

Abstract
Chlamydia trachomatis is the most common sexually transmitted bacterial pathogen and the causative agent of blinding trachoma. Although Chlamydia is protected from humoral immune responses by residing within remodeled intracellular vacuoles, it still must contend with multilayered intracellular innate immune defenses deployed by its host while scavenging for nutrients. Here we provide an overview of Chlamydia biology and highlight recent findings detailing how this vacuole-bound pathogen manipulates host–cellular functions to invade host cells and maintain a replicative niche.
Chlamydia species are intracellular bacterial pathogens that cause urogenital and eye infections. They translocate a staggering number of protein effectors into host cells to co-opt cellular processes.
Chlamydiae are Gram-negative obligate intracellular bacterial pathogens responsible for a range of diseases of clinical and public health importance. For instance, distinct serovariants of Chlamydia trachomatis cause a gamut of diseases, including blinding trachoma (serovars A–C), urogenital tract infections leading to urethritis, cervicitis, and proctitis (serovars D–K), and systemic lymphogranuloma venereum (LGV) disease (serovars L1–L3) (Schachter 1999). In genital tract infections, C. trachomatis primarily replicates within the epithelium of the urethra of men and endocervix of women causing inflammation, edema, and mucosal discharge. Ascending uterine infections can lead to pelvic inflammatory disease (PID), tubal scarring, ectopic pregnancies, and infertility (reviewed in Bebear and de Barbeyrac 2009; Haggerty et al. 2010).
Chlamydia displays a biphasic developmental cycle (Fig. 1). The elementary body (EB) form of the bacteria attaches and invades host-epithelial cells. The nascent phagocytic compartment is rapidly modified by Chlamydia-derived proteins to generate a parasitophorous vacuole termed an inclusion. Within the inclusion, the EB differentiates into a reticulate body (RB), the metabolically active and replicative form of the pathogen. RBs divide by binary fission as the inclusion expands and midway through the infectious cycle begin to asynchronously differentiate back into the EB form. Newly formed EBs are eventually released by cell lysis and/or extrusion to initiate new rounds of infection (reviewed in Dautry-Varsat et al. 2005; Hybiske and Stephens 2007b).
Figure 1.

Life cycle of Chlamydia. Within the first 2 h following internalization into cells, elementary bodies (EBs) fuse to form a nascent inclusion. Between 2 and 6 h postinternalization, EBs begin to differentiate into reticulate bodies (RBs). By 12 h postinfection (hpi) RBs can be observed dividing by binary fission and by 18–24 hpi they peak in numbers. Increasing numbers of RBs differentiate back to EBs around 24 hpi and continue differentiating until lysis or release occurs ∼48–72 hpi depending on the chlamydial species (Hatch 1999).
At all stages of infection, Chlamydia translocates “effector” proteins directly into host membranes and cytoplasm for manipulation of host–cellular functions, including signal transduction pathways (Valdivia 2008). Here we discuss novel strategies used by Chlamydia to generate an environment conducive for intracellular replication and emerging avenues for dissecting chlamydial pathogenic strategies.
CELLULAR BASIS FOR THE INTERACTION OF Chlamydia AND HOST CELLS
Mechanisms of Chlamydia Invasion of Epithelial Cells
Chlamydiae can invade most cultured cells, suggesting that the receptor(s) facilitating invasion are ubiquitous or that multiple receptors can be used. Binding is thought to be a two-step process for some species, involving an initial reversible interaction between the EB and the host cell mediated by heparan sulfate proteoglycans (HSPGs), followed by high-affinity irreversible binding to a secondary receptor (Dautry-Varsat et al. 2005). In addition to heparan sulfate (Chen and Stephens 1994, 1997; Chen et al. 1996; Wuppermann et al. 2001), the mannose receptor, the mannose 6-phosphate receptor, and the estrogen receptor have been proposed to act as host receptors for Chlamydia entry (reviewed in Cocchiaro and Valdivia 2009). Cell surface-exposed protein disulfide isomerase (PDI) has also been shown to play an important role in EB attachment and entry (Abromaitis and Stephens 2009).
Recent studies have revealed an unexpected role for growth factors and their receptors in Chlamydia binding (Elwell et al. 2008; Kim et al. 2011). A genome-wide loss-of-function RNAi screen in Drosophila S2 cells identified a potential role for the platelet-derived growth factor receptor (PDGFR), a transmembrane tyrosine kinase, in Chlamydia binding. In mammalian cells, depletion of PDGFRβ or inhibition with PDGFRβ-neutralizing antibodies reduces bacterial binding (Elwell et al. 2008). In subsequent studies, fibroblast growth factor 2 (FGF2) was shown to bind to EBs, where it appears to function as an HSPG-dependent bridging molecule that enhances interactions between EBs and the FGF receptor (FGFR) (Kim et al. 2011). Remarkably, bacterial binding stimulates production and release of FGF2 through a pathway that is independent of FGFR stimulation although dependent on bacterial protein synthesis and activation of the ERK1/2 signaling pathway (Kim et al. 2011). These events establish a positive-feedback loop that may further propagate bacterial binding and spreading to surrounding cells.
Multiple bacterial adhesins and ligands have been proposed to mediate invasion and their use may vary depending on both the host-cell type and the chlamydial species (Cocchiaro and Valdivia 2009). These include glycosaminoglycan (Menozzi et al. 2002), the major outer membrane protein (MOMP) (Su et al. 1996), OmcB (Fadel and Eley 2007), and PmpD (Mölleken et al. 2010). OmcB from Chlamydia pneumoniae but not from C. trachomatis has an identifiable heparan sulfate-binding domain that, when expressed on the surface of yeast, is necessary for OmcB binding to HSPGs (Moelleken and Hegemann 2008). In the absence of a system for targeted inactivation of Chlamydia genes, it has been difficult to confirm the relative contribution of these putative ligands to Chlamydia attachment and entry.
Chlamydiae entry into nonphagocytic cells is mediated by small GTPase-dependent reorganization of the actin cytoskeleton (reviewed in Hackstadt 2000; Dautry-Varsat et al. 2005; Cocchiaro and Valdivia 2009; Carabeo 2011; Scidmore 2011). Rac1 is required for C. trachomatis entry (Carabeo et al. 2004), whereas Cdc42 and Arf6 are also required for Chlamydophila caviae internalization (Subtil et al. 2004; Balana et al. 2005). Rac1 activation results in the recruitment of the actin regulators WAVE2, Abi-1, and Arp2/3, which are necessary for C. trachomatis-induced actin reorganization (Carabeo et al. 2007). Both chlamydial and host proteins may function synergistically to promote invasion. In the EB, the type III secretion system (a multiprotein needlelike delivery system) (reviewed in Beeckman and Vanrompay 2010), is poised for immediate discharge of effectors on contact with the host cell. At least one early secreted effector, TARP, contributes to bacterial internalization by its ability to directly nucleate actin polymerization through a WH2 actin-binding domain mimic (Jewett et al. 2006) and by recruiting the guanine nucleotide exchange factors (GEFs) Sos1 and Vav2, which activate Rac1 and signal to the actin machinery (Carabeo et al. 2007; Lane et al. 2008). This latter mechanism is only relevant in chlamydial species in which the polymorphic TARP gene (Clifton et al. 2005) can be targeted for tyrosine phosphorylation by Abl, Src, and Syk kinases (Elwell et al. 2008; Jewett et al. 2008; Lane et al. 2008; Mehlitz et al. 2008, 2010). Microinjection of cells with antibodies directed to the actin-binding domain of TARP before infection significantly reduces bacterial invasion, providing the most conclusive support for a direct role of TARP in mediating bacterial entry (Jewett et al. 2010). In addition, several host-tyrosine kinases, including PDGFR, FGFR, and Abl kinase are phosphorylated upon infection, recruited to the site of bacterial attachment, and may function redundantly in entry (Elwell et al. 2008; Kim et al. 2011). Therefore, the entry process is complex, requiring a concerted activation of growth factor receptors, cytoplasmic tyrosine kinases, small GTPases, and TARP to synergistically remodel the actin cytoskeleton and promote bacterial uptake. The actin rearrangements that occur during entry are transient and may be terminated by secreted chlamydial effectors such as CT166, which glucosylates Rac1 (Thalmann et al. 2010), or CT694, which interacts and colocalizes with AHNAK, an actin-binding protein (Hower et al. 2009). Additional host factors that contribute to uptake into nonphagocytic cells include clathrin (Boleti et al. 1999; Hybiske and Stephens 2007a) and cholesterol-rich microdomains (Norkin et al. 2001; Jutras et al. 2003; Stuart et al. 2003; Gabel et al. 2004).
Creating a Unique Niche
Following entry, EBs are sequestered within a membrane-bound compartment, termed the inclusion. The inclusion then quickly dissociates from the endolysosomal pathway, traffics along microtubules to a peri-Golgi location, and avoids fusion with lysosomes (reviewed in Hackstadt 2000). Early studies failed to identify host proteins on the inclusion membrane, leading to a model wherein the inclusion was viewed as an isolated compartment with little to no interactions with other host-cell organelles (reviewed in Fields and Hackstadt 2002). However, recent studies reveal a complex set of interactions between the chlamydial inclusion and host-cell trafficking pathways to facilitate acquisition of essential host-derived nutrients, such as amino acids, lipids, and iron, while limiting detection by the innate immune system. The list of host proteins that associate with the inclusion is rapidly growing; however, the mechanism by which these proteins are recruited to the inclusion has only been elucidated in a few cases.
Transport to the Peri-Golgi Region
Soon after entry, nascent inclusions are transported along microtubules to the microtubule organizing center (MTOC) in a dynein-dependent but dynactin-independent manner (Clausen et al. 1997; Grieshaber et al. 2003; Grieshaber et al. 2006), suggesting that one or more unknown bacterial effectors mimic the cargo-binding activity of dynactin and tether the inclusion to dynein and/or to centrosomes. Src family kinases are required in human-adapted Chlamydia strains for inclusions to migrate to the MTOC and for intracellular growth, even though they are dispensable for binding and entry (Mital and Hackstadt 2011). Recent studies indicate that the inclusion membrane is not homogenous and that microdomains consisting of a subset of inclusion proteins (Incs) (IncB, Inc101, Inc222, and Inc850), active Src family kinases, and cholesterol, associate with centrosomes and dynein (Mital et al. 2010; Mital and Hackstadt 2011). One or more of these Incs may recruit active Src family kinases to the inclusion to promote microtubule nucleation, dynein-dependent movement, and sphingomyelin acquisition (Mital et al. 2010; Mital and Hackstadt 2011). The inclusion maintains a dynein-dependent association with the centrosome throughout the cell cycle, resulting in supernumerary centrosomes, abnormal spindle poles, and segregation defects (Grieshaber et al. 2006; Johnson et al. 2009). These findings are intriguing in light of the association of Chlamydia and human papilloma virus-associated cervical cancer (Wallin et al. 2002).
Interactions with Regulators of Membrane-Trafficking Pathways
The inclusion interacts with a number of host-cell molecules that are involved in membrane trafficking, fusion, and organelle identity. A subset of these are Rab GTPases including Rabs 4, 11, and 14, which are associated with recycling endosomes, and Rabs 1, 6, and 10, which are associated with ER-Golgi traffic (Rzomp et al. 2006; Brumell and Scidmore 2007; Moorhead et al. 2007, 2010; Capmany and Damiani 2010). Rabs 6 and 10 are recruited to inclusions in a species-specific manner, whereas recruitment of Rab1, 4, and 11 appear to be ubiquitous to all Chlamydiae (Rzomp et al. 2003). The C. trachomatis Inc protein CT229 has been identified as a Rab4-interacting protein, although it is not conserved in other strains or serovars that recruit Rab4, suggesting redundancy in Inc protein function (Rzomp et al. 2006). In the case of C. pneumoniae, Cpn0585 is thought to be responsible for the selective recruitment of Rab1, 10, and 11 (Cortes et al. 2007). In some instances, Rab-binding proteins are directly recruited to the inclusion membrane. Bicaudal D1 (BICD1), a Rab6-interacting protein, is recruited in a Rab6-independent manner, suggesting that an Inc protein may directly recruit BICD1 (Moorhead et al. 2007). The recruitment of Rabs and their associated proteins likely promotes selective interaction/fusion with host vesicles containing essential nutrients. Rab6, 11, and 14 play a role in the acquisition of sphingomyelin by the inclusion (Rejman Lipinski et al. 2009; Capmany and Damiani 2010), whereas Rab4 and Rab11 participate in iron acquisition by regulating a slow transferrin recycling pathway intercepted by the inclusion (Rzomp et al. 2003, 2006; Ouellette and Carabeo 2010). Rab6 and Rab11 facilitate sphingomyelin transport to the inclusion by regulating fragmentation of the Golgi into ministacks during infection (Heuer et al. 2009; Rejman Lipinski et al. 2009).
Phosphoinositides are also key determinants of membrane identity and vesicle fusion (Poccia and Larijani 2009), and several proteins associated with phosphoinositol-4-phosphate (PI4P) metabolism are recruited to the inclusion. These include oculocerebrorenal syndrome of Lowe protein 1 (OCRL1), phosphatidylinositol 4-kinase (PI4KIIα), and Arf1 (a GTPase that recruits PI4K enzymes and PI4P-binding proteins to the Golgi) (Moorhead et al. 2010; Elwell et al. 2011). Depletion of OCRL1, PI4KIIα, and Arf1 reduces inclusion formation and the production of infectious progeny, suggesting that generation of PI4P at the inclusion plays an important role in infection (Moorhead et al. 2010). Recruitment of Arf1 is intriguing, as it also regulates actin dynamics and vesicle trafficking at the Golgi (D’Souza-Schorey and Chavrier 2006), and thus may play multiple roles during infection.
In addition to recruiting Rab GTPases, Chlamydia may regulate fusion with host vesicles by recruiting host SNARES, which are key components of intracellular fusion machinery (Sudhof and Rothman 2009). In support of this notion, at least two Golgi-specific SNARES, including Syntaxin 6 and GS15 (Moore et al. 2011; Pokrovskaya et al. 2012), as well as vesicles containing the endocytic SNARES, Vamp3, Vamp7, and Vamp8, are recruited to the inclusion (Delevoye et al. 2008; Paumet et al. 2009). Interestingly, a subset of Incs contains motifs that display similarities to eukaryotic SNAREs, including IncA, CT813, and CT223 (Delevoye et al. 2008; Paumet et al. 2009). It is speculated that these SNARE-like domains pair with host-SNARE proteins on target vesicles to assemble a four-helix bundle that provides the energy to promote fusion of opposing membranes. Indeed, IncA, along with CT813, binds to host SNAREs, Vamp3, 7, and 8, suggesting that host SNAREs may work in concert with IncA to regulate membrane fusion (Delevoye et al. 2004, 2008; Paumet et al. 2009). However, in vitro liposome fusion assays indicate that instead of promoting fusion with endocytic compartments, IncA acts as an inhibitory SNARE to limit fusion with these compartments (Paumet et al. 2009). IncA has also been shown to participate in homotypic fusion of individual inclusions, as naturally occurring IncA-deficient variants or infected cells microinjected with anti-IncA antibodies display an aberrant multilobed inclusion structure (Hackstadt et al. 1999; Delevoye et al. 2004; Xia et al. 2005).
Interactions with Host-Cell Organelles for Acquisition of Host Nutrients
Early studies established a close association of the Chlamydia inclusion with the Golgi apparatus from which it intercepts sphingomyelin and cholesterol-containing exocytic vesicles by a Brefeldin A (BFA)-sensitive vesicular-trafficking pathway (Hackstadt et al. 1996; Carabeo et al. 2003). BFA inhibits activation of Arf1 (a key player in vesicle formation) by targeting its activators, the GEFs GBF1 and BIG1/2 (Claude et al. 1999; Mansour et al. 1999; Yamaji et al. 2000; Kawamoto et al. 2002), which are localized to the cis- and trans-Golgi, respectively, and have distinct functions at the Golgi (Manolea et al. 2008). Recent studies reveal that Chlamydia uses GBF1 (but not BIG1/2) to acquire sphingomyelin, which is necessary for inclusion membrane growth and stability but not bacterial replication (Elwell et al. 2011). Nonvesicular trafficking may also play a major role in lipid acquisition by Chlamydia (Derre et al. 2011; Elwell et al. 2011). Ceramide transfer protein (CERT), a cytosolic protein that transports ceramide from the ER to the trans-Golgi (Hanada 2010), is recruited to the inclusion membrane, possibly by interaction with the inclusion membrane protein IncD (Derre et al. 2011), and is involved in sphingomyelin acquisition (Elwell et al. 2011). Depletion of CERT significantly reduces the production of infectious progeny (Derre et al. 2011; Elwell et al. 2011). It is speculated that CERT mediates the transfer of ceramide from the ER to the inclusion, perhaps at ER-inclusion membrane contact sites, to generate specialized metabolic and/or signaling platforms (Derre et al. 2011). In addition, at least one of the two sphingomyelin synthase enzymes, which converts ceramide to sphingomyelin (SM) (Huitema et al. 2004), is recruited to the inclusion membrane (Elwell et al. 2011), raising the possibility that ceramide may also be converted to SM at the inclusion membrane, thereby creating a localized SM biosynthetic factory at the inclusion membrane. These observations shed some light on the paradox that although host-sphingomyelin biosynthesis is required for progeny production and inclusion biogenesis (van Ooij et al. 2000; Robertson et al. 2009), blocking vesicular transport with BFA has no inhibitory effect on the production of infectious progeny (Hackstadt et al. 1996). Acquisition of sphingomyelin by BFA-sensitive vesicular and nonvesicular pathways may play distinct functional roles, with BFA-sensitive vesicular transport participating in inclusion biogenesis and nonvesicular transport playing a key role in bacterial replication (Elwell et al. 2011). Host glycerophospholipids, such as phosphatidylinositol and phosphatidylcholine, are also acquired through a nonvesicular transport pathway by a process that requires activation of ERK and cytosolic phospholipase A2 (Wylie et al. 1997; Hatch and McClarty 1998; Su et al. 2004).
In addition to the Golgi, the inclusion interacts with other host-cell organelles, including (1) multivesicular bodies (MVBs) (Beatty 2006, 2008; Robertson et al. 2009), which may also serve as a source of sphingolipids and cholesterol, (2) lipid droplets, which are translocated into the inclusion lumen following their capture by the chlamydial proteins Lda1 and/or Lda3 and which may serve as a source of neutral lipids (Kumar et al. 2006; Cocchiaro et al. 2008), (3) mitochondria (Matsumoto et al. 1991; Derre et al. 2007), and (4) lysosomes, which may be a source of essential amino acids derived from host-protein degradation (Ouellette et al. 2011). Together, these interactions (summarized in Fig. 2) likely facilitate acquisition of nutrients for bacterial replication as well as for inclusion membrane stability and expansion.
Figure 2.

Chlamydia and host–cellular interactions. Chlamydia enters host cells by an actin-dependent mechanism that involves both host and bacterial factors. The EBs are enclosed within a membrane-bound inclusion, which becomes modified by insertion of bacterial inclusion proteins (Incs) that prevent fusion with lysosomes and promote interactions with recycling endosomes for iron acquisition. The nascent inclusion is transported along microtubules to the MTOC and EBs transition into RBs. The inclusion expands to accommodate the replicating RBs. During this time, the inclusion interacts, likely via bacterial effectors, with multiple host-cell organelles, including fragmented Golgi ministacks, the ER, lipid droplets, MVBs, and the mitochondria. Delivery of essential host lipids to the inclusion involves vesicular trafficking from Golgi ministacks and MVBs as well as nonvesicular trafficking from lipid droplets and membrane contact sites (MCS) formed between the ER and the inclusion. Although the inclusion does not fuse with lysosomes, Chlamydia may import essential amino acids derived from host-protein degradation within lysosomes.
Exiting the Host Cell
At the end of the developmental cycle, EBs are released by two mutually exclusive mechanisms: cell lysis and extrusion (Hybiske and Stephens 2007b). Cell lysis involves the sequential disruption of the inclusion and cellular membranes by cysteine proteases (Hybiske and Stephens 2007b). In contrast, the extrusion of inclusions requires actin polymerization, myosin, the Rho family GTPase RhoA, and leaves the host cell intact (Hybiske and Stephens 2007b). EB exit from C. trachomatis serovar E-infected cells is accompanied by lysosome-mediated repair of the plasma membrane (Beatty 2007).
MODIFYING THE HOST RESPONSE
Detection of Chlamydia by the Host
As with most bacteria, Chlamydia infections are detected by host pattern recognition receptors (PRRs) that recognize chlamydial LPS via TLR4 (Ingalls et al. 1995; Prebeck et al. 2001, 2003; Heine et al. 2003) and heat shock protein Hsp60 through TLR2 and TLR4 (Kol et al. 1999, 2000; Vabulas et al. 2001; Bulut et al. 2002; Costa et al. 2002; Bulut et al. 2009). TLR2 appears to be the predominant receptor required for an inflammatory response to infection (Prebeck et al. 2001; Darville et al. 2003; O’Connell et al. 2006). Interestingly, TLR2 and its adaptor MyD88 localize to the periphery of the chlamydial inclusion during active infection, suggesting that TLR2 may signal intracellularly during infection (Mackern-Oberti et al. 2006; O’Connell et al. 2006).
In addition to TLRs, cytosolic PRRs recognize chlamydial PAMPS. RNAi-mediated knockdown of NOD1, or its downstream adaptor RIP2, in infected epithelial and endothelial cells ablates expression of inflammatory cytokines (Opitz et al. 2005; Welter-Stahl et al. 2006; Buchholz and Stephens 2008). Similarly, Nod1−/− and Rip2−/− mice show significantly delayed clearance of C. pneumoniae from their lungs (Shimada et al. 2009). Presumably, Nod1 is activated by trace amounts of peptidoglycans (PGNs), and even though PGN components such as N-acetylmuramic acid have not been detected in Chlamydiae above trace levels, the full complement of genes required for PGN biosynthesis and assembly are present in Chlamydiae genomes (Stephens et al. 1998; Kalman et al. 1999; Read et al. 2000). Interestingly, in place of an abundant layer of PGNs, the cell envelope of EBs is comprised of a highly cross-linked latticework of outer membrane proteins (MOMP, OmcA, OmcB, and others) (reviewed in Hatch 1996), many of which are highly immunogenic (Wang et al. 2010) and could possibly be recognized as PAMPs by cytosolic receptors. In addition, infected cells induce type I IFNs, which can activate cell-autonomous resistance mechanisms to Chlamydia (Bernstein-Hanley et al. 2006). The ligands responsible for inducing the expression of type I IFNs are unknown, but the recently reported role for STING in IFN expression (Prantner et al. 2010) during Chlamydia infection point to a role for bacterial nucleic acids as potential ligands.
Antimicrobial molecules such as reactive oxygen species are also synthesized in response to infections (reviewed in D’Autreaux and Toledano 2007), and K+ efflux triggered by Chlamydia infection activates reactive oxygen species (ROS) production (Abdul-Sater et al. 2009; Boncompain et al. 2010; He et al. 2010). ROS production also leads to activation of caspase 1 through the NLRP3-ASC inflammasome and processing of the proinflammatory cytokines IL-1β and IL-18 in Chlamydia-infected cells (Rothermel et al. 1989; Entrican et al. 1999; Lu et al. 2000; Gervassi et al. 2004; Abdul-Sater et al. 2009; He et al. 2010).
Initiating Innate Immune Responses
Early during Chlamydia infection there is an acute localized inflammatory response largely mediated by polymorphonuclear leukocytes (PMNs) and mononuclear leukocytes, recruited by cytokines and chemokines released by infected epithelial cells (Kelly and Rank 1997; Rank et al. 2000, 2008). Proinflammatory cytokines, chemokines, and interferons such as IL-1α, IL-1β, GM-CSF, IL-8, and IFN-α and γ are secreted by Chlamydia-infected cells in in vitro culture systems (Table 1). Activation of MEK-ERK (independent from the p38 and Jun amino-terminal MAPK pathways) and Nod1 signaling pathways, as well as nuclear IL-1α, contribute to cytokine production by infected epithelial cells (Buchholz and Stephens 2007, 2008; Cheng et al. 2008).
Table 1.
Chemokines and cytokines produced during chlamydial infection
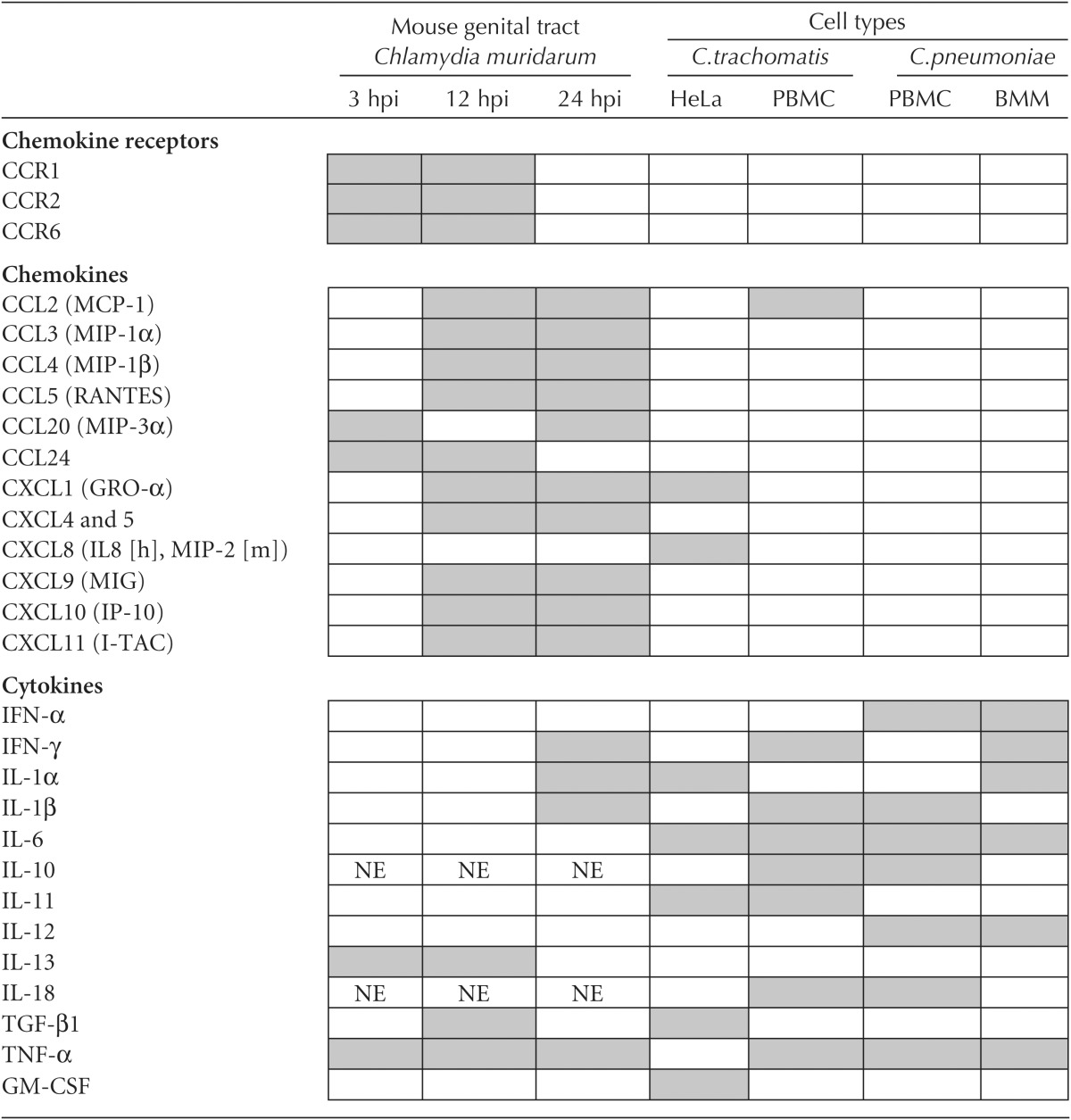
Induced chemokines and cytokines are represented as gray boxes.
Data adapted from Kaukoranta-Tolvanen et al. 1996; Rasmussen et al. 1997; Dessus-Babus et al. 2000; Geng et al. 2000; Netea et al. 2004; Rothfuchs et al. 2004; Rank et al. 2010.
PBMC, peripheral blood mononuclear cell; BMM, murine bone marrow-derived macrophage; IL-8 (h), human CXCL8 homolog; MIP-2 (m), murine CXCL8 homolog; NE, not expressed.
Cytokine/chemokine profiles have also been measured in vivo using a mouse model of genital tract infections with Chlamydia muridarum (Table 1). As early as 3 h postinfection, genes encoding chemokines (CCL20, CCL3, and CCL24) that recruit immature dendritic cells (DCs) are up-regulated, as well as those encoding for TNF-α and C3 (alternative complement). Therefore, early responses prime the site of infection for immature DC infiltration, which is an important step of the adaptive response (Rank et al. 2010). By 12 h postinfection, expression of chemokines that are chemotactic for natural killer (NK) cells (CXCL9-11, CCL2-4, CCL7, CCL8, and CCL12) is significantly induced, which is in agreement with the observed recruitment of NK cells to the site of infection (Tseng and Rank 1998; Rank et al. 2010). Not surprisingly, the proinflammatory cytokines IL-1α, IL-1β, IL-1F6, and IL-1F8 are also expressed as are PMN-recruiting chemokines (CXCL1, CXCL5, and CXCL15) (Rank et al. 2010).
The recruitment of immune cells that mediate innate and adaptive immune responses to Chlamydia infections are, under most circumstances, sufficient for bacterial clearance during primary infections, which may explain why most infections are asymptomatic. However, inflammatory responses resulting from recurring infections or when bacteria access sterile sites (i.e., the upper genital tract) contribute to the detrimental scarring and pathology observed in some infected individuals (Stephens 2003; Darville and Hiltke 2010).
Subversion of Host-Innate Immune Response
Despite the inflammatory response generated by the host during primary Chlamydia infection, in some individuals C. trachomatis infections can persist asymptomatically for months before being spontaneously cleared (Parks et al. 1997; Golden et al. 2000). How infection can persist for so long is not well understood. Chlamydia uses a wide range of strategies that may potentially subvert the host-immune responses, including interfering with the function of the transcription factor NF-κB (p65/RelA), a central regulator of immune responses. NF-κB functions downstream from PPRs and proinflammatory cytokine receptors such as the TLR/IL-1R and tumor necrosis factor (TNF) family of receptors. Signaling is initiated by adaptor proteins belonging to a family of TNF receptor-associated factors (TRAF) such as TRAF2 and TRAF6. In the canonical pathway, a family of IκB proteins maintains NF-κB homo- or heterodimers sequestered in the cytoplasm. Activation of the pathway triggers phosphorylation of IκB and subsequent ubiquitination and proteasome-dependent degradation of IκB proteins culminating in NF-κB release, nuclear translocation, and expression of hundreds of genes, including innate immune response factors (reviewed in Chen 2005).
Chlamydia blocks degradation of the NF-κB retention factor IκBα and nuclear translocation of NF-κB (Lad et al. 2007a). This activity has been attributed to Chlamydia proteins possessing deubiquitinating (DUB) and deneddylating activity, ChlaDub1 and ChlaDub2, which may potentially interfere with host-ubiquitination pathways (Misaghi et al. 2006). Ectopic expression of ChlaDub1 in HeLa cells impaired ubiquitination and degradation of IκBα on stimulation with a variety of NF-κB agonists including TNF-α and IL-1β (Le Negrate et al. 2008). ChlaDub1 did not affect ubiquitination of TRAF2 or TRAF6, or IκBα phosphorylation, indicating that ChlaDub1 activity does not interfere with upstream components of the pathway. However, ChlaDub1 coprecipitates with IκBα suggesting that ChlaDub1 activity may very well be directed at IκBα (Le Negrate et al. 2008).
Chlamydia can also block NF-κB activation directly through the proteolysis of the p65/RelA subunit of NF-κB (Lad et al. 2007a). Initially this activity was attributed to the chlamydial tail-specific (TSP) endoprotease CT441 (Lad et al. 2007a,b). Although CT441 shows robust human p65 proteolytic activity, it is unclear whether CT441 can access the cytoplasm of the host cell where it could engage its target. Instead, a chlamydial protease named CPAF (chlamydial protease-like activity factor) has been recently implicated in the degradation of p65 during infection (Christian et al. 2010). Although CPAF is an extensively characterized protease with numerous potential substrates of importance to innate immunity (reviewed in Zhong 2011), a recent report suggests that several proteins targeted by CPAF, including the p65/RelA subunit of NF-κB, RFX5, Bim, and Puma (discussed below), may not be bona fide CPAF substrates in in vivo settings (Chen et al. 2012).
Chlamydia has also evolved a mechanism to limit the recognition of PAMPs by ensuring the stability of the inclusion during the intracellular stage of infections. This is achieved by reorganizing actin and intermediate filaments at the periphery of the inclusion (Kumar and Valdivia 2008). Disruption of these cytoskeletal elements results in inclusion membrane instability and leakage of inclusion luminal contents into the host-cell cytoplasm. As a result, there is an increase in IL-8 gene transcription (Kumar and Valdivia 2008).
CPAF may also play a role in limiting the presentation to immune cells of chlamydial antigens by major histocompatibility complexes (MHCs). MHC class I and II complexes present bacterial antigens to CD8+ and CD4+ T lymphocytes, respectively (reviewed in Germain 1994). To limit detection by CD8+ and CD4+ T cells, Chlamydia promotes CPAF-dependent degradation of the transcription factors RFX5 and USF-1, two upstream transcriptional regulators of IFN-γ-induced MHC I and II genes (Zhong et al. 1999, 2000). CPAF also stimulates degradation of CD1d, an MHC-like protein complex that stimulates NK and NKT immune cells (Kawana et al. 2007, 2008). In parallel, CPAF also cleaves a subset of chlamydial secreted effectors and inclusion membrane proteins, potentially curtailing their availability as substrates for antigen presentation (Jorgensen et al. 2011). These multiple potential mechanisms used by Chlamydia to dampen immune responses are summarized in Figure 3.
Figure 3.

Current model of chlamydial effector interactions with host-innate immune signaling pathways. (A) TLR2 and MyD88 are trafficked to the inclusion by unknown mechanisms and promote expression of inflammatory cytokines. Nod1 and MEK-ERK MAPK signaling pathways also stimulate inflammatory cytokine gene expression. (B) Chlamydial microbial products and cytokines secreted by recruited immune cells and neighboring infected cells activate Toll-like and cytokine receptors. Receptor signaling activates NF-κB signaling and expression of proinflammatory cytokines/chemokines. Established inclusions impede NF-κB signaling by reversing IκBα ubiquitination through the activity of secreted ChlaDub1 and possibly by CPAF-mediated degradation of p65/RelA. (C) A cytoskeleton cage comprised of actin (red) and intermediate filaments (green) envelops the established inclusion. Remodeling of the cage curtails inclusion contents from activating cytosolic PRRs, possibly the NLRP3-ASC-caspase 1 inflammasome. However, unknown chlamydial agonists can activate the NLRP3-ASC inflammasome and caspase 1-dependent processing of inflammatory cytokines. (D) Additional mechanisms facilitating host survival may include the degradation of BH3-only proteins and PARP (Yu et al. 2010) and blocking death-inducing signaling pathways presumably originating from the TNF family of receptors and cytokine receptors. CPAF activity may also be deployed by Chlamydia to subvert antigen presentation.
Maintaining the Host Cell Alive
Cell death on stimulation by death-inducing ligands secreted by immune cells or in response to cellular cytopathic signals is a common cell-autonomous response to infection (Ying et al. 2006). Chlamydia interferes with multiple proapoptotic pathways as well as potential necrotic cell death to guarantee survival within host cells (Fan et al. 1998; Fischer et al. 2001; Rajalingam et al. 2001; Yu et al. 2010). A prominent mechanism for preventing cell death includes CPAF-mediated degradation of the proapoptotic BH3-only proteins Bad, Bim, and Puma (Fischer et al. 2004; Dong et al. 2005; Ying et al. 2005; Pirbhai et al. 2006). This activity is proposed to be responsible for the reduced activation of Bax and Bak and subsequent block in cytochrome c release observed during infection (Fan et al. 1998; Fischer et al. 2004; Xiao et al. 2004). However, new observations indicate that Bim and Puma may not be targeted for degradation during infection (Chen et al. 2012). These differences will need to be reconciled in the future. In parallel, infection also leads to stabilization of IAP2 (inhibitor of apoptosis protein 2) and up-regulation of the prosurvival factor Mcl-1 (myeoloid cell leukemia) (Rajalingam et al. 2006, 2008; Sharma et al. 2011). Other mechanisms potentially include sequestration of Bad at the inclusion via 14-3-3β, and of protein kinase C-δ (PKCδ) through binding to diacylglycerol-enriched membranes in the vicinity of the inclusion (Scidmore and Hackstadt 2001; Tse et al. 2005; Verbeke et al. 2006).
In addition, Chlamydia contends with the antimicrobial function of IFN-γ, which is secreted by infiltrating immune cells. IFN-γ induces tryptophan depletion by increasing host-indoleamine 2,3-dioxygenase (IDO) expression (reviewed in Taylor and Feng 1991). Tryptophan depletion is detrimental to Chlamydia because most strains are tryptophan auxotrophs. In response to this stress, Chlamydia transitions into quiescent noninfectious aberrant structures termed the “persistent” form (Beatty et al. 1994; Schoborg 2011). Interestingly, cells infected with these chlamydial forms are still resistant to various forms of apoptosis, using unknown mechanisms that are largely independent of CPAF (Dean and Powers 2001; Rodel et al. 2011).
Infection activates the MAPK and PI3K prosurvival signaling pathways. The ERK MAPK signaling pathway appears to be a central regulator for bacterial nutrient acquisition, expression of antiapoptotic factors, and synthesis of proinflammatory cytokines (Su et al. 2004; Buchholz and Stephens 2007; Rajalingam et al. 2008). Epithelial cells display sustained activation of upstream components of the ERK signaling module including RAS and RAF (Su et al. 2004; Gurumurthy et al. 2010; Vignola et al. 2010). However, this activation of ERK is uncoupled from activated RAS and RAF during infection (Gurumurthy et al. 2010; Vignola et al. 2010), suggesting multiple signaling pathways are engaged to further Chlamydia replication and intracellular survival. Avoidance of cell death and promoting self-survival are complex and very likely temporally regulated strategies deployed during infection and reinfection.
GENETICS AND GENOMICS OF Chlamydia
The genomes of a wide range of Chlamydiae serovars and species have been sequenced. Chlamydial genomes are small (<2 Mb), with few repetitive elements and pseudogenes, reflecting their long-term coadaptation to life within eukaryotic hosts (Collingro et al. 2011). Indeed, many of the genes required for the biosynthesis of key metabolites have been lost, presumably as these are now directly acquired from the host. The dearth of genes encoding DNA competence, conjugation, phages, or DNA modification systems in pathogenic Chlamydiae is consistent with that of an organism undergoing very limited genetic exchange (Stephens et al. 1998). Surprisingly, recent observations indicate that Chlamydia can exchange DNA efficiently with other strains. A genomic analysis of clinical Chlamydia isolates provided the first evidence of chimeric strains (Gomes et al. 2007). Such in vivo recombinant strains can have significant clinical implications. For instance, a hypervirulent C. trachomatis strain (L2c) isolated from a patient with severe hemorrhagic proctitis represents a recombination event between serovar L2 and serovar D strains (Somboonna et al. 2011).
In landmark experiments, Robert DeMars provided evidence of DNA exchange among Chlamydiae in the laboratory (Demars et al. 2007; DeMars and Weinfurter 2008). In cells coinfected with C. trachomatis serovars that were naturally resistant to ofloxacin or to rifampin, doubly drug-resistant recombinant Chlamydia arose at ∼104 times higher frequency than that of spontaneous mutations, and the genomes of these recombinants were chimeric recombinants of the parental strains. Whole genome sequencing of recombinants from additional crosses provided evidence that recombination events ranged from 40 kb to 790 kb and that the sizes of recombination sites range from a short 11-bp sequence to a 366-bp stretch (Suchland et al. 2009). The mechanisms underlying the release of DNA, uptake, and recombination are largely unknown.
Introduction of recombinant DNA into Chlamydia has been of limited success until very recently. Early efforts achieved transient transformation of chimeric C. trachomatis (pCT) plasmids (Tam et al. 1994). In later experiments, Chlamydia psittaci was transformed with allelic exchange vectors that targeted the 16S rRNA gene, and the investigators were able to show successful replacement of 16S rRNA loci with the introduced DNA (Binet and Maurelli 2009). Very recently, Clarke and colleagues reported successful stable transformation of C. trachomatis with a pCT shuttle plasmid by calcium chloride-mediated transformation of EBs and selection for resistance to β lactams (Wang et al. 2011). These stable transformants can express a variety of recombinant genes including the green fluorescent protein. Although in its present form the transformation and selection procedures are not very efficient, these experiments represent a significant breakthrough in our ability to experimentally manipulate these organisms.
Despite the lack of molecular genetic tools to manipulate Chlamydiae, significant advances have been made to experimentally address the role of individual genes in pathogenesis. This includes gain-of-function approaches, such as expressing Chlamydia genes in heterologous systems to identify proteins that modulate host functions (Sisko et al. 2006) or to identify proteins that can be secreted by surrogate type III secretion systems (Subtil et al. 2001). Loss-of-function studies have also recently become possible with the implementation of targeting-induced local lesions in genomes (TILLING), an approach used extensively in plant and zebrafish to screen for mutants in genes of interest (reviewed in Stemple 2004). In this method, PCR fragments that span the gene in which one wishes to identify mutations are generated from wild-type and mutagenized strains. DNA heteroduplexes are then generated by annealing these fragments, and if the gene amplified from the mutants strains harbors a nucleotide variant in the target gene, the annealed PCR products will generate a mismatch that can be recognized and cleaved by the endonuclease CEL I (reviewed in Stemple 2004). Kari and colleagues applied TILLING in EMS-mutagenized Chlamydia to identify mutations, including null alleles, in the tryptophan synthase operon trpBA (Kari et al. 2011). Such approaches enable the implementation of reverse genetic approaches by identifying strains with specific mutations. In a complementary approach, a system that uses the concept of “phenotype sequencing” (Harper et al. 2011) has been recently implemented in Chlamydia to perform forward genetic screens. In this approach, the genomes of EMS-derived mutants with similar phenotypes (“granular” plaque morphologies) were sequenced and a common gene (glgB-encoding glycogen branching enzyme) was found to be mutated in a majority of strains (Nguyen and Valdivia 2012). By taking advantage of genetic exchange among Chlamydia strains during coinfections, recombinant strains were generated in which a glgB mutation was the only gene lesion inherited from the parental mutant strain, thus unambiguously linking this mutation to a glycogen granule accumulation plaque morphotype. The ability to screen and select for mutants, mapping the underlying genetic lesions and generating congenic strains by recombination now opens up a broad range of possibilities for the genetic analysis of Chlamydia virulence.
CONCLUDING REMARKS
The obligate intracellular lifestyle of Chlamydia offers a unique opportunity to understand how the host-immune system contends with invading pathogens. Chlamydia species translocate a staggering number of protein effectors into the mammalian host cell to coopt cellular processes. Elucidating the function of these effectors will lead to new insights as to how pathogens subvert their hosts and on the function of the cellular processes they target. Despite the absence of classical genetic tools to study these ancient organisms, recent advances in deep sequencing technologies, the implementation of high-throughput RNAi screens, and comparative genomics approaches have broadened our understanding of the biology of these pathogens. Observations of widespread DNA exchange among Chlamydia organisms in experimental settings coupled with exciting new reports of Chlamydia plasmid transformation techniques will likely lead to major advances in our molecular understanding of chlamydial pathogenesis in the near future.
ACKNOWLEDGMENTS
We thank J.D. Dunn for comments on the manuscript. We apologize to those investigators whose work we were unable to cite because of space constraints. Work in our laboratories is supported by National Institutes of Health (NIH) R01 AI07377 (J.N.E. and C.A.E.) and NIH grants R01AI081694 and R01AI081694-S1 (R.H.V. and R.J.B.). R.H.V. is a Burroughs Wellcome Scholar in the Pathogenesis of Infectious Diseases.
Footnotes
Editors: Pascale Cossart and Stanley Maloy
Additional Perspectives on Bacterial Pathogenesis available at www.perspectivesinmedicine.org
REFERENCES
- Abdul-Sater AA, Koo E, Hacker G, Ojcius DM 2009. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J Biol Chem 284: 26789–26796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abromaitis S, Stephens RS 2009. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog 5: e1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balana ME, Niedergang F, Subtil A, Alcover A, Chavrier P, Dautry-Varsat A 2005. ARF6 GTPase controls bacterial invasion by actin remodelling. J Cell Sci 118: 2201–2210 [DOI] [PubMed] [Google Scholar]
- Beatty WL 2006. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci 119: 350–359 [DOI] [PubMed] [Google Scholar]
- Beatty WL 2007. Lysosome repair enables host cell survival and bacterial persistence following Chlamydia trachomatis infection. Cell Microbiol 9: 2141–2152 [DOI] [PubMed] [Google Scholar]
- Beatty WL 2008. Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect Immun 76: 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty WL, Belanger TA, Desai AA, Morrison RP, Byrne GI 1994. Tryptophan depletion as a mechanism of γ interferon-mediated chlamydial persistence. Infect Immun 62: 3705–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebear C, de Barbeyrac B 2009. Genital Chlamydia trachomatis infections. Clin Microbiol Infect 15: 4–10 [DOI] [PubMed] [Google Scholar]
- Beeckman DS, Vanrompay DC 2010. Bacterial secretion systems with an emphasis on the chlamydial type III secretion system. Curr Issues Mol Biol 12: 17–42 [PubMed] [Google Scholar]
- Bernstein-Hanley I, Coers J, Balsara ZR, Taylor GA, Starnbach MN, Dietrich WF 2006. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci 103: 14092–14097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet R, Maurelli AT 2009. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc Natl Acad Sci 106: 292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boleti H, Benmerah A, Ojcius DM, Cerf-Bensussan N, Dautry-Varsat A 1999. Chlamydia infection of epithelial cells expressing dynamin and Eps15 mutants: Clathrin-independent entry into cells and dynamin-dependent productive growth. J Cell Sci 112 (Pt 10): 1487–1496 [DOI] [PubMed] [Google Scholar]
- Boncompain G, Schneider B, Delevoye C, Kellermann O, Dautry-Varsat A, Subtil A 2010. Production of reactive oxygen species is turned on and rapidly shut down in epithelial cells infected with Chlamydia trachomatis. Infect Immun 78: 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell JH, Scidmore MA 2007. Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol Mol Biol Rev 71: 636–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS 2007. The extracellular signal-regulated kinase/mitogen-activated protein kinase pathway induces the inflammatory factor interleukin-8 following Chlamydia trachomatis infection. Infect Immun 75: 5924–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS 2008. The cytosolic pattern recognition receptor NOD1 induces inflammatory interleukin-8 during Chlamydia trachomatis infection. Infect Immun 76: 3150–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M 2002. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J Immunol 168: 1435–1440 [DOI] [PubMed] [Google Scholar]
- Bulut Y, Shimada K, Wong MH, Chen S, Gray P, Alsabeh R, Doherty TM, Crother TR, Arditi M 2009. Chlamydial heat shock protein 60 induces acute pulmonary inflammation in mice via the Toll-like receptor 4- and MyD88-dependent pathway. Infect Immun 77: 2683–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capmany A, Damiani MT 2010. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14- mediated transport required for bacterial development and replication. PLoS ONE 5: e14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo R 2011. Bacterial subversion of host actin dynamics at the plasma membrane. Cell Microbiol 13: 1460–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Mead DJ, Hackstadt T 2003. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci 100: 6771–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Grieshaber SS, Hasenkrug A, Dooley C, Hackstadt T 2004. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic 5: 418–425 [DOI] [PubMed] [Google Scholar]
- Carabeo RA, Dooley CA, Grieshaber SS, Hackstadt T 2007. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell Microbiol 9: 2278–2288 [DOI] [PubMed] [Google Scholar]
- Chen ZJ 2005. Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol 7: 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Stephens RS 1994. Trachoma and LGV biovars of Chlamydia trachomatis share the same glycosaminoglycan-dependent mechanism for infection of eukaryotic cells. Mol Microbiol 11: 501–507 [DOI] [PubMed] [Google Scholar]
- Chen JC, Stephens RS 1997. Chlamydia trachomatis glycosaminoglycan-dependent and independent attachment to eukaryotic cells. Microb Pathog 22: 23–30 [DOI] [PubMed] [Google Scholar]
- Chen JC, Zhang JP, Stephens RS 1996. Structural requirements of heparin binding to Chlamydia trachomatis. J Biol Chem 271: 11134–11140 [PubMed] [Google Scholar]
- Chen AL, Johnson KA, Lee JK, Sütterlin C, Tan M 2012. CPAF: A chlamydial protease in search of an authentic substrate. PLoS Pathog 8: e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G 2008. Intracellular interleukin-1α mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect Immun 76: 942–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian J, Vier J, Paschen SA, Hacker G 2010. Cleavage of the NF-κB family protein p65/RelA by the chlamydial protease-like activity factor (CPAF) impairs proinflammatory signaling in cells infected with Chlamydiae. J Biol Chem 285: 41320–41327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude A, Zhao BP, Kuziemsky CE, Dahan S, Berger SJ, Yan JP, Armold AD, Sullivan EM, Melancon P 1999. GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. J Cell Biol 146: 71–84 [PMC free article] [PubMed] [Google Scholar]
- Clausen JD, Christiansen G, Holst HU, Birkelund S 1997. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol Microbiol 25: 441–449 [DOI] [PubMed] [Google Scholar]
- Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T 2005. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect Immun 73: 3860–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Valdivia RH 2009. New insights into Chlamydia intracellular survival mechanisms. Cell Microbiol 11: 1571–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH 2008. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci 105: 9379–9384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingro A, Tischler P, Weinmaier T, Penz T, Heinz E, Brunham RC, Read TD, Bavoil PM, Sachse K, Kahane S, et al. 2011. Unity in variety-the pan-genome of the Chlamydiae. Molec Biol Evol 28: 3253–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B 2007. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect Immun 75: 5586–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa CP, Kirschning CJ, Busch D, Durr S, Jennen L, Heinzmann U, Prebeck S, Wagner H, Miethke T 2002. Role of chlamydial heat shock protein 60 in the stimulation of innate immune cells by Chlamydia pneumoniae. Eur J Immunol 32: 2460–2470 [DOI] [PubMed] [Google Scholar]
- Darville T, Hiltke TJ 2010. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis 201 (Suppl 2): S114–S125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darville T, O’Neill JM, Andrews CW Jr, Nagarajan UM, Stahl L, Ojcius DM 2003. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J Immunol 171: 6187–6197 [DOI] [PubMed] [Google Scholar]
- D’Autreaux B, Toledano MB 2007. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824 [DOI] [PubMed] [Google Scholar]
- Dautry-Varsat A, Subtil A, Hackstadt T 2005. Recent insights into the mechanisms of Chlamydia entry. Cell Microbiol 7: 1714–1722 [DOI] [PubMed] [Google Scholar]
- Dean D, Powers VC 2001. Persistent Chlamydia trachomatis infections resist apoptotic stimuli. Infect Immun 69: 2442–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dautry-Varsat A, Subtil A 2004. Conservation of the biochemical properties of IncA from Chlamydia trachomatis and Chlamydia caviae: Oligomerization of IncA mediates interaction between facing membranes. J Biol Chem 279: 46896–46906 [DOI] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A 2008. SNARE protein mimicry by an intracellular bacterium. PLoS Pathog 4: e1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars R, Weinfurter J 2008. Interstrain gene transfer in Chlamydia trachomatis in vitro: Mechanism and significance. J Bacteriol 190: 1605–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars R, Weinfurter J, Guex E, Lin J, Potucek Y 2007. Lateral gene transfer in vitro in the intracellular pathogen Chlamydia trachomatis. J Bacteriol 189: 991–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Pypaert M, Dautry-Varsat A, Agaisse H 2007. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog 3: 1446–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Swiss R, Agaisse H 2011. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog 7: e1002092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessus-Babus S, Knight ST, Wyrick PB 2000. Chlamydial infection of polarized HeLa cells induces PMN chemotaxis but the cytokine profile varies between disseminating and non-disseminating strains. Cell Microbiol 2: 317–327 [DOI] [PubMed] [Google Scholar]
- Dong F, Pirbhai M, Xiao Y, Zhong Y, Wu Y, Zhong G 2005. Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect Immun 73: 1861–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Chavrier P 2006. ARF proteins: Roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7: 347–358 [DOI] [PubMed] [Google Scholar]
- Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN 2008. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4: e1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Jiang S, Kim JH, Lee A, Wittmann T, Hanada K, Melancon P, Engel JN 2011. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathog 7: e1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entrican G, Wilkie R, McWaters P, Scheerlinck J, Wood PR, Brown J 1999. Cytokine release by ovine macrophages following infection with Chlamydia psittaci. Clin Exp Immunol 117: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel S, Eley A 2007. Chlamydia trachomatis OmcB protein is a surface-exposed glycosaminoglycan-dependent adhesin. J Med Microbiol 56: 15–22 [DOI] [PubMed] [Google Scholar]
- Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G 1998. Inhibition of apoptosis in Chlamydia-infected cells: Blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187: 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T 2002. The chlamydial inclusion: Escape from the endocytic pathway. Annu Rev Cell Dev Biol 18: 221–245 [DOI] [PubMed] [Google Scholar]
- Fischer SF, Schwarz C, Vier J, Hacker G 2001. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect Immun 69: 7121–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SF, Harlander T, Vier J, Hacker G 2004. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect Immun 72: 1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel BR, Elwell C, van Ijzendoorn SC, Engel JN 2004. Lipid raft-mediated entry is not required for Chlamydia trachomatis infection of cultured epithelial cells. Infect Immun 72: 7367–7373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Shane RB, Berencsi K, Gonczol E, Zaki MH, Margolis DJ, Trinchieri G, Rook AH 2000. Chlamydia pneumoniae inhibits apoptosis in human peripheral blood mononuclear cells through induction of IL-10. J Immunol 164: 5522–5529 [DOI] [PubMed] [Google Scholar]
- Germain RN 1994. MHC-dependent antigen processing and peptide presentation: Providing ligands for T lymphocyte activation. Cell 76: 287–299 [DOI] [PubMed] [Google Scholar]
- Gervassi A, Alderson MR, Suchland R, Maisonneuve JF, Grabstein KH, Probst P 2004. Differential regulation of inflammatory cytokine secretion by human dendritic cells upon Chlamydia trachomatis infection. Infect Immun 72: 7231–7239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden MR, Schillinger JA, Markowitz L, St Louis ME 2000. Duration of untreated genital infections with Chlamydia trachomatis: A review of the literature. Sex Transm Dis 27: 329–337 [DOI] [PubMed] [Google Scholar]
- Gomes JP, Bruno WJ, Nunes A, Santos N, Florindo C, Borrego MJ, Dean D 2007. Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res 17: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Hackstadt T 2003. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci 116: 3793–3802 [DOI] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Miller N, Hackstadt T 2006. Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic 7: 940–949 [DOI] [PubMed] [Google Scholar]
- Gurumurthy RK, Maurer AP, Machuy N, Hess S, Pleissner KP, Schuchhardt J, Rudel T, Meyer TF 2010. A loss-of-function screen reveals Ras- and Raf-independent MEK-ERK signaling during Chlamydia trachomatis infection. Sci Signal 3: ra21. [DOI] [PubMed] [Google Scholar]
- Hackstadt T 2000. Redirection of host vesicle trafficking pathways by intracellular parasites. Traffic 1: 93–99 [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA 1996. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J 15: 964–977 [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER 1999. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol 1: 119–130 [DOI] [PubMed] [Google Scholar]
- Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB 2010. Risk of sequelae after Chlamydia trachomatis genital infection in women. J Infect Dis 201 (Suppl 2): S134–S155 [DOI] [PubMed] [Google Scholar]
- Hanada K 2010. Intracellular trafficking of ceramide by ceramide transfer protein. Proc Jpn Acad Ser B Phys Biol Sci 86: 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper MA, Chen Z, Toy T, Machado IM, Nelson SF, Liao JC, Lee CJ 2011. Phenotype sequencing: Identifying the genes that cause a phenotype directly from pooled sequencing of independent mutants. PLoS ONE 6: e16517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch TP 1996. Disulfide cross-linked envelope proteins: The functional equivalent of peptidoglycan in chlamydiae? J Bacteriol 178: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch TP 1999. Developmental biology. In Chlamydia: Intracellular biology, pathogenesis, and immunity (ed. Stephens RS), pp. 29–68 American Society for Microbiology, Washington, DC [Google Scholar]
- Hatch GM, McClarty G 1998. Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect Immun 66: 3727–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR 2010. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol 184: 5743–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine H, Muller-Loennies S, Brade L, Lindner B, Brade H 2003. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur J Biochem 270: 440–450 [DOI] [PubMed] [Google Scholar]
- Heuer D, Rejman Lipinski A, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF 2009. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457: 731–735 [DOI] [PubMed] [Google Scholar]
- Hower S, Wolf K, Fields KA 2009. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72: 1423–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC 2004. Identification of a family of animal sphingomyelin synthases. EMBO J 23: 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS 2007a. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infect Immun 75: 3925–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS 2007b. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci 104: 11430–11435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingalls RR, Rice PA, Qureshi N, Takayama K, Lin JS, Golenbock DT 1995. The inflammatory cytokine response to Chlamydia trachomatis infection is endotoxin mediated. Infect Immun 63: 3125–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Fischer ER, Mead DJ, Hackstadt T 2006. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci 103: 15599–15604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Dooley CA, Mead DJ, Hackstadt T 2008. Chlamydia trachomatis Tarp is phosphorylated by Src family tyrosine kinases. Biochem Biophys Res Commun 371: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Miller NJ, Dooley CA, Hackstadt T 2010. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog 6: e1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Tan M, Sutterlin C 2009. Centrosome abnormalities during a Chlamydia trachomatis infection are caused by dysregulation of the normal duplication pathway. Cell Microbiol 11: 1064–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Bednar MM, Amin V, Davis BK, Ting JP, McCafferty DG, Valdivia RH 2011. The Chlamydia protease CPAF regulates host and bacterial proteins to maintain pathogen vacuole integrity and promote virulence. Cell Host Microbe 10: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras I, Abrami L, Dautry-Varsat A 2003. Entry of the lymphogranuloma venereum strain of Chlamydia trachomatis into host cells involves cholesterol-rich membrane domains. Infect Immun 71: 260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman RW, Olinger L, Grimwood J, Davis RW, Stephens RS 1999. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet 21: 385–389 [DOI] [PubMed] [Google Scholar]
- Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, et al. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci 108: 7189–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukoranta-Tolvanen SS, Teppo AM, Laitinen K, Saikku P, Linnavuori K, Leinonen M 1996. Growth of Chlamydia pneumoniae in cultured human peripheral blood mononuclear cells and induction of a cytokine response. Microb Pathog 21: 215–221 [DOI] [PubMed] [Google Scholar]
- Kawamoto K, Yoshida Y, Tamaki H, Torii S, Shinotsuka C, Yamashina S, Nakayama K 2002. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic 3: 483–495 [DOI] [PubMed] [Google Scholar]
- Kawana K, Quayle AJ, Ficarra M, Ibana JA, Shen L, Kawana Y, Yang H, Marrero L, Yavagal S, Greene SJ, et al. 2007. CD1d degradation in Chlamydia trachomatis-infected epithelial cells is the result of both cellular and chlamydial proteasomal activity. J Biol Chem 282: 7368–7375 [DOI] [PubMed] [Google Scholar]
- Kawana K, Matsumoto J, Miura S, Shen L, Kawana Y, Nagamatsu T, Yasugi T, Fujii T, Yang H, Quayle AJ, et al. 2008. Expression of CD1d and ligand-induced cytokine production are tissue specific in mucosal epithelia of the human lower reproductive tract. Infect Immun 76: 3011–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KA, Rank RG 1997. Identification of homing receptors that mediate the recruitment of CD4 T cells to the genital tract following intravaginal infection with Chlamydia trachomatis. Infect Immun 65: 5198–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Jiang S, Elwell CA, Engel JN 2011. Chlamydia trachomatis co-opts the FGF2 signaling pathway to enhance infection. PLoS Pathog 7: e1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol A, Bourcier T, Lichtman AH, Libby P 1999. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest 103: 571–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA 2000. Cutting edge: Heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol 164: 13–17 [DOI] [PubMed] [Google Scholar]
- Kumar Y, Valdivia RH 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH 2006. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol 16: 1646–1651 [DOI] [PubMed] [Google Scholar]
- Lad SP, Li J, da Silva Correia J, Pan Q, Gadwal S, Ulevitch RJ, Li E 2007a. Cleavage of p65/RelA of the NF-κB pathway by Chlamydia. Proc Natl Acad Sci 104: 2933–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lad SP, Yang G, Scott DA, Wang G, Nair P, Mathison J, Reddy VS, Li E 2007b. Chlamydial CT441 is a PDZ domain-containing tail-specific protease that interferes with the NF-κB pathway of immune response. J Bacteriol 189: 6619–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA 2008. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4: e1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, Reed JC 2008. ChlaDub1 of Chlamydia trachomatis suppresses NF-κB activation and inhibits IκBα ubiquitination and degradation. Cell Microbiol 10: 1879–1892 [DOI] [PubMed] [Google Scholar]
- Lu H, Shen C, Brunham RC 2000. Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J Immunol 165: 1463–1469 [DOI] [PubMed] [Google Scholar]
- Mackern-Oberti JP, Maccioni M, Cuffini C, Gatti G, Rivero VE 2006. Susceptibility of prostate epithelial cells to Chlamydia muridarum infection and their role in innate immunity by recruitment of intracellular Toll-like receptors 4 and 2 and MyD88 to the inclusion. Infect Immun 74: 6973–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolea F, Claude A, Chun J, Rosas J, Melancon P 2008. Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi stack and trans-Golgi network, respectively. Mol Biol Cell 19: 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Skaug J, Zhao XH, Giordano J, Scherer SW, Melancon P 1999. p200 ARF-GEP1: A Golgi-localized guanine nucleotide exchange protein whose Sec7 domain is targeted by the drug brefeldin A. Proc Natl Acad Sci 96: 7968–7973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Bessho H, Uehira K, Suda T 1991. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J Electron Microsc (Tokyo) 40: 356–363 [PubMed] [Google Scholar]
- Mehlitz A, Banhart S, Hess S, Selbach M, Meyer TF 2008. Complex kinase requirements for Chlamydia trachomatis Tarp phosphorylation. FEMS Microbiol Lett 289: 233–240 [DOI] [PubMed] [Google Scholar]
- Mehlitz A, Banhart S, Maurer AP, Kaushansky A, Gordus AG, Zielecki J, Macbeath G, Meyer TF 2010. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J Cell Biol 190: 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menozzi FD, Pethe K, Bifani P, Soncin F, Brennan MJ, Locht C 2002. Enhanced bacterial virulence through exploitation of host glycosaminoglycans. Mol Microbiol 43: 1379–1386 [DOI] [PubMed] [Google Scholar]
- Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN 2006. Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol Microbiol 61: 142–150 [DOI] [PubMed] [Google Scholar]
- Mital J, Hackstadt T 2011. Diverse requirements for Src-family tyrosine kinases distinguish chlamydial species. mBio 2: e00031–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J, Miller NJ, Fischer ER, Hackstadt T 2010. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell Microbiol 12: 1235–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moelleken K, Hegemann JH 2008. The Chlamydia outer membrane protein OmcB is required for adhesion and exhibits biovar-specific differences in glycosaminoglycan binding. Mol Microbiol 67: 403–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölleken K, Schmidt E, Hegemann JH 2010. Members of the Pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol Microbiol 78: 1004–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ER, Mead DJ, Dooley CA, Sager J, Hackstadt T 2011. The trans-Golgi SNARE syntaxin 6 is recruited to the chlamydial inclusion membrane. Microbiology 157: 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead AR, Rzomp KA, Scidmore MA 2007. The Rab6 effector Bicaudal D1 associates with Chlamydia trachomatis inclusions in a biovar-specific manner. Infect Immun 75: 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead AM, Jung JY, Smirnov A, Kaufer S, Scidmore MA 2010. Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infect Immun 78: 1990–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Kullberg BJ, Jacobs LE, Verver-Jansen TJ, van der Ven-Jongekrijg J, Galama JM, Stalenhoef AF, Dinarello CA, Van der Meer JW 2004. Chlamydia pneumoniae stimulates IFN-γ synthesis through MyD88-dependent, TLR2- and TLR4-independent induction of IL-18 release. J Immunol 173: 1477–1482 [DOI] [PubMed] [Google Scholar]
- Nguyen BD, Valdivia RH 2012. Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proc Natl Acad Sci 109: 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norkin LC, Wolfrom SA, Stuart ES 2001. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Exp Cell Res 266: 229–238 [DOI] [PubMed] [Google Scholar]
- O’Connell CM, Ionova IA, Quayle AJ, Visintin A, Ingalls RR 2006. Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. Evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. J Biol Chem 281: 1652–1659 [DOI] [PubMed] [Google Scholar]
- Opitz B, Forster S, Hocke AC, Maass M, Schmeck B, Hippenstiel S, Suttorp N, Krull M 2005. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res 96: 319–326 [DOI] [PubMed] [Google Scholar]
- Ouellette SP, Carabeo RA 2010. A functional slow recycling pathway of transferrin is required for growth of Chlamydia. Front Microbiol 1: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL, Carabeo RA 2011. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS ONE 6: e16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks KS, Dixon PB, Richey CM, Hook EW 3rd 1997. Spontaneous clearance of Chlamydia trachomatis infection in untreated patients. Sex Transm Dis 24: 229–235 [DOI] [PubMed] [Google Scholar]
- Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman HA, Subtil A, Rothman JE 2009. Intracellular bacteria encode inhibitory SNARE-like proteins. PLoS ONE 4: e7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G 2006. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J Biol Chem 281: 31495–31501 [DOI] [PubMed] [Google Scholar]
- Poccia D, Larijani B 2009. Phosphatidylinositol metabolism and membrane fusion. Biochem J 418: 233–246 [DOI] [PubMed] [Google Scholar]
- Pokrovskaya ID, Szwedo JW, Goodwin A, Lupashina TV, Nagarajan UM, Lupashin VV 2012. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cell Microbiol 14: 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Darville T, Nagarajan UM 2010. Stimulator of IFN gene is critical for induction of IFN-β during Chlamydia muridarum infection. J Immunol 184: 2551–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prebeck S, Kirschning C, Durr S, da Costa C, Donath B, Brand K, Redecke V, Wagner H, Miethke T 2001. Predominant role of toll-like receptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendritic cells. J Immunol 167: 3316–3323 [DOI] [PubMed] [Google Scholar]
- Prebeck S, Brade H, Kirschning CJ, da Costa CP, Durr S, Wagner H, Miethke T 2003. The Gram-negative bacterium Chlamydia trachomatis L2 stimulates tumor necrosis factor secretion by innate immune cells independently of its endotoxin. Microbes Infect 5: 463–470 [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Al-Younes H, Muller A, Meyer TF, Szczepek AJ, Rudel T 2001. Epithelial cells infected with Chlamydophila pneumoniae (Chlamydia pneumoniae) are resistant to apoptosis. Infect Immun 69: 7880–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Paland N, Hurwitz R, Thieck O, Oswald M, Machuy N, Rudel T 2006. IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog 2: e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Lohmann C, Oswald M, Thieck O, Froelich CJ, Rudel T 2008. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS ONE 3: e3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Bowlin AK, Kelly KA 2000. Characterization of lymphocyte response in the female genital tract during ascending Chlamydial genital infection in the guinea pig model. Infect Immun 68: 5293–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Whittimore J, Bowlin AK, Dessus-Babus S, Wyrick PB 2008. Chlamydiae and polymorphonuclear leukocytes: Unlikely allies in the spread of chlamydial infection. FEMS Immunol Med Microbiol 54: 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Lacy HM, Goodwin A, Sikes J, Whittimore J, Wyrick PB, Nagarajan UM 2010. Host chemokine and cytokine response in the endocervix within the first developmental cycle of Chlamydia muridarum. Infect Immun 78: 536–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, Fierer J, Stephens RS, Kagnoff MF 1997. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest 99: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, et al. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 28: 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejman Lipinski A, Heymann J, Meissner C, Karlas A, Brinkmann V, Meyer TF, Heuer D 2009. Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS Pathog 5: e1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DK, Gu L, Rowe RK, Beatty WL 2009. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathog 5: e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodel J, Grosse C, Yu H, Wolf K, Otto GP, Liebler-Tenorio E, Forsbach-Birk V, Straube E 2011. Persistent Chlamydia trachomatis infection of HeLa cells mediates apoptosis resistance through a CPAF-independent mechanism and induces HMGB1 release. Infect Immun 80: 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel CD, Schachter J, Lavrich P, Lipsitz EC, Francus T 1989. Chlamydia trachomatis-induced production of interleukin-1 by human monocytes. Infect Immun 57: 2705–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuchs AG, Trumstedt C, Wigzell H, Rottenberg ME 2004. Intracellular bacterial infection-induced IFN-γ is critical but not solely dependent on Toll-like receptor 4-myeloid differentiation factor 88-IFN-αβ-STAT1 signaling. J Immunol 172: 6345–6353 [DOI] [PubMed] [Google Scholar]
- Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA 2003. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect Immun 71: 5855–5870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzomp KA, Moorhead AR, Scidmore MA 2006. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect Immun 74: 5362–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J 1999. Infection and disease epidemiology. In Chlamydia: Intracellular biology, pathogenesis, and immunity (ed. Stephens RS), pp. 139–170 American Society for Microbiology, Washington, DC [Google Scholar]
- Schoborg RV 2011. Chlamydia persistence—A tool to dissect Chlamydia-host interactions. Microbes Infect 13: 649–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA 2011. Recent advances in Chlamydia subversion of host cytoskeletal and membrane trafficking pathways. Microbes Infect 13: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T 2001. Mammalian 14-3-3β associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol 39: 1638–1650 [DOI] [PubMed] [Google Scholar]
- Sharma M, Machuy N, Bohme L, Karunakaran K, Maurer AP, Meyer TF, Rudel T 2011. HIF-1α is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell Microbiol 13: 1573–1585 [DOI] [PubMed] [Google Scholar]
- Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, Peterson E, Doherty TM, Underhill D, Crother TR, et al. 2009. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog 5: e1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisko JL, Spaeth K, Kumar Y, Valdivia RH 2006. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol Microbiol 60: 51–66 [DOI] [PubMed] [Google Scholar]
- Somboonna N, Wan R, Ojcius DM, Pettengill MA, Joseph SJ, Chang A, Hsu R, Read TD, Dean D 2011. Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L2) and D lineages. mBio 2: e00045–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemple DL 2004. TILLING—A high-throughput harvest for functional genomics. Nat Rev Genet 5: 145–150 [DOI] [PubMed] [Google Scholar]
- Stephens RS 2003. The cellular paradigm of chlamydial pathogenesis. Trends Microbiol 11: 44–51 [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282: 754–759 [DOI] [PubMed] [Google Scholar]
- Stuart ES, Webley WC, Norkin LC 2003. Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Exp Cell Res 287: 67–78 [DOI] [PubMed] [Google Scholar]
- Su H, Raymond L, Rockey DD, Fischer E, Hackstadt T, Caldwell HD 1996. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc Natl Acad Sci 93: 11143–11148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, McClarty G, Dong F, Hatch GM, Pan ZK, Zhong G 2004. Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J Biol Chem 279: 9409–9416 [DOI] [PubMed] [Google Scholar]
- Subtil A, Parsot C, Dautry-Varsat A 2001. Secretion of predicted Inc proteins of Chlamydia pneumoniae by a heterologous type III machinery. Mol Microbiol 39: 792–800 [DOI] [PubMed] [Google Scholar]
- Subtil A, Wyplosz B, Balana ME, Dautry-Varsat A 2004. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J Cell Sci 117: 3923–3933 [DOI] [PubMed] [Google Scholar]
- Suchland RJ, Sandoz KM, Jeffrey BM, Stamm WE, Rockey DD 2009. Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother 53: 4604–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE 2009. Membrane fusion: Grappling with SNARE and SM proteins. Science 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam JE, Davis CH, Wyrick PB 1994. Expression of recombinant DNA introduced into Chlamydia trachomatis by electroporation. Can J Microbiol 40: 583–591 [DOI] [PubMed] [Google Scholar]
- Taylor MW, Feng GS 1991. Relationship between interferon-γ, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J 5: 2516–2522 [PubMed] [Google Scholar]
- Thalmann J, Janik K, May M, Sommer K, Ebeling J, Hofmann F, Genth H, Klos A 2010. Actin re-organization induced by Chlamydia trachomatis serovar D-evidence for a critical role of the effector protein CT166 targeting Rac. PLoS ONE 5: e9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse SM, Mason D, Botelho RJ, Chiu B, Reyland M, Hanada K, Inman RD, Grinstein S 2005. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: Possible role in the inhibition of host cell apoptosis. J Biol Chem 280: 25210–25215 [DOI] [PubMed] [Google Scholar]
- Tseng CT, Rank RG 1998. Role of NK cells in early host response to chlamydial genital infection. Infect Immun 66: 5867–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H 2001. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276: 31332–31339 [DOI] [PubMed] [Google Scholar]
- Valdivia RH 2008. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol 11: 53–59 [DOI] [PubMed] [Google Scholar]
- van Ooij C, Kalman L, van I, Nishijima M, Hanada K, Mostov K, Engel JN 2000. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell Microbiol 2: 627–637 [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying S, Hansen J, Hacker G, Darville T, Ojcius DM 2006. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog 2: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignola MJ, Kashatus DF, Taylor GA, Counter CM, Valdivia RH 2010. cPLA2 regulates the expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J Biol Chem 285: 21625–21635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin KL, Wiklund F, Luostarinen T, Angstrom T, Anttila T, Bergman F, Hallmans G, Ikaheimo I, Koskela P, Lehtinen M, et al. 2002. A population-based prospective study of Chlamydia trachomatis infection and cervical carcinoma. Int J Cancer J 101: 371–374 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang Y, Lu C, Lei L, Yu P, Zhong G 2010. A genome wide profiling of the humoral immune response to Chlamydia trachomatis infection reveals vaccine candidate antigens expressed in humans. J Immunol 185: 1670–1680 [DOI] [PubMed] [Google Scholar]
- Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN 2011. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7: e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, Philpott D, Kelly KA, Darville T 2006. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol 8: 1047–1057 [DOI] [PubMed] [Google Scholar]
- Wuppermann FN, Hegemann JH, Jantos CA 2001. Heparan sulfate-like glycosaminoglycan is a cellular receptor for Chlamydia pneumoniae. J Infect Dis 184: 181–187 [DOI] [PubMed] [Google Scholar]
- Wylie JL, Hatch GM, McClarty G 1997. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J Bacteriol 179: 7233–7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Suchland RJ, Bumgarner RE, Peng T, Rockey DD, Stamm WE 2005. Chlamydia trachomatis variant with nonfusing inclusions: Growth dynamic and host-cell transcriptional response. J Infect Dis 192: 1229–1236 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Zhong Y, Greene W, Dong F, Zhong G 2004. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect Immun 72: 5470–5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji R, Adamik R, Takeda K, Togawa A, Pacheco-Rodriguez G, Ferrans VJ, Moss J, Vaughan M 2000. Identification and localization of two brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors in a macromolecular complex. Proc Natl Acad Sci 97: 2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Seiffert BM, Hacker G, Fischer SF 2005. Broad degradation of proapoptotic proteins with the conserved Bcl-2 homology domain 3 during infection with Chlamydia trachomatis. Infect Immun 73: 1399–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Fischer SF, Pettengill M, Conte D, Paschen SA, Ojcius DM, Hacker G 2006. Characterization of host cell death induced by Chlamydia trachomatis. Infect Immun 74: 6057–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Schwarzer K, Forster M, Kniemeyer O, Forsbach-Birk V, Straube E, Rodel J 2010. Role of high-mobility group box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect Immun 78: 3288–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G 2011. Chlamydia trachomatis secretion of proteases for manipulating host signaling pathways. Front Microbiol 2: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Fan T, Liu L 1999. Chlamydia inhibits interferon γ-inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J Exp Med 189: 1931–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Liu L, Fan T, Fan P, Ji H 2000. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon γ-inducible major histocompatibility complex class I expression in Chlamydia-infected cells. J Exp Med 191: 1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]