infection in the CNS, with restricted infection of astrocytes. [PubMed] [Google Scholar]
- 28▪.Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4(3):307–318. doi: 10.2174/157016206777709384. Concisely summarizes the mechanism by which neurons are injured in HIV patients and how this damage may contribute to the development of HIV-associated dementia. [DOI] [PubMed] [Google Scholar]
- 29▪.Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186:s193–s198. doi: 10.1086/344528. Describes in detail the molecular mechanisms by which HIV proteins directly damage neurons. [DOI] [PubMed] [Google Scholar]
- 30▪.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev. 2007;8(1):33–44. doi: 10.1038/nrn2040. Reviews the mechanisms that contribute to neuronal damage and repair during HIV infection of the CNS. [DOI] [PubMed] [Google Scholar]
- 31.Barnes DM. Solo actions of AIDS virus coat. Science. 1987;237(4818):971–973. doi: 10.1126/science.3039663. [DOI] [PubMed] [Google Scholar]
- 32.Louboutin JP, Agrawal L, Reyes BA, Van Bockstaele EJ, Strayer DS. Gene delivery of antioxidant enzymes inhibits HIV-1 gp120-induced expression of caspases. Neuroscience. 2012 doi: 10.1016/j. neuroscience.2012.03.061. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Shi Y, Qiao L, et al. Sigma-1 receptor agonists provide neuroprotection against gp120 via a change in bcl-2 expression in mouse neuronal cultures. Brain Res. 2012;1431:13–22. doi: 10.1016/j.brainres.2011.10.053. [DOI] [PubMed] [Google Scholar]
- 34.Sabatier JM, Vives E, Mabrouk K, et al. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65(2):961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotoxicity Res. 2009;16(3):205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- 36.Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-n-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37(3):373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- 37.Norman JP, Perry SW, Reynolds HM, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One. 2008;3(11):e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hui L, Chen X, Bhatt D, et al. Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem. 2012 doi: 10.1111/j.1471-4159.2012.07764. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang JR, Mukerjee R, Bagashev A, et al. HIV-1 Tat protein promotes neuronal dysfunction through disruption of microRNAs. J Biol Chem. 2011;286(47):41125–41134. doi: 10.1074/jbc.M111.268466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maragos WF, Tillman P, Jones M, et al. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience. 2003;117(1):43–53. doi: 10.1016/s0306-4522(02)00713-3. [DOI] [PubMed] [Google Scholar]
- 41.Alirezaei M, Watry DD, Flynn CF, et al. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J Neurosci. 2007;27(41):11047–11055. doi: 10.1523/JNEUROSCI.2733-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W, Galey D, Mattson MP, Nath A. Molecular and cellular mechanisms of neuronal cell death in HIV dementia. Neurotoxicity Res. 2005;8(1–2):119–134. doi: 10.1007/BF03033824. [DOI] [PubMed] [Google Scholar]
- 43.Lu SM, Tremblay ME, King IL, et al. HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One. 2011;6(9):e23915. doi: 10.1371/journal.pone.0023915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh KK, Nathamu S, Adame A, et al. Expression of mannose binding lectin in HIV-1-infected brain: implications for HIV-related neuronal damage and neuroAIDS. Neurobehav HIV Med. 2011;3:41–52. doi: 10.2147/NBHIV.S19969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mothobi NZ, Brew BJ. Neurocognitive dysfunction in the highly active antiretroviral therapy era. Curr Opin Infect Dis. 2012;25(1):4–9. doi: 10.1097/QCO.0b013e32834ef586. [DOI] [PubMed] [Google Scholar]
- 46.Langford D, Marquie-Beck J, De Almeida S, et al. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J Neurovirol. 2006;12(2):100–107. doi: 10.1080/13550280600713932. [DOI] [PubMed] [Google Scholar]
- 47▪.Letendre S. Central nervous system complications in HIV disease: HIV-associated neurocognitive disorder. Top Antiviral Med. 2011;19(4):137–142. Summarizes the current understanding of the risks factors for developing HIV-associated neurocognitive disorders (HAND), the impacts of antiretroviral therapy on HAND and how HAND is assessed in the clinic. [PMC free article] [PubMed] [Google Scholar]
- 48.Everall IP, Hansen LA, Masliah E. The shifting patterns of HIV encephalitis neuropathology. Neurotoxicity Res. 2005;8(1–2):51–61. doi: 10.1007/BF03033819. [DOI] [PubMed] [Google Scholar]
- 49.Petito CK, Roberts B. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 1995;146(5):1121–1130. [PMC free article] [PubMed] [Google Scholar]
- 50.Masliah E, Ge N, Achim CL, Wiley CA. Cytokine receptor alterations during HIV infection in the human central nervous system. Brain Res. 1994;663(1):1–6. doi: 10.1016/0006-8993(94)90456-1. [DOI] [PubMed] [Google Scholar]
- 51.Masliah E, Ge N, Achim CL, Hansen LA, Wiley CA. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51(6):585–593. doi: 10.1097/00005072-199211000-00003. [DOI] [PubMed] [Google Scholar]
- 52.Crews L, Patrick C, Achim CL, Everall IP, Masliah E. Molecular pathology of neuro-AIDS (CNS-HIV) Int J Mol Sci. 2009;10(3):1045–1063. doi: 10.3390/ijms10031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed F, Tessarollo L, Thiele C, Mocchetti I. Brain-derived neurotrophic factor modulates expression of chemokine receptors in the brain. Brain Res. 2008;1227:1–11. doi: 10.1016/j.brainres.2008.05.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanders VJ, Everall IP, Johnson RW, Masliah E. Fibroblast growth factor modulates HIV coreceptor CXCR4 expression by neural cells. HNRC Group J Neurosci Res. 2000;59(5):671–679. doi: 10.1002/(SICI)1097-4547(20000301)59:5<671::AID-JNR10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 55.Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1 associated neurodegeneration. Crit Rev Neurobiol. 1996;10(1):57–67. doi: 10.1615/critrevneurobiol.v10.i1.30. [DOI] [PubMed] [Google Scholar]
- 56.Masliah E, Ge N, Achim CL, Deteresa R, Wiley CA. Patterns of neurodegeneration in HIV encephalitis. J NeuroAIDS. 1996;1(1):161–173. doi: 10.1300/j128v01n01_08. [DOI] [PubMed] [Google Scholar]
- 57.Mucke L, Masliah E, Campbell IL. Transgenic models to assess the neuropathogenic potential of HIV-1 proteins and cytokines. Curr Top Microbiol Immunol. 1995;202:187–205. doi: 10.1007/978-3-642-79657-9_13. [DOI] [PubMed] [Google Scholar]
- 58.Zauli G, Secchiero P, Rodella L, et al. HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem. 2000;275(6):4159–4165. doi: 10.1074/jbc.275.6.4159. [DOI] [PubMed] [Google Scholar]
- 59.Nath A, Anderson C, Jones M, et al. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol (Oxford, England) 2000;14(3):222–227. doi: 10.1177/026988110001400305. [DOI] [PubMed] [Google Scholar]
- 60.Lopez OL, Smith G, Meltzer CC, Becker JT. Dopamine systems in human immunodeficiency virus-associated dementia. Neuropsych Neuropsychol Behav Neurol. 1999;12(3):184–192. [PubMed] [Google Scholar]
- 61.Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB. Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem. 2001;78(4):874–889. doi: 10.1046/j.1471-4159.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- 62.Fox L, Alford M, Achim C, Mallory M, Masliah E. Neurodegeneration of somatostatin-immunoreactive neurons in HIV encephalitis. J Neuropathol Exp Neurol. 1997;56(4):360–368. doi: 10.1097/00005072-199704000-00004. [DOI] [PubMed] [Google Scholar]
- 63.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367(6459):188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 64.Campbell IL, Abraham CR, Masliah E, et al. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA. 1993;90(21):10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Campbell IL, Stalder AK, Chiang CS, et al. Transgenic models to assess the pathogenic actions of cytokines in the central nervous system. Mol Psychiatr. 1997;2(2):125–129. doi: 10.1038/sj.mp.4000225. [DOI] [PubMed] [Google Scholar]
- 66.Budka H, Costanzi G, Cristina S, et al. Brain pathology induced by infection with the human immunodeficiency virus (HIV) A histological, immunocytochemical, and electron microscopical study of 100 autopsy cases. Acta Neuropathologica. 1987;75(2):185–198. doi: 10.1007/BF00687080. [DOI] [PubMed] [Google Scholar]
- 67.Everall IP, Luthert PJ, Lantos PL. Neuronal loss in the frontal cortex in HIV infection. Lancet. 1991;337(8750):1119–1121. doi: 10.1016/0140-6736(91)92786-2. [DOI] [PubMed] [Google Scholar]
- 68.Wiley CA, Masliah E, Morey M, et al. Neocortical damage during HIV infection. Ann Neurol. 1991;29(6):651–657. doi: 10.1002/ana.410290613. [DOI] [PubMed] [Google Scholar]
- 69.Langford TD, Everall IP, Maslaih E. Neuronal injury, white matter disease and neurotrophic factors. In: Gendelman HE, Everall IP, Grant I, et al., editors. The Neurology of AIDS. 3. Oxford University Press; UK: 2012. pp. 619–632. [Google Scholar]
- 70.Gray F, Adle-Biassette H, Chretien F, Lorin De La Grandmaison G, Force G, Keohane C. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol. 2001;20(4):146–155. [PubMed] [Google Scholar]
- 71.Gray F, Haug H, Chimelli L, et al. Prominent cortical atrophy with neuronal loss as correlate of human immunodeficiency virus encephalopathy. Acta Neuropathologica. 1991;82(3):229–233. doi: 10.1007/BF00294450. [DOI] [PubMed] [Google Scholar]
- 72.Everall IP, Heaton RK, Marcotte TD, et al. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder HNRC Group HIV Neurobehavioral Research Center. Brain Pathol (Zurich, Switzerland) 1999;9(2):209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Masliah E, Heaton RK, Marcotte TD, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders HNRC Group The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42(6):963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- 74.Everall IP, Gray F, Masliah E. Neuronal injury and apoptosis. In: Gendelman H, Lipton S, Epstein L, Swindells S, editors. Neurology of AIDS. Chapman and Hall; NY, USA: 1988. pp. 261–274. [Google Scholar]
- 75.Wiley CA, Achim CL, Hammond R, et al. Damage and repair of DNA in HIV encephalitis. J Neuropathol Exp Neurol. 2000;59(11):955–965. doi: 10.1093/jnen/59.11.955. [DOI] [PubMed] [Google Scholar]
- 76.Adle-Biassette H, Chretien F, Wingertsmann L, et al. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25(2):123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- 77.Noorbakhsh F, Ramachandran R, Barsby N, et al. MicroRNA profiling reveals new aspects of HIV neurodegeneration: caspase-6 regulates astrocyte survival. FASEB J. 2010;24(6):1799–1812. doi: 10.1096/fj.09-147819. [DOI] [PubMed] [Google Scholar]
- 78.Encinas M, Iglesias M, Liu Y, et al. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75(3):991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- 79.Asensio VC, Campbell IL. Chemokines and viral diseases of the central nervous system. Adv Virus Res. 2001;56:127–173. doi: 10.1016/S0065-3527(01)56006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langford D, Masliah E. Role of trophic factors on neuroimmunity in neurodegenerative infectious diseases. J Neurovirol. 2002;8(6):625–638. doi: 10.1080/13550280290100996. [DOI] [PubMed] [Google Scholar]
- 81.Spehar T, Strand M. Molecular mimicry between HIV-1 gp41 and an astrocyte isoform of alpha-actinin. J Neurovirol. 1995;1(5–6):381–390. doi: 10.3109/13550289509111028. [DOI] [PubMed] [Google Scholar]
- 82.Scheidegger P, Weiglhofer W, Suarez S, et al. Signalling properties of an HIV-encoded angiogenic peptide mimicking vascular endothelial growth factor activity. Biochem J. 2001;353(Pt 3):569–578. doi: 10.1042/0264-6021:3530569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nyagol J, De Falco G, Lazzi S, et al. HIV-1 Tat mimetic of VEGF correlates with increased microvessels density in AIDS-related diffuse large B-cell and Burkitt lymphomas. J Hematopathol. 2008;1(1):3–10. doi: 10.1007/s12308-008-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy PM. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat Immunol. 2001;2(2):116–122. doi: 10.1038/84214. [DOI] [PubMed] [Google Scholar]
- 85.Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Mol Med Today. 2000;6(9):365–372. doi: 10.1016/S1357-4310(00)01775-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lalani AS, Mcfadden G. Evasion and exploitation of chemokines by viruses. Cytokine Growth Factor Rev. 1999;10(3–4):219–233. doi: 10.1016/s1359-6101(99)00018-0. [DOI] [PubMed] [Google Scholar]
- 87.Barnea A, Roberts J, Ho RH. Evidence for a synergistic effect of the HIV-1 envelope protein gp120 and brain-derived neurotrophic factor (BDNF) leading to enhanced expression of somatostatin neurons in aggregate cultures derived from the human fetal cortex. Brain Res. 1999;815(2):349–357. doi: 10.1016/s0006-8993(98)01098-1. [DOI] [PubMed] [Google Scholar]
- 88.Corasaniti MT, Bagetta G, Rotiroti D, Nistico G. The HIV envelope protein gp120 in the nervous system: interactions with nitric oxide, interleukin-1beta and nerve growth factor signalling, with pathological implications in vivo and in vitro. Biochem Pharmacol. 1998;56(2):153–156. doi: 10.1016/s0006-2952(98)00044-6. [DOI] [PubMed] [Google Scholar]
- 89.Bagetta G, Corasaniti MT, Aloe L, et al. Intracerebral injection of human immunodeficiency virus type 1 coat protein gp120 differentially affects the expression of nerve growth factor and nitric oxide synthase in the hippocampus of rat. Proc Natl Acad Sci USA. 1996;93(2):928–933. doi: 10.1073/pnas.93.2.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peruzzi F, Gordon J, Darbinian N, Amini S. Tat-induced deregulation of neuronal differentiation and survival by nerve growth factor pathway. J Neurovirol. 2002;8(Suppl 2):91–96. doi: 10.1080/13550280290167885. [DOI] [PubMed] [Google Scholar]
- 91.Boven LA, Middel J, Portegies P, Verhoef J, Jansen GH, Nottet HS. Overexpression of nerve growth factor and basic fibroblast growth factor in AIDS dementia complex. J Neuroimmunol. 1999;97(1–2):154–162. doi: 10.1016/s0165-5728(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 92.Albrecht D, Garcia L, Cartier L, et al. Trophic factors in cerebrospinal fluid and spinal cord of patients with tropical spastic paraparesis, HIV, and Creutzfeldt–Jakob disease. AIDS Res Hum Retroviruses. 2006;22(3):248–254. doi: 10.1089/aid.2006.22.248. [DOI] [PubMed] [Google Scholar]
- 93.Saarelainen T, Vaittinen S, Castren E. trkB-receptor activation contributes to the kainate-induced increase in BDNF mRNA synthesis. Cell Mol Neurobiol. 2001;21(4):429–435. doi: 10.1023/A:1012775808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu X, Yao H, Peng F, Callen S, Buch S. PDGF-mediated protection of SH-SY5Y cells against Tat toxin involves regulation of extracellular glutamate and intracellular calcium. Toxicol Appl Pharmacol. 2009;240(2):286–291. doi: 10.1016/j.taap.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peng F, Dhillon NK, Yao H, Zhu X, Williams R, Buch S. Mechanisms of platelet-derived growth factor-mediated neuroprotection – implications in HIV dementia. Eur J Neurosci. 2008;28(7):1255–1264. doi: 10.1111/j.1460-9568.2008.06444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peng F, Dhillon N, Callen S, et al. Platelet-derived growth factor protects neurons against gp120-mediated toxicity. J Neurovirol. 2008;14(1):62–72. doi: 10.1080/13550280701809084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ. TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ. 2009;16(12):1681–1693. doi: 10.1038/cdd.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sacktor N, Nakasujja N, Skolasky RL, et al. HIV subtype D is associated with dementia, compared with subtype A, in immunosuppressed individuals at risk of cognitive impairment in Kampala, Uganda. Clin Infect Dis. 2009;49(5):780–786. doi: 10.1086/605284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Satishchandra P, Nalini A, Gourie-Devi M, et al. Profile of neurologic disorders associated with HIV/AIDS from Bangalore, south India (1989–1996) Indian J Med Res. 2000;111:14–23. [PubMed] [Google Scholar]
- 100.Shankar SK, Mahadevan A, Satishchandra P, et al. Neuropathology of HIV/AIDS with an overview of the Indian scene. Indian J Med Res. 2005;121(4):468–488. [PubMed] [Google Scholar]
- 101.Yepthomi T, Paul R, Vallabhaneni S, et al. Neurocognitive consequences of HIV in southern India: a preliminary study of clade C virus. J Int Neuropsychol Soc. 2006;12(3):424–430. doi: 10.1017/s1355617706060516. [DOI] [PubMed] [Google Scholar]
- 102.Clifford DB, Mitike MT, Mekonnen Y, et al. Neurological evaluation of untreated human immunodeficiency virus infected adults in Ethiopia. J Neurovirol. 2007;13(1):67–72. doi: 10.1080/13550280601169837. [DOI] [PubMed] [Google Scholar]
- 103.Gupta JD, Satishchandra P, Gopukumar K, et al. Neuropsychological deficits in human immunodeficiency virus type 1 clade C-seropositive adults from south India. J Neurovirol. 2007;13(3):195–202. doi: 10.1080/13550280701258407. [DOI] [PubMed] [Google Scholar]
- 104.Riedel D, Ghate M, Nene M, et al. Screening for human immunodeficiency virus (HIV) dementia in an HIV clade C-infected population in India. J Neurovirol. 2006;12(1):34–38. doi: 10.1080/13550280500516500. [DOI] [PubMed] [Google Scholar]
- 105.Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol. 2008;63(3):366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- 106.Campbell GR, Watkins JD, Loret EP, Spector SA. Differential induction of rat neuronal excitotoxic cell death by human immunodeficiency virus type 1 clade B and C tat proteins. AIDS Res Hum Retroviruses. 2011;27(6):647–654. doi: 10.1089/aid.2010.0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nath A, Hauser KF, Wojna V, et al. Molecular basis for interactions of HIV and drugs of abuse. J Acquired Immune Deficiency Syndr. 2002;31(Suppl 2):S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- 108.Ferris MJ, Mactutus CF, Booze RM. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev. 2008;32(5):883–909. doi: 10.1016/j.neubiorev.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Langford D, Adame A, Grigorian A, et al. Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J Acquir Immune Defic Syndr. 2003;34(5):467–474. doi: 10.1097/00126334-200312150-00004. [DOI] [PubMed] [Google Scholar]
- 110.Chana G, Everall IP, Crews L, et al. Cognitive deficits and degeneration of interneurons in HIV+ methamphetamine users. Neurology. 2006;67(8):1486–1489. doi: 10.1212/01.wnl.0000240066.02404.e6. [DOI] [PubMed] [Google Scholar]
- 111.Turchan J, Anderson C, Hauser KF, et al. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci. 2001;2:3. doi: 10.1186/1471-2202-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cass WA, Harned ME, Peters LE, Nath A, Maragos WF. HIV-1 protein Tat potentiation of methamphetamine-induced decreases in evoked overflow of dopamine in the striatum of the rat. Brain Res. 2003;984(1–2):133–142. doi: 10.1016/s0006-8993(03)03122-6. [DOI] [PubMed] [Google Scholar]
- 113.Maragos WF, Young KL, Turchan JT, et al. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J Neurochem. 2002;83(4):955–963. doi: 10.1046/j.1471-4159.2002.01212.x. [DOI] [PubMed] [Google Scholar]
- 114.Conant K, St Hillaire C, Anderson C, Galey D, Wang J, Nath A. Human immunodeficiency virus type 1 Tat and methamphetamine affect the release and activation of matrix-degrading proteinases. J Neurovirol. 2004;10(1):21–28. doi: 10.1080/13550280490261699. [DOI] [PubMed] [Google Scholar]
- 115.Langford D, Grigorian A, Hurford R, Adame A, Crews L, Masliah E. The role of mitochondrial alterations in the combined toxic effects of human immunodeficiency virus Tat protein and methamphetamine on calbindin positive-neurons. J Neurovirol. 2004;10(6):327–337. doi: 10.1080/13550280490520961. [DOI] [PubMed] [Google Scholar]
- 116.Theodore S, Cass WA, Nath A, Steiner J, Young K, Maragos WF. Inhibition of tumor necrosis factor-alpha signaling prevents human immunodeficiency virus-1 protein Tat and methamphetamine interaction. Neurobiol Dis. 2006;23(3):663–668. doi: 10.1016/j.nbd.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 117.Qi L, Gang L, Hang KW, et al. Programmed neuronal cell death induced by HIV-1 tat and methamphetamine. Microsc Res Tech. 2011;74(12):1139–1144. doi: 10.1002/jemt.21006. [DOI] [PubMed] [Google Scholar]
- 118.Bandaru VV, Patel N, Ewaleifoh O, Haughey NJ. A failure to normalize biochemical and metabolic insults during morphine withdrawal disrupts synaptic repair in mice transgenic for HIV-gp120. J Neuroimmune Pharmacol. 2011;6(4):640–649. doi: 10.1007/s11481-011-9289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fitting S, Xu R, Bull C, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177(3):1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, et al. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia. 2008;56(13):1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu S, Sheng WS, Lokensgard JR, Peterson PK. Morphine potentiates HIV-1 gp120-induced neuronal apoptosis. J Infect Dis. 2005;191(6):886–889. doi: 10.1086/427830. [DOI] [PubMed] [Google Scholar]
- 122.Suzuki M, El-Hage N, Zou S, et al. Fractalkine/CX3CL1 protects striatal neurons from synergistic morphine and HIV-1 Tat-induced dendritic losses and death. Mol Neurodegen. 2011;6:78. doi: 10.1186/1750-1326-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hauser KF, El-Hage N, Buch S, et al. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1(1):98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- 124.Zou S, Fitting S, Hahn YK, et al. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain. 2011;134(Pt 12):3616–3631. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Avdoshina V, Biggio F, Palchik G, Campbell LA, Mocchetti I. Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia. 2010;58(13):1630–1639. doi: 10.1002/glia.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Podhaizer EM, Zou S, Fitting S, et al. Morphine and gp120 toxic interactions in striatal neurons are dependent on HIV-1 strain. J Neuroimmune Pharmacol. 2011 doi: 10.1007/s11481-011-9326-z. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tan IL, McArthur JC. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs. 2012;26(2):123–134. doi: 10.2165/11597770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 128.Gelman BB, Nguyen TP. Synaptic proteins linked to HIV-1 infection and immunoproteasome induction: proteomic analysis of human synaptosomes. J Neuroimmune Pharmacol. 2010;5:92–102. doi: 10.1007/s11481-009-9168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]