

Abstract
Two brefeldin A (BFA)-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors, 200-kDa BIG1 and 190-kDa BIG2, were copurified from bovine brain cytosol associated with >670-kDa macromolecular complexes. When observed by immunofluorescence in HeLa S3 and HepG2 cells, endogenous BIG1 and coexpressed BIG2 were distributed in a punctate pattern throughout the cytosol, and also concentrated in the perinuclear region, where endogenous BIG1 and BIG2 each partially colocalized with Golgi-specific 58K protein and γ-adaptin. On Western blot analysis, both BIG1 and BIG2 were clearly more abundant in the cytosol than in the microsomal fractions. After density gradient centrifugation of a microsomal fraction, BIG1 and BIG2 were recovered in the same fraction as β-COP, a marker for Golgi membranes. When cytosol from HeLa S3 cells was subjected to gel filtration and fractions were analyzed by Western blotting, the largest percentages of both BIG1 and BIG2 were detected in fractions containing proteins with a molecular mass of >670 kDa. Western blotting using anti-peptide antibodies specific for BIG1 or BIG2 demonstrated that ≈70% of BIG2 was immunoprecipitated along with 100% of BIG1 by the anti-BIG1 IgG, and ≈75% of BIG1 was coprecipitated with 100% of BIG2 by the anti-BIG2 IgG. All observations were consistent with the conclusion that significant fractions of BIG1 and BIG2 exist as components of the same macromolecular complexes in bovine brain cytosol and are similarly localized in cultured cells.
Vesicular protein transport between intracellular compartments is initiated by the recruitment of cytosolic coat proteins to the membrane site at which a vesicle will be formed. ADP-ribosylation factors (ARFs) are ≈20-kDa GTPases that regulate several vesicular trafficking pathways in eukaryotic cells (1, 2). Based on amino acid sequence, molecular size, and phylogenetic analysis, six mammalian ARFs fall into three classes (ARFs 1, 2, and 3 in class I, ARFs 4 and 5 in class II, and ARF 6 in class III) (3), among which ARF1 is perhaps the most studied. ARF1 is required for the formation of two types of Golgi vesicles, the coatomer (or COP)-coated (4, 5) and clathrin-coated (6, 7) vesicles that bud from membranes and are involved in both retrograde and anterograde transport. The inactive GDP-bound form of ARF1 is located in the cytosol, whereas GTP-bound ARF1 is active and associates with Golgi membranes. The alternation of ARF1 between active and inactive forms is controlled by two types of regulatory proteins, guanine nucleotide-exchange proteins (GEPs) that catalyze the replacement of GDP with GTP, and GTPase-activating proteins that accelerate the hydrolysis of bound GTP to GDP (reviewed in ref. 8).
Brefeldin A (BFA) is a fungal metabolite that blocks protein secretion and causes apparent disintegration of the Golgi complex structure, at least in part, by inhibition of GEP activation of ARF1 (1, 2). Thus far, four families of mammalian ARF GEPs have been distinguished, based on molecular structure and size and sensitivity to BFA. All ARF GEPs contain a so-called Sec7 domain that is responsible for activation of ARF1 (9–11). Proteins of the ≈47-kDa cytohesin family (9, 12, 13) comprise an N-terminal coiled-coil segment, a central Sec7 domain, and a C-terminal pleckstrin homology domain, which binds phospholipid. The EFA6 family, which acts preferentially on ARF6 (14), has a molecular structure comprising central Sec7 and pleckstrin homology domains like the cytohesin family, and also N- and C-terminal proline-rich regions with a C-terminal coiled-coil. The BIG1/BIG2 (15, 16) and GBF1 families (17), which are closely related to yeast Sec7 (18) and Gea1/Gea2 (19), respectively, are proteins of ≈200 kDa with central Sec7 domains. Of these ARF GEPs, the last two families (BIG1/BIG2 and Gea1/Gea2) are inhibited by BFA (11, 15, 16, 19). BIG1 (20) and GBF1 (17) were identified in Golgi membranes. BIG1 (11) exhibits GEP activity toward ARFs 1 and 3 (much less with ARF5), whereas GBF1 acts on ARF5 and much less on ARFs 1 and 3 (17). The ≈200-kDa BIG1 and ≈190-kDa BIG2 were initially copurified from bovine brain cytosol on the basis of their BFA-sensitive ARF activities (21). The purified BIG1 and BIG2 behaved as molecules of ≈670 kDa on gel-filtration, indicating that they were components of the same or different molecular complexes of similar size. Here, we report that, although both BIG1 and BIG2 in intact cells were demonstrable in the Golgi apparatus, in broken cell preparations, they were largely cytosolic and were, at least in part, associated with the same macromolecular complexes in both cytosol and microsomal fractions (including Golgi membranes).
Materials and Methods
Cell Culture.
HepG2 cells and HeLa S3 cells, purchased from American Type Culture Collection, were grown in DMEM (GIBCO) with 25 mM glucose, 10% FCS, 1 mM pyruvate, 25 mM Hepes, penicillin G (100 units/ml), and streptomycin (100 μg/ml) at 37°C with an atmosphere of 5% CO2/95% air. HepG2 cells were grown in dishes coated with collagen (Type I).
BIG1 and BIG2 Antibodies.
Peptides CSQPPEQELGINKQ and RLKHSQAQSK, corresponding, respectively, to amino acids 1837–1849 of BIG1 (with the underlined cysteine added to facilitate coupling) and amino acids 232–241 of BIG2, were coupled to keyhole limpet hemocyanin. Anti-BIG1 and anti-BIG2 antibodies were raised in rabbits and affinity-purified with the peptides used for immunization coupled to AminoLink coupling gel (Pierce), according to the manufacturer's instructions.
Western Blotting.
All procedures were carried out at 4°C. To prepare cytosol, cells were washed three times with ice-cold PBS, scraped into buffer A [25 mM Hepes⋅NaOH (pH 7.4), 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), leupeptin and aprotinin (each 10 μg/ml), and TPCK and TLCK (each 1 μg/ml)] containing 0.25 M sucrose, and homogenized with 10 strokes in a Dounce tissue grinder (Wheaton Scientific). The homogenate was centrifuged (1,000 × g for 3 min), the supernatant was centrifuged (8,000 × g for 10 min), and that supernatant was centrifuged at 105,000 × g for 1.5 hr. The final supernatant and precipitate are referred to as cytosol and microsomes, respectively. Microsomes were homogenized in 0.5 ml of buffer A. Samples of HeLa and HepG2 cytosol (each 100 μg of protein) and microsomes (HeLa, 10 μg, and HepG2, 15 μg) were subjected to SDS/PAGE in 6% gel and transferred to poly(vinylidene difluoride) (PVDF) membrane, which was incubated with anti-BIG1 (0.5 μg/ml) and/or anti-BIG2 IgG (1 μg/ml), followed by goat anti-rabbit IgG conjugated to horseradish peroxidase (Promega) and detection using Super Signal Chemiluminescent substrate (Pierce). Immunoreactive bands were quantified by Personal Densitometer SI (Molecular Dynamics).
Immunoprecipitation.
Cytosol and microsomes were prepared from HepG2 cells as described above, except that microsomes were homogenized in buffer A containing 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS). CHAPS and BSA were added to samples of cytosol to final concentrations of 0.5% (wt/vol) and 0.1% (wt/vol), respectively, and BSA was added to samples of microsomes to a final concentration of 0.1% (wt/vol). Samples of cytosol (2.6 mg of protein) and microsomes (0.4 mg) were used for immunoprecipitation. As a control for reactions with anti-BIG1 and anti-BIG2 antibodies, the appropriate immunizing peptide was added (100 μg of BIG1 or 300 μg of BIG2), followed by anti-BIG1 (2.5 μg) and/or anti-BIG2 IgG (5 μg). After incubation for 1 hr, 60 μl of a 50% slurry of protein A Sepharose CL-4B in PBS (Amersham Pharmacia) were added, and the mixture was incubated overnight. Beads were washed five times with ice-cold PBS containing 0.5% CHAPS. Proteins bound to beads were eluted by boiling in SDS/PAGE loading buffer, separated by SDS/PAGE in 6% gel, transferred to PVDF membranes, and detected using chemiluminescent substrate.
For titration of anti-BIG1 or anti-BIG2 IgG, samples of cytosol (30 μg) from HepG2 cells were incubated with different amounts of each IgG, 0.1% (wt/vol) BSA, and 60 μl of protein A-Sepharose CL-4B (50% slurry in PBS) overnight. After centrifugation, the supernatant was transferred to another tube, and beads were washed five times with ice-cold PBS containing 0.5% CHAPS. Proteins bound to beads were eluted and analyzed by Western blotting, followed by quantification of immunoreactive proteins by densitometry. The supernatant from the first immunoprecipitation was further incubated with 3.75 μg of anti-BIG1 or anti-BIG2 IgG and 60 μl of protein A-Sepharose CL-4B (50% slurry in PBS) overnight. Beads were washed five times with ice-cold PBS containing 0.5% CHAPS. Proteins bound to beads were eluted and immunoreactive proteins were quantified by densitometry.
Immunofluorescence Microscopy.
HepG2 cells and HeLa cells were grown on 2-well culture slides with or without collagen-coating, respectively, for 24 hr, and further incubated with or without BFA, 10 μg/ml, in culture medium, followed by fixation with 4% paraformaldehyde in PBS for 20 min at room temperature. After washing with PBS, cells were permeabilized in PBS containing 0.05% Triton X-100 for 4 min, washed with PBS, incubated with blocking buffer (PBS containing 5% horse serum, 5% goat serum, and 6% BSA), and incubated overnight at 4°C with primary antibodies diluted in the blocking buffer. Anti-BIG1 and anti-BIG2 IgGs were used at final concentrations of 2 and 5 μg/ml, respectively. To assess specificity of reactions with anti-BIG1 and anti-BIG2 IgGs, the appropriate immunizing peptide was added at a final concentration of 100 (BIG1) or 300 μg/ml (BIG2). Mouse monoclonal antibodies to Golgi 58K protein (clone no. 58K-9) and γ-adaptin (AP-1: clone no. 100/3), purchased from Sigma, were diluted 1:50 and 1:500, respectively, with blocking buffer. After washing with PBS, cells were incubated with fluorescein isothiocyanate-labeled anti-rabbit IgG and Texas Red-labeled anti-mouse IgG antibodies (Vector Laboratories), which had been diluted 1:200 in the blocking buffer, for 1 hr and washed with PBS. Coverslips were mounted in Vectashield (Vector Laboratories) and inspected with a confocal microscope (Leica model TCS4D/DMIRBE).
Fractionation of Microsomes.
All procedures were carried out at 4°C. Microsomes (1.5 mg of protein) prepared from HepG2 cells were homogenized with six strokes in 4 ml of buffer A containing 1.4 M sucrose in a Dounce tissue grinder (Wheaton). The homogenates were loaded at the bottom of a discontinuous sucrose gradient, composed of successive layers of 2 ml of buffer A containing 1.15, 0.85, 0.6, and 0.25 M sucrose, followed by centrifugation at 175,000 × g for 2 hr. Fractions from the 1.15/0.85, 0.85/0.6 and 0.6/0.25 M interfaces were defined as heavy Golgi, intermediary Golgi, and light Golgi fractions, respectively (22). To each fraction (2 ml) was added 0.1 mg BSA (using a solution of 1 mg/ml), followed by 0.2 ml of 100% (wt/vol) trichloroacetic acid. Precipitated proteins were dissolved in 200 μl of loading buffer, and samples (40 μl) were subjected to SDS/PAGE in 6% gel, transferred to PVDF membranes, and reacted with anti-BIG1 (0.5 μg/ml) or anti-BIG2 (1 μg/ml) IgG, or anti-β-COP antibody, followed by detection as described above. Monoclonal anti-β-COP antibody (clone no. maD from Sigma), was diluted 1:200, and goat anti-mouse IgG conjugated to horseradish peroxidase (Promega) was used as a secondary antibody.
Mammalian Expression Vector with BIG2 cDNA.
pcDNA3.1/BIG2-Myc, which encodes a fusion protein of BIG2 with a C-terminal myc tag, was constructed as follows. To remove a TAG stop codon from BIG2 cDNA, two oligonucleotides, 5′-CAGCAGCACTGTCACCAGTGTGGC-3′ and 5′-GGCCGCCACACTGGTGACAGTGCTGCTGGTAC-3′, were used. Underlined sequences are KpnI or NotI restriction sites. The complementary oligonucleotides were mixed, heated at 100°C for 5 min, and cooled to room temperature before ligation to pcDNA3.1/Myc-His(+) (Invitrogen) that had been digested with KpnI and NotI (named pcDNA3.1/Myc-KN). BIG2 cDNA in pBluescript SK(−) (16) was digested with KpnI, and the excised fragment was ligated to the KpnI site of pc DNA3.1/Myc-KN (named pcDNA3.1/BIG2-Myc-KN).
To remove an ATG preceding the initiation codon, PCR was performed using BIG2 cDNA as template, forward primer 5′-GCTAGCCATGCAGGAGAGCCAGACC-3′ including the NheI site (underline) and reverse primer 5′-TGTGCCCGTATGCGATGAGTTTCTG-3′. The PCR product was cloned into PCR-TOPO vector (Invitrogen) and sequenced. The excised fragment of the cloned PCR product with NheI was ligated to SpeI and NheI sites of pcDNA3.1/BIG2-Myc-KN (named pcDNA3.1/BIG2-Myc).
Transfection.
Cells were incubated in 2-well culture slides for 24 hr at 37°C in 5% CO2/95% air before transfection with pcDNA3.1/BIG2-Myc, using Geneporter (Gene Therapy Systems, San Diego) according to the manufacturer's instructions. After incubation for 24 hr, cells were fixed and prepared for immunofluorescence analysis. Mouse monoclonal anti-myc antibodies (Invitrogen) were diluted 1:1,000.
Fractionation of HeLa Cells, Gel Filtration, and Analysis.
HeLa cells (3 × 108 cells) were washed three times with ice-cold PBS, collected by scraping, suspended in 6 ml of PBS with 0.5 mM AEBSF, 1 mM DTT, and protease inhibitors (soybean and lima bean trypsin inhibitors, leupeptin, and aprotinin, each 1 μg/ml), and homogenized with 10 strokes of a Dounce tissue grinder. The homogenate was centrifuged (150,000 × g for 1.25 hr) and cytosol, concentrated to 1.4 ml (16 mg protein) with Centricon-10 (Millipore), was applied to a column (1.2 × 88 cm; Vt = 99 ml) of Sepharose CL-6B, equilibrated and eluted with 20 mM Tris⋅HCl (pH 8.0), containing 0.25 M sucrose, 2 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 1 mM NaN3, 1 mM DTT, and 0.5 mM AEBSF. Samples (45 μl) of fractions (0.96 ml) were subjected to SDS/PAGE in 8% gel and reacted with affinity-purified anti-BIG1 and anti-BIG2 IgGs; immunoreactive BIG1 and BIG2 were quantified by densitometry.
Results
Immunoreaction of Proteins with Anti-BIG1 and/or BIG2 IgGs.
When proteins immunoprecipitated by anti-BIG1 or anti-BIG2 IgGs from HepG2 cell cytosol were reacted on Western blot with the precipitating antibody, single immunoreactive bands with mobilities, respectively, of proteins of 200 or 190 kDa were observed. Simultaneous immunoprecipitation using both antibodies, followed by Western blot analysis with the same two antibodies revealed the two proteins with differing mobilities. Immunoprecipitation with anti-BIG1 and/or anti-BIG2 IgG in the presence of the peptide used for immunization resulted in no detectable immunoreactive BIG1 or BIG2 on Western blots (data not shown). Molecular weights calculated from the deduced amino acid sequences of BIG1 and BIG2 cDNAs are 208,634 and 201,964, respectively, i.e., slightly higher than the apparent molecular weights on SDS/PAGE, which are similar for the native and recombinant (synthesized in Sf9 cells) BIG1 or BIG2 (data not shown).
Localization of BIG1 and BIG2.
Confocal laser scanning microscopy was used for localization of BIG1 and BIG2 in HepG2 cells with anti-BIG1 and anti-BIG2 IgGs. Both BIG1 and BIG2 were detected in the perinuclear region and in a punctate distribution throughout the cytosol, and addition of the immunizing peptide largely prevented the reaction (data not shown). To assess independently the intracellular localization of BIG1 and BIG2, cytosolic and microsomal fractions were prepared from HeLa and HepG2 cells. Samples of cytosol and microsomal proteins were subjected to SDS/PAGE and transferred to PVDF membranes. Western blot analysis showed that both BIG1 and BIG2 were largely cytosolic, although both were detected also in the microsomal fractions, relatively more in HepG2 than HeLa cells (Fig. 1A). To determine the relative amounts of BIG1 or BIG2 in the cytosol and microsomal fractions, samples of cytosolic proteins from HepG2 cells were subjected to SDS/PAGE, followed by Western blotting using anti-BIG1 or anti-BIG2 IgG (Fig. 1B). After quantification of immunoreactive protein by densitometry, standard curves for cytosolic BIG1 and BIG2 were prepared. Immunoreactive BIG1 or BIG2 was proportional to the amount of cytosolic protein up to 15–20 μg. The amount of BIG1 in 15 μg of microsomal protein corresponded to that in 4.2 μg of cytosolic protein, whereas BIG2 in 15 μg of microsomal protein was equivalent to that in 9 μg of cytosolic protein (Fig. 1B). Thus, the total amounts of BIG1 and BIG2 in the cytosolic protein fractions were, respectively, about 24 and 11 times those in microsomal proteins.
Figure 1.

Western blot analysis of BIG1 and BIG2 in the cytosolic and microsomal fractions from HeLa and HepG2 cells. (A) Samples of cytosol (C, 100 μg of proteins) or microsomes (M, 10 μg from HeLa, 15 μg from HepG2 cells) were subjected to SDS/PAGE and transferred to PVDF membrane, followed by reaction with anti-BIG1 (Left) or anti-BIG2 (Right) IgG. (B) The indicated amounts of proteins from M or C fractions of HepG2 cells were subjected to SDS/PAGE in 6% gel and transferred to PVDF membrane, followed by Western blotting using anti-BIG1 or anti-BIG2 IgG. Immunoreactive proteins were quantified by densitometry. On standard curves for cytosolic BIG1 and BIG2, positions of the M samples (15 μg) are indicated. Total C and M protein was, respectively, 3.9 and 0.39 mg for HeLa, and 16 and 2.4 mg for HepG2 cells. Similar data were obtained in four independent experiments.
Subcellular Localization of BIG1 and BIG2.
To define more precisely the subcellular distribution of BIG1 and BIG2, the microsomal fraction (1.7 mg protein) from HepG2 cells was centrifuged on a discontinuous sucrose density gradient. Proteins in selected fractions were separated by SDS/PAGE and reacted with anti-BIG1 and anti-BIG2 IgGs to reveal single bands of about 200 and 190 kDa, respectively, in the heavy Golgi fraction (Fig. 2). β-COP was found in the same fraction, consistent with the association of both BIG1 and BIG2 with Golgi membranes.
Figure 2.

Subcellular fractionation of microsomes from HepG2 cells. Microsomal proteins (1.7 mg) from HepG2 cells were fractionated on a discontinuous sucrose density gradient. Proteins from the 0.6/0.25, 0.85/0.6, and 1.15/0.85 interfaces were precipitated with trichloroacetic acid and separated by SDS/PAGE, followed by Western blotting using anti-BIG1 or anti-BIG2 IgG or anti-β-COP antibodies, as indicated. Observations were replicated three times.
The distribution of native BIG1 and BIG2 in intact cells as evaluated by immunostaining using the specific anti-peptide IgGs and antibodies against Golgi-specific 58K protein or γ-adaptin (AP-1). BIG1 was partially colocalized with 58K or AP-1 in a perinuclear region and also distributed throughout the cells in a punctate pattern (Fig. 3). The pattern of BIG2 staining was quite similar to that of BIG1, i.e., partial colocalization with 58K or AP-1 near the nucleus and in the rest of the cell (Fig. 3). Similar results were obtained in analogous experiments with HeLa cells (not shown). It appears that Golgi membranes are a predominant site of concentration of both BIG1 and BIG2 in the perinuclear region.
Figure 3.
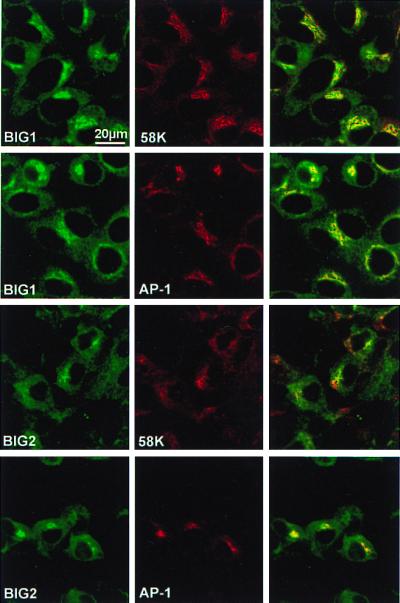
Golgi localization of BIG1 and BIG2. HepG2 cells were reacted with rabbit anti-BIG1 or anti-BIG2 IgG and mouse anti-58K or -AP-1 antibodies, as indicated, followed by FITC-labeled anti-rabbit IgG and Texas Red-labeled anti-mouse IgG. In the third panel of each row, images of the preceding two panels are superimposed. Observations were replicated with other samples of cells, five times for BIG1 and three times for BIG2.
Double immunofluorescence microscopy of HeLa and HepG2 cells transfected with mammalian expression vector, pcDNA3.1/BIG2-Myc, revealed endogenous BIG1 (labeled with FITC), for the most part, colocalized with recombinant BIG2 (Texas Red staining), suggesting very similar distribution of endogenous BIG1 and overexpressed BIG2 in both types of cells (Fig. 4).
Figure 4.

Colocalization of endogenous BIG1 and overexpressed BIG2. HeLa and HepG2 cells were transfected with pcDNA3.1/BIG2-Myc encoding a fusion protein of BIG2 with a C-terminal myc tag. After 24 hr, cells were fixed, permeabilized, and incubated with rabbit anti-BIG1 IgG and mouse anti-myc antibodies (to detect BIG2), as indicated, followed by FITC-labeled anti-rabbit IgG and Texas Red-labeled anti-mouse IgG. In the third panel of each row, images of the preceding two panels are superimposed. Observations were replicated twice.
Identification of BIG1 and BIG2 in the Same Protein Complexes.
BIG1 and BIG2 were initially copurified with protein complexes of >670 kDa (21). It was unclear, however, whether the two proteins were associated with the same or different macromolecular complexes. To investigate this question, cytosol from HeLa cells was filtered on Sepharose CL-6B, followed by quantification of immunoreactive BIG1 and BIG2 in each fraction by Western blotting using anti-BIG1 or anti-BIG2 IgG and densitometry. Although their elution patterns were not identical, the largest percentages of both proteins were in fractions containing molecules of >670 kDa (Fig. 5), consistent with the possibility that BIG1 and BIG2 are components of the same, or different but quite similar, complexes.
Figure 5.

Fractionation of BIG1 and BIG2 on Sepharose CL-6B. Cytosol (16 mg protein) from HeLa cells was fractionated on Sepharose CL-6B. Samples of fractions were subjected to SDS/PAGE, followed by Western blotting using anti-BIG1 and anti-BIG2 IgGs. Immunoreactive bands were quantified by densitometry; amounts of BIG1 and BIG2 are recorded as percentage of the total of each recovered. Elution positions of standard proteins are indicated. Th, thyroglobulin (669 kDa); Fe, ferritin (440 kDa); Ca, catalase (232 kDa); Al, aldolase (158 kDa). Data from a replicate experiment were similar.
To determine whether BIG1 and BIG2 existed in the same protein complexes, cytosolic and microsomal fractions from HepG2 cells were subjected to immunoprecipitation using anti-BIG1 or anti-BIG2 IgG. When the immunoprecipitated proteins were analyzed by Western blotting with anti-BIG1 IgG, a single reactive band of ≈200 kDa was detected among the anti-BIG2 IgG-precipitated proteins, as well as the anti-BIG1 IgG-precipitated proteins (Fig. 6). Similarly, a 190-kDa band reactive with anti-BIG2 IgG was present on Western blots of proteins precipitated using anti-BIG1 or anti-BIG2 IgG (Fig. 6). No bands were observed when immunoprecipitation was performed in the presence of the peptide that had been used for immunization (Fig. 6). Thus, it appears that both BIG1 and BIG2 were present together in at least some of the macromolecular complexes from both cytosol and microsomal fractions.
Figure 6.

Immunoprecipitation of BIG1 and BIG2 complexes from HepG2 cells. Samples of proteins, cytosol (C) 2.6 mg, or microsomes (M) 0.4 mg, were immunoprecipitated (IP) by anti-BIG1 or anti-BIG2 IgG with or without the peptide used for immunization, as indicated. Immunoprecipitated proteins were separated by SDS/PAGE, and reacted on Western blots (WB) with anti-BIG1 or anti-BIG2 IgG. Data were similar in three experiments.
To establish the amount of anti-BIG1 or anti-BIG2 IgG needed to immunoprecipitate completely BIG1 or BIG2 from 30 μg of HepG2 cells cytosolic proteins, proteins precipitated by different amounts of antibodies were separated by SDS/PAGE and anti-BIG1 or anti-BIG2 IgG immunoreactive bands were quantified by densitometry. BIG1 or BIG2 in 30 μg of cytosolic proteins were completely immunoprecipitated by more than 2.5 μg of the appropriate IgG (Fig. 7A). After immunoprecipitation without or with anti-BIG1 or anti-BIG2 IgG (3.75 μg), the supernatants were subjected to a second immunoprecipitation using anti-BIG1 or anti-BIG2 IgG. Western blot analysis of immunoprecipitated proteins using anti-BIG1 IgG demonstrated immunoreactive BIG1 among cytosolic proteins that had been precipitated by anti-BIG2 IgG, as well as in the remaining supernatant. Similarly, BIG2 was present among proteins precipitated by anti-BIG1 IgG and in the supernatant (Fig. 7B). Neither BIG1 nor BIG2 was detected on second immunoprecipitation with the same antibody. Densitometry showed that total BIG1 and BIG2 were, respectively, 4 and 3.4 times the amounts remaining in the supernatant after immunoprecipitation with the opposite antibody. Thus, 70–75% of total BIG1 and BIG2 in the cytosol of HepG2 cells appeared to be components of the same protein complexes.
Figure 7.

Immunoprecipitation of protein complexes containing BIG1 and BIG2. (A) Proteins immunoprecipitated from samples of cytosolic proteins (30 μg) from HepG2 cells with the indicated amounts of anti-BIG1 or anti-BIG2 IgG were analyzed by Western blotting using anti-BIG1 or anti-BIG2 IgG, followed by quantification of immunoreactive protein by densitometry. (B) Samples of HepG2 cell cytosolic proteins (30 μg) were incubated first without or with anti-BIG1 or anti-BIG2 IgG (3.75 μg) and protein A beads. After centrifugation, the supernatant was incubated a second time with anti-BIG1 or anti-BIG2 IgG and protein A beads. Immunoprecipitated proteins were analyzed by SDS/PAGE and Western blotting (WB) using anti-BIG1 or anti-BIG2 IgG, and quantified by densitometry. Data were similar in three experiments.
Effect of BFA on Localization of BIG1 and BIG2.
When HepG2 cells were incubated with BFA for 1 or 10 min, BIG1 and BIG2 remained clustered in the perinuclear region, and the distribution of 58K was relatively little altered by BFA (Fig. 8). After BFA addition, however, β-COP was rapidly dispersed through the cytosol (Fig. 8).
Figure 8.

Effect of BFA on intracellular distribution of BIG1 and BIG2. HepG2 cells were incubated without or with BFA (10 μg/ml) for 0, 1, or 10 min, fixed, permeabilized, and incubated with anti-BIG1 or anti-BIG2 IgG, or rabbit anti-β-COP or mouse anti-58K antibodies, as indicated, followed by FITC-labeled anti-rabbit IgG or Texas Red-labeled anti-mouse IgG. Data were similar in three experiments.
Discussion
ARFs, which are components of several vesicular trafficking pathways, are converted from an inactive GDP-bound form to an active GTP-bound form by GEPs. In mammals, six ARFs and at least four families of ARF GEPs have been identified to date. The intracellular functions of individual GEPs are likely determined by their interactions with specific ARFs and with other proteins that modify their activity or intracellular localization, or are themselves modified by the GEP interaction. ARF1, which is cytosolic in the inactive state and associated with membranes when active, can recruit coat proteins to the Golgi membrane to initiate vesicle formation (1, 2). In vitro, all ARF GEPs can apparently activate ARF1. An important question that remains unresolved is which GEPs are responsible for ARF1 activation in the Golgi of the intact cell. Evidence to date is that ARF1 activation in the Golgi is inhibited by BFA (23, 24) and the BFA-sensitive, 200-kDa BIG1, when overexpressed as a fusion protein, was localized in the Golgi apparatus (20), as were endogenous BIG1 and BIG2 in our studies.
The results of immunofluorescent staining and Western blotting of cytosolic and microsomal fractions reported here indicate that the BFA-sensitive BIG1 and BIG2 were colocalized with the Golgi marker proteins, 58K and AP-1, in the perinuclear region. Like the 58K protein (25), they were not dispersed from the Golgi by BFA treatment that causes release of ARF and β-COP, which depends on ARF for membrane association, at least during vesicle formation (1). Thus, the Golgi association of BIG1 and BIG2 appears not to involve ARF. Another ARF GEP of large molecular mass, GBF1 (17), which resembles yeast Gea1/Gea2, is also present in the Golgi apparatus; it, however, has BFA-resistant GEP activity that is greater toward ARF5 than ARF1. ARF5 associates with the Golgi, but its role in Golgi function remains unclear. Views about GEP interactions with ARF6 are contradictory (26, 27). Langille et al. (28) recently provided clarification by addressing directly the limitations inherent in overexpression experiments, and in drawing conclusions about physiological specificity from in vitro guanine nucleotide-binding assays. They demonstrated convincingly that endogenous ARF6 and cytohesin-3 (GRP1) were rapidly recruited to plasma membrane ruffles of cultured cells exposed to insulin or epidermal growth factor (28). In addition, overexpression of GRP1 or cytohesin-1 increased the fraction of GTP-bound endogenous ARF6 (28). All of their observations are consistent with a role for ARF6, and probably one or more cytohesins, in an endosomal membrane recycling pathway (29).
BIG1 and BIG2 are, to date, the only BFA-sensitive GEPs purified from mammalian tissue (21). On subcellular fractionation, as reported here, they were largely cytosolic, consistent with their original identification in bovine brain cytosol (21). BIG1 and BIG2 were copurified associated with molecules of >670 kDa, suggesting that the native proteins might exist as components of the same or very similar macromolecular complexes. We provide here direct evidence that BIG1 and BIG2 do exist together as components of the same protein complexes in the cytosol and in microsomal fractions containing Golgi membranes. In the cytosol, 70–75% of total BIG1 and BIG2 were associated with the same immunoprecipitated protein complexes. Whether the remaining cytosolic BIG1 and BIG2 (25–30% of the total) are associated with other proteins in different kinds of complexes is among many questions that need to be addressed, as is, for example, the physical state of BIG1 and BIG2 in the Golgi membrane. Yeast Gea1, a 160-kDa protein of 1,408 amino acids, was present in a 500- to 600-kDa complex (19), and GBF1, the mammalian homologue of yeast Gea1/Gea2 (30), may also exist in multi-protein complexes.
The yeast homologue of BIG1 and BIG2, the 230-kDa Sec7 protein (Sec7p), has a highly acidic domain near the N terminus that may interact with lipids or proteins on the cytoplasmic surface of Golgi membranes (18), and participate in Sec7 movement between cytosol and membranes. Alignment of amino acid sequences of the N-terminal one-third of BIG1 and BIG2 shows segments of 79, 30, and 89% identity with the acidic region of Sec7p in the first (16). Although BIG1 and BIG2 lack similar acidic regions, BIG1 is reported to have a Golgi-localization signal in the N-terminal third of the protein (20). The mechanism of the membrane association of BIG1 and BIG2 remains among many unanswered questions.
Coiled-coil structures are known to participate in protein–protein interactions. Recombinant cytohesin-2 (ARNO), intact or lacking the C-terminal pleckstrin homology domain, was dimeric, but the protein from which the coiled-coil region had also been deleted was not (9). If the cytohesin-2 coiled-coil region is involved in dimerization, cytohesins 1, -3, and -4 (31) and EFA6 (14), all of which include coiled-coil regions, may also exist in cells as dimers. Cytohesins or EFA6 might also interact with unidentified proteins in larger complexes. BIG1 and BIG2, unlike the cytohesin and EFA6 families, lack coiled-coil regions, and it will be important to learn just how they interact, whether directly or by means of intermediary molecules, to identify the other components, and, of course, to establish their stoichiometry in the complexes. It is clear that ARF activation is not the only function of the multidomain GEP molecules, and all ARF GEPs probably operate within the architecture of multiprotein structures that regulate and coordinate their diverse activities and conformations to control chemical and mechanical processes with the temporal and spatial precision that is critical for cell vitality.
Evidence is provided here for the existence of a multimolecular complex containing BIG1 and BIG2 that is present in cytosol and microsomal fractions, probably in Golgi membranes. Identification of the remaining components and quantification of their stoichiometry is expected to contribute to understanding the role of the protein complex in membrane trafficking, the significance of the rather puzzling existence of two very similar GEPs in the same complex, and the structural-functional interactions of the complex with other molecules in the cytosol and Golgi apparatus.
Acknowledgments
We thank Carol Kosh for expert secretarial assistance.
Abbreviations
- ARF
ADP-ribosylation factor
- GEP
guanine nucleotide-exchange protein
- BFA
brefeldin A
- PVDF
poly(vinylidene difluoride)
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- AEBSF
4-(2-aminoethyl)benzenesulfonyl fluoride
- AP-1
γ-adaptin
References
- 1.Rothman J E, Wieland F T. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 2.Springer S, Spang A, Schekman R. Cell. 1999;97:145–198. doi: 10.1016/s0092-8674(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 3.Tsuchiya M, Price S R, Tsai S-C, Moss J, Vaughan M. J Biol Chem. 1991;266:2772–2777. [PubMed] [Google Scholar]
- 4.Donaldson J G, Cassel D, Kahn R A, Klausner R D. Proc Natl Acad Sci USA. 1992;89:6408–6412. doi: 10.1073/pnas.89.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer D J, Helms J B, Beckers C J, Orci L, Rothman J E. J Biol Chem. 1993;268:12083–12089. [PubMed] [Google Scholar]
- 6.Stamnes M A, Rothman J E. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- 7.Dell'Angelica E C, Mullins C, Bonifacino J S. J Biol Chem. 1999;274:7278–7285. doi: 10.1074/jbc.274.11.7278. [DOI] [PubMed] [Google Scholar]
- 8.Moss J, Vaughan M. J Biol Chem. 1998;273:21431–21434. doi: 10.1074/jbc.273.34.21431. [DOI] [PubMed] [Google Scholar]
- 9.Chardin P, Paris S, Antonny B, Robineau S, Béraud-Dufour S, Jackson C L, Chabre M. Nature (London) 1996;384:481–484. doi: 10.1038/384481a0. [DOI] [PubMed] [Google Scholar]
- 10.Sata M, Donaldson J G, Moss J, Vaughan M. Proc Natl Acad Sci USA. 1998;95:4204–4208. doi: 10.1073/pnas.95.8.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morinaga N, Adamik R, Moss J, Vaughan M. J Biol Chem. 1999;274:17417–17423. doi: 10.1074/jbc.274.25.17417. [DOI] [PubMed] [Google Scholar]
- 12.Meacci E, Tsai S-C, Adamik R, Moss J, Vaughan M. Proc Natl Acad Sci USA. 1997;94:1745–1748. doi: 10.1073/pnas.94.5.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klarlund J K, Guilherme A, Holik J J, Virbasius J V, Chawla A, Czech M P. Science. 1997;275:1927–1930. doi: 10.1126/science.275.5308.1927. [DOI] [PubMed] [Google Scholar]
- 14.Franco M, Peters P J, Boretto J, Donselaar E, Neri A, D'Souza-Schorey C, Chavrier P. EMBO J. 1999;18:1480–1491. doi: 10.1093/emboj/18.6.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morinaga N, Moss J, Vaughan M. Proc Natl Acad Sci USA. 1997;94:12926–12931. doi: 10.1073/pnas.94.24.12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Togawa A, Morinaga N, Ogasawara M, Moss J, Vaughan M. J Biol Chem. 1999;274:12308–12315. doi: 10.1074/jbc.274.18.12308. [DOI] [PubMed] [Google Scholar]
- 17.Claude A, Zhao B-P, Kuziemsky C E, Dahan S, Berger S J, Yan J-P, Arnold A D, Sullivan E M, Melançon P. J Cell Biol. 1999;146:71–84. [PMC free article] [PubMed] [Google Scholar]
- 18.Achstetter T, Franzusoff A, Field C, Schekman R. J Biol Chem. 1988;263:11711–11717. [PubMed] [Google Scholar]
- 19.Peyroche A, Paris S, Jackson C L. Nature (London) 1996;384:479–481. doi: 10.1038/384479a0. [DOI] [PubMed] [Google Scholar]
- 20.Mansour S L, Skaug J, Zhao X-H, Giordano J, Scherer S W. Proc Natl Acad Sci USA. 1999;96:7968–7973. doi: 10.1073/pnas.96.14.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morinaga N, Tsai S-C, Moss J, Vaughan M. Proc Natl Acad Sci USA. 1996;93:12856–12860. doi: 10.1073/pnas.93.23.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrenreich J H, Bergeron J J M, Siekevitz P, Palade G E. J Cell Biol. 1973;59:45–72. doi: 10.1083/jcb.59.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helms J B, Rothman J E. Nature (London) 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- 24.Donaldson J G, Finazzi D, Klausner R D. Nature (London) 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- 25.Gao Y, Alvarez C, Nelson D, Sztul E. J Biol Chem. 1998;273:33825–33834. doi: 10.1074/jbc.273.50.33825. [DOI] [PubMed] [Google Scholar]
- 26.Franco M, Boretto J, Robineau S, Monier S, Goud B, Chardin P, Chavrier P. Proc Natl Acad Sci USA. 1998;95:9926–9931. doi: 10.1073/pnas.95.17.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank S, Upender S, Hansen S H, Casanova J E. J Biol Chem. 1998;273:23–27. doi: 10.1074/jbc.273.1.23. [DOI] [PubMed] [Google Scholar]
- 28.Langille S E, Patki V, Klarlund J K, Buxton J M, Holik J J, Chawla A, Corvera S, Czech M P. J Biol Chem. 1999;274:27099–27104. doi: 10.1074/jbc.274.38.27099. [DOI] [PubMed] [Google Scholar]
- 29.Radhakrishna H, Donaldson J G. J Cell Biol. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mansour S J, Herbrick J-A, Scherer S W, Melançon P. Genomics. 1998;54:323–327. doi: 10.1006/geno.1998.5563. [DOI] [PubMed] [Google Scholar]
- 31.Ogasawara M, Kim S-C, Adamik R, Togawa A, Ferrans V J, Takeda K, Kirby H, Moss J, Vaughan M. J Biol Chem. 2000;275:3221–3230. doi: 10.1074/jbc.275.5.3221. [DOI] [PubMed] [Google Scholar]