

Abstract
Desformylflustrabromine (dFBr; 1) and desformylflustrabromine-B (dFBr-B; 2) have been previously isolated from natural sources, and the former has been demonstrated to be a novel and selective positive allosteric modulator of α4β2 nicotinic acetylcholine (nACh) receptors. The present study describes the synthesis of water-soluble salts of 1 and 2, and confirms and further investigates the actions of 1 and 2 using two-electrode voltage clamp recordings.
Keywords: Nicotinic cholinergic receptors, Allosteric modulators
Over the past decade or so, nicotinic acetylcholine (nACh) receptors have been targeted for the development of agents with potential for the treatment of certain cardiovascular and neurological (e.g., Parkinson’s disease, schizophrenia, Alzheimer’s disease) disorders, memory and learning, appetite control, and pain.1,2 These receptors are pentameric ion channel receptors composed of heteromeric or homomeric assemblies and, although numerous subtypes have been identified, the most prevalent in brain are the α4β2 and the homomeric α7 nACh receptors.1 A few recently developed ligands can differentiate between the latter two subtypes, but few (if any) have been shown to selectively target α4β2 or any other nACh receptor subtype.2 A novel strategy for the modulation of nACh receptor subtypes is the identification of allosteric modulators that interact at receptors at a site distinct from that of the endogenous ligand; they modulate the receptor protein via a non-competitive mechanism. Negative allosteric modulators of nACh receptors have received some attention (reviewed1,3–5); however, relatively little work has been published on positive nACh receptor allosteric modulators (or non-competitive agonists),1,3,6,7 and it is not yet known precisely how they work.8,9 Nevertheless, several distinct binding domains for examples of the latter agents already have been proposed from docking studies using models of α3β4, αβ4β2, and α7 nACh receptors.1,10
Recently, desformylflustrabromine (dFBr; 1) was identified as a positive allosteric modulator of nACh receptors. dFBr (1), desformylflustrabromine-B (dFBr-B; 2), and at least a dozen additional indolic alkaloids have been isolated and characterized from the marine bryo-zoan Flustra foliacea.11–13
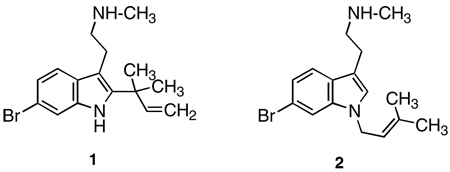 |
In particular, dFBr (1) and dFBr-B (2) (Ki = 3400 nM and >50,000 nM, respectively) lack appreciable affinity for α4β2 receptors, and bind with >3500-fold lower affinity at this receptor subtype than the standard agonist ligand (−)-nicotine (Ki < 1 nM); dFBr displays even lower affinity (Ki > 50,000 nM) for α7 nACh receptors.14 However, it has been further demonstrated with several heteromeric (i.e., α3β2, α3β4, α4β2, α4β4) and the homomeric α7 nACh receptors expressed in Xenopus oocytes, that dFBr (1), but not dFBr-B (2), selectively increases the whole cell current obtained when co-applied with the endogenous agonist ACh in α4β2-containing preparations.15 Single channel studies with dFBr are consistent with the hypothesis that it increases the channel-opening probability, perhaps by increasing the ratio of the rate constants for channel opening and closing.15 As such, dFBr offers a novel tool for the investigation of nACh receptors, and serves as a lead structure for the development of selective agents with potential therapeutic application.
To date, studies with dFBr (1) and dFBr-B (2) have employed DMSO solutions of their water-insoluble free bases as isolated directly from the marine organism. The purpose of the present study was (a) to identify a convenient synthetic route to 1 and 2, and to prepare their water-soluble salts, and (b) to confirm, and further examine, their actions at α4β2 and α7 nACh receptors.
Tryptamine 7 (Scheme 1) was a key intermediate in the synthesis of both 1 and 2. Although 7 has been previously reported,16 a different method of preparation was employed here. In the present study, 6-bromoindole was used in a Speeter/Anthony reaction and the resulting glyoxylamide was conveniently reduced to amine 6 using DMEA-alane, and then converted to 7 by treatment with (Boc)2O.
Scheme 1.

Reagents and conditions: (a) oxalyl chloride, Et2O, rt; (b) MeNH2/H2O, rt; (c) DMEA-alane/THF, Δ; (d) (Boc)2O, Et3N, DMF; (e) i—tBuOCl/THF, −78 °C; ii—prenyl 9-BBN, THF, −78 °C; (f) i—TFA, CH2Cl2, 30 min, rt; ii—HCl/Et2O; (g) i—NaH, DMF, 0 °C; ii—(Me)2C=CHACH2Cl.18
Compound 1 was subsequently obtained from 7 by reaction with prenyl 9-BBN17 followed by deprotection, whereas the anion generated from 7 using NaH was alkylated with 1-chloro-3-methyl-2-butene to afford 2 after deprotection (Scheme 1). Both 1 and 2 were obtained in moderately good yields and were isolated as their water-soluble hydrochloride salts.
Proton NMR spectra were obtained for dFBr (1) and dFBr-B (2) as their free bases (for comparison with their literature11,14 spectra) and as their salts. In general, the spectra of the free bases were consistent with those reported except that the N-CH3 signal for 1 appeared as a singlet at δ 2.4318 rather than at δ 3.39.11 A small signal at the latter position was evident in the spectra of 1 but appeared to be that from deuterated MeOH used as solvent.
Sala et al.15 identified a potentiating action of dFBr onβ2-containing nACh receptors. We evaluated the actions of synthetic dFBr (1) and dFBr-B (2) as their HCl salts at concentrations ranging from 1 nM to 100 µM on human α7 and α4β2 nACh receptors expressed in Xenopus oocytes with two-electrode voltage clamp techniques.19
Application of 100 µM ACh produced a near maximal response in α7 receptors (Fig. 1). Co-application of >0.1 µM dFBr (1) and 100 µM ACh inhibited the response to ACh. No potentiation of α7 responses was observed over the range of dFBr (1) concentrations tested. A dose–response curve obtained from multiple experiments identical to that shown in Figure 1A is plotted in Figure 1B. Peak amplitudes of currents obtained from α7 receptors were inhibited by dFBr (1) (IC50 = 44 ± 1 µM). The low potency for dFBr inhibition is consistent with its low affinity for the receptors.11 Our data demonstrate an inhibitory action of dFBr (1) on α7 receptors; Sala et al.15 also observed slight inhibition of α7 receptors by dFBr (1).
Figure 1.

dFBr (1) effects on α7 nACh receptors. dFBr (1 nM to 100 µM) was co-applied with 100 µM ACh to Xenopus oocytes expressing human α7 nACh receptors. Responses were obtained using two-electrode voltage clamp at a −60 mV holding potential. Peak amplitudes were normalized to the responses obtained using 100 µM ACh in the absence of dFBr (1). (A) Currents elicited by 100 µM ACh at dFBr (1) concentrations of 0, 0.1, and 1.0 µM. (B) Concentration/ response curve for dFBr inhibition of peak currents elicited by 100 µM ACh (IC50 = 44 µM).
On α4β2 receptors, co-application of 100 µM ACh and dFBr (1) produced a biphasic response over the concentration range tested (Fig. 2). Responses elicited by 100 µM ACh were potentiated by dFBr (1) (0.001–100 µM), although an overlapping inhibition of the response was observed at concentrations >10 µM dFBr (1), producing a bell shaped dose–response curve. Similar results were obtained when the slope of the rise time of the response was plotted rather than peak amplitudes (data not shown). The IC50 value estimated for the inhibitory component of this curve is 150 ± 0.2 µM. This value is 44-fold higher than that reported by Peters et al.11 (3.4 µM) for inhibition of [3H]epibatidine binding at rat α4β2 receptors. Similar to our data, Sala et al.15 did not report inhibition of ACh responses at these concentrations of dFBr but observed potentiation at 3 µM. While Peters’ data showed displacement of [3H]epibatidine binding it is not clear if this is due to competitive or non-competitive effects. If dFBr binding to an allosteric potentiation site reduces the affinity of epibatidine, then dFBr could appear as an inhibitor in a binding assay and as a potentiator of ACh responses in a functional assay. Unlike Sala et al.,15 we observe inhibition of 100 µM ACh responses at high (>10 µM) dFBr concentrations. However, the current traces obtained at these concentrations were not typical of those resulting from displacement of agonist. Displacement of agonist typically produces a decrease in overall response amplitude with little change in response profiles. In our study, high dFBr concentrations produced currents with a greater increase in the apparent rate and extent of desensitization rather than a simple decrease in overall response amplitudes (see Fig. 2A). This is particularly evident when comparing equal amplitude responses in the presence of dFBr (Fig. 2A, c and d); trace c (1 µM dFBr—a sub-inhibiting dFBr concentration) shows a relatively slow apparent rate of desensitization, while trace d (10 µM—an inhibiting dFBr concentration) shows a much sharper peak with a faster apparent rate of desensitization. If inhibition were due to displacement of ACh binding, these two response profiles would be expected to be similar. We also typically observed rebound currents on washout of ACh/dFBr (1) (Fig. 3). These currents are commonly observed with open-channel block due to removal of the channel blocker prior to dissociation of the agonist from the receptor. The rebound currents are particularly evident in the 10 µM responses as shown in Figure 3. Our data support the hypothesis that open-channel block contributes to inhibition of ACh responses by dFBr. Similar effects have been observed for other potentiating ligands including physostigmine and levamisole. 20,21 Thus, the inhibition observed at high dFBr (1) concentrations may not be related to the inhibition of [3H]epibatidine binding observed by Peters et al.11 but might occur via other mechanisms. Additional studies will be necessary to fully explain inhibition of [3H]epibatidine binding at potentiating dFBr (1) concentrations and the inhibition of response amplitudes at higher dFBr (1) concentrations.
Figure 2.

dFBr (1) effects on α4β2 nACh receptors. (A) Responses were obtained using two-electrode voltage clamp on Xenopus oocytes expressing human α4β2 nACh receptors. The responses elicited by each concentration of ACh/dFBr are indicated by the letters above the current trace. The time of drug exposure is indicated by the horizontal bar above the traces. Trace c (1 µM dFBr) produced a maximum potentiation of the peak response. Reduced peak amplitudes are seen at concentrations >1 µM dFBr (trace d). Peak amplitudes for each trace are: a. 14 µA, b. 24 µA, c. 37 µA, d. 25 µA. (B) Dose/response curve for dFBr applied in the presence of 100 µMACh on human α4β2 receptors. Peak amplitudes were normalized to peak currents obtained from 100 µM ACh in the absence of dFBr. Identical experimental conditions were used in A and B. The half-maximal potentiation observed is 120 nM (IC50 = 150 µM). Maximum potentiation was 295% of the control trace (n > 4, 13 oocytes).
Figure 3.

Rebound currents evident on washout of ACh and dFBr. Current traces were elicited by co-application of 100 µM ACh and dFBr (10 and 30 µM). Experimental conditions are identical to those shown in Figure 2. All three responses were obtained from a single oocyte. The horizontal black bar above the traces indicates the presence of ACh (a) or ACh + dFBr (b). Rebound currents are particularly evident in trace b.
The potentiating component of the dose–response curve obtained by co-application of dFBr (1) and 100 µM ACh produced a half-maximal potentiation at a concentration of 120 ± 0.6 nM dFBr (1) although the true potency is likely obscured by the inhibitory component of the dose–response curve (Fig. 2B). A maximum potentiation of 295 ± 67% of the control trace was observed at approximately 3 µM dFBr (1). This is similar to the 250% potentiation observed by Sala et al.15 To determine the effect of holding potential on potentiation by dFBr (1), voltage clamp experiments were performed using holding potentials varying from −80 to −10 mV. No effect of membrane potential on dFBr (1) potentiation of α4β2 receptors was observed. A comparison of ACh dose–response curves in the presence and absence of 1 µMdFBr (Fig. 4) showed a decrease in the apparent EC50 (24 ± 1 µM to 12±1 µM) for ACh and a corresponding increase in the maximal response obtained, suggesting that potentiating effects may be due to binding of dFBr (1) to a site distinct from the agonist binding site as has been proposed for other ACh potentiating ligands.
Figure 4.

Acetylcholine concentration/response curve in the presence and absence of dFBr (1). Responses were obtained using two-electrode voltage clamp on Xenopus oocytes expressing human α4β2 receptors. Peak amplitudes were normalized to peak currents obtained in the presence of 100 µM ACh in the absence of dFBr. A comparison of EC50 values with and without dFBr shows a shift in potency of ACh for α4β2 receptors from 24 µM (without dFBr) to 12 µM (with dFBr). dFBr increased the maximal response by 225% when compared to the maximal peak amplitudes in the absence of dFBr. The data shown reflect at least four repeated evaluations for each point on a minimum of six oocytes.
dFBr-B (2) was evaluated for its effects on human α7 and α4β2 nACh receptors (Fig. 5). Co-application of 2 with 100 µM ACh produced responses that were inhibited when compared to those obtained using 100 µM ACh alone (IC50 = 14 ± 1 µM). Inhibited response profiles were similar to those resulting from co-application of dFBr (1) on α7 receptors; currents were decreased in amplitude with no alteration in the apparent rate of desensitization. In contrast, co-application of dFBr-B (2) with 100 µM ACh produced a slight potentiation of responses (although the observed increases were not statistically significant) followed by inhibition of the responses (IC50 = 209 ± 4 µM). Inhibiting concentrations of dFBr-B (2) produced responses showing increased apparent desensitization rates and decreased peak currents similar to those obtained for high concentrations of dFBr (1) and dFBr-B (2) on α4β2 receptors (Fig. 5B). These data suggest that 1 and 2 may inhibit α4β2 responses by a similar mechanism. Since both 1 and 2 alter apparent desensitization rates on α4β2 but not α7 receptors their mechanism of action might be different on each receptor subtype.
Figure 5.

dFBr-B (2) effects on (A) α4β2 and (B) α7 nACh receptors. Currents elicited by 100 µM ACh in the presence of dFBr-B (2) using two-electrode voltage clamp. dFBr-B (2) was co-applied at varying concentrations with 100 µM ACh to Xenopus oocytes expressing human α4β2 or human α7 nACh receptors. (C) Concentration/response curve showing the normalized response to co-application of dFBr-B (2) and 100 µM ACh on human α7 nACh receptors. (D) Concentration/response curve of the normalized response to co-application of dFBr-B (2) and 100 µM ACh on human α4β2 nACh receptors. Peak amplitudes were normalized to peak currents elicited by 100 µM ACh in the absence of dFBr-B (2). Data reflect at least four repeated evaluations for each point on a minimum of five oocytes.
The synthetic HCl salts of dFBr (1) and dFBr-B (2) evaluated in this study behaved similarly to the compounds obtained from F. foliacea. Two primary differences were observed. First, while Sala et al.15 observed only potentiation, we observed both potentiation and inhibition of peak amplitudes by dFBr (1) on α4β2 receptors and inhibition only on α7 receptors. IC50 values for inhibition of acetylcholine responses on α7 receptors agree with the Ki values determined by Peters et al.11 in competition binding experiments. IC50 values obtained on α4β2 receptors were significantly higher than those obtained by Peters for inhibition of [3H]epibatidine binding and did not produce the decrease in overall responses typical of competitive antagonists. The change in response profiles, particularly the apparent loss of the slow desensitizing phase and the presence of rebound currents on washout of acetylcholine/dFBr, supports the hypothesis that inhibition is due to open-channel block at higher dFBr concentrations. In addition to the inhibitory effects of dFBr, we also observed an increased potency for potentiation and a slight increase in the maximal amount of potentiation obtained compared to data presented by Sala et al.15 While these differences are not easily explained they could be due to a difference in solubility between the synthetic HCl salt used in our study and the compound purified from F. foliacea used by Sala. Sala et al.15 utilized DMSO to solubilize dFBr due to its low solubility in buffer, while we used a more soluble HCl salt that may have permitted better dissolution of the compound. Decreased availability of dFBr due to DMSO solubilization or decreased concentrations of dFBr due to solubility problems would be reflected in an apparent decrease in potency for potentiation and may have prevented observation of inhibition. This conclusion is consistent with the higher potency and maximal potentiation we observed. In our preparation, dFBr showed potentiating effects at concentrations as low as 30 nM, whereas Sala et al.15 reported no potentiation below 3 µM. The rapid perfusion system used in the present study may also have contributed to these differences. We use a modified Xenopus oocytes recording chamber that permits solution changes that are significantly faster than those used by Sala et al.15 This system more accurately detects peak amplitudes prior to desensitization since solution exchange takes place within 200 ms. Slower perfusion and exchange rates would result in smaller response amplitudes due to receptor desensitization, and less potentiation would be observed at lower dFBr concentrations.
The water-soluble HCl salt of dFBr (1) examined in this study provides a novel, subtype selective modulator of nACh receptors. We have confirmed that dFBr (1), but not dFBr-B (2), is capable of increasing responses on α4β2 receptors when co-applied with ACh. This modulation is due to an increase in efficacy of ACh and not simply a shift in EC50 value. Effects are observed at dFBr concentrations ranging from 30 nM to 100 µM although inhibition of peak amplitudes and alterations in desensitization rates are observed at concentrations >3 µM. dFBr-B (2) inhibited ACh-induced responses both on α7 and α4β2 receptors. Although dFBr-B (2) does not significantly potentiate responses to ACh on α4β2 receptors, our data show that it possesses a similar capacity for inhibiting response amplitudes and altering apparent desensitization rates at higher concentrations. Hence, similar effects produced by 1 and 2 might be due to structural features associated with the parent N-methyl-6-bromotryptamine moiety common to both, whereas the potentiating effects of dFBr (1) appear related to structural features not shared by these two agents. The selectivity of dFBr (1) forβ2-containing nACh receptors makes it an ideal lead molecule for future studies involving selective modulation of α4β2 nACh receptors and a possible candidate for the treatment of diseases such as autism or Alzheimer’s disease in which α4β2 nACh receptors are decreased in some brain regions. Selective, potentiating ligands would produce increased response amplitudes at nicotinic synapses without the loss of synaptic control or desensitization that results from the use of nicotinic agonists.
Footnotes
Note added in proof
While our manuscript was being reviewed, Lindel et al., Organic Lett., 2007, 9, 283, published an alternative synthesis of desformylflustrabromine.
References and notes
- 1.Arias HR. Neurochem. Int. 2000;36:595. doi: 10.1016/s0197-0186(99)00154-0. [DOI] [PubMed] [Google Scholar]
- 2.Glennon RA. Progress in Medicinal Chemistry. Vol. 42. Amsterdam: Elsevier Science; 2004. p. p 55. [DOI] [PubMed] [Google Scholar]
- 3.Pereira EFR, Hilmas C, Santos MD, Alkondon M, Maelicke A, Albuqerque EX. J. Neurobiol. 2002;53:479. doi: 10.1002/neu.10146. [DOI] [PubMed] [Google Scholar]
- 4.Arias HR. J. Neurosci. Res. 1998;52:369. doi: 10.1002/(SICI)1097-4547(19980515)52:4<369::AID-JNR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 5.Arias HR, Bhumireddy P, Bouzat C. Int. J. Biochem. Cell Biol. 2006;38:1254. doi: 10.1016/j.biocel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Albuquerque EX, Santos MD, Alkondon M, Pereira EF, Maelicke A. Alzheimer Dis. Assoc. Disord. 2001;15:S19. doi: 10.1097/00002093-200108001-00004. [DOI] [PubMed] [Google Scholar]
- 7.Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffmann WE, Piotrowski DW, Groppi VE, Allaman G, Ogier R, Bertrand S, Bertrand D, Arneric SP. J. Neurosci. 2005;25:4396. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samochocki M, Hofle A, Fehrenbacher A, Jostock R, Ludwig J, Christner C, Radina M, Zerlin M, Ullmer C, Pereira EFR, Lubbert H, Albuquerque EX, Maelicke A. J. Pharmacol. Exp. Ther. 2003;305:1024. doi: 10.1124/jpet.102.045773. [DOI] [PubMed] [Google Scholar]
- 9.Smulders CJGM, Zwart R, Bermudez I, van Kleef RGDM, Groot-Kormelink PJ, Vijerberg HPM. Eur. J. Pharmacol. 2005;509:97. doi: 10.1016/j.ejphar.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 10.Iorga B, Herlem D, Barre E, Guillou C. J. Mol. Model. 2006;12:366. doi: 10.1007/s00894-005-0057-z. [DOI] [PubMed] [Google Scholar]
- 11.Peters L, Konig GM, Terlau H, Wright AD. J. Nat. Prod. 2002;65:1633. doi: 10.1021/np0105984. [DOI] [PubMed] [Google Scholar]
- 12.Peters L, Wright AD, Krock A, Konig GM. J. Chem. Ecol. 2004;30:1165. doi: 10.1023/b:joec.0000030270.65594.f4. [DOI] [PubMed] [Google Scholar]
- 13.Lysek N, Rachor E, Lindel T. Verlag Zeit. Naturforsch. 2002;1056 doi: 10.1515/znc-2002-11-1218. www.znaturforsch.com. [DOI] [PubMed] [Google Scholar]
- 14.Peters L, Wright AD, Kehraus S, Gündisch D, Tilotta MC, König GM. Planta Med. 2004;70:883. doi: 10.1055/s-2004-832610. [DOI] [PubMed] [Google Scholar]
- 15.Sala F, Mulet J, Reddy KP, Bernal JA, Wikman P, Valor LM, Peters L, Konig GM, Criado M, Sala S. Neurosci. Lett. 2005;373:144. doi: 10.1016/j.neulet.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Burm BEA, Meijler MM, Korver J, Wanner MJ, Koomen G-J. Tetrahedron. 1998;54:6135. [Google Scholar]
- 17.Schkeryantz JM, Woo JCG, Siliphaivanh P, Depew KM, Danishefsky JJ. J. Am. Chem. Soc. 1999;121:11964. [Google Scholar]
- 18.Compounds analyzed within 0.4% of theory for C, H, N where indicated. The free base of dFBr (1) was obtained as a pale yellow oil by deprotection of 8 with TFA at rt: 1H NMR (CD3OD) δ 1.55 (s, 6H, 2CH3), 2.43 (s, 3H, N-CH3), 2.71–2.80 (m, 2H, CH2), 2.95–3.04 (m, 2H, CH2), 5.11 (dd, J = 1.2, 10.5 Hz, 1H, vinylic H), 5.13 (dd, J = 1.2, 17.4 Hz, 1H, vinylic H), 6.18 (dd, J = 10.5, 17.4 Hz, 1H, vinylic H), 7.08 (dd, J = 1.8, 8.4 Hz, 1H, ArH), 7.39 (d, J = 8.4 Hz, 1H, ArH), 7.47 (d, J = 1.8 Hz, 1H, ArH). 1·HCl (off-white solid; mp 211–214 °C); 1H NMR (DMSO-d6) δ 1.49 (s, 6H, 2CH3), 2.61 (s, 3H, N-CH3), 2.90–3.01 (m, 2H, CH2), 3.01–3.11 (m, 2H, CH2), 5.10 (dd, J = 1.2, 17.7 Hz, 1H, vinylic H), 5.11 (dd, J = 1.2, 10.8 Hz, 1H, vinylic H), 6.14 (dd, J = 10.8, 17.7, 1H, vinylic H), 7.13 (dd, J = 1.8, 8.4 Hz, 1H, ArH), 7.46–7.54 (m, 2H, ArH), 8.54 (br s, 2H, H2O, HCl), 10.79 (br s, 1H, NH); Anal. for C16H21Br N2·HCl·H2O. dFBr-B was obtained as a pale yellow oil from 9; 1H NMR (CD3OD) δ 1.79 (br s, 3H, CH3), 1.86 (br s, 3H, CH3), 2.42 (s, 3H, NCH3), 2.83–2.89 (m, 2H, CH2), 2.90–2.97 (m, 2H, CH2), 4.68 (br d, J = 6.9 Hz, 2H, allylic CH2), 5.35 (m, 1H, vinylic H), 7.05 (br s, 1H, ArH), 7.14 (dd, J = 1.8, 8.7 Hz, 1H, ArH), 7.47 (d, J = 8.7 Hz, 1H, ArH), 7.49 (d, J = 1.8 Hz, 1H, ArH). 2·HCl (creamy-white solid; mp 161–163 °C); 1H NMR (DMSO-d6) δ 1.73 (s, 3H, CH3), 1.82 (s, 3H, CH3), 2.57 (s, 3H, N-CH3), 2.98–3.07 (m, 2H, CH2), 3.07–3.18 (m, 2H, CH2), 4.73 (br d, J = 6.6 Hz, 2H, allylic CH2), 5.32 (m, 1H, vinylic H), 7.18 (dd, J = 1.8, 8.7 Hz, 1H, ArH), 7.26 (s, 1H, ArH), 7.58 (d, J = 8.7 Hz, 1H, ArH), 7.67 (d, J = 1.8 Hz, 1H, ArH), 8.82 (br s, 2H, NH, HCl). Anal. for C16H21N2·HCl.Compound 5, prepared by reaction of oxalyl chloride with 3 followed by stirring with H2NMe (40% in water) (off-white solid; mp 249–252 °C, dec), has been prepared by a different method,16 but no melting point was reported. Compound 6, prepared by DMEA-alane (0.5 Min toluene) reduction of 5 and purified by column chromatography (CH2Cl2/MeOH = 9:1→ CH2Cl2/MeOH/NH4OH = 9:1:0.1) (off-white solid, mp112–114 °C), although reported as a salt obtained by a different synthetic route,16 has not been previously isolated as its free base.Compound 7: Reaction of di-tert-butyl dicarbonate, Et3N, and 6 in DMF gave a crude product which was purified by column chromatography (hexane/EtOAc = 2:1) to afford 7 (white solid, mp 120–124 °C).Compound 8, from tBuOCl in THF with 7 and Et3N in THF at −78 °C under N2, followed by addition of prenyl 9-BBN,18 was purified by column chromatography (hexane/EtOAc = 7:1) to give a white foam.Compound 9, from the anion of 7 (NaH/DMF) at 0 °C under N2, treated with 1-chloro-3-methyl-2-butene in DMF; the resulting oil was purified by a column chromatography (hexane/EtOAc = 5:1) to give a homogeneous, colorless oil.
- 19.The cDNAs for human α7 and α4β2 receptors were obtained from Dr. Jon Lindstrom’s laboratory. Subunit cDNA was cloned into a pBud-CE4.1 (Invitrogen, CA) vector prior to mRNA synthesis. Xenopus laevis frogs and frog food were purchased from Xenopus Express (Homosassa, FL). Ovarian lobes were surgically removed from X. laevis frogs and washed twice in Ca+2-free Barth’s buffer (82.5 mM NaCl/2.5 mM KCl/1 mM MgCl2/5 mM Hepes, pH 7.4) then gently shaken with 1.5 mg/ml collagenase (Sigma type II, Sigma–Aldrich) for 1 h at 20–25 °C. Stage IV oocytes were selected for microinjection. Synthetic cRNA transcripts for human α7 and α4β2 were prepared using the mMESSAGE mMACHINE™ High Yield Capped RNA Transcription Kit (Ambion, TX). Oocytes were injected with a total of 50 nL cRNA at a concentration of 0.2 ng/nL in appropriate subunit ratios then incubated at 19 °C for 24 to 72 h prior to their use in voltage clamp experiments. Recordings were made using an automated two-electrode voltage-clamp system incorporating an OC-725C oocyte clamp amplifier (Warner Instruments, CT) coupled to a computerized data acquisition (Datapac 2000, RUN technologies) and autoinjection system (Gilson). Recording and current electrodes with resistance 1–4MΩ were filled with 3 M KCl. Details of the chambers and methodology employed for electrophysiological recordings have been described earlier.21 Oocytes were held in a vertical flow chamber of 200-µL volume and perfused with ND-96 recording buffer (96 mM NaCl/2 mM KCl/1.8 mM CaCl2/1 mM MgCl2/5 mM Hepes; pH 7.4) at a rate of 20 mL/min. Test compounds were dissolved in ND-96 buffer and injected into the chamber at a rate of 20 mL/min using the Gilson auto-sampler injection system. For dFBr (1) and dFBr-B (2) experiments, compounds were co-applied with 100 µM ACh. Data analysis: Concentration/response curves were fit by nonlinear curve fitting and GraphPad Prism Software (San Diego, CA) using standard built-in algorithms. For IC50 determinations, data were fit to a single site competition model. For the potentiation/inhibition curves obtained for α4β2 modulation by dFBr ·HCl, the data were fit to a bell shaped dose–response equation. This equation simultaneously fits both the potentiation and inhibitory effects to give an EC50 for the potentiation component and an IC50 for the inhibitory component. The accuracy by which this algorithm separates these two constants is dependent on the amount of difference between them. In the case of dFBr·HCl and dFBr-B·HCl, this difference is about 33-fold making it difficult to fully separate the two values.
- 20.Rauniyar V, Hall DG. J. Am. Chem. Soc. 2004;126:4518. doi: 10.1021/ja049446w. [DOI] [PubMed] [Google Scholar]
- 21.Joshi PR, Suryanarayanan A, Schulte MK. J. Neurosci. Methods. 2004;132:69. doi: 10.1016/j.jneumeth.2003.09.002. [DOI] [PubMed] [Google Scholar]