

Abstract
Hematopoietic stem and progenitor cell (HSPC) phenotype and function can change in response to infectious challenge. These changes can be mediated by cytokines, interferons (IFNs), and pathogen-associated molecules, via TLR, and are thought to promote tailored immune responses for particular pathogens. Here we investigated the signals that activate HSPCs during ehrlichiosis, a disease characterized by profound hematopoietic dysfunction in both humans and mice. In a mouse model of ehrlichiosis we observed that infection-induced proliferation of bone marrow HSPCs was dependent on IFNγ signaling, and was partially dependent on MyD88. However, MyD88 was not required in HSPCs for their expansion during infection, as similar frequencies of MyD88-deficient and wild type HSPCs proliferated in mixed bone marrow chimeric mice. MyD88-deficient mice exhibited low serum and bone marrow concentration of IFNγ compared to wild type mice. We next identified CD4 T cells as the primary cells producing IFNγ in the bone marrow, and demonstrated a non-redundant role for CD4-derived IFNγ in increased HSPCs. Using mixed bone marrow chimeric mice, we identified a requirement for MyD88 in CD4 T cells for increased T-bet expression, optimal IFNγ production, and CD4 T cell proliferation. Our data demonstrate an essential role for CD4 T cells in mediating HSPC activation in response to bacterial infection, and illustrate a novel role for MyD88 signaling in CD4 T cells in this process. These findings further support the idea that IFNγ production is essential for HSPC activation and hematopoietic responses to infection.
Introduction
The hematopoietic system is maintained by the hematopoietic stem cell (HSC), a cell that can self-renew and differentiate into all cells of the blood and immune systems. Hematopoietic stress, brought about by inflammation or injury, induces the enhanced production of cells in the bone marrow, in part, by activating HSCs (1). The influence of inflammatory factors in modulating hematopoiesis has been observed in a number of different models, including endotoxemia and arthritis (2, 3), but the molecular processes utilized in HSCs and progenitor cells during inflammation are not well-characterized. Understanding the mechanisms that drive HSC differentiation and self-renewal, particularly during infection and inflammation, are essential to our understanding of both pathological hematopoietic deficiencies and mechanisms of host defense.
The direct stimulation of hematopoietic progenitors by pathogen-associated molecules was first demonstrated by Nagai et al. (4), who showed that myeloid cells could be generated from hematopoietic progenitors via TLR and MyD88-dependent signaling. Related studies of vaccinia virus infection demonstrated that the TLR9 ligand, CpG, can act directly on common lymphoid progenitors (CLP) to drive dendritic cell production, at the expense of lymphopoiesis (5). Candida albicans was shown to direct the production of myeloid cells in mice, via TLR2, which required intact MyD88-signaling (6, 7). The TLR adaptor protein, MyD88 has also been implicated in the maintenance of monocytes, as was shown during Listeria monocytogenes infection (8). Thus, host responses to a wide variety of pathogens involve the infection-induced modification of hematopoiesis via direct TLR- and MyD88-mediated signaling.
In addition to their capacity to directly sense pathogens via TLRs, hematopoietic stem and progenitor cells (HSPCs) can also respond to inflammatory cytokines and interferons produced during infeciton. We and others have demonstrated a critical role for IFNγ in activating HSCs during infection (9). Intrinsic IFNγR-mediated signals were essential for functional myelopoiesis during infection with ehrlichia (10) and lymphocytic choriomeningitits virus (LCMV) (11). IFNγ also has been shown to play a role in the emergence of a unique hematopoietic progenitor cell population during Plasmodium chaubudi infection (12). These findings demonstrate a novel role for IFNγ in promoting immune responses during infection through its direct action on hematopoietic progenitors.
In this study we have addressed which cells are responsible for driving IFNγ–mediated changes in hematopoiesis during ehrlichial infection. Ehrlichia muris is a tick-transmitted, obligate intracellular pathogen, closely related to the causative agent of human monocytic ehrlichiosis (HME), E. chaffeensis. HME is characterized by profound hematopoietic dysfunction and important clinical manifestations of the disease are severe anemia and thrombocytopenia (13). The ehrlichia do not encode genes for LPS or peptidoglycan synthesis (14), and the cellular host mechanisms by which these organisms elicit innate inflammation are currently unknown. Bone marrow-derived dendritic cells (BMDC) from TLR-deficient and caspase-1-deficient mice responded to E. muris, and generated normal amounts of IL-12p40 in vitro. However, MyD88-deficient BMDCs produced less IL12-p40 in response to E. muris infection (15), suggesting an important role for MyD88-signaling in production of IL-12, and/or IL-23, in response to ehrlichial infection, although the pathway in which MyD88 is required during ehrlichial infection is not yet known. We also noted that in the absence of the adaptor molecule MyD88, E. muris infected mice exhibited increased susceptibility to infection, which was correlated with significantly reduced IFNγ production. These findings prompted our investigation of how MyD88-deficiency impacted hematopoietic activity in response to ehrlichial infection. MyD88 signaling was not required in HSPCs for their expansion; rather, MyD88-signaling within CD4 T cells was essential for the production of IFNγ. These studies are relevant to our understanding of how hematopoiesis is modulated during infection and inflammation, and point to an important role for MyD88-dependent mechanisms within T lymphocytes in regulating the functional capacity of hematopoietic progenitors.
Materials and Methods
Mice
C57BL/6 mice and the following transgenic and gene-targeted strains were obtained from the Jackson Laboratory (Bar Harbor, ME): IFNγR1-deficient (B6.129S7-Ifngr1tm1AgtJ; described in this study as IFNγR-deficient), IFNγ-deficient (B6.129S7-Ifngtm1Ts/J), MyD88-deficient (B6.129P2(SJL)-Myd88tm1Defr/J), C3H/HeJ, C3H/HeOUJ, CD4-deficient (B6.129S2-Cd4tm1Mak/J), β-act EGFP (B6-Tg(CAG-EGFP)31Osb/LeySopJ), and a CD45 congenic strain (B6.SJL-Ptprca Pepcb/BoyJ). TLR9-deficient mice were kindly provided by Dr. S. Swain (Trudeau Institute, Saranac Lake, NY) and TLR2-deficient mice were kindly provided by Dr. T. Sellati (Albany Medical College, Albany, NY). All mice were bred in the Animal Resources Facility at Albany Medical College under microisolater conditions.
Bacteria
Mice were infected, via intraperitoneal injection, between 6 and 12 weeks of age, with 50,000 E. muris bacteria obtained from infected mouse splenocytes, as previously described (10).
PCR quantification for bacterial burden
DNA from 2 × 106 splenic cells was extracted using DNAzol (Molecular Research Center, Cincinnati, OH). The number of bacterial copies was assayed using a real-time quantitative probe-based PCR that measured the copy number of the bacterial dsb gene (which encodes a thiodisulfide oxidoreductase gene) (16, 17).
Flow cytometry and antibodies
Bone marrow mononuclear cells were harvested and prepared as previously described (10). The antibodies used for flow cytometry included the following: biotin-conjugated lineage markers specific for CD3 (clone 17A2), CD11b (M1/70), Gr-1 (RB6-8C5), Ter119 (Ly-76) and CD45R (RA3-6B2), Alexa700-CD45.2 (104), and eFluor450-streptavidin, allophycocyanin (APC)-CD3 (17A2)(all from eBiosciences, SanDiego, CA), and phycoerythrin (PE)-cychrome-7 (Cy7)-Sca-1 (D7), PE-Scal-1 (D7), peridinin chlorophyll protein (PerCP)-Cy5.5-CD45.2 (104), PE-CD45.1 (RMV-7), APC- c-Kit (2B8), APC-Cy7-streptavidin, Pacific Blue (PB)-CD4 (RM4-5), fluorescein isothiocyanate (FITC)-CD4 (RM4-5), FITC-CD3 (17A2), PE-CD8 (53-6.7), PerCP-Cy5.5-CD8 (53-6.7), FITC-B220 (RA3-6B2), PB-CD19 (6D5), PE-Cy7-NK1.1 (PK136), APC-IFNγ (XMG1.2), FITC-CD11b (M1/70), APC-CD11c (N418), PE-CD115 (AFS98), APC-Ly6C (HK1.4), PB-Ly6C (HK1.4), PerCP-Cy5.5-Ly6G (1A8), PE-Cy7-MHC II (AF6-120.1), PE-GR1 (RB6-8C5), APC-Cy7-F4/80 (CI: A3-1), FITC-F4/80 (CI: A3-1) and PE-CD68 (FA-11) (all from Biolegend, SanDiego, CA), and V500-CD45.2 (104) (from BD Biosciences) PE-conjugated CD1d tetramer was a kind gift from Dr. Elizabeth Leadbetter (Trudeau Institute, Saranac Lake, NY). Unstained cells were used as negative controls to establish the flow cytometer voltage settings, and single-color positive controls were used to adjust the instrument compensation. The flow cytometric data were acquired using an LSR II flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (TreeStar, Ashland, OR).
In vitro hematopoietic progenitor cell assays
Mice were administered recombinant IFNγ (PeproTech, Rocky Hill, NJ) via intravenous injection (10μg/ 200 μL) on days 8, 9 and 10 post-infection. Bone marrow cells were harvested at day 11 post-infection, plated at 2.0 × 104 per 35-mm tissue culture dish, in duplicate, and cultured in methocellulose media (MethoCult™ GF M3434, Stem Cell Technologies, Vancouver, BC, Canada). After incubation for 7 days at 37°C in 5% CO2, colonies derived from multipotential granulocyte, erythroid, macrophage, and megakaryocyte progenitors (GEMM), granulocyte-macrophage progenitors (GM), macrophage progenitors (M), and granulocyte progenitors (G) were scored.
Intracellular cytokine staining
Bone marrow (femur and tibia) and splenic single cell suspensions were prepared, and erythrocytes were removed by a brief hypotonic lysis. A total of 2×106 cells were plated in a 96 well plate and Fc receptors were blocked by incubation with anti-CD16/32 monoclonal antibody for 20 minutes on ice. Cells were then incubated on ice for 30 minutes with specific antibodies to stain for surface proteins. Cells were washed and then fixed and permeabilized in Fix/Perm buffer (BD Biosciences). Intracellular IFNγ was detected by incubating cells in Wash/Perm buffer (BD Biosciences) with anti-IFNγ antibody (clone: XMG1.2) for 30 minutes on ice. Cells were washed two times in Wash/Perm buffer, resuspended in simple wash buffer (SWB) and analyzed on an LSR II (BD Biosciences).
Nuclear staining for T-bet
Bone marrow and splenic cells were prepared and surface staining was performed as described. Thereafter, cells were processed fixed and permeabilized and then stained with PE-Cy7-conjugated anti-T-bet (4B10) antibody(buffers and antibodies from eBioscience). Protocols were following the manufacture’s instructions. Cells were analyzed on an LSR II and data was analyzed using FlowJo software.
In vivo proliferation assay
Mice were administrated with BrdU (1mg per mouse; i.p.) 6 hrs prior to harvest at day 11 post-infection. Cells were surface stained as described above, and then fixed and permeabilized in Fix/Perm buffer (BD Biosciences) and Fix/Perm buffer containing DMSO, respectively. Cells were incubated with DNase I (Sigma) at 37 °C for 1 hr, and then incubated with FITC-conjugated anti-BrdU antibody (PRB-1; eBiosciences) or PE (Bu20a; Biolegend)-conjugated anti-BrdU antibody. Flow cytometry was performed using an LSR II flow cytometer, and data were analyzed using FlowJo software.
Serum and bone marrow cytokine measurements
Bone marrow homogenates were made in the presence of NP-40 and proteinase inhibitors. Serum and bone marrow homogenate cytokine assessment was performed using the Luminex platform (Bio-Rad) according to the manufacturer’s instructions. Total protein concentration in bone marrow homogenate was measured by the BCA kit (Pierce, Rockford, IL) according to the manufacturer’s instruction. Cytokines in bone marrow were calculated as pg/mg of total protein.
Generation of mixed bone marrow chimeras
To generate mixed wild type and MyD88-deficient chimeric mice, CD45 congenic mice were lethally irradiated (950 RADs, administered in 2 doses, 3 hours apart). Irradiated mice received a total of 5 × 106 bone marrow cells derived from β-ACT-EGFP and MyD88-deficient mice. Mice were screened for chimerism at 4-6 weeks and infected with E. muris at 7 weeks post-reconstitution. To generate chimeras where CD4 cells were unable to produce IFNγ (referred to as CD4Ifng-/-), IFNγ-deficient mice were lethally irradiated and reconstituted with 2.5 × 106 bone marrow cells from each CD4-deficient and IFNγ-deficient mice. Control chimeric mice (referred to as CD4Ifng+/+) were generated by irradiating wild type C57BL/6 mice that were reconstituted with equal numbers of CD4-deficient and wild type bone marrow cells (total 5 × 106 cells)
Statistical analyses
Statistical analyses were performed with a two-way ANOVA or a Student’s t-test, as indicated, using Prism GraphPad Software (LaJolla, CA); a P value of < 0.05 was considered to be significant.
Results
IFNγ and MyD88 signaling contribute to E. muris-induced expansion of HSPCs
We previously identified IFNγ as a major mediator of hematopoietic function during E. muris infection, and demonstrated that IFNγ promoted the production of mature myeloid cells (10). These studies led us to address whether bacterial ligands contributed to direct activation of HSPCs, as has been shown in other models of infection (4). Similar to a previous report, we found no changes in bacterial infection or immunity (data not shown) and similar frequencies of Lin-negative, Sca-1+, c-Kit+ (LSK) cells in mice deficient in TLR2 or TLR4 (Supp. Fig. 1A and B and (15)), consistent with the fact that E. muris lacks the genes required for the synthesis of LPS or peptidoglycan. The primary TLR for bacterial CpG DNA is TLR9 (18), which we predicted would play an important role in the induction of IFNγ production during ehrlichiosis. However, we found that bacterial infection was similar in TLR9-deficient mice and C57BL/6 mice, and expansion of the LSK population, greatly enriched for HSPCs, was similar in frequency and number in both strains after E. muris infection (Supp. Fig. 1C). In contrast, we found that mice deficient in the TLR adaptor molecule, MyD88, exhibited reduced frequencies and numbers of LSK cells, as compared to C57BL/6 mice (Fig. 1A-C). To test whether the increase in LSK cells was accompanied by cell proliferation, we measured BrdU-incorporation in progenitor cells in response to E. muris infection. Mice were injected with BrdU 6 hours prior to sacrifice, and we found that the BrdU incorporation was significantly higher in the LSK population of wild type mice, relative to IFNγR-deficient, and MyD88-deficient (Fig. 1D and E). Although LSK cells in MyD88-deficient mice exhibited reduced BrdU-incorporation, relative to wild type mice, the proliferative defect in the former strain was of lower magnitude relative to that observed in the absence of IFNγR-mediated signaling. Increased proliferation may represent an increase in Sca-1 expression on Lin-negative, cKit+, Sca-1-negative myeloid progenitors, as well as increased proliferation of Sca-1+ cells. These data suggest that MyD88-signaling partially mediates the infection-induced increase of this progenitor cell population during infection.
Figure 1. Expansion of bone marrow LSK cells requires MyD88-signaling and IFNγ-signaling.
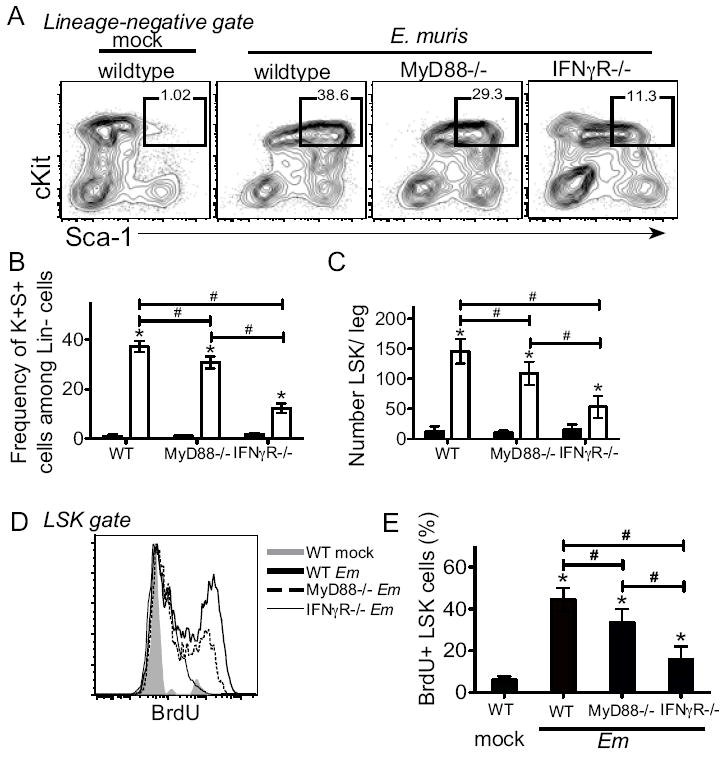
Wild type C57BL/6 mice, MyD88-deficient, and IFNγR-deficient mice were infected with E. muris. Bone marrow cells were purified and examined for surface expression of lineage markers, c-Kit, and Sca-1. (A) Lineage-negative cells were analyzed for expression of c-Kit+, Sca-1+. Square gates are drawn on Lin-negative cells expression both c-Kit and Sca-1 (LSK) in mock-infected (left column) and E. muris-infected (right column) mice on day 11 post-infection. Numbers above the LSK gate represent the frequency of c-Kit+ Sca-1+ cells among total Lin-negative cells, and the average frequencies are shown in panel B. (C) Numbers of LSK cells in one leg is shown for mock- (filled bars) and E.muris-infected (open bars) mice. (D) LSK cells were analyzed for BrdU incorporation in mock-infected (gray, filled) and E.muris infected C57BL/6 (dark line), MyD88-deficient (dashed line), and IFNγR-deficient (thin line) mice. (E) The frequency of BrdU+ LSK cells is shown. The data represent the mean and standard deviation of the data. At least four mice are examined for each group and the experiment was performed two independent times. The asterisks reflect significant differences between mock and infected groups and # reflect differences between different strains of mice; * or # p < 0.05.
Intrinsic MyD88-signaling is not required for progenitor cell expansion
To test whether MyD88-signaling was required in progenitor cells for the infection-induced increase in LSK cells, we generated radiation-induced mixed bone marrow chimeric mice that contained similar numbers of wild type cells, derived from β-ACT-GFP mice (CD45.2; GFP+), and MyD88-deficient donor cells (CD45.2; GFP-). Seven weeks post-reconstitution, chimeric mice were infected with E. muris. On day 11 post-infection, the frequency of BM LSK cells induced by E. muris infection in the chimeric mice was equivalent to what we observed in wild type infected mice, but was higher relative to MyD88-deficient mice (Fig. 2A and B). We hypothesized that if MyD88-signaling was intrinsically required for the infection-induced increase in LSK progenitor cells, we would observe a reduced frequency of MyD88-deficient LSK cells, relative to wild type LSK cells, in E. muris infected chimeric mice. However, the LSK population contained equivalent frequencies of wild type and MyD88-deficient cells (Fig. 2C and D). Moreover, in chimeric mice we observed similar proliferation of both wild type and MyD88-deficient bone marrow LSK cells during infection (Fig. 2E and F). These data reveal that MyD88 signaling in progenitor cells is not required for the infection-induced expansion the progenitor population. We also observed significantly higher bacterial infection in splenocytes from E. muris-infected mice that lacked MyD88, as compared to wild type controls and mixed chimeric mice on day 11 post-infection (Fig. 2G). The increased bacterial burden observed in MyD88-deficient mice was similar to what was observed in mice that lack IFNγ, suggesting a defect in IFNγ production or signaling in the absence of MyD88. Although bacteria can be detected in the bone marrow, burden is much lower relative to the spleen. Bacterial burden diminished by day 15 post-infection in all strains, however, it was slightly elevated in MyD88-deficient mice relative to wild type mice.
Figure 2. Intrinsic MyD88-signaling is not required for LSK expansion during E. muris infection.
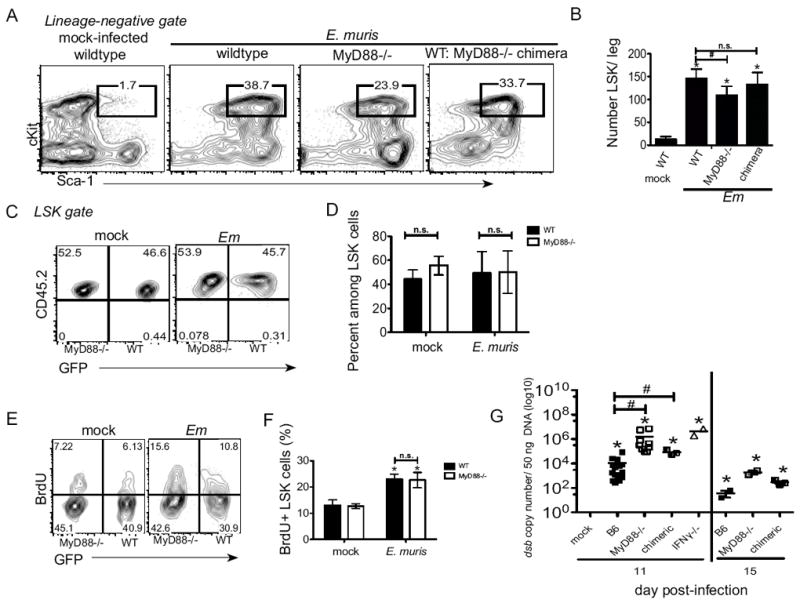
Bone marrow from wild type B6 mice, MyD88-deficient, and mixed chimeric (WT: MyD88-deficient) mice was harvested from mock- and E. muris-infected mice. (A) Representative flow cytometric plots of bone marrow LSK cells on day 11 post-infection are shown. Numbers above the gated region represent the frequency of c-Kit+ Sca-1+ cells among total Lin-negative cells. (B) The absolute number of LSK cells per leg is shown for wildtype, MyD88-deficient, and mixed chimeric mice on day 11 post-infection. (C) The LSK population in mixed chimeric mice was further analyzed for the frequency of wild type (CD45.2+; GFP+) and MyD88-deficient (CD45.2+;GFP-) cells. (D) The average frequency and standard deviation of wild type (filled bars) and MyD88-/- (open bars) cells among the LSK population is shown. (E) CD45.2+ LSK cells were also analyzed for BrdU incorporation and GFP expression. Numbers in the flow cyotmetry plots represent represent the frequency of cells in each quadrant. (F) The frequency of BrdU+ cells among wildtype (filled bars) and MyD88-deficient cells in chimeric mice are shown in mock and E. muris infected mice. (G) Bacteria infection was evaluated by quantitative-PCR in spleen tissues of E. muris-infected C57BL/6, MyD88-deficient, mixed wild type:MyD88-deficient chimeric, and IFNγ-deficient mice. Data represents the copy number of the bacterial dsb gene per 50 ng of splenic DNA on day 11 post-infection. The asterisks reflect significant differences between mock and infected groups and # reflect differences between different strains of mice; * or # p < 0.05.
MyD88-signaling promotes IFNγ production during ehrlichiosis
MyD88-signaling can augment IFNγ production, as has been shown in murine models of tularemia, legionella infection, and chlamydia (19-21). As intrinsic MyD88 signaling was not required for the expansion of the LSK population, we next addressed whether MyD88 signaling was required for IFNγ production during E. muris infection. IL-12p70 was also measured because it is known to induce IFNγ production (22). Both IFNγ and IL-12p70 concentrations were significantly increased in the serum of infected mice, relative to mock-infected mice, by day 11 post-infection (Fig. 3A). In comparison with E. muris-infected wild type mice, MyD88-deficient mice had significantly reduced amounts of IFNγ and IL-12p70 in sera on day 11 post-infection. Serum concentrations of IFNγ and IL-12p70 in mixed bone marrow chimeric mice were similar to concentrations observed in wild type mice. Both MyD88-deficient and wild type mice expressed comparable amounts of serum monocyte chemoattractant protein (MCP-1), IL-10, IL-6, and TNFα in mock-infected and E. muris-infected mice (data not shown), indicating that IFNγ and/or IL-12p70 production might require MyD88 signaling.
Figure 3. Altered cytokine expression in E. muris infection in the absence of MyD88-signaling.
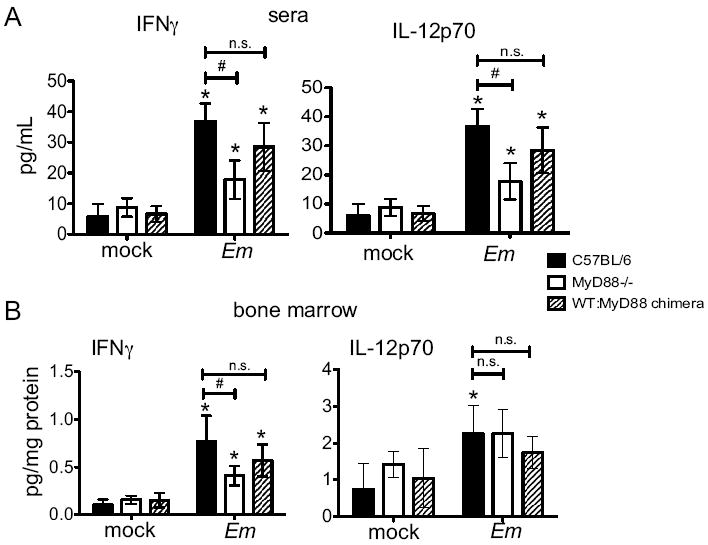
(A) Serum IFNγ and IL-12p70 were evaluated in C57BL/6, MyD88-deficient, and mixed chimeric mice on day 11 post-E.muris infection. (B) Bone marrow homogenate was analyzed for IFNγ and IL-12p70 on day 11 post-infection. Statistically significant differences are indicated as follows: # represents differences between strains of mice, and * represent differences between mock and infected groups; n.s. indicates no significance. Data were from two separate experiments, and at least three mice were assayed within each group.
To address whether changes in serum cytokine concentrations were reflected by similar changes within the bone marrow, where HSPCs reside, we next measured cytokine concentrations in bone marrow homogenates. As was observed in the sera, significantly increased IFNγ was observed in wild type bone marrow following infection, relative to MyD88-deficient mice; the mixed bone marrow chimeric mice exhibited an intermediate concentration of IFNγ on day 11 post-infection (Fig. 3B). IL-12p70 concentration was increased in wild type mice on day 11 post-infection, however, no significant differences were observed between wild type mice and MyD88-deficient mice after infection. Thus, MyD88 plays a role in regulating the production of IFNγ in response to ehrlichia infection.
IFNγ contributes to LSK expansion, macrophage colony formation, and monopoiesis
To directly test whether reduced LSK expansion in MyD88-deficient mice was due to decreased IFNγ production during ehrlichiosis, we administered recombinant IFNγ to MyD88-deficient mice on days 8, 9, and 10 post-infection. Relative to E. muris infected MyD88-deficient mice, administration of rIFNγ resulted in an increase in the frequency and number of LSK cells, such that rIFNγ treated MyD88-deficient mice resembled E. muris-infected wild type mice (Fig. 4A and B). As we previously demonstrated that infection-induced LSK cells contained increased myeloid potential (23), we next determined the functional significance of IFNγ–induced changes to progenitor cell phenotype by quantitating hematopoietic progenitors in the bone marrow. Infection elicited an increase in primitive granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM) and granulocyte, macrophage (CFU-GM) colonies in both wild type and MyD88-deficient mice (Fig. 4C), indicating MyD88-signaling is not required for an increase in primitive progenitors. The number of granulocyte colonies was unchanged in infected mice, however, an increase in macrophage colonies (CFU-M) was observed. Whereas MyD88-deficient mice exhibited significantly reduced CFU-M, relative to wild type mice, rIFNγ treatment of MyD88-deficient mice resulted in an increase in CFU-M after infection. In addition to the IFNγ-dependent increase in macrophage progenitors we observed increased monocytes (CD11b+ Ly6Chi) in the bone marrow after infection, a process dependent on IFNγ (Fig. 4D and E). Increased monopoiesis did not require intrinsic MyD88-signaling as equal numbers of wild type and MyD88-deficient monocytes were observed in mixed chimeric mice (Fig. 4F and G). These data demonstrate that MyD88 contributes to increased LSK cells, and specifically increased monopoiesis, in an IFNγ-dependent manner.
Figure 4. Monopoiesis is independent of intrinsic MyD88-signaling but dependent on IFNγ during E. muris infection.
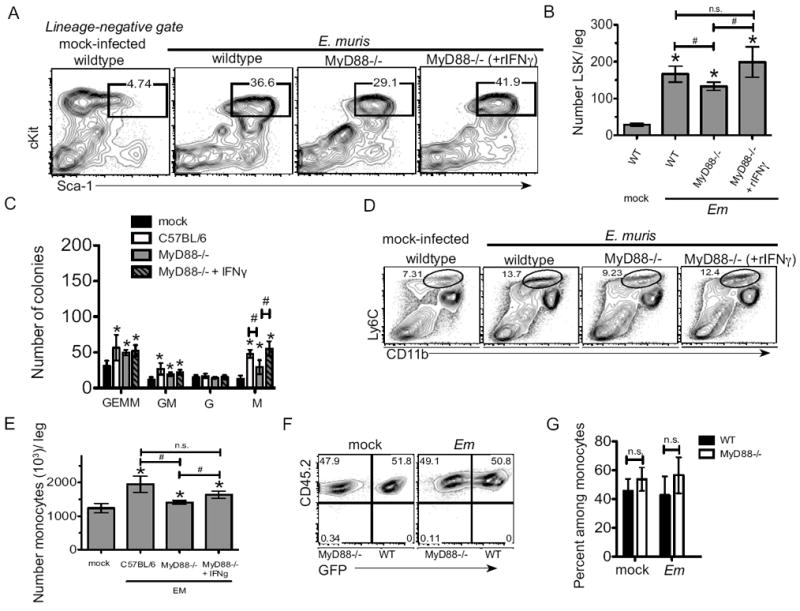
Bone marrow from wild type mice, MyD88-deficient mice, MyD88-deficient mice administrated with rIFNγ (i.v.) mice was harvested from mock- and/or E. muris-infected mice at day 11 post infection. (A) Representative flow cytometric plots of bone marrow LSK cells are shown. Numbers above the gated region represent the frequency of c-Kit+ Sca-1+ cells among total Lin-negative cells. (B) The absolute number of LSK cells per leg is shown for wildtype mice, MyD88-deficient mice, and MyD88-deficient mice received rIFNγ. (C) The number of colonies derived from bone marrow cells of wild type mice, MyD88-deficient mice, and MyD88-deficient mice received rIFNγ is shown. Bone marrow cells were cultured in semisolid methocellulose media for 7 days, and multipotent granulocyte/erythroid/macrophage (GEMM), granulocyte/macrophage (GM), granulocyte (G), and macrophage (M) colonies were scored. (D) Representative flow cytometric plots of bone marrow monocytes (CD11b+ Ly6Chi) in wild type mice, MyD88-deficient mice and MyD88-deficient mice that received rIFNγ are shown. Numbers above the gated region represent the frequency of CD11b+ Ly6Chi cells among total bone marrow cells. (E) The absolute number of monocytes per leg is shown for each group. (F) The frequency of wild type (CD45.2+; GFP+) and MyD88-deficient (CD45.2+;GFP-) bone marrow monocytes in radiation-induced mixed chimeric mice is shown. Numbers in the gated region represent the frequency among total monocytes. (G) The average frequency and standard deviation of wild type (filled bars) and MyD88-deficient (open bars) cells among bone marrow monocytes in mixed chimeric mice is shown. The asterisks reflect significant differences between mock and infected groups and # reflect differences between different strains of mice; * or # p < 0.05.
Identification of the source of IFNγ in the bone marrow during infection
IFNγ can activate dormant HSCs (24), can induce Sca-1 expression (25), and contributes to infection-induced monopoiesis in models of bacterial and viral infection (11, 23); however, the source(s) of IFNγ within the bone marrow during infection have not been defined. Expression of Ifng in the bone marrow is increased significantly on day 11 post-infection (23), and we demonstrated here that total IFNγ protein was also significantly increased at this time point. As the increase in IFNγ occurs concomitantly with phenotypic and functional activation of HSCs (23), we sought to identify the cell population(s) responsible for the production of IFNγ at this time. Populations of cells were purified from the bone marrow of mock- or E. muris-infected wild type and MyD88-deficient mice on day 11 post-infection, and expression of Ifng was quantitated (Supp. Fig 2). We observed relatively high Ifng expression in CD4 T cells from E. muris-infected mice, but very little expression in other cell populations. This increase was markedly reduced in the absence of MyD88 signaling. To address the functional significance of the increase in Ifng in CD4 T cells during infection, we performed intracellular cytokine staining (ICCS) for IFNγ on CD4 T cells directly ex vivo. Within the bone marrow of wild type mice we found that nearly 30% of CD4 T cells produced IFNγ during infection (Fig. 5A). The frequency of IFNγ+ CD4 T cells was significantly higher in the bone marrow, relative to the spleens of infected mice. In the absence of MyD88 signaling, however, frequencies of IFNγ+ CD4 T cells were significantly reduced in response to ehrlichia infection. CD8 T cells in E. muris infected mice also produced higher amounts of IFNγ as compared to CD8 T cells obtained from mock-infected mice (Fig. 5B). In contrast to CD4 T cells, however, the increased CD8 T cell-derived IFNγ in E. muris-infected mice was not dependent on MyD88-mediated signals. As the frequencies of IFNγ+ CD4 T cells were much higher than anticipated, we sought to validate our ICCS assay by examining IFNγ production in mice that lacked the Ifng and Ifngr genes. IFNγ was not detected in IFNγ-deficient mice, as expected (Supp. Fig. 3), but we observed significantly increased IFNγ in mice lacking the Ifngr. Increased IFNγ expression in CD4 T cells in IFNγR-deficient mice was not unexpected, based on previous reports that IFNγ production is elevated in the absence of IFNγR-mediated signaling (26). Furthermore, CD4 T cells exhibited little to no IFNγ staining in the absence of permeabilization demonstrating that the IFNγ detected in our assay was produced and not simply bound to the surface of the IFNγ+ cell.
Figure 5. Increased IFNγ expression in CD4 T lymphocytes requires MyD88.
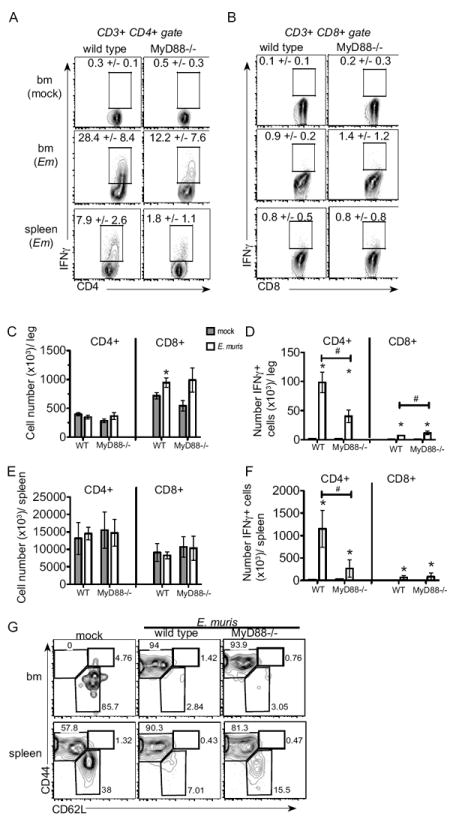
ICCS was performed directly ex vivo. Bone marrow and spleen cells were harvested from mock-infected and E. muris-infected C57BL/6 and MyD88-deficient mice on day 11 post-infection. CD3+ CD4+ (A) and CD3+ CD8+ (B) cells were analyzed for IFNγ expression in mock-infected bone marrow (top row), E. muris-infected bone marrow (middle row) and infected spleens (bottom row). Representative flow plots are shown for C57BL/6 mice (left column) and MyD88-deficient mice (right column) and the numbers above the gated region represent the average frequency and standard deviation of IFNγ + cells among CD4 T cells. Total numbers (C) and numbers of IFNγ+ CD4 and CD8 T (D) cells in the bone marrow are shown. Total numbers (E) and numbers of IFNγ+ CD4 and CD8 T (F) cells in the spleen are shown. Average numbers and the standard deviation are shown; gray bars represent mock-infected mice and open bars represent mice infected with E. muris. (G) Expression of CD44 and CD62L on CD3+ CD4+ IFNγ+ T cells in the bone marrow (upper row) and spleen (bottom row) in the wild type mock and E. muris-infected wild type and MyD88-deficient mice are shown. Numbers represent the frequencies within each gated region. Statistically significant differences are shown; # indicates differences between strains of mice, whereas * represent differences between mock and infected groups. At least 5 mice were assayed for each group in two independent experiments.
Although MyD88 signaling can modulate T cell migration (20), similar numbers of CD4 T cells were detected within the bone marrow of mock- and E. muris-infected mice, and no differences were observed between wild type and MyD88-deficient mice (Fig. 5C). In contrast, E. muris infection elicited an increase in CD8 T cells within the bone marrow that was independent of MyD88-signaling. Despite the increase in CD8 T cells in the bone marrow the number of IFNγ+ CD8 T cells was much lower than IFNγ+ CD4 T cells (Fig. 5D). The number of splenic CD4 and CD8 T cells was also similar between mock- and E. muris-infected wild type and MyD88-deficient mice (Fig. 5E), although, we observed significantly higher numbers of IFNγ+ CD4 T cells in the spleens of infected wild type mice (Fig. 5F). In the bone marrow in both wild type and MyD88-deficient mice IFNγ+ CD4 T cells exhibited a highly activated phenotype (CD44hi CD62Llo) (Fig. 5G). In the spleen, however, wild type IFNγ+ producing CD4 T cells exhibited a more activated phenotype, as compared to MyD88-deficient IFNγ+ CD4 T cells. Thus, the bone marrow contains a more activated pool of CD4 T cells. These data demonstrate that MyD88-signaling was partially required for CD4 T cell activation and production of IFNγ.
Although IFNγ can be produced by cells of different lineages, it was not detectable in NK cells in the bone marrow (Supp. Fig 4A and B). In contrast, IFNγ was detected in NKT cells in both wild type mice and MyD88-deficient mice (Supp. Fig 4C), however, E. muris infection did not induce a significant change in NKT cell IFNγ production in either wild type or MyD88-deficient mice. The number of NKT cells was similar in all groups of mice, whereas a decrease in NK cell number was noted in response to infection, and in the bone marrow of MyD88-deficient mice (data not shown). It has been demonstrated that myeloid cells can produce IFNγ during invasive group A Streptococcus infection (27), thus we also performed ICCS on neutrophils, monocytes, and macrophages in the bone marrow. We observed a low amount of IFNγ expression in neutrophils and macrophages, however this was likely due to surface-bound IFNγ as we observed similar IFNγ staining in the absence of permeablization (Supp. Fig 4D and F). IFNγ was not detected in monocytes and immature myeloid cells (Supp. Fig 4E). Similar negative data were obtained for splenic NK cells, NKT cells, and myeloid cells (data not shown). Thus, we have identified CD4 T cells as critical producers of IFNγ during ehrlichia infection, and a requirement for MyD88-signaling for this response.
CD4 T cell-derived IFNγ is essential for bone marrow LSK expansion during infection
To test whether CD4 T cell-derived IFNγ was essential for LSK cell expansion during infection, we generated mixed bone marrow chimeric mice wherein CD4 T cells were unable to produce IFNγ (CD4Ifng-/-). A small percentage of T cells can persist even after lethal irradiation (28), thus IFNγ-deficient mice were used as recipient mice, to ensure that all CD4 T cells were unable to produce IFNγ. IFNγ-deficient mice were irradiated and reconstituted with equal numbers of bone marrow cells from CD4-deficient, and IFNγ-deficient mice; thus, in the chimeric mice, all cells, other than CD4 T cells, were able to produce IFNγ. Control chimeric mice in which CD4 T cells were sufficient for IFNγ (CD4Ifng+/+) were also generated. After E. muris infection reduced frequencies and lower numbers of LSK cells were detected in the bone marrow of CD4Ifng-/- mice (Fig 6. A-C). Although it is currently unclear if local production of IFNγ in the bone marrow is essential for LSK expansion, these data demonstrate that CD4 T cells play a non-redundant role in promoting the optimal expansion of HSPCs via IFNγ in response to an intracellular bacterial infection.
Figure 6. CD4 T cell-derived IFNγ is required for LSK expansion during bacterial infection.
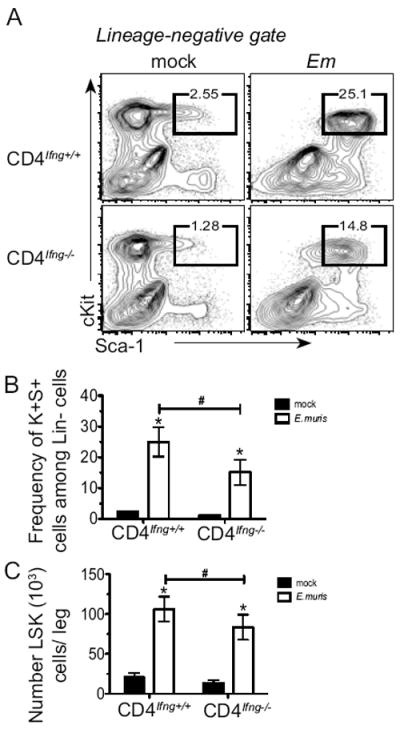
Radiation-induced mixed bone marrow chimeric mice were generated in which CD4 T cells were unable to secrete IFNγ (CD4Ifng-/-); control chimeric mice contained wildtype CD4 T cells (CD4Ifng+/+). (A) Representative flow cytometric plots of lineage-negative cells that were analyzed for expression of c-Kit+, Sca-1+ in mock- and E. muris-infected chimeric mice on day 11 post-infection. The frequency of c-Kit+ Sca-1+ cells among total Lin-negative cells is shown above each gate, and the average frequencies are shown in panel B. (C) Numbers of LSK cells in one leg is shown for mock- (filled bars) and E.muris-infected (open bars) chimeric mice. Statistically significant differences are shown; # indicates differences between strains of mice, whereas * represent differences between mock and infected groups. This data represents one experiment with between 3 and 7 mice in each experimental group.
MyD88 signaling is required in CD4 T cells for robust expression of IFNγ and cell proliferation
To determine whether MyD88-signaling regulates CD4 T cell function via an intrinsic mechanism, we generated mixed chimeric mice using wild type and MyD88-deficient bone marrow. Donor wild type cells were obtained from βACT-GFP mice, which facilitated the detection of both donor wild type (CD45.2; GFP+) and MyD88-deficient T cells (CD45.2+; GFP-negative). This strategy also allowed us to distinguish wild type (ie., radioresistant) host-derived T cells (CD45.1+, GFP-negative). Routine screening at four weeks post-transplantation, and analysis of bone marrow at the time of infection revealed normal numbers of T cells, relative to non-irradiated mice, and equal percentages of donor wild type and MyD88-deficient cells in chimeras (data not shown). Following infection, reduced frequencies of IFNγ-producing cells were detected among MyD88-deficient CD4 T cells, relative to wild type cells, in both the bone marrow and the spleen (Fig. 7A and C). We also observed that wild type CD4 T cells exhibited increased BrdU incorporation, relative to MyD88-deficient CD4 T cells, in both the bone marrow and spleen (Fig. 7B and D). As MyD88-signaling is well-documented to contribute to activation of APCs, which may also contribute to CD4 T cell function, we also measured expression of MHC class II on populations of APCs in the mixed bone marrow chimeric mice. Very similar class II surface expression was detected on both wild type and MyD88-deficient dendritic cells, macrophages, and B cells in both the bone marrow and spleen (data not shown).
Figure 7. Intrinsic MyD88 signaling is required for CD4 T cell production of IFNγ production.
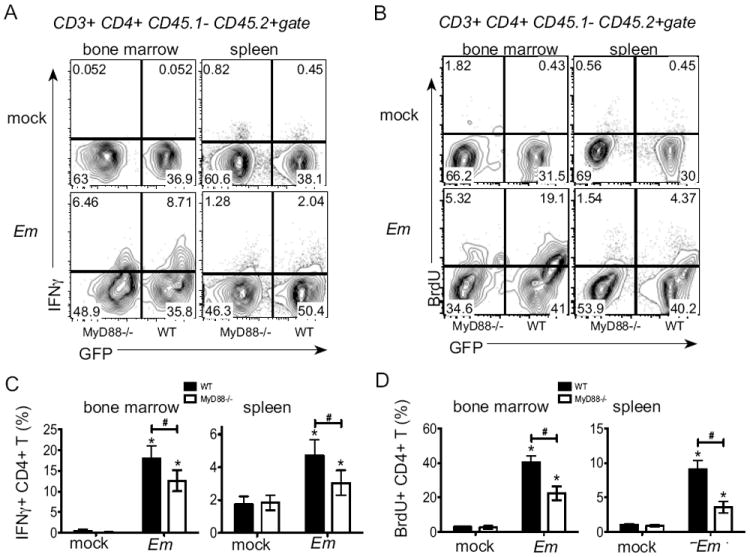
ICCS was performed on bone marrow and spleen cells from mixed chimeric mice (wild type: MyD88-deficient) on day 11 post-infection. (A) Representative flow plots of CD3+ CD4+ CD45.1- CD45.2+ cells. IFNγ and GFP expression are shown, and the numbers on the plots represent the frequency of cells in each quadrant. (B) Cells were gated as in A and examined for BrdU. (C) Percentage of IFNγ+ cells among CD4 T cells is shown for wildtype (filled bars) and MyD88-/- cells (open bars) in bone marrow and spleen of mock and E.muris infected chimeric mice. (D) Percentage of BrdU+ cells among wild type (filled bar) and MyD88-deficienct (open bar) CD4 T cells in bone marrow and spleen are shown. Statistically significant differences are shown; # indicates differences between strains of mice, whereas * represent differences between mock and infected groups. This experiment was performed two times and experimental groups contained 3-5 mice.
MyD88 signaling contributes to T-bet expression in CD4 T cells during infection
T-bet is a critical transcription factor that regulates IFNγ expression by T cells (29), thus we determined whether MyD88 signaling was required for T-bet expression in response to infection. Following E. muris infection, T-bet expression was significantly increased in CD4 T cells in the bone marrow and spleen; MyD88 signaling was required for high T-bet expression in CD4 T cells and relatively higher T-bet expression was observed in bone marrow CD4 T cells, as compared to splenic CD4 T cells (Fig. 8A and B). Moreover, we found that T-bet expression was significantly higher in wild type CD4 T cells, as compared to MyD88-deficient CD4 T cells (Fig. 8C and D), in E. muris infected mixed chimeric mice, demonstrating an intrinsic requirement for MyD88 in increasing T-bet expression. These data establish that intrinsic MyD88 signaling in CD4 T cells regulates T-bet expression. Thus, our data together identify an intrinsic defect in MyD88-deficient CD4 T cell function that results in reduced HSPC activation and proliferation in response to a bacterial infection.
Figure 8. Intrinsic MyD88 signaling contributes to infection-induced increase in T-bet expression in CD4 T cells.
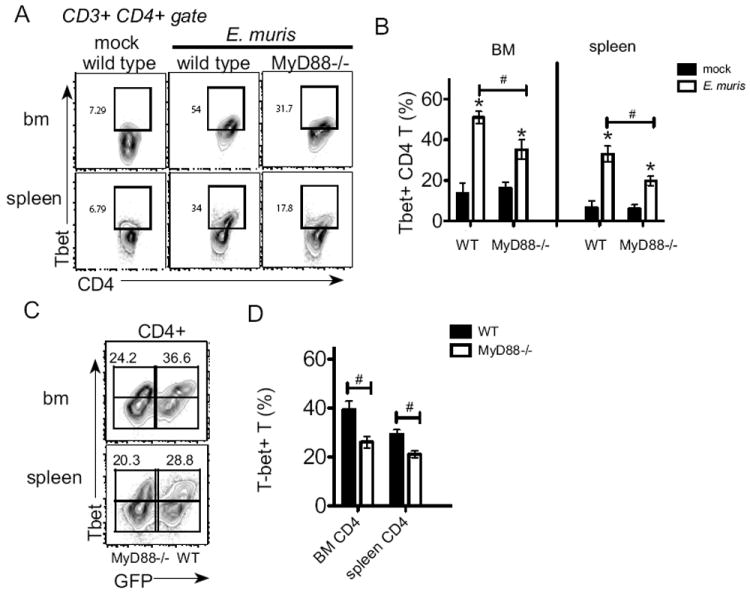
Intracellular staining was performed on bone marrow and spleen T lymphocytes to detect T-bet. (A) Representative flow plots of CD3+ CD4+ cells analyzed for expression of CD4 and T-bet; numbers on the plots represent the frequency of cells in each quadrant. (B) The average frequency of T-bet+ CD4 T cells in the bone marrow and spleen is shown. (C) Intracellular staining for T-bet was performed on bone marrow from mixed chimeric mice (wild type (GFP-positive): MyD88-deficient) on day 11 post-infection. CD3+ CD4+ cells were analyzed for expression of GFP and T-bet. Numbers represent the percentage of T-bet+ cells among wild type (GFP-positive) or MyD88-/- cells (GFP-negative) in the bone marrow and spleen of E.muris infected chimeric mice. (D) The average frequencies of T-bet+ CD4 T cells is shown for wild type (filled bar) and MyD88-deficienct (open bar) CD4 T cells in bone marrow and spleen are shown. Statistically significant differences are shown; # indicates differences between strains of mice, whereas * represent differences between mock and infected groups. This experiment was performed one time and experimental groups contained 5-7 mice.
Discussion
In the present study we found that intrinsic MyD88 signaling contributed to the infection-induced increase in LSK cells by driving IFNγ production by CD4 T cells. Furthermore, MyD88 signaling was not required in HSPCs for their increase or proliferation during E. muris infection. This finding reinforces the idea that cytokine-mediated signaling is an important component of HSPC activation during infection. As LSK expansion is not completely ablated in the absence of IFNγ, we propose that additional cytokines may impact HSPC activation and proliferation including TNFα and IL-6, as has been reported in vitro (30), either alone or in concert with IFNγ. Our data suggest that direct sensing of pathogens by stem and progenitor cells is not absolutely essential to drive progenitor cell expansion and differentiation. Thus, progenitor cells can respond to infectious agents at sites distant from infection due to the action of soluble factors and activated cells, which may be particularly important for host defense against intracellular pathogens. Extracellular bacteria and fungi may be more able to directly activate progenitor cells that express TLRs. In contrast, hematopoietic responses to intracellular pathogens, such as E. muris, may rely on cytokine-mediated activation. The cytokine cues present during different infections, and in response to particular hematopoietic stresses, likely act to shape the hematopoietic response at the stem and progenitor cell level to gear cell production towards generating particular cell types.
In response to adjuvants such as CFA and Alum production of neutrophils is increased, at the expense of lymphopoiesis, a process termed “emergency granulopoiesis” (1, 31). Alum-elicited emergency granulopoiesis depends on IL-1β and TNFα (1, 32). In contrast to Alum, the primary mediator of LPS-induced emergency granulopoiesis is granulocyte-colony stimulating factor (G-CSF), and it occurs independent of IL-1β-mediated signaling (33). Thus, different adjuvants operate via unique mechanisms to elicit granulocyte production, and the production of granulocytes is likely particularly important for control of extracellular bacterial pathogens. However, myelopoiesis is increased in response to intracellular bacteria and virus infections as well. Ehrlichial infection is characterized by robust IFNγ response and significantly increased monocytes, supporting an important role for IFNγ in eliciting monopoiesis. In fact, we observed a critical role for IFNγ in driving increased blood monocytes during ehrlichiosis (10) and bone marrow monocytes, and in the absence of intact IFNγ-signaling E. muris infection elicited a more profound increase in immature myeloid cells and neutrophils reminiscent of Alum or LPS-induced emergency granulopoiesis (1, 10, 33). DeBruin et al. demonstrated that IFNγ can augment M-CSF signaling in myeloid progenitors while simultaneously inhibiting G-CSF signaling, thus driving monopoiesis at the expense of granulopoiesis during LCMV infection (11). Here we demonstrate that one mechanism accounting for reduced macrophage progenitors and monocytes in MyD88-deficient mice is reduced IFNγ production in response to E. muris infection. We envision that the capacity of the hematopoietic system to generate monocytes may be critical for resolution and containment of ehrlichial infection, and other intracellular pathogens. How pathogens might alter cytokine responses to avoid eliciting particular hematopoietic programs is an important question for future investigation.
Previously we, and others, have reported that infection-induced activation of HSPCs required IFNγ signaling (23, 24). Although multiple types of cells including T cells, NK cells, and NK T cells are able to produce IFNγ in vivo, the source(s) of IFNγ that drives LSK expansion in the bone marrow during E. muris infection had not been defined. Our data show that CD4 T cells are the major producers for IFNγ in bone marrow and that a very high frequency of IFNγ+ CD4 T cells is present in this tissue. Although IFNγ+ CD8 T cells were also significantly increased during E. muris infection, the total numbers of IFNγ+ CD8 T cells were minor compared to the CD4 T cell population. The striking frequency of IFNγ+ CD4 T cells within the bone marrow was nearly triple what was observed in the spleen. It will be interesting to determine if robust IFNγ production by CD4 T cells in the bone marrow is common to many different infections or is unique to ehrlichial infection and infections that elicit robust production of monocytes. Another outstanding question is whether CD4 T cells are activated within the bone marrow or traffic to the bone marrow from peripheral sites, which will be an important area of future research. IFNγ is also responsible for inducing severe blood pathologies, including anemia and thrombocytopenia, during ehrlichiosis and other intracellular infections such as Toxoplasma gondii infection (34). The observation of profound hematopoietic dysfunction in HME patients, combined with our data demonstrating that IFNγ drives many similar changes in the murine model of ehrlichiosis, suggests that a unique feature of HME pathogenesis is the overwhelming activation of a Th1 CD4 T cell response within the bone marrow.
The mechanisms by which E. muris elicits an IFNγ response are not yet clear, however we found a critical role for MyD88-signaling in CD4 T cells for robust production of IFNγ. It is also important to note that MyD88-signaling in non-hematopoeitic cells, such as mesenchymal stem cells and other stromal cells in the bone marrow, may contribute to optimal HSPC responses during ehrlichiosis. Here we demonstrate an intrinsic requirement for MyD88 in the infection-induced increase in T-bet expression in CD4 T cells. MyD88 is an essential member of the IL-1R/TLR superfamily and mediates TLR, IL-18R and IL-1βR-signaling (as reviewed in (35)). It is unclear if the CD4 T cell response observed in response to ehrlichial infection is antigen-specific, or if it occurs via non-specific activation, such as what can occur during Salmonella typhimurium infection when IL-18 is abundant (36). As CD4 T cells do express TLRs, at least at the transcriptional level, it is possible that the reduced IFNγ production by CD4 T cells is due to impaired TLR signaling. However, the requirement for TLR signaling in mediating infection-induced LSK expansion is not supported by our other data, thus we favor the possibility that diminished responses to IL-18 in MyD88-deficient CD4 T cells accounts for reduced IFNγ production by these cells.
T cell intrinsic defects in the absence of MyD88 have been reported in other infection models, most notably with LCMV. In a model of LCMV induced neuropathology, MyD88 was shown to play a CD4 T cell intrinsic role in driving pathogenesis in the brain (37). In the absence of MyD88 signaling mice were protected from neuroinflammation and significantly fewer IFNγ+ CD4 T cells were observed in the brain. It was also shown that MyD88-dependent CD4 T cell function was not due to IL-18 or IL-1β suggesting a novel mechanism by which MyD88-signaling can regulate CD4 T cell function. In addition, the authors demonstrated that CD4 defects were independent of antigen presenting cells, similar to what we observe in our studies. T cell intrinsic defects in CD8 T cells were also observed during LCMV infection, and the primary defect was seen in survival during clonal expansion (38). MyD88-deficient antigen-specific CD8 T cells were able to proliferate similarly to wild type cells, but exhibited impaired survival. The authors suggested that MyD88 dependent pathways were linked to the inflammatory signals specific to LCMV infection although they were unable to identify the specific signals involved. In a model of Toxoplasma gondii infection, T cell intrinsic MyD88 was also found to be critical for controlling disease (39). Interestingly, the impact of MyD88 signaling was not through IL-1R or IL-18-R, supporting the possibility that direct TLR stimulation of T cells during toxoplasmosis is important for T cell effector function. Thus, MyD88 may operate in T cells in a variety of pathways that may depend on the infectious agent and inflammatory cytokines present in particular tissues during infection.
NKT cells have been reported to be activated by E. muris (40) and were shown to regulate host immunity in response to infection with a related strain of ehrlichia, Ixodes ovatus ehrlichia (IOE) (17). We did not observe a significant difference in NKT cell production of IFNγ in wild type and MyD88-deficient mice indicating the decreased IFNγ observed in MyD88-deificient mice is not due to a defect in NKT cell function. In fact, we observed slightly, although not statistically significant, increased frequencies of IFNγ+ NKT cells in the absence of MyD88 signaling. This is in agreement with the observation that serum IFNγ concentrations were not significantly reduced in the absence of NKT cells as shown during IOE infection (17). Taken together, we conclude that CD4 T cells are the major IFNγ producing cells in the bone marrow during E. muris infection.
The total number of CD4 T cells in spleen and bone marrow are comparable between wild type mice and MyD88-deficient mice during infection, suggesting MyD88-signaling is not involved in T cell trafficking. In a model of Aspergillus fumigatus infection of the lung, CD4 T cells were found to traffic normally to the lung in the absence of MyD88 (41), supporting the idea that MyD88 is not required for proper trafficking of lymphocytes. This is in contrast to Chlamydia muridarum infection, where MyD88 signaling was critical for recruitment of CD4 T cells into the genital tract during infection (20). This difference in MyD88-dependent CD4 T cell migration may be due to specific requirements for migration into lymphoid versus non-lymphoid tissues, such as the genital tract. Another possibility is that these differences are due to pathogen-specific TLR ligands.
The importance of TLR-mediated signaling in controlling T cell function is primarily thought to occur via MyD88-signaling in APCs. MyD88 signaling in BMDCs was important for IL-12p40 production in E. muris infection in vitro (15). However, our in vivo observation that very similar amounts of IL-12p70 were generated in the bone marrow of wildtype and MyD88-deficient mice in response to E. muris infection, suggest that differences in IL-12-p40 do not affect IL-12, but may impact IL-23 production. We observed similar activation of APCs in mixed chimeric mice, supporting our conclusion that the primary defect in control of bacteria in MyD88-deficient mice, is due to the intrinsic defect in CD4 T cells. However, we cannot rule out the possibility that APC function is compromised in the absence of MyD88. Interestingly, it is unclear what pathways require MyD88-signaling, and the MyD88-dependent responses likely differ between cells of the innate and adaptive immune cells. It was recently reported that Ehrlichia chaffeensis, the agent of HME, induces inflammatory responses in monocytes through MyD88, ERK, and NF-kB, but did not require TLRs, the adaptor TIR-domain-containing adapter-inducing IFNβ, IL-18R, or IL-1βR (42). These data suggest the existence of previously unrecognized pathogen associated molecular patterns and cognate receptors.
Our data support a model whereby cytokines, in particular IFNγ, produced by mature hematopoietic cells can act locally, directly and/or indirectly, to promote differentiation of HSPCs. During ehrlichial infection MyD88-mediated signals act directly in CD4 T helper cells to promote their activation and ability to secrete IFNγ. Regulation of hematopoiesis by T helper cells during infection raises important questions about how hematopoietic function is maintained in the face of chronic infections where CD4 T cells are increased (or decreased, as in HIV infection) or exhibit a more activated phenotype. Thus, our findings highlight a previously unappreciated role for MyD88 in modulating hematopoietic function.
Supplementary Material
Literature Cited
- 1.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, Bovio I, Akira S, Kumagai Y, Moldawer LL. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oduro KA, Jr, Liu F, Tan Q, Kim CK, Lubman O, Fremont D, Mills JC, Choi K. Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood. 2012;120:2203–2213. doi: 10.1182/blood-2011-11-391342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welner RS, Pelayo R, Nagai Y, Garrett KP, Wuest TR, Carr DJ, Borghesi LA, Farrar MA, Kincade PW. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood. 2008;112:3753–3761. doi: 10.1182/blood-2008-04-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yanez A, Murciano C, O’Connor JE, Gozalbo D, Gil ML. Candida albicans triggers proliferation and differentiation of hematopoietic stem and progenitor cells by a MyD88-dependent signaling. Microbes Infect. 2009;11:531–535. doi: 10.1016/j.micinf.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Yanez A, Megias J, O’Connor JE, Gozalbo D, Gil ML. Candida albicans induces selective development of macrophages and monocyte derived dendritic cells by a TLR2 dependent signalling. PLoS One. 2011;6:e24761. doi: 10.1371/journal.pone.0024761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serbina NV, Hohl TM, Cherny M, Pamer EG. Selective expansion of the monocytic lineage directed by bacterial infection. Journal of Immunology. 2009;183:1900–1910. doi: 10.4049/jimmunol.0900612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends in immunology. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, Borjesson DL, Winslow GM. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-gamma signaling. Journal of Immunology. 2011;186:1032–1043. doi: 10.4049/jimmunol.1001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood. 2012;119:1543–1554. doi: 10.1182/blood-2011-07-367706. [DOI] [PubMed] [Google Scholar]
- 12.Belyaev NN, Brown DE, Diaz AI, Rae A, Jarra W, Thompson J, Langhorne J, Potocnik AJ. Induction of an IL7-R(+)c-Kit(hi) myelolymphoid progenitor critically dependent on IFN-gamma signaling during acute malaria. Nat Immunol. 2010;11:477–485. doi: 10.1038/ni.1869. [DOI] [PubMed] [Google Scholar]
- 13.Paddock CD, Childs JE. Ehrlichia chaffeensis: a prototypical emerging pathogen. Clin Microbiol Rev. 2003;16:37–64. doi: 10.1128/CMR.16.1.37-64.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin M, Rikihisa Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. INfection and Immunity. 2003;71:5324–5331. doi: 10.1128/IAI.71.9.5324-5331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh YS, Koo JE, Biswas A, Kobayashi KS. MyD88-dependent signaling contributes to host defense against ehrlichial infection. PLoS One. 2010;5:e11758. doi: 10.1371/journal.pone.0011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect Immun. 2009;77:4061–4069. doi: 10.1128/IAI.01550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stevenson HL, Crossley EC, Thirumalapura N, Walker DH, Ismail N. Regulatory roles of CD1d-restricted NKT cells in the induction of toxic shock-like syndrome in an animal model of fatal ehrlichiosis. INfection and Immunity. 2008;76:1434–1444. doi: 10.1128/IAI.01242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 19.Pietras EM, Miller LS, Johnson CT, O’Connell RM, Dempsey PW, Cheng G. A MyD88-dependent IFNgammaR-CCR2 signaling circuit is required for mobilization of monocytes and host defense against systemic bacterial challenge. Cell Res. 2011;21:1068–1079. doi: 10.1038/cr.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagarajan UM, Sikes J, Prantner D, Andrews CW, Jr, Frazer L, Goodwin A, Snowden JN, Darville T. MyD88 deficiency leads to decreased NK cell gamma interferon production and T cell recruitment during Chlamydia muridarum genital tract infection, but a predominant Th1 response and enhanced monocytic inflammation are associated with infection resolution. INfection and Immunity. 2011;79:486–498. doi: 10.1128/IAI.00843-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sporri R, Joller N, Albers U, Hilbi H, Oxenius A. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. Journal of Immunology. 2006;176:6162–6171. doi: 10.4049/jimmunol.176.10.6162. [DOI] [PubMed] [Google Scholar]
- 22.Seder RA, Gazzinelli R, Sher A, Paul WE. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10188–10192. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacNamara KC, Jones M, Martin O, Winslow GM. Transient activation of hematopoietic stem and progenitor cells by IFNgamma during acute bacterial infection. PLoS One. 2011;6:e28669. doi: 10.1371/journal.pone.0028669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells. 2007;25:1339–1347. doi: 10.1634/stemcells.2006-0644. [DOI] [PubMed] [Google Scholar]
- 26.Rottman M, Catherinot E, Hochedez P, Emile JF, Casanova JL, Gaillard JL, Soudais C. Importance of T cells, gamma interferon, and tumor necrosis factor in immune control of the rapid grower Mycobacterium abscessus in C57BL/6 mice. INfection and Immunity. 2007;75:5898–5907. doi: 10.1128/IAI.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumura T, Ato M, Ikebe T, Ohnishi M, Watanabe H, Kobayashi K. Interferon-gamma-producing immature myeloid cells confer protection against severe invasive group A Streptococcus infections. Nat Commun. 2012;3:678. doi: 10.1038/ncomms1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson BE, McNiff JM, Matte C, Athanasiadis I, Shlomchik WD, Shlomchik MJ. Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood. 2004;104:1565–1573. doi: 10.1182/blood-2004-01-0328. [DOI] [PubMed] [Google Scholar]
- 29.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P, Nelson S, Bagby GJ, Siggins R, 2nd, Shellito JE, Welsh DA. The lineage-c-Kit+Sca-1+ cell response to Escherichia coli bacteremia in Balb/c mice. Stem Cells. 2008;26:1778–1786. doi: 10.1634/stemcells.2007-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, Tenen DG. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nature immunology. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 32.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. 2004;199:47–58. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, Heikenwalder M, Manz MG. Cutting Edge: LPS-Induced Emergency Myelopoiesis Depends on TLR4-Expressing Nonhematopoietic Cells. Journal of Immunology. 2012;188:5824–5828. doi: 10.4049/jimmunol.1103253. [DOI] [PubMed] [Google Scholar]
- 34.Mullarky IK, Szaba FM, Kummer LW, Wilhelm LB, Parent MA, Johnson LL, Smiley ST. Gamma interferon suppresses erythropoiesis via interleukin-15. INfection and Immunity. 2007;75:2630–2633. doi: 10.1128/IAI.01836-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzgerald KA, O’Neill LA. The role of the interleukin-1/Toll-like receptor superfamily in inflammation and host defence. Microbes and infection / Institut Pasteur. 2000;2:933–943. doi: 10.1016/s1286-4579(00)00396-8. [DOI] [PubMed] [Google Scholar]
- 36.McSorley SJ, Srinivasan A. Interview with Dr. Stephen J. McSorley and Ms. Aparna Srinivasan regarding Pivotal Advance: secondary exposure to LPS suppresses CD4+ T cells and exacerbates murine typhoid. Interview by Helene F. Rosenberg. Journal of Leukocyte Biology. 2007;81:401–402. doi: 10.1189/jlb.1306194. [DOI] [PubMed] [Google Scholar]
- 37.Zhou S, Kurt-Jones EA, Cerny AM, Chan M, Bronson RT, Finberg RW. MyD88 intrinsically regulates CD4 T-cell responses. Journal of virology. 2009;83:1625–1634. doi: 10.1128/JVI.01770-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, Wherry EJ, Kaech SM, Turka LA. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. Journal of Immunology. 2008;181:3804–3810. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, Turka LA. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3855–3860. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, 3rd, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 41.Rivera A, Ro G, Van Epps HL, Simpson T, Leiner I, Sant’Angelo DB, Pamer EG. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity. 2006;25:665–675. doi: 10.1016/j.immuni.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Miura K, Matsuo J, Rahman MA, Kumagai Y, Li X, Rikihisa Y. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-kappaB but not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or toll-like receptors. INfection and Immunity. 2011;79:4947–4956. doi: 10.1128/IAI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.