

Abstract
A variety of DNA vaccine prime and recombinant viral boost immunization strategies have been developed to enhance immune responses in humans, but inherent limitations to these strategies exist. There is still an overwhelming need to develop safe and effective approaches that raise broad humoral and T cell-mediated immune responses systemically and on mucosal surfaces. We have developed a novel mucosal immunization regimen that precludes the use of viral vectors yet induces potent T cell responses. Using hepatitis B surface antigen (HBsAg), we observed that vaccination of Balb/c mice with an intramuscular HBsAg-DNA vaccine prime followed by an intranasal boost with HBsAg protein encapsulated in biologically inert liposomes enhanced humoral and T cell immune responses, particularly on mucosal surfaces. Intranasal live virus challenge with a recombinant vaccinia virus expressing HBsAg revealed a correlation between T cell immune responses and protection of immunized mice. A shortened immunization protocol was developed that was successful in both adult and neonatal mice. These results support the conclusion that this new approach is capable of generating a T helper type 1 biased, broad spectrum immune response, specifically at mucosal surfaces. The success of this design may provide a safe and effective vaccination alternative for human use.
Keywords: Vaccination, Mucosa, T cells, Cytokines, Antibodies
The development of various vaccines has significantly improved the quality of human life resulting in drastic decreases in morbidity and mortality (1, 2). While the strategies employed to develop these vaccines have proven greatly effective, they have not been sufficient to combat pathogens that demonstrate unique pathogenesis or which require multiple components of the immune system for their elimination (3–6). The vast majority of data indicate that new, innovative vaccine strategies are required to combat the array of pathogens for which preventative and therapeutic avenues are lacking. Development of robust, practical vaccines for sexually transmitted diseases (e.g. human immunodeficiency virus, herpes simplex virus, Chlamydia trachomatis), new and re-emerging pathogens (e.g. avian influenza virus, antibiotic resistant tuberculosis), and bioterrorism threats (e.g. anthrax, smallpox) remains a challenge (6–9).
Currently, only limited numbers of vaccines are safe and effective in raising protective immunity at mucosal surfaces, the most common entry portal of pathogens (10). Vaccines are also limited in their ability to raise robust cell-mediated immune responses that are usually required to eliminate intracellular infections (11, 12). One new approach is a heterologous vaccine regimen involving a DNA prime followed by an attenuated live or non-replicating virus boost. This prime and boost vaccination strategy has been employed to overcome the disappointingly ineffective induction of immune responses displayed by DNA immunization alone in non-human primates and humans (4, 13–18). The advantages of this approach include a synergistic effect on the induction of immune responses and the generation of a robust T cell mediated immune response (3, 4, 12–16, 19–25).
Limitations to the use of viral vectors are apparent. Viral vectors are usually not administered intranasally to raise mucosal immune responses due to the concern that they could result in localization to the central nervous system (26). Instead, generation of mucosal immune responses usually requires the inclusion of potentially toxic adjuvants delivered with a protein component of the vaccine (27–29). Development of immune responses to the viral vector raise concerns over widespread and repeated use of viral vectors (4, 30). Furthermore, recent results using a non-replicating adenoviral-based HIV vaccine led to halting of two clinical trials (31–36). Not only had the vaccine failed to prevent infection or reduce viral load, but further analysis suggested that the vaccine might have actually increased susceptibility to infection (31–36). The failure of the vaccine raises the question as to whether vaccines that only induce cell-mediated responses will prove effective without increasing risk to the vaccinated individual (31–36).
To address these issues, we have developed a new heterologous immunization strategy that relies on a DNA vaccine prime followed by a liposome-encapsulated protein boost. Both of these components are safe, easily manufactured and have demonstrated capability in the development of vaccines (37–39).
Using the model antigen, hepatitis B virus surface antigen (HBsAg), that is well-characterized and commercially available, we demonstrated that this protocol was highly effective in inducing systemic B and T cell-mediated responses and robust mucosal immune responses when the boost was delivered intranasally. The advantage of this protocol was that it not only induced a broad spectrum of systemic and mucosal immune responses, but also induced protective immune responses that were associated with elevated levels of antigen-specific CD8+ T cells in lungs of vaccinated mice. We also demonstrated a marked ability of the protocol to induce responses in HBsAg-nonresponder and neonatal mice, and the ability to induce potent responses after delivery of the components in a shortened time span.
This newly developed immunization protocol demonstrates all of the hallmarks of a successful vaccination regimen and may be an ideal candidate for the formulation of new preventative and therapeutic vaccines.
Materials and Methods
Immunogens
HBsAg vaccine (Engerix™-B; GSK Biologicals, HBsAg adw subtype), HBsAg-DNA vaccine pRc/CMV-HBs(S) ayw subtype (40) (Aldevron, Fargo, ND), purified recombinant HBsAg protein (ayw subtype, Advanced Immunochemical, San Diego, CA) and the Ld-restricted peptide of HBsAg (amino acids 28–39, i.e., S28–39, IPQSLDSWWTSL-OH, Biopeptide Co., San Diego, CA) were purchased from commercial sources.
Large multi-lamellar liposomes were prepared at the following lipid concentrations: Phosphatidylcholine/cholesterol/dicetyl phosphate 7/3/0.5 mole %. Dicetyl phosphate was dissolved in chloroform/5% ethanol and sonicated prior to the addition of phosphatidylcholine and cholesterol. Lipids were dried in a Labconco rotary evaporator for one hour and traces of chloroform were removed by freeze-drying with a Freezone 4.5 Freeze Dry System overnight. The lipid film was hydrated with antigen at a concentration of 125–300 μg/ml in 10 mM HEPES-buffer, 150 mM NaCl, pH 7.4 (HBS), and filtered through a 0.2 μm nylon filter. The mixture was vortexed thoroughly and allowed to sit for 1 hour and then vortexed again to ensure the formation of multilamellar vesicles. The resultant liposomes were then subjected to three freeze-thaw cycles (1 cycle = freezing for one hour and thawing for one hour at room temperature). The size of the liposomes was measured with a N4 MD Submicron Particle Size Analyzer (Coulter Electronics). The ζ-potential was measured using Zeta-Puls ζ-potential analyzer (Brookhaven Instruments) in 5 mM HEPES buffer, 1.0 mM NaCl, pH 7.4.
Mice and immunizations
Pathogen free, barrier maintained female Balb/c mice (H-2d) and female C57BL/6 (H-2b) mice 6–7 weeks of age were obtained from Harlan (Indianapolis, IN). All mice were maintained under specific-pathogen-free conditions. After one week of acclimation, mice were anesthetized with a ketamine/xylazine mixture, and administered the appropriate amount of DNA vaccine intramuscularly or HBsAg-liposomes or empty liposomes (10 to 50 μl total dose per mouse per time point) in both nostrils. All animals were housed in sterile microisolator cages and had no evidence of spontaneous infection. Animals were maintained in accordance with the guidelines of the Institutional Animal Care and Use Committees (IACUC) of both the University of Massachusetts Medical School and BRM, and in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 1996).
Sample collection
Blood samples were collected by snipping the end of the tail and collecting blood by capillary action into clot activating Microvette® microtubes (Sarstedt, Newton, NC). Vaginal washes were performed by instilling 2×40 μl of sterile saline intravaginally to anesthetized mice, flushing the cavity, and collecting the wash with a pipet tip. This procedure was repeated twice. Vaginal washes were preserved with a 20% solution (volume:volume) of 2.2% protease inhibitor cocktail (Sigma, St. Louis, MO) in PBS.
Measurement of antibody responses
HBsAg-specific antibody responses were measured using ELISA assays. Assays included measurements for total Ig, IgG, IgG1 and IgG2a (three layers consisting of HBsAg, sample, and anti-Ig-HRP detecting antibodies), and mucosal IgG and IgA (five layers consisting of anti-mouse IgG or IgA capture, sample, HBsAg, rabbit anti-HBsAg, and goat anti-rabbit Ig-HRP detecting antibody). Antibodies and mouse Ig isotype standards were purchased from BD Biosciences (San Diego, CA), Novus Biologicals (Littleton, CO) and Southern Biotech (Birmingham, AL). High binding ELISA plates were purchased from Costar. Assays were developed and optimized based on the conditions recommended by BD Biosciences using 10μg/ml (50μl per well) of recombinant HBsAg in binding buffer to coat the plates for direct assays, and 2–5μg/ml (100μl per well) for the antibody sandwich layers. Checkerboard titrations were performed to optimize the signal:noise ratio for each assay. ELISAs were developed using Sure-Blue™ TMB microwell peroxidase substrate (KPL, Gaithersburg, MD) and 2 N sulfuric acid to stop the reaction. Plates were read using a Multiskan Ascent plate reader (Thermo Electron Corp., Mountain View, CA). Results are expressed either as OD at 450 nm (total IgG and IgA assays) or as μg/ml of IgG (quantitative assays for IgG, IgG1, and IgG2a).
Virus and viral challenge
A recombinant vaccinia virus expressing HBsAg (vHBs4) was kindly provided by Dr. Bernard Moss (41). Virus was grown in NIH L929 cells and titrated in Vero cells. Anesthetized mice received 2 to 5 × 105 PFU of vHBs4 intranasally in a volume of 50 μl.
Virus titration
The number of PFU of vaccinia virus was determined by plaque assay using 10-fold serial dilutions of a 10% homogenate of lungs taken from individual mice. Results were expressed as the geometric mean titers, i.e., the arithmetic averages of the logs for five separate animals titrated for virus individually plus or minus the SEM. Titers reported are log10 PFU (42).
Intracellular cytokine staining assay (ICS)
Intracellular staining for IFN-γ was performed on cells using commercial reagents and protocols (Cytofix/Cytoperm kit including GolgiPlug; Pharmingen, San Diego, CA). Briefly, mononuclear cells were first stimulated in vitro for 5 hours at 37° C in complete medium with the Ld-restricted HBsAg T cell epitope S28–39 or with no stimulus in the presence of GolgiPlug. Cells were then harvested and incubated with Fc block (anti-mouse CD16/CD32 unlabeled) followed by anti-CD8α-PerCP and anti-CD4-FITC. Cells were then permeabilized with Cytofix/Cytoperm and incubated with anti-mouse IFN-γ-PE in the presence of PermWash buffer. Appropriate isotype control antibodies were also included to determine background staining levels. All staining procedures were performed in 96-well U-bottom plates using standard immunfluorescence staining protocols. Freshly stained samples were analyzed on a BD Biosciences LSR II using FlowJo software. At least 200,000 events were typically analyzed.
ELISpot assays
ELISpot assays for IFN-γ and IL-4 release were performed using antibody pairs and reagents from BD Pharmingen, and 96 well ELISpot plates from Millipore Corp. (Hopkington, MA). Samples (1×106 and 5×105 cells per well) were incubated with either recombinant HBsAg protein (starting at a 10mM concentration), Con A (2.5 μg/ml) or anti-mouse CD3α (10 μg/ml) for positive controls or were left untreated or incubated with irrelevant antigen (negative controls) for 1.5 days in a 37°C incubator (5% CO2). All samples were tested in duplicate. ELISpot assays were developed according to manufacturer’s directions. Plates were scanned and digitally quantified to determine the number of spots per well and size distribution of the spots (CTL, Cleveland, OH).
Plaque reduction neutralization assay
Two serum pools were used for these assays: a pool obtained from mice immunized with the heterologous immunization protocol, and hyperimmune anti-vaccinia virus specific reference serum (positive control). Serial 2-fold dilutions of serum were combined with 300 PFU of either recombinant vHBs4 or wild type vaccinia virus (WR strain) as a control. After incubation for 1 hour at 37°C, the virus was added to 6 well plates containing 80% confluent Vero cells. Cells were incubated for 2 days at 37°, overlaid with 1% Seakem agarose containing normal cell growth media and neutral red stain, incubated for an additional day and analyzed for the number of plaques per well to determine the plaque inhibition efficiency of sample sera.
Statistical analysis
Student’s t tests were performed to compare statistical differences between groups. Probability (p) values less than 0.05 were considered to be significant, and p values less than 0.01 were considered to be highly significant.
Results
Development of a DNA and liposome based heterologous immunization protocol
We first performed a series of experiments to optimize the conditions for immunization. Dose, route of administration (intramuscular; i.m. vs intranasal; i.n.), sequence of delivery (DNA and liposome components), the composition, electric charge and the size (0.2 to 4 microns) of the liposomes were all evaluated (data not shown). A negatively-charged liposome of 1 to 4 micron size was determined to be the chosen vehicle for encapsulation of HBsAg to raise optimal immune responses. Based on these data, we devised the heterologous immunization protocol shown in Figure 1. Critical features of this protocol include intramuscular injection of DNA, intranasal administration of liposome-encapsulated antigen, and order of administration of the components. Two rounds of DNA immunization were chosen to ensure successful inoculation into the quadriceps. The time interval between immunizations is described below. Measurement of immune responses occurred between 2 and 8 weeks after the last immunization.
Figure 1.
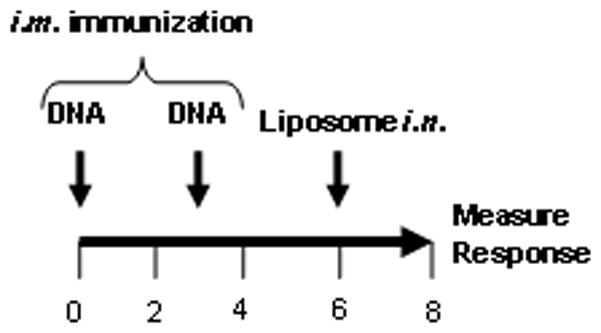
Prime boost schedule for the heterologous vaccine protocol. Naïve mice are immunized with 2 doses of HBsAg-DNA delivered intramuscularly (i.m.; 10–100μg/dose) followed by one dose of HBsAg-liposomes delivered intranasally (i.n.; 3 or 15 μg/dose). DNA immunizations may be delivered in 3 weeks (long protocol) or 3 days (short protocol). Liposomes are delivered 2–4 weeks after the last DNA immunization. Maximal immune responses are detected beginning two weeks following the final immunization.
Heterologous immunization induces synergistic HBsAg-specific IgG responses
We determined whether i.m. immunization with HBsAg-DNA (prime) followed by i.n. delivery of HBsAg-liposome (boost) would induce greater antibody responses than the individual components alone. Mice were either immunized with the individual components of the heterologous vaccine (2x i.m. with HBsAg-DNA or 1x i.n. with HBsAg-liposome), or were immunized first with 2x i.m. HBsAg-DNA on weeks 0 and 6 followed by i.n. immunization with HBsAg-liposome on week 10. As shown in Figure 2, the individual components induced weak HBsAg-specific total serum antibody responses. However, DNA immunization followed by i.n. boosting with HBsAg-liposomes markedly increased the total HBsAg-specific antibody responses. Quantitative ELISA assays were used to determine the amount of specific IgG in the sera. HBsAg-specific IgG levels after two rounds of HBsAg-DNA immunization or one round of administration of HBsAg-liposomes averaged 43.2μg/ml and 1μg/ml of antibody, respectively. In contrast, HBsAg-specific serum IgG levels were 248μg/ml after the combined heterologous immunization protocol, demonstrating a clear synergistic effect (p< 0.01).
Figure 2.
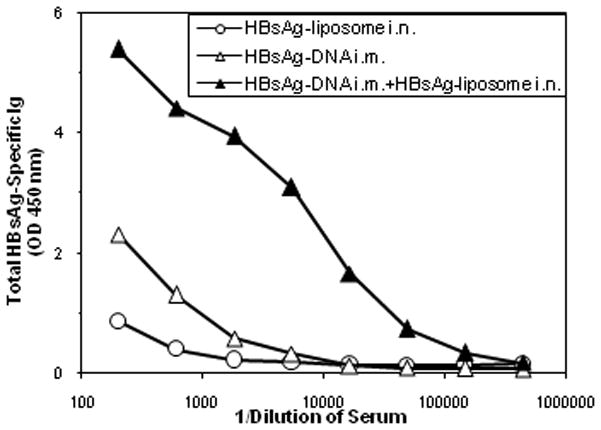
Heterologous immunization generates synergistic serum HBsAg-specific Ig responses. Groups of female Balb/c mice were vaccinated with either HBsAg-DNA i.m. (2x 100 μg/dose) alone on weeks 0 and 6, HBsAg-liposome delivered intranasally once (15μg/50μl) on week 10 or HBsAg-DNA (delivered i.m on weeks 0 and 6) followed by HBsAg-liposomes (3 μg/dose; i.n.) on week 10. Total HBsAg-specific Ig responses were measured using antigen-specific ELISA assays 4 weeks after the last immunization. The plotted data represent the mean responses in pooled sera from 8 mice per group.
A similar pattern was observed when the HBsAg-liposome boost was delivered i.m. HBsAg-specific serum IgG levels after two rounds of HBsAg-DNA immunization or one round of administration of HBsAg-liposomes averaged 27μg/ml and <1μg/ml of antibody respectively. HBsAg-specific serum IgG levels were 424μg/ml after the combined heterologous immunization protocol. This synergy was only observed when DNA immunization preceded HBsAg-liposome immunization (p<0.01). Reversing the order of the immunizations revealed no synergy in the antibody responses (data not shown).
Intranasal inoculation volume was also evaluated for its role in antibody responses. We observed no statistical difference among the antibody responses of different groups of mice receiving equivalent amounts of HBsAg-liposomes delivered in 20, 30, 40 or 50μl volumes (p> 0.5). Only mice receiving a 10μl inoculation volume showed a significantly lower antibody response (p< 0.05).
The heterologous immunization regimen induces robust responses in non-responder C57BL/6 mice
It has been previously shown that C57BL/6 mice are non-responsive to the HBsAg protein vaccine, however DNA vaccination can prime antigen-specific CD8+ T cells successfully (43). To determine whether the heterologous immunization protocol could overcome this unresponsiveness, the following experiment was performed. One group of C57BL/6 mice was immunized with two rounds of HBsAg-liposome delivered i.n. (Group A, n=5). As expected, these mice failed to generate a detectable level of anti-HBsAg serum IgG. These mice and a second group of naïve mice (Group B, n=8) were then immunized with two rounds of HBsAg-DNA delivered i.m. Serum IgG levels in both groups of mice in response to DNA vaccination were comparable (12.8 μg/ml and 8.0 μg/ml, for Groups A and B, respectively). Each group was then further boosted with HBsAg-liposomes delivered intranasally. Surprisingly, both groups of C57BL/6 mice demonstrated equivalent high levels of anti-HBsAg serum IgG (Group A, 238μg/ml; Group B, 310μg/ml). The results from this experiment illustrate three points: 1) heterologous immunization can stimulate high antibody responses in a historically non-responsive mouse strain; 2) the order of immunization is critical for the induction of serum IgG responses; 3) synergistic antibody responses are observed after heterologous immunization.
Heterologous immunization generates a Th1-biased immune response
A Th1-biased response is the objective for vaccines that are effective at controlling intracellular pathogens such as viruses. The ratio of antigen-specific IgG1 and IgG2a antibodies and cytokine profiling can be used to characterize the Th bias of an immune response. IgG1:IgG2a ratios ≤ 0.5 indicate a Th1-biased immune response, while a ratio of ≥ 2.0 indicates a Th2-biased immune response. Ratios between 0.5 and 2.0 indicate a mixed response (44, 45). To evaluate the contribution from each vaccine component, the IgG1:IgG2a ratio obtained after administration of individual components of the heterologous protocol was determined. Figure 3 shows the ratios of HBsAg-specific IgG1:IgG2a following i.m. immunization with HBsAg-DNA, or with HBsAg-liposomes delivered either i.n. or i.m. The ratios following immunization with a commercial human HBsAg vaccine, which is known to result in Th2-biased responses in humans, is also shown for comparison. The ratios following one or two homologous immunizations are also shown. As predicted from published data, the human vaccine and the HBsAg-DNA resulted in Th2 and Th1-biased immune responses respectively when administered i.m. HBsAg-liposomes gave a strong Th2-biased response when administered i.n. and a mixed response when administered i.m.
Figure 3.
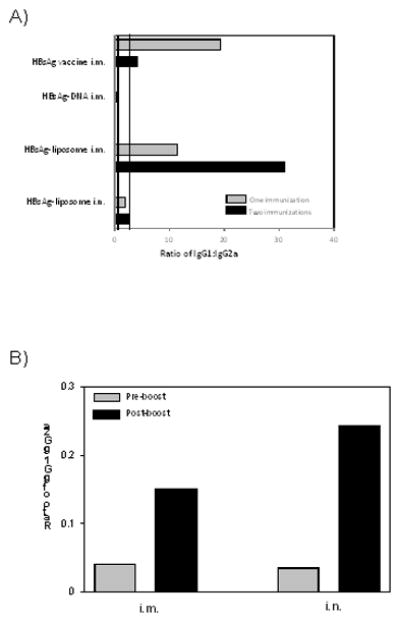
Heterologous immunization induces IgG1: IgG2a ratios indicative of a Th1-biased immune response. Groups of 5 female Balb/c mice were vaccinated with a human HBV vaccine, the indicated individual components of the immunization regimen. All immunizations were given at three week intervals. IgG1 to IgG2a ratios were determined from quantitative measurements of HBsAg-specific serum IgG1 and IgG2a measured two weeks after the last immunization on pools of sera from 5 mice. Grey bars represent the IgG1: IgG2a ratios from mice that received one immunization. Black bars are the ratios from mice that received two immunizations. Two vertical lines at 0.5 and at 2.0 demarcate three different patterns of antibody responses. A ratio of 0.5 or less indicates a Th1 polarized response. A ratio of 2.0 or more indicates a Th2 polarized response. Ratios between 0.5 and 2.0 indicate a mixed or balanced response.
The IgG1 to IgG2a ratio was obtained after heterlogous immunization with DNA followed by liposome-encapsulated antigen. As demonstrated in Figure 3, a Th1-biased response was observed following immunization with HBsAg-DNA (pre-boost, gray bars). Notably, a Th1 response was still observed after a boost with HBsAg-liposomes administered either i.n. or i.m. two weeks after immunization (IgG1 to IgG2a ratio is less than 0.3). These results demonstrate that the Th1-biased response that is established by the DNA immunization is not diverted to either a mixed or a Th2-biased response following the liposome boost.
To confirm T helper type of the immune responses, we performed ELISpot assays to compare cytokine responses from naïve mice, mice immunized with 2 doses of antigen encapsulated in liposome (homologous) or mice that received a heterologous immunization. Spleen cells isolated from mice immunized with the heterologous protocol predominantly produced IFN-γ (Th1-biased), while those isolated from mice immunized with the homologous protocol predominantly expressed IL-4 (Th2-biased; data not shown). An automatic Lincoplex assay for testing cytokine profiles not only confirmed high levels of IFN-γ in the serum of mice immunized with the heterologous protocol but also detected elevated IL-12 levels in these same samples (data not shown). Interestingly, IL-12 has been shown to be one of the crucial cytokines for induction of systemic and mucosal T cell responses (46–49). Taken together, these results suggest that the heterologous immunization protocol induces a Th1-biased response that should be associated with high, antigen-specific CD8 T cell activity for clearance of virally infected cells.
Heterologous immunization induces mucosal IgA and IgG responses
The levels of IgA and IgG following heterologous immunization with an intranasal boost with HBsAg-liposomes were determined in the serum, and in vaginal and lung washes. Figure 4 shows compiled results from two separate experiments. Circulating serum IgA was detected at comparable levels in both experiments. Equivalent or nearly equivalent levels of secretory IgA were also detected in vaginal washes and lung washes. In addition, substantial levels of lung IgG were also observed. IgA was also observed in a more limited number of samples from nasal washes and from saliva collected ≥ 4 weeks following the i.n. HBsAg-liposome boost (data not shown). However, only low or marginal levels of mucosal antibodies were detected when the liposome HBsAg vaccine was delivered by the i.m. route (data not shown). The heterologous immunization protocol is therefore effective at inducing antibody responses at both local (lung) and distant (vagina) mucosal sites.
Figure 4.
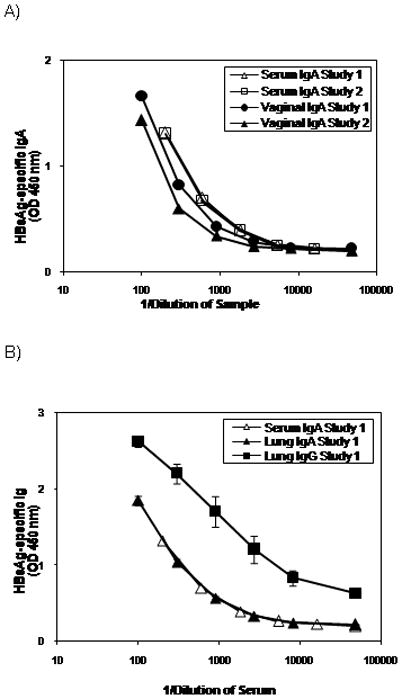
Heterologous immunization induces mucosal IgA and IgG responses. Groups of 6 female Balb/c mice were immunized i.m. on weeks 0 and 4 with 100μg of HBsAg-DNA and i.n. on week 8 with 15μg of HBsAg-liposomes. Serum and vaginal wash samples were collected on week 12, and lung washes were collected on week 14. A, Serum and vaginal IgA responses from two independent experiments. Results show the HBsAg-specific IgA levels determined using a five-layer sandwich ELISA assay. The results are from sample pools using equal volumes of serum or vaginal washes from six mice per group. Negligible (< 2 SD from levels in naïve mice) HBsAg-specific IgA was detected from the following treatment groups: (1) mice that received DNA immunization alone, (2) mice that received HBsAg-liposome alone, and (3) mice that received DNA immunization followed by HBsAg-liposome delivered i.m.(data not shown). B, HBsAg-specific IgA and IgG from the lungs. Results show the HBsAg-specific IgA and IgG levels determined using a five-layer sandwich ELISA assays. Results shown are the mean ± SEM from six mice per group. The serum IgA levels are shown again in this panel for reference.
Both long and abbreviated DNA immunization regimens are equally effective at priming for the HBsAg-liposome boost to raise HBsAg-specific IgG responses
In our initial experiments (i.e., Figure 1), immunizations were performed at intervals of 4–6 weeks. We later observed comparable results when those intervals were reduced to 3 weeks (data not shown). We therefore tested whether the DNA priming portion of the heterologous immunization could be further reduced from weeks to days without compromising the immune responses generated. Figure 5 shows a comparison between heterologous immunizations in which the DNA was given at an interval of 2 days (short protocol) versus an interval of 3 weeks (long protocol). Two rounds of DNA injection were used to ensure immunization in the small leg muscle mass of the mouse. The short interval between DNA injections precludes a prime-boost induction of immune responses. After i.n. boosting with HBsAg-liposomes, both protocols were shown to be equally effective in eliciting HBsAg-specific serum IgG responses, as well as comparable mucosal IgA responses (data not shown).
Figure 5.
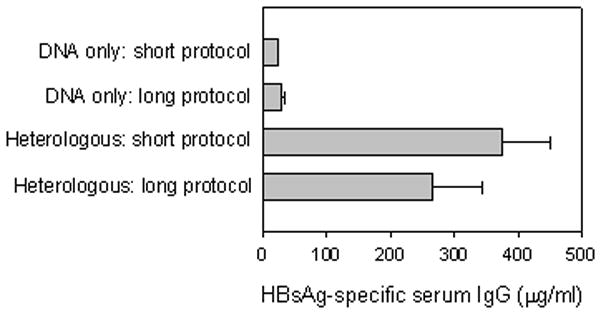
Short (3 days) and long (3 weeks) DNA priming regimens are both effective at priming for antibody responses. Groups of female Balb/c mice were immunized with 50μg of HBsAg-DNA i.m. on days 0 and 2 (short protocol) or 100μg of HBsAg-DNA i.m. on day 0 and week 3 (long protocol) followed by 15μg of HBsAg-liposomes i.n. 3 weeks after the last DNA immunization. Antibody responses were measured 2 weeks after the liposome immunization. Average HBsAg-specific IgG responses (μg/ml) +/− SEM are shown. From top to bottom, n= 8, 6, 8, and 12 per group.
In addition, we tested whether the shortened protocol could be used to induce immune responses in neonatal mice. Neonates were injected i.m. with 50μg of DNA at 2 days of age followed by intranasal administration of 20μl HBsAg-liposomes three weeks later. Total IgG antibody responses were measured 2 weeks after the last immunization. Similar to those results obtained in adult mice, we observed a synergistic robust response after DNA and liposome administration (331.9μg IgG /per ml) and a minimal response after DNA immunization alone (0.7μg/ml IgG). These differences were highly significant (p=0.002; n=6).
The adjuvant role of liposomes in the heterologous immunization protocol
HBsAg is known to be a moderately strong immunogen containing both T and B cell epitopes (50–54). Liposomal protein vaccines alone have demonstrated only weak to moderate immune stimulating effect (55, 56). The contribution of the liposome element of the booster immunization to immunogenicity in the heterologous immunization protocol was evaluated in two independent experiments (Table 1). Pair-wise comparisons of HBsAg protein versus HBsAg-liposomes showed that HBsAg protein alone contributed a significant boosting effect when it was given after priming with HBsAg-DNA (p<0.001). Further enhancement was observed upon encapsulation of the HBsAg protein within liposomes. Encapsulated protein induced greater levels of HBsAg-specific serum IgG when compared with DNA priming alone (p<0.001) and DNA priming followed by a non-encapsulated protein boost (p<0.01).
Table 1.
HBsAg-specific antibody responses are enhanced by liposomes (Long protocol)
| Group* | Prime | Boost | Ag-specific IgG (μg/ml)^ | Comparison | p value |
|---|---|---|---|---|---|
| A1 | HBsAg-DNA | None | 23.9 ± 8.8 | A1 vs. A2 | <0.001 |
| A2 | HBsAg-DNA | HBsAg | 109.5 ± 17.6 | A2 vs. A3 | <0.01 |
| A3 | HBsAg-DNA | HBsAg-liposome | 374.8 ± 64.4 | A3 vs. A1 | <0.001 |
| B1 | HBsAg-DNA | None | 28.6 ± 5.1 | B1 vs. B2 | <0.001 |
| B2 | HBsAg-DNA | HBsAg | 203.4 ± 43.9 | B2 vs. B3 | <0.01 |
| B3 | HBsAg-DNA | HBsAg-liposome | 618.0 ± 99.6 | B3 vs. B1 | <0.001 |
Two independent experiments A and B were performed with three sub-group comparisons in each experiment (1, 2, and 3).
HBsAg-specific serum IgG was evaluated using a quantitative ELISA assay on individual mouse serum samples. Experiments A and B contained 8 (N = 8) and 6 (N = 6) mice per group, respectively.
Heterologous immunization provides protection against intranasally delivered, live recombinant vaccinia virus expressing HBsAg without causing immunopathology
An important objective of our vaccine development research is to increase the levels of antigen specific T cell responses that are important in the control of intracellular pathogens. We evaluated the protective immunity conferred by the heterologous immunization protocol. Since Balb/c mice are not susceptible to infection with human hepatitis B virus, we used a recombinant vaccinia virus expressing HBsAg (vHBs4) to evaluate protective immunity in Balb/c mice.
The first challenge experiment applied the long protocol (1, 3, and 6 week as immunization intervals), high DNA vaccine priming (100μg) and lower vHBs4 dose for challenge (2×105 PFU). Three different groups were evaluated: naïve mice (Group A), mice that received two i.m. immunizations with HBsAg-DNA followed by i.n. immunization with empty liposomes (Group B), and mice that received two i.m. immunizations with HBsAg-DNA followed by one i.n. immunization with HBsAg-liposomes (Group C). Since vHBs4 is an attenuated recombinant vaccinia virus with a thymidine kinase (TK) gene disruption, it usually does not cause death in mice. A dose of 2 × 105 PFU of recombinant vHBs4 was administered i.n. two weeks after the HBsAg-liposome boost. Viral titers in the lung and the frequency of CD8+IFN-γ+ cells detected after ex vivo stimulation with an immunodominant HBsAg CTL peptide were measured five days post infection. The results are shown in Figure 6.
Figure 6.
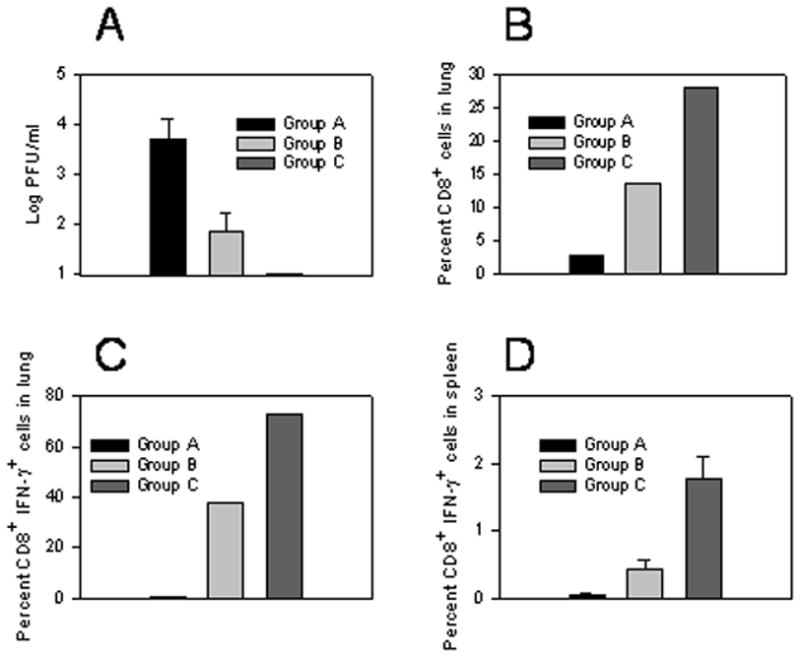
Heterologous immunization induces protective immune responses and high frequencies of CD8+IFN-γ+ T cells. Groups of 5 female Balb/c mice were either left untreated (Group A), were immunized with HBsAg-DNA + empty liposomes (Group B) or were immunized with HBsAg-DNA+HBsAg-liposomes (Group C) using the long immunization protocol. 2 weeks after the last immunization, mice were infected intranasally with 2×105 PFU of vHBs4. 5 days after infection, lungs from 4 mice per group were harvested for determination of viral titers (A), while the final lung was analyzed for total frequency of CD8+ T cells in the mononuclear population (B) and frequency of antigen-specific CD8+IFN-γ+ T cells (C). Individual spleens (4/group +/− SEM) were also analyzed for antigen-specific CD8+IFN-γ+ T cells (D, A vs. B: p=0.01; A vs. C: p=0.0005; B vs. C: p<0.004). All analyses were performed as described in the Materials and Methods. For C and D, cells incubated without peptide showed less than 0.1% positive staining for IFN-γ within the CD8 population (data not shown).
Viral titers in the lungs of mice that received the HBsAg-DNA immunization and empty liposomes were significantly reduced compared to naïve mice (Figure 6A, Group A versus Group B). All naïve mice had detectable virus in the lungs (3.72 +/− 0.4 log PFU) while 3 out of 4 mice immunized with DNA and empty liposomes had detectable but significantly lower viral titers (1.86 +/− 0.4 log PFU; p=0.01). In comparison, no detectable virus could be recovered from the lungs of mice that were immunized with both HBsAg-DNA and HBsAg-liposomes (Figure 6A, Group C), indicating complete clearance of virus.
High percentages of CD8+ T cells were observed in the lungs of mice immunized with both DNA and HBsAg-liposomes (~28% of viable lymphocytes; Figure 6B, Group C). In comparison, naïve mice or mice immunized solely with DNA demonstrated CD8+ T cell frequencies of ~3% and ~12%, respectively in the lung (Figure 6B, Groups A and B). To determine whether the CD8+ T cells were antigen-specific, we performed an intracellular cytokine staining assay to determine the frequency of CD8+ T cells that expressed IFN-γ. The challenge with vHBs4 revealed highly significant differences between groups. Mice immunized with the DNA prime and liposome-HBsAg boost protocol demonstrated high frequencies of antigen-specific CD8+IFN-γ+ T cells in the lung (>70% of CD8 T cells; Figure 6C) and also systemically in the spleen, albeit on a much lower scale (1.79± 0.31%, Figure 6D). The DNA portion of the heterologous protocol contributed to induction of these responses as immunization with DNA alone only induced antigen-specific CD8+IFN-γ+ T cells in the lung (38% of cells) and in the spleen (0.44% of the cells). In contrast, naïve mice exhibited few antigen-specific CD8+IFN-γ+ T cells in the lung and spleen.
Reduced viral titers at 5 days post viral challenge was also observed in experiments using the shortened immunization protocol (2 × DNA priming within 3 days), a lower DNA vaccine dose (10μg) and higher vHBs4 challenge dose (5 x105 PFU). Mice in one experiment were either left untreated (naïve), or were immunized with HBsAg-DNA + empty liposomes or HBsAg-DNA + HBsAg-liposomes (15μg HBsAg). While immunization of mice with HBsAg-DNA + empty liposomes significantly reduced viral titers in the lung (3.06+/−0.08 log), heterologous immunization with HBsAg-DNA + HBsAg-liposomes reduced viral titers even further (1.7+/−0.26 log), in comparison to naïve group (4.95+/−0.08 log). This demonstrates that significantly better protection was achieved in mice immunized with both the DNA and liposome components of the vaccine as compared to mice immunized with HBsAg DNA + empty liposome vector (p< 0.01, n=5).
We also analyzed antigen specific T cells responses in immunized mice prior to viral infection. Balb/c mice were immunized with either the DNA portion or HBsAg-liposome portion of the vaccine, or were immunized with both HBsAg-DNA and HBsAg-liposomes. Pooled lung samples from all mice were tested for antigen specific CD4+IFN-γ+ and CD8+IFN-γ T cells 2 weeks after the final immunization. Significantly higher frequencies of CD8+IFN-γ T cells (5.7%, n=5) were observed in the lungs of mice primed with DNA and boosted with liposome vaccine, but not in the lungs of mice immunized solely with DNA or liposome vaccine (0.5%, 0.4%, n=5). No significant differences in CD4+IFN-γ+ T cells were observed in any of the groups. These data suggest that whereas no antigen specific T cells were detected in the lungs of mice immunized by DNA or liposome vaccine alone, antigen specific T cells were induced in the lung at elevated levels in mice immunized using the heterologous immunization protocol.
Importantly, no immunopathology was observed in the lungs of mice immunized with this strategy and challenged with the 5 × 105 PFU dose of vHBs4, highlighting the safety of this approach (data not shown).
Neutralizing antibodies do not account for clearance of virus
Current commercial HBsAg vaccines usually raise a Th-2 type antibody response and the mechanism of protection is primarily through neutralizing antibodies. Due to the lack of mouse models for HBV infection to help dissect the protective mechanism, a pulmonary infection with recombinant vaccinia virus expressing HBsAg antigen (vHBs4) was designed to evaluate the role of T cell-mediated responses in the mice immunized with our protocol. Since the expressed HBsAg is foreign protein that is neither integral to nor incorporated into the recombinant vaccinia viral particle, it is unlikely that neutralizing antibodies are responsible for immune protection against the vHBs4 virus. To demonstrate that neutralizing antibodies were not responsible for clearance of virus in this model, we performed an in vitro plaque reduction neutralization assay. Sera from mice that received the heterologous immunization protocol did not neutralize either recombinant vHBs4 or wild type vaccinia virus even at the highest concentrations of serum used (dilution 1:25 of whole serum). In contrast, a serum pool composed of known anti-vaccinia antibodies neutralized both viruses. A 96% reduction in plaque formation was observed when this serum was diluted 1:200. This in vitro data strongly supports the conclusion that neutralization activity is not responsible for the observed protection in this experimental system, but T cell mediated immunity plays a protective role against virus infection in the lung.
Discussion
We have developed a heterologous immunization protocol consisting of immunization with DNA vaccine encoding antigen followed by administration of antigen encapsulated in liposomes. Our approach combines liposome encapsulation, specific delivery to mucosal sites, and the use of a heterologous immunization regimen. To the best of our knowledge, this is the first report of a heterologous immunization protocol combining systemic priming with DNA and a mucosal boost with liposome-encapsulated protein. The enhanced boosting effect seen after use of protein encapsulated in liposomes rather than a simple (not encapsulated) protein boost suggests that the liposome acts as both a delivery vehicle for the protein antigen and as a potent vaccine adjuvant. Unlike other heterologous immunization protocols, this regimen was specifically designed for enhanced safety by excluding the use of live viral vectors and/or potentially toxic adjuvants. The unique benefits of this protocol include induction of Th1-biased, humoral and cellular immune responses on mucosal surfaces and flexibility in delivery of individual components and induction of responses in “non-responder” animals and neonates.
While heterologous immunization using the intranasal boost induced strong systemic antibody responses, we also observed induction of antigen-specific IgA and IgG on multiple mucosal surfaces such as respiratory tracts and the remote vaginal cavity. However, we were unsuccessful in enhancing immune responses after delivery of liposome-HBsAg by the vaginal route (data not shown) supporting the general belief that although immune responses can be detected in the vagina they are not directly generated in the vaginal mucosa (57). In contrast, heterologous immunization with an intramuscularly-delivered liposome boost induced strong systemic responses, but only induced marginal mucosal immunity. Robust antigen-specific CD8+ T cell responses were also induced after heterologous immunization as revealed by intracellular cytokine-staining assays prior to and after viral challenge. These CD8+ T cell responses provided enhanced protection from a mucosal virus challenge. To our best knowledge, this is the first time that a liposome-based vaccine has been shown to enhance protective CD8+ T cell responses in mucosal sites of mice primed with DNA vaccines.
It is well established that homologous immunization with an intramuscular DNA vaccine induces a T helper type 1 (Th-1) biased immune response which may favor T cell mediated immune responses (44, 45). We observed similar results confirmed by IgG subclass profiles and enhanced IFN-γ production by CD8+ HBsAg-specific T cells. On the other hand, homologous immunization with intranasally delivered liposomal HBsAg induced greater levels of IgG1 than IgG2a antibodies, suggesting a T helper Type 2 (Th-2) biased response. The combination of intramuscular DNA priming followed by an intranasal liposomal HBsAg boost maintained Th-1-type IgG subclass and cytokine profiles, suggesting that initial priming with DNA vaccine determines the ultimate T helper type, in agreement with others’ findings (4, 16, 19, 21). The maintenance of Th-1-biased responses suggests that this protocol will be useful for any situation where induction of robust CD8+ T cell responses is desired.
Other important experiments highlight the strengths of this vaccination protocol. C57BL/6 mice, typically described as non-responders to HBsAg, can be primed by DNA vaccine against HBsAg (43). C57BL/6 mice were vaccinated using the heterologous immunization protocol with an intranasal liposomal-HBsAg boost. Surprisingly, we also observed synergistic induction of robust systemic antibody responses. It will be interesting to determine whether this vaccine approach may also induce responses in the approximately 5 to 10% healthy vaccine recipients that fail to respond to the currently available HBV vaccine (40, 58).
We also compared long and short DNA priming protocols. We observed no significant differences in antibody titers after either protocol, suggesting there is flexibility in timing of delivery of the vaccine components and that a single DNA vaccine prime may be sufficient. The long protocol consists of a prime and a boost with the same DNA vaccine at an interval of three weeks. In the short protocol, we employed intramuscular injection of DNA on day 0 and day 2 for technical reasons to ensure successful inoculation of DNA into the small target area (quadricep muscle). However, this short interval between injections virtually precludes the development of a prime and boost scenario. Since intramuscular administration of vaccines in humans is more efficient simply due to the abundance of muscle tissue and ease of administration, the success of the short protocol (one prime) in comparison with the long protocol (two primes) in mice suggests the feasibility of applying only one prime with DNA vaccine and one boost with liposome vaccine in clinical trials. This short protocol may prove ideal for emerging or pandemic threats with the potential to rapidly raise protective immunity against mucosally transmitted pathogens. It will be very interesting and significant to further investigate if the three week interval between the prime and boost used in the shortened protocol can be further decreased to less than 3 weeks without compromising immune responses and protection. Finally, the demonstrated success of the shortened protocol in both adult and neonatal mice can greatly broaden the application potential of this heterologous immunization regimen by which rapid protective immunity can be generated.
Our data demonstrate the feasibility of eliciting mucosal T cell responses that are primarily responsible for protective immunity using a heterologous immunization protocol. Protective immunity was demonstrated by the complete clearance of virus from the lung, enhanced frequencies of CD8+ IFN-γ cells prior to viral challenge, and the exclusion of neutralizing antibodies as a mechanism of protection for this specially designed challenge model.
Additional success in raising broad and effective protective immunity was also seen in HSV-2 and influenza virus A (H1N1) models by applying this prime and boost immunization protocols (data not shown).
This novel heterologous immunization approach using a DNA and liposome-based protein vaccine is safe and effective in raising enhanced immune responses and immune protection especially at mucosal surfaces. Recent studies show that humoral and cellular immune responses at the systemic immune level can be augmented in mice by priming with a hepatitis B DNA vaccine and boosting subcutaneously with a complex of HBsAg protein and alum hydroxide(59, 60). Delivery of liposomal HBsAg vaccine intranasally using our strategy raised both systemic and mucosal immune responses. Although HBsAg was used as a model antigen for development of the vaccine, the regimen was designed for application to a variety of pathogens. The robust immune responses observed in mice encourage further investigation in larger animal models including non-human primates. In addition, studies investigating the relationship between enhanced mucosal antibody responses and immune protection from other pathogens are underway.
Acknowledgments
We thank Dr. Bernard Moss (NIAID, NIH) for kindly providing the vHBs4 virus. We also thank Hong Cao and Robert Breen for their technical assistance.
Abbreviations used in this paper
- HBsAg
hepatitis B virus surface antigen
- vHBs
a recombinant vaccinia virus expressing HBsAg
- ICS
intracellular cytokine staining assay
- i.m.
intramuscular
- i.n.
intranasal
References
- 1.Hilleman MR. Personal reflections on twentieth century vaccinology. Southeast Asian J Trop Med Public Health. 2003;34:244–248. [PubMed] [Google Scholar]
- 2.Krieg AM. Toll-free vaccines? Nat Biotechnol. 2007;25:303–305. doi: 10.1038/nbt0307-303. [DOI] [PubMed] [Google Scholar]
- 3.Levine MM, Sztein MB. Vaccine development strategies for improving immunization: the role of modern immunology. Nat Immunol. 2004;5:460–464. doi: 10.1038/ni0504-460. [DOI] [PubMed] [Google Scholar]
- 4.Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Hilleman MR. Overview of the needs and realities for developing new and improved vaccines in the 21st century. Intervirology. 2002;45:199–211. doi: 10.1159/000067911. [DOI] [PubMed] [Google Scholar]
- 7.Hilleman MR. Overview: past and future of immunologic intervention in the pathogenesis, prophylaxis and therapeusis of hepatitis B. J Gastroenterol Hepatol. 2002;17(Suppl):S449–451. doi: 10.1046/j.1440-1746.17.s4.8.x. [DOI] [PubMed] [Google Scholar]
- 8.Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the challenge for immunology. Nat Immunol. 2006;7:449–455. doi: 10.1038/ni1343. [DOI] [PubMed] [Google Scholar]
- 9.Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don’t know, what we should know. Nat Med. 2004;10:806–810. doi: 10.1038/nm0804-806. [DOI] [PubMed] [Google Scholar]
- 10.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 11.Seder RA, Hill AV. Vaccines against intracellular infections requiring cellular immunity. Nature. 2000;406:793–798. doi: 10.1038/35021239. [DOI] [PubMed] [Google Scholar]
- 12.Robinson HL, Montefiori DC, Johnson RP, Manson KH, Kalish ML, Lifson JD, Rizvi TA, Lu S, Hu SL, Mazzara GP, Panicali DL, Herndon JG, Glickman R, Candido MA, Lydy SL, Wyand MS, McClure HM. Neutralizing antibody-independent containment of immunodeficiency virus challenges by DNA priming and recombinant pox virus booster immunizations. Nat Med. 1999;5:526–534. doi: 10.1038/8406. [DOI] [PubMed] [Google Scholar]
- 13.Schneider J, Gilbert SC, Blanchard TJ, Hanke T, Robson KJ, Hannan CM, Becker M, Sinden R, Smith GL, Hill AV. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat Med. 1998;4:397–402. doi: 10.1038/nm0498-397. [DOI] [PubMed] [Google Scholar]
- 14.Amara RR, Villinger F, Altman JD, Lydy SL, O’Neil SP, Staprans SI, Montefiori DC, Xu Y, Herndon JG, Wyatt LS, Candido MA, Kozyr NL, Earl PL, Smith JM, Ma HL, Grimm BD, Hulsey ML, Miller J, McClure HM, McNicholl JM, Moss B, Robinson HL. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001;292:69–74. doi: 10.1126/science.1058915. [DOI] [PubMed] [Google Scholar]
- 15.Shiver JW, Fu TM, Chen L, Casimiro DR, Davies ME, Evans RK, Zhang ZQ, Simon AJ, Trigona WL, Dubey SA, Huang L, Harris VA, Long RS, Liang X, Handt L, Schleif WA, Zhu L, Freed DC, Persaud NV, Guan L, Punt KS, Tang A, Chen M, Wilson KA, Collins KB, Heidecker GJ, Fernandez VR, Perry HC, Joyce JG, Grimm KM, Cook JC, Keller PM, Kresock DS, Mach H, Troutman RD, Isopi LA, Williams DM, Xu Z, Bohannon KE, Volkin DB, Montefiori DC, Miura A, Krivulka GR, Lifton MA, Kuroda MJ, Schmitz JE, Letvin NL, Caulfield MJ, Bett AJ, Youil R, Kaslow DC, Emini EA. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–335. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 16.Moore AC, Hill AV. Progress in DNA-based heterologous prime-boost immunization strategies for malaria. Immunol Rev. 2004;199:126–143. doi: 10.1111/j.0105-2896.2004.00138.x. [DOI] [PubMed] [Google Scholar]
- 17.Kirman JR, Seder RA. DNA vaccination: the answer to stable, protective T-cell memory? Curr Opin Immunol. 2003;15:471–476. doi: 10.1016/s0952-7915(03)00068-2. [DOI] [PubMed] [Google Scholar]
- 18.Liu MA, Ulmer JB. Human clinical trials of plasmid DNA vaccines. Adv Genet. 2005;55:25–40. doi: 10.1016/S0065-2660(05)55002-8. [DOI] [PubMed] [Google Scholar]
- 19.Schneider J, Gilbert SC, Hannan CM, Degano P, Prieur E, Sheu EG, Plebanski M, Hill AV. Induction of CD8+ T cells using heterologous prime-boost immunisation strategies. Immunol Rev. 1999;170:29–38. doi: 10.1111/j.1600-065x.1999.tb01326.x. [DOI] [PubMed] [Google Scholar]
- 20.Hanke T, McMichael AJ, Mwau M, Wee EG, Ceberej I, Patel S, Sutton J, Tomlinson M, Samuel RV. Development of a DNA-MVA/HIVA vaccine for Kenya. Vaccine. 2002;20:1995–1998. doi: 10.1016/s0264-410x(02)00085-3. [DOI] [PubMed] [Google Scholar]
- 21.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, Vuola JM, Blanchard TJ, Gothard P, Watkins K, Hannan CM, Everaere S, Brown K, Kester KE, Cummings J, Williams J, Heppner DG, Pathan A, Flanagan K, Arulanantham N, Roberts MT, Roy M, Smith GL, Schneider J, Peto T, Sinden RE, Gilbert SC, Hill AV. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–735. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 22.Robinson HL. Prime boost vaccines power up in people. Nat Med. 2003;9:642–643. doi: 10.1038/nm0603-642. [DOI] [PubMed] [Google Scholar]
- 23.Tritel M, Stoddard AM, Flynn BJ, Darrah PA, Wu CY, Wille U, Shah JA, Huang Y, Xu L, Betts MR, Nabel GJ, Seder RA. Prime-boost vaccination with HIV-1 Gag protein and cytosine phosphate guanosine oligodeoxynucleotide, followed by adenovirus, induces sustained and robust humoral and cellular immune responses. J Immunol. 2003;171:2538–2547. doi: 10.4049/jimmunol.171.5.2538. [DOI] [PubMed] [Google Scholar]
- 24.Tan GS, McKenna PM, Koser ML, McLinden R, Kim JH, McGettigan JP, Schnell MJ. Strong cellular and humoral anti-HIV Env immune responses induced by a heterologous rhabdoviral prime-boost approach. Virology. 2005;331:82–93. doi: 10.1016/j.virol.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Casimiro DR, Bett AJ, Fu TM, Davies ME, Tang A, Wilson KA, Chen M, Long R, McKelvey T, Chastain M, Gurunathan S, Tartaglia J, Emini EA, Shiver J. Heterologous human immunodeficiency virus type 1 priming-boosting immunization strategies involving replication-defective adenovirus and poxvirus vaccine vectors. J Virol. 2004;78:11434–11438. doi: 10.1128/JVI.78.20.11434-11438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemiale F, Kong WP, Akyurek LM, Ling X, Huang Y, Chakrabarti BK, Eckhaus M, Nabel GJ. Enhanced mucosal immunoglobulin A response of intranasal adenoviral vector human immunodeficiency virus vaccine and localization in the central nervous system. J Virol. 2003;77:10078–10087. doi: 10.1128/JVI.77.18.10078-10087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eriksson K, Holmgren J. Recent advances in mucosal vaccines and adjuvants. Curr Opin Immunol. 2002;14:666–672. doi: 10.1016/s0952-7915(02)00384-9. [DOI] [PubMed] [Google Scholar]
- 28.Stambas J, Brown SA, Gutierrez A, Sealy R, Yue W, Jones B, Lockey TD, Zirkel A, Freiden P, Brown B, Surman S, Coleclough C, Slobod KS, Doherty PC, Hurwitz JL. Long lived multi-isotype anti-HIV antibody responses following a prime-double boost immunization strategy. Vaccine. 2005;23:2454–2464. doi: 10.1016/j.vaccine.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 29.Fujihashi K, Koga T, van Ginkel FW, Hagiwara Y, McGhee JR. A dilemma for mucosal vaccination: efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine. 2002;20:2431–2438. doi: 10.1016/s0264-410x(02)00155-x. [DOI] [PubMed] [Google Scholar]
- 30.Barouch DH, Pau MG, Custers JH, Koudstaal W, Kostense S, Havenga MJ, Truitt DM, Sumida SM, Kishko MG, Arthur JC, Korioth-Schmitz B, Newberg MH, Gorgone DA, Lifton MA, Panicali DL, Nabel GJ, Letvin NL, Goudsmit J. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172:6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 31.HIV vaccine failure prompts Merck to halt trial. Nature. 2007;449:390. doi: 10.1038/449390c. [DOI] [PubMed] [Google Scholar]
- 32.STEP study: disappointing, but not a failure. Lancet. 2007;370:1665. doi: 10.1016/S0140-6736(07)61702-4. [DOI] [PubMed] [Google Scholar]
- 33.Cohen J. AIDS research. Did Merck’s failed HIV vaccine cause harm? Science. 2007;318:1048–1049. doi: 10.1126/science.318.5853.1048. [DOI] [PubMed] [Google Scholar]
- 34.Cohen J. AIDS research. Promising AIDS vaccine’s failure leaves field reeling. Science. 2007;318:28–29. doi: 10.1126/science.318.5847.28. [DOI] [PubMed] [Google Scholar]
- 35.Ledford H. HIV vaccine may raise risk. Nature. 2007;450:325. doi: 10.1038/450325a. [DOI] [PubMed] [Google Scholar]
- 36.Steinbrook R. One step forward, two steps back--will there ever be an AIDS vaccine? N Engl J Med. 2007;357:2653–2655. doi: 10.1056/NEJMp0708117. [DOI] [PubMed] [Google Scholar]
- 37.O’Hagan DT. Recent developments in vaccine delivery systems. Curr Drug Targets Infect Disord. 2001;1:273–286. doi: 10.2174/1568005014606008. [DOI] [PubMed] [Google Scholar]
- 38.Alving C. Theoretical basis for development of liposomes as carrieres of vaccines. Elsevier Science B.V; 1998. [Google Scholar]
- 39.Alving CR, Koulchin V, Glenn GM, Rao M. Liposomes as carriers of peptide antigens: induction of antibodies and cytotoxic T lymphocytes to conjugated and unconjugated peptides. Immunol Rev. 1995;145:5–31. doi: 10.1111/j.1600-065x.1995.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 40.Davis HL, Michel ML, Whalen RG. DNA-based immunization induces continuous secretion of hepatitis B surface antigen and high levels of circulating antibody. Hum Mol Genet. 1993;2:1847–1851. doi: 10.1093/hmg/2.11.1847. [DOI] [PubMed] [Google Scholar]
- 41.Moss B, Smith GL, Gerin JL, Purcell RH. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature. 1984;311:67–69. doi: 10.1038/311067a0. [DOI] [PubMed] [Google Scholar]
- 42.Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. 2001;2:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 43.Schirmbeck R, Bohm W, Ando K, Chisari FV, Reimann J. Nucleic acid vaccination primes hepatitis B virus surface antigen-specific cytotoxic T lymphocytes in nonresponder mice. J Virol. 1995;69:5929–5934. doi: 10.1128/jvi.69.10.5929-5934.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feltquate DM, Heaney S, Webster RG, Robinson HL. Different T helper cell types and antibody isotypes generated by saline and gene gun DNA immunization. J Immunol. 1997;158:2278–2284. [PubMed] [Google Scholar]
- 45.Pertmer TM, Roberts TR, Haynes JR. Influenza virus nucleoprotein-specific immunoglobulin G subclass and cytokine responses elicited by DNA vaccination are dependent on the route of vector DNA delivery. J Virol. 1996;70:6119–6125. doi: 10.1128/jvi.70.9.6119-6125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belyakov IM, Derby MA, Ahlers JD, Kelsall BL, Earl P, Moss B, Strober W, Berzofsky JA. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc Natl Acad Sci U S A. 1998;95:1709–1714. doi: 10.1073/pnas.95.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belyakov IM, Ahlers JD, Clements JD, Strober W, Berzofsky JA. Interplay of cytokines and adjuvants in the regulation of mucosal and systemic HIV-specific CTL. J Immunol. 2000;165:6454–6462. doi: 10.4049/jimmunol.165.11.6454. [DOI] [PubMed] [Google Scholar]
- 48.Belyakov IM, Ahlers JD, Brandwein BY, Earl P, Kelsall BL, Moss B, Strober W, Berzofsky JA. The importance of local mucosal HIV-specific CD8(+) cytotoxic T lymphocytes for resistance to mucosal viral transmission in mice and enhancement of resistance by local administration of IL-12. J Clin Invest. 1998;102:2072–2081. doi: 10.1172/JCI5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staats HF, Bradney CP, Gwinn WM, Jackson SS, Sempowski GD, Liao HX, Letvin NL, Haynes BF. Cytokine requirements for induction of systemic and mucosal CTL after nasal immunization. J Immunol. 2001;167:5386–5394. doi: 10.4049/jimmunol.167.9.5386. [DOI] [PubMed] [Google Scholar]
- 50.Schirmbeck R, Melber K, Kuhrober A, Janowicz ZA, Reimann J. Immunization with soluble hepatitis B virus surface protein elicits murine H-2 class I-restricted CD8+ cytotoxic T lymphocyte responses in vivo. J Immunol. 1994;152:1110–1119. [PubMed] [Google Scholar]
- 51.Schirmbeck R, Melber K, Mertens T, Reimann J. Selective stimulation of murine cytotoxic T cell and antibody responses by particulate or monomeric hepatitis B virus surface (S) antigen. Eur J Immunol. 1994;24:1088–1096. doi: 10.1002/eji.1830240512. [DOI] [PubMed] [Google Scholar]
- 52.Schirmbeck R, Melber K, Mertens T, Reimann J. Antibody and cytotoxic T-cell responses to soluble hepatitis B virus (HBV) S antigen in mice: implication for the pathogenesis of HBV-induced hepatitis. J Virol. 1994;68:1418–1425. doi: 10.1128/jvi.68.3.1418-1425.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schirmbeck R, Wild J, Reimann J. Similar as well as distinct MHC class I-binding peptides are generated by exogenous and endogenous processing of hepatitis B virus surface antigen. Eur J Immunol. 1998;28:4149–4161. doi: 10.1002/(SICI)1521-4141(199812)28:12<4149::AID-IMMU4149>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 54.Schirmbeck R, Deml L, Melber K, Wolf H, Wagner R, Reimann J. Priming of class I-restricted cytotoxic T lymphocytes by vaccination with recombinant protein antigens. Vaccine. 1995;13:857–865. doi: 10.1016/0264-410x(94)00038-o. [DOI] [PubMed] [Google Scholar]
- 55.Chikh G, Schutze-Redelmeier MP. Liposomal delivery of CTL epitopes to dendritic cells. Biosci Rep. 2002;22:339–353. doi: 10.1023/a:1020151025412. [DOI] [PubMed] [Google Scholar]
- 56.Childers NK, Miller KL, Tong G, Llarena JC, Greenway T, Ulrich JT, Michalek SM. Adjuvant activity of monophosphoryl lipid A for nasal and oral immunization with soluble or liposome-associated antigen. Infect Immun. 2000;68:5509–5516. doi: 10.1128/iai.68.10.5509-5516.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11:S45–53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 58.Zuckerman JN. Nonresponse to hepatitis B vaccines and the kinetics of anti-HBs production. J Med Virol. 1996;50:283–288. doi: 10.1002/(SICI)1096-9071(199612)50:4<283::AID-JMV1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 59.Xiao-wen H, Shuhan S, Zhen-lin H, Jun L, Lei J, Feng-juan Z, Ya-nan Z, Ying-jun G. Augmented humoral and cellular immune responses of a hepatitis B DNA vaccine encoding HBsAg by protein boosting. Vaccine. 2005;23:1649–1656. doi: 10.1016/j.vaccine.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Li DA, Hey Y, Wang F, Guo YJ, Yang F, Zhou Q, Sun SH. Proteomic analysis of augmented immune responses in mouse by prime-and-boost immunization strategy with DNA vaccine coding HBsAg and rHBsAg protein. Vaccine. 2007;25:8146–8153. doi: 10.1016/j.vaccine.2007.09.037. [DOI] [PubMed] [Google Scholar]