

Abstract
PMEL is a pigment cell-specific protein responsible for the formation of fibrillar sheets within the pigment organelle, the melanosome. The fibrillar sheets serve as a template upon which melanins polymerize as they are synthesized. The PMEL fibrils are required for optimal pigment cell function, as animals that either lack PMEL expression or express mutant PMEL variants show varying degrees of hypopigmentation and pigment cell inviability. The PMEL fibrils have biophysical properties of amyloid, a protein fold that is frequently associated with neurodegenerative and other diseases. However, PMEL is one of a growing number of non-pathogenic amyloid proteins that contribute to the function of the cell and/or organism that produces them. Understanding how PMEL generates amyloid in a non-pathogenic manner might provide insights into how to avoid toxicity due to pathological amyloid formation. In this review we summarize and reconcile data concerning the fate of PMEL from its site of synthesis in the endoplasmic reticulum to newly formed melanosomes and the role of distinct PMEL subdomains in trafficking and amyloid fibril formation. We then discuss how its progression through the secretory pathway into the endosomal system might allow for the regulated and non-toxic conversion of PMEL to an ordered amyloid polymer.
Introduction
Melanosomes are membrane-bound organelles in pigment cells in which melanin pigments are synthesized and stored (Delevoye et al., 2011; Sitaram and Marks, 2012). In vertebrate pigment cells that make predominantly black and brown eumelanins, melanins polymerize within ellipsoidal melanosomes upon intralumenal fibrils or fibrillar sheets that run the length of the organelle (Birbeck, 1963; Moyer, 1966; Seiji et al., 1963). Often referred to as “the melanosome matrix”, it is now clear that these proteinaceous fibrils are composed predominantly – if not entirely – of proteolytic fragments of a single pigment cell-specific protein, PMEL. This remarkable protein – which has been referred to in the literature by many names, including Pmel17, Silver, SILV, gp100, and ME20, or by reference to the apparent molecular mass of its full-length or prominent proteolytic products (p100, p85, p95, p115, etc.) – is initially synthesized as an integral membrane glycoprotein in the endoplasmic reticulum (ER). Through post-translational modifications, proteolytic processing steps and precisely timed oligomerization, the glycoprotein becomes transformed into fibrillar structures that laterally assemble into sheets. The fibrils have biophysical features of amyloid, making PMEL a prime model for a class of proteins called functional amyloids (Blanco et al., 2012; Fowler et al., 2007). In this review, we detail the experimental evidence that PMEL underlies the melanosome fibrils and update (relative to an earlier review on the same topic (Theos et al., 2005b)) our current understanding of PMEL processing and trafficking to melanosomes in pigment cells. We focus on the amyloid properties of PMEL and its new role as a model for distinguishing functional from pathological amyloid, discussing those characteristics of PMEL amyloid formation that might underlie its lack of toxicity and how alterations in this pathway might result in pathology.
What is amyloid?
The amyloid fold is a cross β-pleated sheet structure in which the β sheets stack upon themselves in a plane perpendicular to that of the sheets themselves (Dobson, 2003; Greenwald and Riek, 2010). Amyloid is characterized by the formation of stable fibrils that are insoluble in detergents, can be detected by electron microscopy, bind to specific dyes such as thioflavins and Congo Red, and are highly resistant to protease digestion (Chiti and Dobson, 2006). Historically, amyloid formation has been associated with disease, particularly neurodegeneration as observed in Alzheimer Disease (AD) and Parkinson Disease (PD) and the prion-associated spongiform encephalopathies, in which specific proteins misfold into an amyloid conformation (Buxbaum and Linke, 2012); indeed, many proteins can adopt an amyloid fold “by mistake” (Goldschmidt et al., 2010). However, there is a growing body of evidence pointing to an evolutionarily conserved use of the amyloid fold in functional processes such as adaptation to and protection from environmental stresses (Blanco et al., 2012; Iconomidou et al., 2000; Uptain and Lindquist, 2002; Wickner et al., 2004), concentration of biologically active peptides (Maji et al., 2009), organization of extracellular environments (Chapman et al., 2002; Kenney et al., 2002), establishment and maintenance of long-term memory in the central nervous system (Majumdar et al., 2012), and others. It is therefore important to understand (1) what makes some proteins acquire amyloidogenic properties and how this might be associated with disease, (2) the similarities and differences among pathological and functional amyloids, and (3) the conformational changes that direct amyloidogenic proteins towards either a benign or pathological role in cells and organisms.
Fibrils within melanosomes
Melanosomes are generated in the skin and hair within epidermal melanocytes and in the eye within choroidal melanocytes and retinal, iris and ciliary body pigment epithelia. Eye pigment cells retain their melanosomes, and a body of evidence indicates that melanosome formation in the retinal pigment epithelia is largely limited to pre- and early post-natal development (Lopes et al., 2007). By contrast, melanosomes in skin melanocytes are constantly generated and transferred to neighboring keratinocytes in a process that is poorly understood (Van Den Bossche et al., 2006). Melanosomes in skin melanocytes that generate predominantly red and yellow pigments, or pheomelanins, are fundamentally distinct in content and morphology from those that make predominantly black and brown eumelanins, and harbor neither PMEL nor intralumenal fibrils (Furumura et al., 1998; Moyer, 1966; Simon et al., 2009). This review will thus focus only on melanosomes that generate predominantly eumelanins in the eye and skin.
Melanosome formation can be divided into four stages of development (Figure 1). Stage I and II melanosomes, or premelanosomes, lack pigment. The onset of melanin synthesis allows for the conversion of stage II melanosomes to pigmented stage III and ultimately stage IV melanosomes (Seiji et al., 1963). This onset correlates with the delivery of melanogenic enzymes, such as Tyrosinase (TYR) (Hirobe, 1982; Huizing et al., 2001; Novikoff et al., 1968; Theos et al., 2005a), the Tyrosinase-related protein 1 (TYRP1) (Raposo et al., 2001; Vijayasaradhi et al., 1991; Vijayasaradhi et al., 1995) and likely DOPAchrome tautomerase (DCT) (Kushimoto et al., 2001), and of transporters such as OCA2/ pink-eyed dilute protein (Puri et al., 2000; Sitaram et al., 2009) and the copper transporter ATP7A (Setty et al., 2008), that modify the lumenal environment of the melanosome to favor melanin synthesis. These melanosome components are delivered to fully formed stage II and III melanosomes from early endosomes by membrane transport mechanisms that are just beginning to be understood (reviewed in (Sitaram and Marks, 2012)). The resulting melanin precursors then polymerize on the fibrillar matrix, resulting in the thickening and blackening of the fibrillar sheets in stage III and their ultimate masking in stage IV (Seiji et al., 1963). The deposition of melanin onto fibrillar sheets allows for the concentration and retention of pigment into a single functional unit.
Figure 1. Morphology of different stage melanosomes.

Shown are representative electron micrographs of melanosomes of stages I, II, III and IV from thin sections of human MNT-1 melanoma cells preserved by high pressure freezing and freeze substitution. Note the short fibrils (arrows) emerging from intralumenal vesicles (ILVs; arrowheads) in stage I, the parallel arrays of non-pigmented fibrillar sheets in stage II, the melanized fibrils in stage III, and the complete masking of intralumenal contents in stage IV. Scale bar, 200 nm. The figure is adapted from Figures 1 and 3 in (Hurbain et al., 2008), copyright 2008, National Academy of Sciences, U.S.A. A modified form of this figure was also shown in (Watt et al., 2010).
The fibrils begin to form in stage I melanosomes, which are vacuolar structures with few intralumenal membrane vesicles (ILVs; Figure 1) and patches of flat, electron dense clathrin-containing coats on the cytoplasmic leaflet of the limiting membrane (Hurbain et al., 2008; Raposo et al., 2001; Seiji et al., 1963). Unlike later stage melanosomes which are segregated from the endosomal system in melanocytes, stage I melanosomes are accessible to internalized cargoes and correspond to sorting endosomes within the classical endocytic pathway (Raposo et al., 2001). ILVs form by invagination of the endosomal limiting membrane. In vacuolar endosomes of conventional cell types, ubiquitylated growth factor receptors and other cargoes become sequestered on ILVs, which accumulate in late endosomal multivesicular bodies (MVBs) and are eventually degraded upon fusion of the MVBs with lysosomes (Babst, 2011). However, PMEL accumulates on the ILVs in stage I melanosomes (Berson et al., 2001; Raposo et al., 2001) and is destined for fibril formation instead of degradation. The melanosome fibrils begin to form and elongate in association with the ILVs, and as the fibrils grow, the ILVs become “pushed” to the periphery of the organelle (Hurbain et al., 2008). Concomitantly, the fibrils assemble into sheets, generating the classic striated melanosome morphology as observed by electron microscopy and distending the organelle into its ellipsoid shape (Figure 1) (Hurbain et al., 2008). Ellipsoidal melanosomes that are enriched in sheet-like fibrils but lack pigment are referred to as stage II melanosomes, and are no longer accessible to endocytic cargoes – marking the first clear separation between the late endosomal/ lysosomal pathway from the melanosomal pathway (Raposo et al., 2001).
PMEL as the biogenetic component of the fibrils
Orlow and colleagues were the first to characterize components of the “melanosome matrix”. They isolated melanosome-enriched subcellular fractions from melanocytes, and used differential detergent extraction and antibody screening to identify proteolytic fragments of PMEL that were enriched in the insoluble fibrillar fraction (Orlow et al., 1993; Zhou et al., 1994). Later experiments using well-characterized antibodies identified the predominant PMEL species within stage II melanosome-enriched subcellular fractions as corresponding to proteolytic fragments of the lumenal domain (Berson et al., 2003; Harper et al., 2008; Hoashi et al., 2006; Kushimoto et al., 2001; Watt et al., 2009). Using immunoelectron microscopy, Raposo and colleagues showed that PMEL was indeed detected on stage I and II melanosomes and was associated with the fibrils (Raposo et al., 2001), supporting earlier qualitative results (Lee et al., 1996). In addition, PMEL immunodetection was reduced as melanization increased (Raposo et al., 2001), suggesting that PMEL becomes buried in melanins, as had been previously inferred from biochemical analyses (Donatien and Orlow, 1995). Numerous studies have now shown that PMEL expression is both necessary and sufficient for fibril formation. Ectopic expression of PMEL in non-pigment cell types that do not normally express fibrils results in the production of fibrillar arrays that are morphologically similar to those observed in melanocytes (Figure 2a, b) (Berson et al., 2001; Berson et al., 2003), whereas naturally occurring mutations that prevent PMEL delivery to melanosomes (Theos et al., 2006a; Zhou et al., 1994) or loss of PMEL expression by gene ablation (Hellström et al., 2011) result in a loss of fibrils from melanocytes (see Figure 3). Finally, a purified recombinant fragment of PMEL – corresponding to the largest fragment associated with melanosome fibrils (see below) – is capable of very efficiently forming fibrillar structures when allowed to refold in native buffers (Figure 2c) (Fowler et al., 2006; Watt et al., 2009); other fragments of PMEL do so less effectively (McGlinchey et al., 2011; McGlinchey et al., 2009) as discussed later in more detail. Together, these data convincingly show that PMEL is necessary and sufficient to generate, and likely the sole protein component of, melanosome fibrils.
Figure 2. PMEL generates fibrils in melanocytic cells, transfected non-melanocytic cells, and in recombinant form.
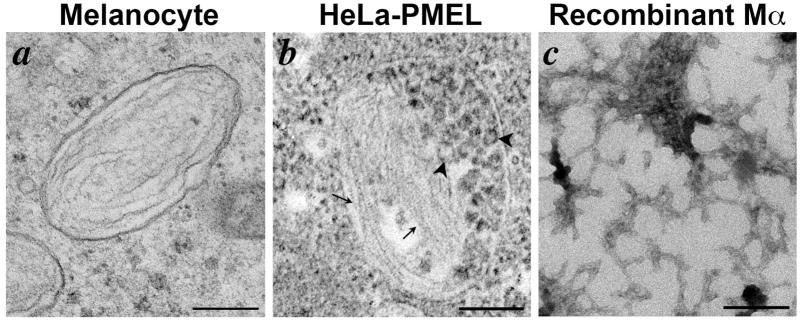
Shown are electron micrographs of fibrils generated by PMEL in three settings. a, thin section of human MNT-1 melanoma cells preserved by high pressure freezing and freeze substitution, highlighting a fibrillar stage II melanosome. b, thin section of transiently transfected non-melanocytic HeLa cells expressing a human PMEL transgene and fixed chemically with glutaraldehyde and formaldehyde. Note the fibrillar arrays (arrows) within a multivesicular body containing numerous intralumenal vesicles (ILVs; arrowheads). c, whole mount analysis of fibrils generated by dilution of purified recombinant PMEL-Mα fragment (tagged at the C-terminus with hexahistidine and purified from bacterial inclusion bodies by affinity chromatography in 6M guanidine-HCl) into a physiological buffer at neutral pH. Note the lattice-like fibrillar network. Scale bars, 200 nm. Panel a is from Figure 3 in (Hurbain et al., 2008), copyright 2008, National Academy of Sciences, U.S.A. Panel b is from (Berson et al., 2003), copyright 2003, The Rockefeller University Press. A modified form of this figure was shown in (Watt et al., 2010).
Figure 3. PMEL is necessary for fibril formation and melanosome shape.
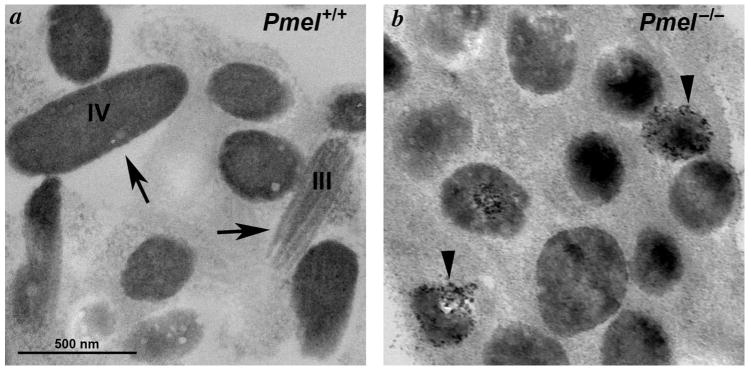
Shown are electron microscopy analyses of primary melanocytes from C57BL/6 wild-type mice (a, Pmel+/+) and Pmel−/− mice (b), showing the morphology of the melanosomes in the presence and absence of PMEL. Note the loss of the oblong shape and the fibrillar morphology, and the irregular deposition of melanin deposits in melanosomes from Pmel−/− melanocytes. Similar data were shown in (Hellström et al., 2011).
PMEL fibrils are amyloid
The melanosome fibrils are insoluble in non-ionic detergents and are highly stable (Berson et al., 2003; Hoashi et al., 2006; Orlow et al., 1993). Moreover, as will be discussed later, PMEL maturation to the fibrillar form requires a number of proteolytic processing steps from an integral membrane precursor (Berson et al., 2001; Berson et al., 2003; Kummer et al., 2009; Kushimoto et al., 2001; van Niel et al., 2011). The similarity of these characteristics with those of the formation of Aβ amyloid from the amyloid precursor protein (APP) in Alzheimer disease was noted early on (Berson et al., 2003; Kelly and Balch, 2003). Accordingly, Fowler et al (Fowler et al., 2006) showed that isolated bovine retinal melanosomes bound to amyloidogenic dyes in a pattern that overlapped with that of labeling for PMEL. Moreover, they showed that fibrils consisting of recombinant PMEL fragments isolated from bacteria were amyloid based on binding to amyloidogenic dyes, characteristic X-ray diffraction, circular dichroism and attenuated total reflectance Fourier transform infrared spectra, and electron microscopy analyses (Fowler et al., 2006). These data strongly support the notion that PMEL is a functional amyloid protein.
What function do the fibrils serve?
PMEL is an extraordinarily well conserved protein; for example, human and teleost fish orthologues share >40% amino acid identity throughout most domains (Theos et al., 2005b). What is the evolutionary advantage for the melanocyte to promote the formation of a potentially hazardous protein aggregate?
One possible function for the fibrils is to sequester highly reactive oxidative intermediates that are generated during melanin synthesis (Simon et al., 2009). Such intermediates could potentially oxidize and thus damage melanosome protein contents, melanosome membrane integrity, and/or cytosolic contents within melanocytes. In support of a role in melanin sequestration, PMEL has been shown to accelerate the polymerization of the intermediates 5,6-dihydroxyindole-2-carboxylic acid and 5,6-dihydroxyindole into melanins (Chakraborty et al., 1996; Fowler et al., 2006; Lee et al., 1996). Intriguingly, Fowler and colleagues noted the chemical similarity of these melanin intermediates with the amyloid dye thioflavin T, and found that several amyloid fibrils, but not non-amyloid collagen fibrils, were similarly capable of accelerating the conversion of 5,6-dihydroxyindole to insoluble melanin (Fowler et al., 2006). These studies suggest that the PMEL amyloid fold might have evolved to detoxify oxidative intermediates and thereby protect melanocytes from oxidative damage. Consistent with this notion, the hypomorphic PMEL mutant silver mouse, which expresses a truncated form of PMEL that is unable to form fibrils due to protein mislocalization (Martínez-Esparza et al., 1999; Theos et al., 2006a), shows premature graying of coat color on some backgrounds (Dunn and Thigpen, 1930), correlating with reduced survival of hair bulb melanocytes (Quevedo et al., 1981). A strain on cell viability upon loss of fibrils is further supported by delayed growth in culture of immortalized melanocytes derived from silver mice relative to wild-type immortalized melanocytes (Spanakis et al., 1992), although it is not clear whether this phenotype is a direct result of the silver mutation, a consequence of an undefined stress induced by cell culture, or whether it is replicated in Pmel−/− melanocytes (Hellström et al., 2011)). Complete loss of PMEL expression in Pmel−/− mice also causes a modest dilution of coat color pigmentation and impaired integrity of the melanosomal membrane in electron microscopy analyses (Figure 3) (Hellström et al., 2011). In both silver and Pmel−/− mice, the loss of coat color is more pronounced in the background of mutations at the Tyrp1 locus, encoding the melanogenic enzyme TYRP1 (Dunn and Thigpen, 1930; Hellström et al., 2011; Lamoreux et al., 2010), perhaps suggesting that PMEL is most effective in detoxifying intermediates that accumulate in the absence of TYRP1. Finally, a recent study has shown that polymorphisms in the PMEL promoter region are associated with vitiligo in Chinese populations and with reduced PMEL expression in vitiligous lesions (Tang et al., 2012). Together, these data support the notion that PMEL fibrils protect melanocytes from toxicity.
A second potential function for the PMEL fibrils is to concentrate melanins to facilitate intracellular and intercellular transport. For example, aggregation of melanin polymers onto a network of PMEL fibrillar sheets might facilitate efficient melanin transfer from epidermal melanocytes to neighboring keratinocytes by at least one potential mechanism. The mechanism by which melanin transfer occurs is debated (Van Den Bossche et al., 2006), but one model proposes that melanins are secreted – by fusion of the melanosome limiting membrane with the melanocyte plasma membrane – into the tightly enclosed space between the melanocyte dendrite and the keratinocyte; subsequent engulfment of free melanin by the keratinocyte would then complete the transfer. By this mechanism, aggregation of melanins into a single large particle would provide a more efficient package for phagocytic uptake than would endocytosis of many small, irregular-sized aggregates (Watt et al., 2010). Moreover, the presence of a protease-resistant amyloid core (Schraermeyer and Dohms, 1996) might make this structure more stable within keratinocytes. In the retinal pigment epithelia, melanosomes temporarily fuse with phagosomes that harbor internalized outer photoreceptor segments, likely providing a means to scavenge free radicals from oxidized photoreceptor membranes (Schraermeyer, 1995; Schraermeyer et al., 1999). In these cells, aggregation of melanins into a large particle with a sheet-like matrix would allow for rapid reformation of melanosomes following fusion with phagolysosomes. Consistently, melanosomes in the retinal pigment epithelia are more severely compromised than those in skin melanocytes of Pmel−/− mice (Hellström et al., 2011).
One might have imagined PMEL fibrils playing a similar role in regulating organelle segregation during transient interactions between endosomes and maturing melanosomes within melanocytes (Delevoye et al., 2009). However, the effective segregation of melanosomal and lysosomal cargoes in melanocytes from silver (Theos et al., 2006a) and Pmel−/− (Hellström et al., 2011) mice suggests that this is not the case, and that PMEL is not required in general to segregate the melanosomal and endosomal systems.
PMEL structure/ function relationships
PMEL is a type I transmembrane glycoprotein, meaning that it is anchored to membranes by a single membrane spanning domain with its bulky, glycosylated N-terminal region facing the extracellular space/ lumen of organelles and its short C-terminus facing the cytoplasm. In humans, alternative splicing leads to the generation of four distinct primary PMEL protein products, the largest of which consists of 645 residues (reviewed in (Theos et al., 2005b)). For the purpose of this review we will focus on the long form. The large lumenal domain can be divided into a short signal peptide, which is excised from the mature protein, and four major subdomains based on primary sequence and homology to known structural domains (Figure 4a). Other groups have subdivided the lumenal domain into additional subdomains (Hoashi et al., 2006), but we will use the simpler architecture that we originally proposed (Theos et al., 2005b).
Figure 4. PMEL Structure and proteolytic processing during fibril formation.

a, schematic structure of PMEL showing the individual domain structure, glycosylation of the “mature” form, and major cleavage products. NTR, PKD, RPT and KLD domains are defined in the text; SP, signal peptide; Cyt., cytoplasmic domain. The transmembrane domain (light blue) is indicated by association with the lipid bilayer. The two products of PC cleavage, Mα and Mβ, are indicated, as is the CTF; cleavage sites are indicated by dotted red lines. N-linked oligosaccharides are indicated as green branched structures; O-linked oligosaccharides in the RPT domain are indicated by green unbranched structures; sialic acid residues are indicated in red. Amino acid residue numbers are shown and correspond to those of the longest human PMEL isoform. b, model of the PMEL PKD domain, threaded onto the structure of the PKD domain of the Saccharophagus degradans cellulose-binding protein using the Phyre2 server (Kelley and Sternberg, 2009). The image was generated using MacPyMOL (http://www.pymol.org/). The N- and C-terminal residues within the model are highlighted in red, the single conserved cysteine is highlighted in yellow, and the four residues corresponding to those deleted in the Smoky chicken PMEL allele are highlighted in blue. c, proteolytic processing and maturation of PMEL prior to fibril formation, showing the P1, P2, Mα-Mβ, isoforms and subsequent cleavage products CTF and MβN. Colors of domains are as in panel a. Cleavage by PC and S2P are indicated by red arrows. d, model for PMEL domain composition within fibrils and fibrillar sheets in stage I and II melanosomes. Colors of domains are as in panel a. Note the emergence of fibrils from small ILVs (pale yellow). e, cleavages and model for assembly of PMEL fragments into fibrils in stage I melanosomes. Mα fragments separate from MβN, and oligomerize. The oligomers are further processed into MαN, PKD/NTR and MαC fragments; note that the precise sites of cleavage have not yet been determined, and it is not yet clear whether NTR-derived fragments are present within the fibrils. In this hypothetical model, PKD-NTR fragments form the core of the fibril and MαC fragments assemble onto them. These fibrils would then assemble laterally into sheets in stage II melanosomes.
The N-terminal region (NTR) immediately follows the signal peptide (SP) – which is removed cotranslationally by signal peptidase – and spans amino acid residues 25–216. The NTR lacks homology to known protein domains, except for a similarly placed region in a homologous protein, GPNMB (Theos et al., 2005b). The NTR contains three highly conserved consensus N-glycosylation sites and three cysteine residues that might participate in disulfide bonding. C-terminal to the NTR is a domain of ~90 amino acids with homology to a repeated domain in the Polycystic Kidney Disease associated protein, polycystin 1 (PKD1), and hence referred to as the PKD domain. This domain lacks glycosylation sites and is predicted to adopt a β-sheet conformation like other characterized PKD domains (Figure 4b) (Bycroft et al., 1999). A single highly conserved cysteine residue is present on the last predicted β-sheet strand. A short linker region between the NTR and the PKD domain is required for efficient PMEL folding, trafficking and amyloid formation (Leonhardt et al., 2010). Following and partially overlapping with the PKD domain is the repeat (RPT) domain, which consists of a varying number (10 in the most prominent human PMEL isoforms) of imperfect direct repeats of a 13-residue sequence rich in glutamic acid, proline, and serine/ threonine residues. This domain is highly modified by O-glycosylation in the mature protein (Harper et al., 2008; Valencia et al., 2007).
The major fibrillogenic proteolytic fragment of PMEL, Mα (see next section), comprises the NTR, PKD and RPT domains (Figure 4a, c). The remaining region of the lumenal domain contains a cysteine-rich region referred to as the Kringle-like domain (KLD) because the spacing of the cysteine residues resembles that of the Kringle cysteine knot structure that often functions in protein-protein interactions (Cao et al., 2002). The KLD also harbors an N-linked glycosylation site that is essential for effective folding and secretion (Hoashi et al., 2010) and additional cysteine residues that might form disulfide bonds with the NTR or PKD cysteines. The KLD is flanked by linker regions that have been referred to as GAP2 and GAP3 by Hearing and colleagues (Hoashi et al., 2006). GAP3 links the KLD with the 26-residue transmembrane domain, which is followed by a 45-residue cytoplasmic domain. The NTR, PKD and KLD domains are highly conserved throughout vertebrate evolution (Theos et al., 2005b), and although the sequence of the RPT, transmembrane, and cytoplasmic domains is not as conserved, the general features of these domains are (Theos et al., 2005b).
How does each of these domains contribute to PMEL function? Their role in PMEL trafficking and fibril formation in vivo has been addressed by a number of studies in which PMEL variants lacking distinct domains have been expressed in HeLa cells or other non-pigment cell types. In such cells, the trafficking features of PMEL (described more below) are largely conserved and PMEL is capable of forming fibrils (albeit with less efficiency than in melanocytic cells and not appropriately compartmentalized). By this approach, the PKD domain is essential for proper PMEL processing and intracellular trafficking to ILVs of premelanosome-like multivesicular endosomes, both prerequisites for fibril formation (Hoashi et al., 2006; Theos et al., 2006b). Moreover, the RPT domain is dispensable for localization to multivesicular endosomes but is required in HeLa cells – and likely other non-pigment cell types such as CHO – for the generation of amyloid fibrils within these compartments (Hoashi et al., 2006; Theos et al., 2006b). O-glycosylation of the RPT domain has also been suggested to be required for fibril formation (Valencia et al., 2007), supported by the lysosomal degradation of PMEL expressed in CHO cell variants that fail to extend O-linked oligosaccharides (Harper et al., 2008). The functions of the NTR and KLD in trafficking and fibril formation are more controversial, likely due to the use of distinct domain boundaries in different studies that might impact protein folding (e.g. see (Leonhardt et al., 2010)). The NTR is clearly important for ultimate fibril formation, and at least part of the KLD is required for protein folding (Hoashi et al., 2006; Theos et al., 2006b). One caveat of these studies is that fibrillar melanosomes do not segregate from the endocytic pathway in non-pigment cells, and so fibrils form and accumulate aberrantly within multivesicular late endosomes (Berson et al., 2001) (see Figure 2b). This might influence the ability of protofibrils to resist proteolysis or to assemble into sheets. The use of a PMEL-negative human melanoma cell line for such studies (Leonhardt et al., 2011; Leonhardt et al., 2010) will likely circumvent this problem in future analyses.
Two groups have used bacterially expressed PMEL fragments to define the subdomains that are capable of forming amyloid. Consistent with the requirement for the RPT domain in amyloid formation in vivo, McGlinchey et al. showed that prolonged incubation of a recombinant peptide corresponding to a His6-tagged human PMEL RPT domain resulted in amyloid fibril formation under acidic conditions (McGlinchey et al., 2009); at neutral pH, RPT fibrils failed to form and preformed fibrils dissolved (McGlinchey et al., 2009; Pfefferkorn et al., 2010). Similar results were obtained with PMEL RPT domains from other species, despite a lack of primary sequence conservation (McGlinchey et al., 2011), and NMR spectroscopy revealed a parallel β-sheet structure, similar to that of Aβ amyloid, composed of non-uniform RPT segments (Hu et al., 2011; McGlinchey et al., 2011). While these authors argued that pH-dependent formation of RPT domain amyloid was consistent with the acid pH of melanosomes, the data fail to account for a number of features of melanosome biology that raise concerns about the significance of the RPT as the PMEL amyloid core in vivo. First, the recombinant form of the RPT domain isolated from bacteria lacks the extensive O-linked glycosylation that modifies this domain in vivo and that seems to be required for amyloid fibril accumulation in mammalian cells (Harper et al., 2008; Valencia et al., 2007). The modification of serine and threonine residues by these highly charged oligosaccharide side chains would likely interfere with the assembly of a closely packed cross-beta sheet that forms the structural basis for amyloid. Second, pH increases as melanosomes mature (Raposo et al., 2001), and TYR is largely inactive at acid pH (Wang and Hebert, 2006) suggesting that mature melanosomes might be near neutral pH; hence, the dissolution of RPT fibrils at neutral pH would likely destroy the fibrils before they were significantly bound to melanins. Third, fibrils can be isolated from subcellular fractions of early stage melanosomes at neutral pH (Berson et al., 2003; Kushimoto et al., 2001; Watt et al., 2009), indicating that they do not dissolve like the RPT fibrils do in vitro. Finally, the kinetics of recombinant RPT fibril formation are seemingly incompatible with the rapid kinetics of melanosome fibril formation in vivo; it takes several weeks to form fibrils initially, and several days to form fibrils if “seeded” with preformed fibrils (McGlinchey et al., 2009). Thus, while an interesting in vitro experimental observation, it seems unlikely that the RPT domain forms the core of the PMEL amyloid.
By contrast, Watt et al found that His6-tagged recombinant fragments corresponding to the NTR and PKD domain both had amyloid properties in vitro that were more consistent with experimental observations in vivo (Watt et al., 2009). Like full-length recombinant Mα (Fowler et al., 2006; Watt et al., 2009), these fragments isolated under denaturing conditions from bacteria formed amyloid within seconds to minutes of dilution into non-denaturing buffers of varying pH, as evaluated by amyloid dye binding, detergent insolubility, X-ray diffraction and electron microscopy analyses. Under similar conditions, the RPT domain did not form amyloid (Watt et al., 2009). Moreover, whereas deletion of the PKD or NTR domains from Mα diminished amyloid dye binding, deletion of the RPT domain did not. Protease-resistant fragments obtained from Mα fibrils contained regions corresponding to the NTR and/or PKD domain, whereas the RPT domain was completely digested by limited protease treatment. Unlike the RPT domain fibrils characterized by McGlinchey and colleagues, the NTR and PKD fibrils were stable at neutral pH, and the morphology of the PKD fibrils resembled those of amyloid protofibrils observed in stage I premelanosomes (Watt et al., 2009). Finally, a fragment of the PKD domain was identified in detergent-insoluble fibril fractions isolated from melanocytic cells (van Niel et al., 2011; Watt et al., 2009). These data support the notion that the PKD and/or NTR domains form the PMEL amyloid core in vivo. Because the PKD domain is unmodified by glycosylation in vivo and is predicted to fold into a beta sheet-rich conformation that might be primed for incorporation into the cross β-sheet amyloid conformation (Figure 4b) (Greenwald and Riek, 2010), we favor the model that the PKD is the physiological core of PMEL and that interactions with regions of the NTR might facilitate a conformational shift to the amyloid form. Rather than functioning in amyloid assembly per se, acidification of early stage melanosomes is needed for the proteolytic processing events that generate the amyloidogenic Mα precursor (Berson et al., 2001) (see below) and we speculate might be additionally required for local PKD domain unfolding to initiate amyloid formation – a function that is not needed for the bacterial recombinant proteins that are isolated under denaturing conditions. We further speculate that the RPT domain functions in regulating the timing of this unfolding event and might additionally participate in regulating the assembly of fibrils into sheets and protecting them from lysosomal proteases within mature melanosomes.
PMEL synthesis, glycosylation and proteolytic processing
To avert toxicity, functional amyloid must be formed under tightly controlled conditions. In order to gain insight into these circumstances and when and how PMEL undergoes its transformation from a typical transmembrane glycoprotein to an insoluble amyloid, it is essential to understand the mechanisms controlling PMEL biosynthesis, maturation and trafficking.
PMEL is co-translationally translocated into the endoplasmic reticulum (ER) and modified by signal peptide cleavage and by addition of four N-linked core oligosaccharides (Berson et al., 2001; Kwon et al., 1987; Maresh et al., 1994a; Maresh et al., 1994b), generating a “precursor 1” (P1) form (Figure 4c). PMEL is slowly exported from the ER, likely due to slow folding of the large lumenal domain (Berson et al., 2001); consequently, the unmodified P1 form is the most prominent form detected by immunoblotting using antibodies to the N- or C-termini (Berson et al., 2001; Harper et al., 2008; Kushimoto et al., 2001). PMEL exit is facilitated by a C-terminal valine residue (Theos et al., 2006a) that likely engages the COPII coat machinery for formation of Golgi-bound vesicles (Nufer et al., 2002). In the Golgi, the core N-linked oligosaccharides are further modified, such that the oligosaccharide on N81 is maintained in high mannose form and the others are modified to the complex type (Berson et al., 2001; Hoashi et al., 2010; Maresh et al., 1994b). In addition, the central “RPT” region of PMEL (see below) is modified extensively by terminally sialylated O-linked oligosaccharides (Figure 4a, c) (Harper et al., 2008; Valencia et al., 2007). Depending on the sialic acid linkages, these latter modifications make mammalian forms of PMEL detectable by the widely used monoclonal antibody HMB45 (Chiamenti et al., 1996; Harper et al., 2008; Valencia et al., 2007). The fully modified full-length form of PMEL has been referred to as precursor 2 (P2; Figure 4c).
P2 is short-lived, and is subject to a number of proteolytic cleavages that generate the “mature” forms of PMEL (Figure 4c–e). P2 is proteolytically cleaved within an acidic compartment – either the trans Golgi network (TGN) (Leonhardt et al., 2011) or early endosomes (Theos et al., 2006b), perhaps depending on the cell line analyzed. The cleavage occurs at a dibasic Lys-Arg sequence within the PMEL lumenal domain by furin or a related protease of the proprotein convertase (PC) family of enzymes (Berson et al., 2001; Berson et al., 2003). This produces two fragments, a large lumenal mature α (Mα) fragment and a smaller integral membrane mature β (Mβ fragment), that remain at least temporarily tethered by disulfide bonds (Figure 4a, c) (Berson et al., 2001; Berson et al., 2003). The dibasic residues on Mα are removed, likely by carboxypeptidase E (Fricker, 1988), to generate the Mα form that has been sequenced (Maresh et al., 1994b). Because Mα remains covalently bound to the integral membrane Mβ subunit, PC cleavage is not sufficient to release the fibrillogenic Mα fragment from the membrane to allow for fibril formation. A second proteolytic cleavage in the lumenal juxtamembrane region liberates Mα and the bound lumenal region of Mβ (MβN) from the membrane (Kummer et al., 2009). This cleavage is mediated by a “site 2 protease” (S2P; Figure 4c), occurs independently of but subsequent to PC cleavage (Kummer et al., 2009) (and our unpublished data), and in transfected HeLa cells requires a metalloproteinase such as a disintegrin and metalloproteinase (ADAM) 10 or ADAM17 (Kummer et al., 2009). It is not yet clear whether the ADAMs are also required for site 2 cleavage in melanocytic cells or whether they mediate site 2 cleavage directly. Cleavage of PMEL by both the PC and S2P is required for Mα release and fibril formation; inhibition of either cleavage results in the formation of non-fibrillar aggregates and loss of amyloid fibrils (Berson et al., 2003; Kummer et al., 2009). Interestingly, juxtamembrane cleavage is also required for the secretion of a small fraction of PMEL (Berson et al., 2001; Esclamado et al., 1986; Maresh et al., 1994a; Maresh et al., 1994b; Vennegoor et al., 1988), most of which is cleaved by the PC and includes Mα bonded to MβN (Hoashi et al., 2010). The sequence requirements within the PMEL juxtamembrane region for ectodomain shedding/ secretion and S2P cleavage/ fibril formation are distinct (Hoashi et al., 2010; Kummer et al., 2009), suggesting that they are mediated by distinct proteases and/or occur in distinct compartments.
While S2P cleavage liberates Mα-MβN fragments into the lumen of premelanosomes to initiate fibril formation, it also generates a membrane-bound C-terminal fragment or CTF (Figure 4c) (Kummer et al., 2009). The CTF is a substrate for yet another protease, γ-secretase (Figure 4c) (Kummer et al., 2009; van Niel et al., 2011). γ-secretase is a multi-subunit intramembrane protease that cleaves transmembrane domain-containing substrates with a short lumenal extension – such as those produced by sheddases – at a site within their membrane-spanning segments (Prox et al., 2012). This liberates short intracellular fragments, many of which are involved in intracellular signaling pathways (Fortini, 2002). In the case of PMEL, cleavage by γ-secretase is required for CTF degradation (Kummer et al., 2009; van Niel et al., 2011) and its segregation from the melanosomal pathway into the degradative late endosomal/ lysosomal system (van Niel et al., 2011). While the cytoplasmic fragments of other γ-secretase substrates have been shown to initiate signal transduction cascades (Fortini, 2002), there is no current evidence to suggest a role for the PMEL intracellular fragment in cell signaling. Other melanosomal proteins including TYR, TYRP1 and DCT are also targets of γ-secretase, and loss of γ-secretase function results in TYR mistrafficking and loss of pigmentation (Wang et al., 2006).
The progressive cleavage of PMEL by a PC, S2P and γ-secretase is reminiscent of the series of proteolytic cleavages of APP by BACE1 and γ-secretase that generate the amyloidogenic Aβ fragment in Alzheimer Disease (Prox et al., 2012). Indeed, proteolysis-mediated amyloidogenic conversion is observed in many pathologic amyloidogenic proteins, such as for gelsolin in familial amyloidosis of Finnish type (Chen et al., 2001) and perhaps for α-synucleins in Parkinson Disease (Choi et al., 2011; Kim et al., 2003; Levin et al., 2009), and in functional amyloids such as polypeptide pro-hormones in mammals (Maji et al., 2009). Proteolysis by similar classes of proteases can also interfere with amyloid formation, as exemplified by the cleavage of APP (Prox et al., 2012) and PrPc (Vincent et al., 2001) to non-amyloidogenic fragments by ADAM10. Thus, proteolysis to fibrillogenic fragments – or proteolytic degradation of such fragments – is a commonly used theme in regulating amyloid formation. Nonetheless, given that appropriately cleaved secreted PMEL Mα fragments are isolated from soluble fractions of cell supernatants (Hoashi et al., 2010; Maresh et al., 1994b), proteolytic processing is necessary but not sufficient to drive PMEL amyloid conversion.
While intact Mα fragments are fibrillogenic and can be detected within fibril-enriched subcellular fractions (Berson et al., 2003; Harper et al., 2008; Kummer et al., 2009), the major PMEL fragments detected in these fractions from pigment cells are products of further proteolytic maturation (Figure 4d–e) (Chiamenti et al., 1996; Harper et al., 2008; Hoashi et al., 2006; Kushimoto et al., 2001; Watt et al., 2009). These fragments are also observed when PMEL is ectopically expressed in non-pigment cells (Harper et al., 2008; Hoashi et al., 2006) and likely arise by Mα proteolysis by lysosomal proteases which are present within melanosomes (Diment et al., 1995; Novikoff et al., 1968; Raposo et al., 2001). While this proteolytic maturation does not seem to be required to form amyloid protofibrils in vitro (Watt et al., 2009), it might be required for the formation of fully mature fibrils and the proper lateral association of fibrils into sheets (Figure 4d–e) (Hurbain et al., 2008).
PMEL trafficking to premelanosomes
The sequence of PMEL processing described above reflects the segregation of processing enzymes and ultimate ordered oligomerization of PMEL into fibrils within sequential compartments of the secretory and endocytic pathways (Figure 5). PMEL synthesis, core N-linked oligosaccharide addition and trimming, and disulfide bond formation occur in the ER, and O-linked oligosaccharide addition and processing of N- and O-linked oligosaccharides occur in the Golgi and TGN, as for all known glycoproteins of the secretory and endocytic pathways. The site of PC cleavage is debated. Our group showed that PC cleavage of PMEL in MNT-1 human melanoma cells or transfected non-pigment HeLa cells is inhibited by agents that disrupt acidification (Berson et al., 2001) and can be mediated by furin only when it is massively overexpressed (Theos et al., 2006b). Furin is normally restricted to the TGN, but when overexpressed a substantial cohort is found in endosomes (Bosshart et al., 1994). Moreover, surface expressed PMEL in MNT-1 or HeLa cells was not PC modified (our unpublished results), and PMEL mutants that accumulate at the plasma membrane of HeLa cells (see below) are largely uncleaved (Theos et al., 2006a). By contrast, Leonhardt and colleagues reported conflicting results, suggesting that secretory forms or surface-exposed PMEL were PC cleaved in a different human melanoma cell line (Leonhardt et al., 2011). It is not yet clear whether these conflicting results reflect differing experimental approaches or cell-type specific differences. Nonetheless, PC cleavage clearly precedes further maturation in premelanosomes.
Figure 5. PMEL trafficking to stage I and II melanosomes.

a, schematic of the trafficking pathway for PMEL from its site of synthesis in the endoplasmic reticulum (ER) through the Golgi and trans Golgi network (TGN) and the plasma membrane, ultimately to stage I and II melanosomes. Red arrows depict trafficking steps, and PMEL domains are colored as in Figure 4. The predominant processed PMEL isoform is indicated within each organelle. The blue arrow indicated PMEL CTF trafficking to lysosomes. Also shown are the clathrin-associated adaptors AP-2 – which facilitates PMEL endocytosis from the plasma membrane – and AP-1 and AP-3 – which associate with separate domains of early endosomes/ stage I melanosomes. Tyrosinase and Tyrp1 are delivered to maturing stage III and IV melanosomes via transport carriers that emerge from these domains. b, enlarged schematic of stage I melanosomes emphasizing processing of PMEL by S2P and subsequent ESCRT-independent sorting of (i) PMEL-Mα to ILVs, from which fibrils emanate, and (ii) CTF to an ESCRT- and γ-secretase dependent degradation pathway in lysosomes.
From the TGN, PMEL accumulates in early endosomes/ stage I premelanosomes (Raposo et al., 2001), but likely does so by clathrin-mediated endocytosis after first traversing the plasma membrane (Figure 5). PMEL is robustly internalized from the plasma membrane (Chen et al., 2012; Theos et al., 2006a) by virtue of a di-leucine-based consensus internalization signal in the cytoplasmic domain (Theos et al., 2006a). This signal likely mediates internalization by binding to the AP-2 plasma membrane clathrin adaptor, as AP-2 depletion results in PMEL accumulation at the plasma membrane and interferes with its localization to melanosomes (Robila et al., 2008). Similarly, a natural truncation of the PMEL cytoplasmic domain in the silver mouse (Martínez-Esparza et al., 1999) blocks internalization (Theos et al., 2006a) and a similar truncation introduced into human PMEL blocks localization to endosomes (Lepage and Lapointe, 2006). Whereas ectopically expressed PMEL in HeLa cells accesses early endosomes effectively by an independent pathway that does not require cytoplasmic domain targeting sequences (Theos et al., 2006b), such a pathway is likely not prominent in melanocytes, as melanosomes in silver melanocytes (Theos et al., 2006a) or AP-2-depleted melanoma cells (Robila et al., 2008) are severely depleted of PMEL. In wild-type melanocytes, the subsequent accumulation of PMEL in vacuolar endosomes/ stage I melanosomes (Raposo et al., 2001) likely explains the cofractionation of PMEL with AP-1 adaptors (Valencia et al., 2006), which themselves are localized primarily to early endosomes in melanocytes; depletion of AP-1 had no effect on PMEL maturation or fibril formation (Delevoye et al., 2009).
Within vacuolar domains of early endosomes, PMEL is preferentially sorted to and becomes enriched in ILVs (Figure 5) (Berson et al., 2001; Raposo et al., 2001; Theos et al., 2006b; van Niel et al., 2011). Only the Mα fragment that has been segregated from CTF fragments preferentially accumulates on the ILVs, as is evident from the paucity of labeling for the PMEL C-terminus on ILVs (Raposo et al., 2001; van Niel et al., 2011). Whereas ILV sorting for cargoes such as activated epidermal growth factor receptors has been extensively studied and shown to be dependent on cargo ubiquitylation and sequential cargo hand-off by a series of ESCRT complexes (Babst, 2011; Hanson and Cashikar, 2012), selective accumulation of PMEL on ILVs is independent of both ubiquitylation and the ESCRT machinery (Theos et al., 2006b; Truschel et al., 2009). Rather, Mα associates with ILVs using a mechanism that is dependent on its PKD and NTR domains (Theos et al., 2006b) and the tetraspanin protein, CD63 (Figure 5) (van Niel et al., 2011). Interestingly, whereas Mα associates with ILVs in an ESCRT-independent manner, CTF fragments are degraded in a lysosomal pathway that requires the ESCRT machinery (van Niel et al., 2011). CD63 seems to be critical to segregate Mα and CTF into distinct sorting pathways since depletion of CD63 from MNT-1 melanoma cells results in the ESCRT-dependent degradation of both domains (van Niel et al., 2011). CD63 likely functions to generate microdomains on the limiting membrane of vacuolar endosomes that favor PMEL-Mα sequestration and subsequent ILV formation.
As discussed earlier, fibrils begin to emanate and elongate from the ILVs within stage I melanosomes (Figures 4d, 5) (Hurbain et al., 2008). ILVs are essential for fibril formation, as the impaired ILV formation induced by CD63 depletion completely abrogates amyloidogenesis (van Niel et al., 2011) and PMEL deletion mutants that fail to associate with ILVs also cannot form fibrils (Theos et al., 2006b) (although interpretation of this latter result is complicated by their inefficient proteolytic maturation). How association with ILVs favors amyloid conversion of Mα is not clear. It is possible that interactions of lumenal subdomains with lipid headgroups that are enriched on ILVs induces local unfolding that promotes amyloid conversion, as has been previously shown in vitro for amyloid conversion of the prion protein (Wang et al., 2007) and islet amyloid polypeptide (Jean et al., 2010). Another possibility is that separation of PMEL-Mα from the MβN fragment is sufficient to drive the amyloid conversion, and that the ILVs merely serve as a mechanism to enhance this separation. Indeed, either the continued association of Mα with MβN (Hoashi et al., 2010) or the absence of association with ILVs (our unpublished data) might explain the solubility of secreted PMEL-Mα. Finally, it remains possible that an as yet undiscovered conversion factor associates constitutively with the ILVs. Resolution of these mechanisms will require in vitro reconstitution with purified components in their native state. The maturation of the fibrils to sheets within premelanosomes correlates with a loss of ILVs, likely by their fusion with the limiting membrane, and with maturation to stage II melanosomes (Hurbain et al., 2008). Protofibrils obtained with recombinant PMEL lumenal domain do not form the fibrillar sheets, suggesting that factors associated with melanosome maturation, including low lumenal pH, ILV association or proteolytic maturation, are necessary for lateral assembly of the fibrils. Intriguingly, this maturation step also correlates with the segregation of the melanosome pathway from the endosomal pathway, as stage II melanosomes are no longer accessible to endocytosed cargoes (Raposo et al., 2001). However, PMEL is not required for the segregation of these two pathways, as melanocytes derived from Pmel−/− or silver mice that lack melanosomal PMEL or fibrils altogether nevertheless segregate melanosomal cargoes from late endosomal/ lysosomal cargoes (Hellström et al., 2011; Theos et al., 2006a). How this segregation is established and how both stage II melanosomes and late endosomes derive from functionally and morphologically similar compartments is still not clear. It is possible that the small melanosomal protein, MART-1, and the G protein-coupled receptor, OA1, play roles in this segregation (Aydin et al., 2012; Giordano et al., 2009; Giordano et al., 2011; Hoashi et al., 2005).
Averting amyloid toxicity
How does PMEL amyloid avoid the toxicity associated with pathological amyloid? Several features of the PMEL life cycle might contribute to its lack of toxicity. One likely important feature is the sequestration of PMEL amyloid formation within stage I melanosomes. This compartmentalization might prevent the exposure of potentially harmful amyloidogenic intermediates from cellular proteins that might otherwise be subject to denaturation by exposed hydrophobic interactions (Klein et al., 2001). A related mechanism might be the sequestration of amyloidogenic PMEL-Mα to the ILVs; this would allow specific concentration of the amyloidogenic species to favor fibril elongation, and exclude the majority of other cellular proteins that do not specifically concentrate on ILVs. Besides compartmentation, PMEL might be non-toxic due to the kinetics of amyloid conversion. Denatured recombinant PMEL-Mα, NTR and PKD fragments form amyloid almost immediately upon dilution into native buffers, and much more rapidly than conventional pathological amyloids (Fowler et al., 2006; Watt et al., 2009). If transient denaturation and rapid amyloid formation similarly occurs in vivo, as suggested by the rapid incorporation of the Mα fragment into detergent insoluble fractions in melanocytes, it would minimize exposure of potentially toxic oligomeric intermediates (perhaps amyloid seeds) and sequester amyloidogenic species into non-toxic fibrils. A third mechanism to avert toxicity might be the “protection” of a PKD/NTR domain amyloid core by binding to the highly hydrophilic RPT domain region (Figure 4d, e). The RPT clearly associates with fibrils in cells (Kushimoto et al., 2001; Raposo et al., 2001), and might prevent association of other lumenal contents with the growing fibrils. Lastly, lateral association of the fibrils into sheets (Hurbain et al., 2008) might also protect the amyloid core from association with other lumenal contents and avert toxicity. These potential detoxification mechanisms are not mutually exclusive, and might cooperate with each other in vivo to minimize potential damage of PMEL amyloid.
If mechanisms are in place to avert PMEL-associated toxicity, one would predict that mutations that interfere with these mechanisms might result in pathology. Indeed, studies of naturally occurring PMEL mutations in animals such as chicken (Kerje et al., 2004), horse (Brunberg et al., 2006), cattle (Jolly et al., 2008; Kühn and Weikard, 2007; Schmutz and Dreger, 2012), dog (Clark et al., 2006), and perhaps zebrafish (Schonthaler et al., 2005) are associated with more severe hypopigmentation than that observed in Pmel−/− mice and suggest that aberrant fibril formation might be deleterious. These mutations vary in nature from point mutations and small deletions/insertions to large truncations, and their phenotypic consequences range in severity from mild hypopigmentation, such as in smoky chickens (Kerje et al., 2004), to a complete loss of eumelanin in Dominant White chickens (Keeling et al., 2004; Kerje et al., 2004) and Silver horses (Brunberg et al., 2006). The latter two severe phenotypes are inherited as dominant mutations, suggesting a gain-of-function loss of pigmentation, and at least the Dominant White mutation (found in White Leghorn chickens in the United States) is associated with a loss of pigmented melanocytes in feathers (Hamilton, 1940). Interestingly, both Dominant White chickens and Silver horses have mutations in or near the PMEL transmembrane domain (Brunberg et al., 2006; Kerje et al., 2004), far from the amyloidogenic domains. Analyses of epitope-tagged Dominant White chicken PMEL (Kuliawat and Santambrogio, 2009) or of human PMEL engineered with an orthologous mutation (hPMELinsWAP; (Watt et al., 2011) expressed in non-pigment cells shows that the Dominant White mutation does not influence PMEL maturation, processing or trafficking, and surprisingly has at most a minor effect on association with distinct membrane microdomains. Moreover, hPMELinsWAP is capable of forming fibrils when expressed in HeLa cells (Watt et al., 2011). However, both hPMELinsWAP and human PMEL with the Silver horse mutation induce aberrantly assembled fibrillar sheets and a loss of pigmentation when expressed in melanocytes (Watt et al., 2011) (Figure 6). This phenotype correlates with altered biophysical properties of the PMEL transmembrane domain (Watt et al., 2011). Interestingly, the mildly hypopigmenting smoky allele arose as a revertant of the severely hypopigmented Dominant White allele and disrupts the PKD domain (Figure 4b; deleted amino acids in smoky are highlighted) (Kerje et al., 2004); engineering this mutation into hPMELinsWAP blocks trafficking to fibril-forming compartments and renders PMEL incapable of forming fibrils (Watt et al., 2011). This indicates that the negative consequences of the Dominant White mutation are blocked by interfering with fibril formation, and support the notion that this and other dominant mutations generate fibrils or fibril intermediates with toxic properties.
Figure 6. The Dominant White PMEL variant generates aberrant fibrils.

Immunoelectron microscopy analysis of mouse melanocytes expressing wild-type human PMEL (wild-type; a, b) or PMEL-TMinsWAP, harboring an insertion in the transmembrane domain identical to that of the chicken Dominant White allele [TMinsWAP (Dom. White); c, d]. Ultrathin cryosections were immunogold labeled for human PMEL. Note the effective formation of fibrils within stage I melanosomes by both isoforms (a, c). Whereas wild-type human PMEL becomes incorporated into mature fibrillar sheets that are quickly coated with melanins and inaccessible to antibodies in stage II/III melanosomes (b), the TMinsWAP variant forms sheets in stage II-like melanosomes that do not become melanized and that remain accessible to antibody labeling (d). Adapted from Figure 6 of (Watt et al., 2011).
Perspectives
Our extensive understanding of PMEL trafficking and processing and the availability of both loss-of-function and gain-of-function mutants makes PMEL an outstanding model to understand the generation of functional amyloid in mammals and to differentiate this process from the generation of pathological amyloid. While we have learned a great deal over the last 12 years, a number of key questions remain. The most fundamental questions revolve around the conformational change that initiates amyloid formation within stage I melanosomes. How is this conformational change initiated? Is protein unfolding a key step, and if so, what induces unfolding? What is the role of the ILVs – do they template amyloid conversion, provide a source of PMEL monomers for amyloid fibril elongation, or merely serve as a convenient site for fibril accumulation? What are the mechanisms leading to the formation of the ILVs and the accumulation of PMEL-Mα on them, and how do these mechanisms rely on CD63? How do the tertiary and quaternary structures of PMEL on precursor membranes contribute to the conformational change, and what are the respective roles of the different subdomains? Other questions revolve around how the amyloid “switch” is prevented in earlier secretory compartments prior to arrival at the stage I melanosome. Is PMEL in some way “tethered” to block the exposure of the amyloidogenic domain, and is the tether released by proteolytic processing? Does the KLD contribute to tethering the amyloidogenic domain until its removal in stage I melanosomes? What is the role of the RPT domain and its sialylated O-linked glycans? Finally, the new finding that several PMEL mutants might generate pathological amyloid warrants further investigation. Why are these mutants deleterious to pigment cells or pigmentation – is there a change in the kinetics of amyloid formation or in the lateral assembly of fibrils into sheets? Does it affect melanin binding to the matrix? What is the toxic species – the pre-amyloid oligomers or the mature amyloid fibrils? Are they actually toxic to the survival of melanocytes, and if so, why? Rather, do they merely disturb the integrity of the melanosome and interfere with melanization, and if so, how? Resolution of these issues will require a number of new experimental systems, including (1) better in vitro reconstitution systems with correctly modified and folded PMEL isoforms, (2) live cell imaging modalities, such as FRET and FLIM, to measure changes in inter-domain contacts during the progression of PMEL from the Golgi to stage I/ II melanosomes, (3) imaging methods such as RUSH (Boncompain et al., 2012) to follow PMEL from its site of synthesis to stage II melanosomes or to monitor the segregation of stage I/ II melanosomes from the endocytic pathway, and (4) transgenic animal models to study the impact of natural PMEL mutants. Further analyses of this fascinating pigment cell-specific protein are sure to provide new avenues of research and new insights into amyloidoses.
Acknowledgments
The authors would like to thank lab members past and present who have contributed to our understanding of PMEL, and we apologize to authors whose work we neglected to cite. We particularly thank Kathryn Ferguson (University of Pennsylvania, Philadelphia, PA) for assistance with molecular modeling of the PMEL PKD domain in Figure 4. This work was supported by National Institutes of Health grants R01 AR048155 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and F31 GM081917 and T32 GM007229 from the National Institute of General Medical Sciences, Fondation ARC pour la Recherche sur le Cancer, Institut Curie and Centre National de la Recherche Scientifique.
References
- Aydin IT, Hummler E, Smit NP, Beermann F. Coat color dilution in mice because of inactivation of the melanoma antigen MART-1. Pigment Cell Melanoma Res. 2012;25:37–46. doi: 10.1111/j.1755-148X.2011.00910.x. [DOI] [PubMed] [Google Scholar]
- Babst M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr Opin Cell Biol. 2011;23:452–457. doi: 10.1016/j.ceb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Harper D, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–533. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbeck MSC. Electron microscopy of melanocytes: the fine structure of hair-bulb premelanosomes. Annals NY Acad Sci. 1963;100:540–547. doi: 10.1111/j.1749-6632.1963.tb42871.x. [DOI] [PubMed] [Google Scholar]
- Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. 2012;20:66–73. doi: 10.1016/j.tim.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F. Synchronization of secretory protein traffic in populations of cells. Nat Methods. 2012;9:493–498. doi: 10.1038/nmeth.1928. [DOI] [PubMed] [Google Scholar]
- Bosshart H, Humphrey J, Deignan E, Davidson J, Drazba J, Yuan L, Oorschot V, Peters PJ, Bonifacino JS. The cytoplasmic domain mediates localization of furin to the trans-Golgi network en route to the endosomal/lysosomal system. J Cell Biol. 1994;126:1157–1172. doi: 10.1083/jcb.126.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunberg E, Andersson L, Cothran G, Sandberg K, Mikko S, Lindgren G. A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet. 2006;7:46. doi: 10.1186/1471-2156-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN, Linke RP. A molecular history of the amyloidoses. J Mol Biol. 2012;421:142–159. doi: 10.1016/j.jmb.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL, Chothia C. The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J. 1999;18:297–305. doi: 10.1093/emboj/18.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Cao R, Veitonmäki N. Kringle structures and antiangiogenesis. Curr Med Chem Anticancer Agents. 2002;2:667–681. doi: 10.2174/1568011023353705. [DOI] [PubMed] [Google Scholar]
- Chakraborty AK, Platt JT, Kim KK, Kwon BS, Bennett DC, Pawelek JM. Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur J Biochem. 1996;236:180–188. doi: 10.1111/j.1432-1033.1996.t01-1-00180.x. [DOI] [PubMed] [Google Scholar]
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, JHS Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Huff ME, Matteson J, Page LJ, Phillips R, Kelly JW, Balch WE. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca2+ stabilization. EMBO J. 2001;20:6277–6287. doi: 10.1093/emboj/20.22.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chalouni C, Tan C, Clark R, Venook R, Ohri R, Raab H, Firestein R, Mallet W, Polakis P. The melanosomal protein PMEL17 as a target for antibody drug conjugate therapy in melanoma. J Biol Chem. 2012;287:24082–24091. doi: 10.1074/jbc.M112.361485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamenti AM, Vella F, Bonetti F, Pea M, Ferrari S, Martignoni G, Benedetti A, Suzuki H. Anti-melanoma monoclonal antibody HMB-45 on enhanced chemiluminescence-western blotting recognizes a 30–35 kDa melanosome-associated sialated glycoprotein. Melanoma Res. 1996;6:291–298. doi: 10.1097/00008390-199608000-00003. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Choi DH, Kim YJ, Kim YG, Joh TH, Beal MF, Kim YS. Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J Biol Chem. 2011;286:14168–14177. doi: 10.1074/jbc.M111.222430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LA, Wahl JM, Rees CA, Murphy KE. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci USA. 2006;103:1376–1381. doi: 10.1073/pnas.0506940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, Giordano F, Marks MS, Raposo G. The biogenesis of melanosomes. In: Borovansky J, Riley P, editors. Melanins & Melanosomes: Biosynthesis, Biogenesis, Physiological, and Pathological Functions. Weinheim, Germany: Wiley-VCH Verlag and Co; 2011. pp. 247–294. [Google Scholar]
- Delevoye C, Hurbain I, Tenza D, Sibarita JB, Uzan-Gafsou S, Ohno H, Geerts WJC, Verkleij AJ, Salamero J, Marks MS, et al. AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J Cell Biol. 2009;187:247–264. doi: 10.1083/jcb.200907122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diment S, Eidelman M, Rodriguez GM, Orlow SJ. Lysosomal hydrolases are present in melanosomes and are elevated in melanizing cells. J Biol Chem. 1995;270:4213–4215. doi: 10.1074/jbc.270.9.4213. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Donatien PD, Orlow SJ. Interaction of melanosomal proteins with melanin. Eur J Biochem. 1995;232:159–164. doi: 10.1111/j.1432-1033.1995.tb20794.x. [DOI] [PubMed] [Google Scholar]
- Dunn LC, Thigpen LW. The silver mouse: a recessive color variation. J Heredity. 1930;21:495–498. [Google Scholar]
- Esclamado RM, Gown AM, Vogel AM. Unique proteins defined by monoclonal antibodies specific for human melanoma. Am J Surg. 1986;152:376–385. doi: 10.1016/0002-9610(86)90308-9. [DOI] [PubMed] [Google Scholar]
- Fortini ME. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673–684. doi: 10.1038/nrm910. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Fricker LD. Carboxypeptidase E. Annu Rev Physiol. 1988;50:309–321. doi: 10.1146/annurev.ph.50.030188.001521. [DOI] [PubMed] [Google Scholar]
- Furumura M, Sakai C, Potterf SB, Vieira WD, Barsh GS, Hearing VJ. Characterization of genes modulated during pheomelanogenesis using differential display. Proc Natl Acad Sci USA. 1998;95:7374–7378. doi: 10.1073/pnas.95.13.7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano F, Bonetti C, Surace EM, Marigo V, Raposo G. The Ocular Albinism type 1 (OA1) G-protein coupled receptor functions with MART-1 at early stages of melanogenesis to control melanosome identity and composition. Hum Mol Genet. 2009;18:4530–4545. doi: 10.1093/hmg/ddp415. [DOI] [PubMed] [Google Scholar]
- Giordano F, Simoes S, Raposo G. The ocular albinism type 1 (OA1) GPCR is ubiquitinated and its traffic requires endosomal sorting complex responsible for transport (ESCRT) function. Proc Natl Acad Sci USA. 2011;108:11906–11911. doi: 10.1073/pnas.1103381108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Hamilton H. A study of the physiological properties of melanophores with special reference to their role in feather coloration. Anat Rec. 1940;78:525–548. [Google Scholar]
- Hanson P, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol. 2012;28:337–362. doi: 10.1146/annurev-cellbio-092910-154152. [DOI] [PubMed] [Google Scholar]
- Harper DC, Theos AC, Herman KE, Tenza D, Raposo G, Marks MS. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of pmel17 that have been proteolytically processed in endosomes. J Biol Chem. 2008;283:2307–2322. doi: 10.1074/jbc.M708007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström AR, Watt B, Fard SS, Tenza D, Mannström P, Narfström K, Ekesten B, Ito S, Wakamatsu K, Larsson J, et al. Inactivation of the Pmel gene alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011;7:e1002285. doi: 10.1371/journal.pgen.1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirobe T. Origin of melanosome structures and cytochemical localizations of tyrosinase activity in differentiating epidermal melanocytes of newborn mouse skin. J Exp Zool. 1982;224:355–363. doi: 10.1002/jez.1402240308. [DOI] [PubMed] [Google Scholar]
- Hoashi T, Muller J, Vieira WD, Rouzaud F, Kikuchi K, Tamaki K, Hearing VJ. The repeat domain of the melanosomal matrix protein Pmel17/gp100 is required for the formation of organellar fibers. J Biol Chem. 2006;281:21198–22208. doi: 10.1074/jbc.M601643200. [DOI] [PubMed] [Google Scholar]
- Hoashi T, Tamaki K, Hearing VJ. The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J. 2010;24:916–930. doi: 10.1096/fj.09-140921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoashi T, Watabe H, Muller J, Yamaguchi Y, Vieira WD, Hearing VJ. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J Biol Chem. 2005;280:14006–14016. doi: 10.1074/jbc.M413692200. [DOI] [PubMed] [Google Scholar]
- Hu KN, McGlinchey RP, Wickner RB, Tycko R. Segmental polymorphism in a functional amyloid. Biophys J. 2011;101:2242–2450. doi: 10.1016/j.bpj.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Sarangarajan R, Strovel E, Zho Y, Gahl WA, Boissy RE. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell. 2001;12:2075–2085. doi: 10.1091/mbc.12.7.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurbain I, Geerts WJC, Boudier T, Marco S, Verkleij A, Marks MS, Raposo G. Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc Natl Acad Sci USA. 2008;105:19726–19731. doi: 10.1073/pnas.0803488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iconomidou VA, Vriend G, Hamodrakas SJ. Amyloids protect the silkmoth oocyte and embryo. FEBS Lett. 2000;479:141–145. doi: 10.1016/s0014-5793(00)01888-3. [DOI] [PubMed] [Google Scholar]
- Jean L, Lee CF, Lee C, Shaw M, Vaux DJ. Competing discrete interfacial effects are critical for amyloidogenesis. FASEB J. 2010;24:309–317. doi: 10.1096/fj.09-137653. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Wills JL, Kenny JE, Cahill JI, Howe L. Coat-colour dilution and hypotrichosis in Hereford crossbred calves. New Zealand Vet J. 2008;56:74–77. doi: 10.1080/00480169.2008.36812. [DOI] [PubMed] [Google Scholar]
- Keeling L, Andersson L, Schutz KE, Kerje S, Fredriksson R, Carlborg O, Cornwallis CK, Pizzari T, Jensen P. Feather-pecking and victim pigmentation. Nature. 2004;431:645–646. doi: 10.1038/431645a. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJE. Protein structure prediction on the web: a case study using the Phyre server. Nat Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kelly JW, Balch WE. Amyloid as a natural product. J Cell Biol. 2003;161:461–462. doi: 10.1083/jcb.200304074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JM, Knight D, Wise MJ, Vollrath F. Amyloidogenic nature of spider silk. Eur J Biochem. 2002;269:4159–4163. doi: 10.1046/j.1432-1033.2002.03112.x. [DOI] [PubMed] [Google Scholar]
- Kerje S, Sharma P, Gunnarsson U, Kim H, Bagchi S, Fredriksson R, Schutz K, Jensen P, von Heijne G, Okimoto R, et al. The Dominant white, Dun and Smoky color variants in chicken are associated with insertion deletion polymorphisms in the PMEL17 gene. Genetics. 2004;168:1507–1518. doi: 10.1534/genetics.104.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Sung JY, Um JW, Hattori N, Mizuno Y, Tanaka K, Paik S, Kim J, Chung KC. Parkin cleaves intracellular alpha-synuclein inclusions via the activation of calpain. J Biol Chem. 2003;278:41890–41899. doi: 10.1074/jbc.M306017200. [DOI] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Kühn C, Weikard R. An investigation into the genetic background of coat colour dilution in a Charolais × German Holstein F2 resource population. Animal Genet. 2007;38:109–113. doi: 10.1111/j.1365-2052.2007.01569.x. [DOI] [PubMed] [Google Scholar]
- Kuliawat R, Santambrogio L. A mutation within the transmembrane domain of melanosomal protein Silver (Pmel17) changes lumenal fragment interactions. Eur J Cell Biol. 2009;88:653–667. doi: 10.1016/j.ejcb.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Maruyama H, Huelsmann C, Baches S, Weggen S, Koo EH. Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J Biol Chem. 2009;284:2296–2306. doi: 10.1074/jbc.M808904200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushimoto T, Basrur V, Valencia J, Matsunaga J, Vieira WD, Ferrans VJ, Muller J, Appella E, Hearing VJ. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc Natl Acad Sci USA. 2001;98:10698–10703. doi: 10.1073/pnas.191184798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Halaban R, Kim GS, Usack L, Pomerantz S, Haq AK. A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol Biol Med. 1987;4:339–355. [PubMed] [Google Scholar]
- Lamoreux ML, Delmas V, Larue L, Bennett DC. The Colors of Mice: A Model Genetic Network. Oxford, UK: Wiley-Blackwell; 2010. [Google Scholar]
- Lee ZH, Hou L, Moellmann G, Kuklinska E, Antol K, Fraser M, Halaban R, Kwon BS. Characterization and subcellular localization of human Pmel 17/silver, a 100-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J Invest Dermatol. 1996;106:605–610. doi: 10.1111/1523-1747.ep12345163. [DOI] [PubMed] [Google Scholar]
- Leonhardt RM, Vigneron N, Rahner C, Cresswell P. Proprotein convertases process Pmel17 during secretion. J Biol Chem. 2011;286:9321–9337. doi: 10.1074/jbc.M110.168088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt RM, Vigneron N, Rahner C, Van den Eynde BJ, Cresswell P. Endoplasmic reticulum (ER)-export, subcellular distribution and fibril formation by PMEL17 requires an intact N-terminal domain junction. J Biol Chem. 2010;285:16166–16683. doi: 10.1074/jbc.M109.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepage S, Lapointe R. Melanosomal targeting sequences from gp100 are essential for MHC class II–restricted endogenous epitope presentation and mobilization to endosomal compartments. Cancer Res. 2006;66:2423–2432. doi: 10.1158/0008-5472.CAN-05-2516. [DOI] [PubMed] [Google Scholar]
- Levin J, Giese A, Boetzel K, Israel L, Högen T, Nübling G, Kretzschma rH, Lorenzl S. Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp Neurol. 2009;215:201–208. doi: 10.1016/j.expneurol.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Lopes VS, Wasmeier C, Seabra MC, Futter CE. Melanosome maturation defect in Rab38-deficient retinal pigment epithelium results in instability of immature melanosomes during transient melanogenesis. Mol Biol Cell. 2007;18:3914–3927. doi: 10.1091/mbc.E07-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan MR, Li L, Choi EM, Kannan K, Guo F, et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell. 2012;148:515–529. doi: 10.1016/j.cell.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Marken JS, Neubauer M, Aruffo A, Hellström I, Hellström KE, Marquardt H. Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA and Cell Biology. 1994a;13:87–95. doi: 10.1089/dna.1994.13.87. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Wang WC, Beam KS, Malacko AR, Hellström I, Hellström KE, Marquardt H. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch Biochem Biophys. 1994b;311:95–102. doi: 10.1006/abbi.1994.1213. [DOI] [PubMed] [Google Scholar]
- Martínez-Esparza M, Jiménez-Cervantes C, Bennett DC, Lozano JA, Solano F, García-Borrón JC. The mouse silver locus encodes a single transcript truncated by the silver mutation. Mammalian Genome. 1999;10:1168–1171. doi: 10.1007/s003359901184. [DOI] [PubMed] [Google Scholar]
- McGlinchey RP, Shewmaker F, Hu KN, McPhie P, Tycko R, Wickner RB. The repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J Biol Chem. 2011;286:8385–8393. doi: 10.1074/jbc.M110.197152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey RP, Shewmaker F, McPhie P, Monterroso B, Thurber K, Wickner RB. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc Natl Acad Sci USA. 2009;106:13731–13736. doi: 10.1073/pnas.0906509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer FH. Genetic variations in the fine structure and ontogeny of mouse melanin granules. Am Zoologist. 1966;6:43–66. doi: 10.1093/icb/6.1.43. [DOI] [PubMed] [Google Scholar]
- Novikoff AB, Albala A, Biempica L. Ultrastructural and cytochemical observations on B-16 and Harding-Passey mouse melanomas. The origin of premelanosomes and compound melanosomes. J Histochem Cytochem. 1968;16:299–319. doi: 10.1177/16.5.299. [DOI] [PubMed] [Google Scholar]
- Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J Cell Sci. 2002;115:619–628. doi: 10.1242/jcs.115.3.619. [DOI] [PubMed] [Google Scholar]
- Orlow SJ, Zhou BK, Boissy RE, Pifko-Hirst S. Identification of a mammalian melanosomal matrix glycoprotein. J Invest Dermatol. 1993;101:141–144. doi: 10.1111/1523-1747.ep12363626. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn CM, McGlinchey RP, Lee JC. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc Natl Acad Sci USA. 2010;107:21447–21452. doi: 10.1073/pnas.1006424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prox J, Rittger A, Saftig P. Physiological functions of the amyloid secretases ADAM10, BACE1, and presenlilin. Exp Brain Res. 2012;217:331–341. doi: 10.1007/s00221-011-2952-0. [DOI] [PubMed] [Google Scholar]
- Puri N, Gardner JM, Brilliant MH. Aberrant pH of melanosomes in Pink-Eyed Dilution (p) mutant melanocytes. J Invest Dermatol. 2000;115:607–613. doi: 10.1046/j.1523-1747.2000.00108.x. [DOI] [PubMed] [Google Scholar]
- Quevedo WC, Fleischmann RD, Dyckman J. Premature loss of melanocytes from hair follicles of light (Blt) and silver (si) mice. In: Seiji M, editor. Phenotypic expression in pigment cells. Tokyo: Tokyo Univ. Press; 1981. pp. 177–184. [Google Scholar]
- Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–823. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robila V, Ostankovitch M, Altrich-Vanlith ML, Theos AC, Drover S, Marks MS, Restifo N, Engelhard VH. MHC class II presentation of gp100 epitopes in melanoma cells requires the function of conventional endosomes and is influenced by melanosomes. J Immunol. 2008;181:7843–7852. doi: 10.4049/jimmunol.181.11.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz SM, Dreger DL. Interaction of MC1R and PMEL alleles on solid coat colors in Highland cattle. Anim Genet. 2012 doi: 10.1111/j.1365–2052.2012.02361. In press. [DOI] [PubMed] [Google Scholar]
- Schonthaler HB, Lampert JM, von Lintig J, Schwarz H, Geisler R, Neuhauss SC. A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev Biol. 2005;284:421–436. doi: 10.1016/j.ydbio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Schraermeyer U. Transport of endocytosed material into melanin granules in cultured choroidal melanocytes of cattle--new insights into the relationship of melanosomes with lysosomes. Pigment Cell Res. 1995;8:209–214. doi: 10.1111/j.1600-0749.1995.tb00665.x. [DOI] [PubMed] [Google Scholar]
- Schraermeyer U, Dohms M. Detection of a fine lamellar gridwork after degradation of ocular melanin granules by cultured peritoneal macrophages. Pigment Cell Res. 1996;9:248–254. doi: 10.1111/j.1600-0749.1996.tb00114.x. [DOI] [PubMed] [Google Scholar]
- Schraermeyer U, Peters S, Thumann G, Kociok N, Heimann K. Melanin granules of retinal pigment epithelium are connected with the lysosomal degradation pathway. Exp Eye Res. 1999;68:237–245. doi: 10.1006/exer.1998.0596. [DOI] [PubMed] [Google Scholar]
- Seiji M, Fitzpatrick TM, Simpson RT, Birbeck MSC. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature. 1963;197:1082–1084. doi: 10.1038/1971082a0. [DOI] [PubMed] [Google Scholar]
- Setty SRG, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature. 2008;454:1142–1146. doi: 10.1038/nature07163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JD, Peles D, Wakamatsu K, Ito S. Current challenges in understanding melanogenesis: bridging chemistry, biological control, morphology and function. Pigment Cell Melanoma Res. 2009;22:563–579. doi: 10.1111/j.1755-148X.2009.00610.x. [DOI] [PubMed] [Google Scholar]
- Sitaram A, Marks MS. Mechanisms of protein delivery to melanosomes in pigment cells. Physiology. 2012;27:85–99. doi: 10.1152/physiol.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram A, Piccirillo R, Palmisano I, Harper DC, Dell’Angelica EC, Schiaffino MV, Marks MS. Localization to mature melanosomes by virtue of cytoplasmic dileucine motifs is required for human OCA2 function. Mol Biol Cell. 2009;20:1464–1477. doi: 10.1091/mbc.E08-07-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanakis E, Lamina P, Bennett DC. Effects of the developmental colour mutations silver and recessive spotting on proliferation of diploid and immortal mouse melanocytes in culture. Development. 1992;114:675–680. doi: 10.1242/dev.114.3.675. [DOI] [PubMed] [Google Scholar]
- Tang XF, Zhang Z, Hu DY, Xu AE, Zhou HS, Sun LD, Gao M, Gao TW, Gao XH, Chen HD, et al. Association analyses identify three susceptibility loci for vitiligo in the Chinese Han population. J Invest Dermatol. 2012 doi: 10.1038/jid.2012.320. In press. [DOI] [PubMed] [Google Scholar]
- Theos AC, Berson JF, Theos SC, Herman KE, Harper DC, Tenza D, Sviderskaya EV, Lamoreux ML, Bennett DC, Raposo G, et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol Biol Cell. 2006a;17:3598–3612. doi: 10.1091/mbc.E06-01-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Tenza D, Martina JA, Hurbain I, Peden AA, Sviderskaya EV, Stewart A, Robinson MS, Bennett DC, Cutler DF, et al. Functions of AP-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 2005a;16:5356–5372. doi: 10.1091/mbc.E05-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Truschel ST, Raposo G, Marks MS. The Silver locus product Pmel17/ gp100/ Silv/ ME20: Controversial in name and in function. Pigment Cell Res. 2005b;18:322–336. doi: 10.1111/j.1600-0749.2005.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Truschel ST, Tenza D, Hurbain I, Harper DC, Berson JF, Thomas PC, Raposo G, Marks MS. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev Cell. 2006b;10:343–354. doi: 10.1016/j.devcel.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truschel ST, Simoes S, Setty SRG, Harper DC, Tenza D, Thomas PC, Herman KE, Sackett SD, Cowan DC, Theos AC, et al. ESCRT-1 function is required for Tyrp1 transport from early endosomes to the melanosome limiting membrane. Traffic. 2009;10:1318–1336. doi: 10.1111/j.1600-0854.2009.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol. 2002;56:703–741. doi: 10.1146/annurev.micro.56.013002.100603. [DOI] [PubMed] [Google Scholar]
- Valencia JC, Rouzaud F, Julien S, Chen KG, Passeron T, Yamaguchi Y, Abu-Asab M, Tsokos M, Costin GE, Yamaguchi H, et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J Biol Chem. 2007;282:11266–11280. doi: 10.1074/jbc.M608449200. [DOI] [PubMed] [Google Scholar]
- Valencia JC, Watabe H, Chi A, Rouzaud F, Chen KG, Vieira WD, Takahashi K, Yamaguchi Y, Berens W, Nagashima K, et al. Sorting of Pmel17 to melanosomes through the plasma membrane by AP1 and AP2: evidence for the polarized nature of melanocytes. J Cell Sci. 2006;119:1080–1091. doi: 10.1242/jcs.02804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bossche K, Naeyaert JM, Lambert J. The quest for the mechanism of melanin transfer. Traffic. 2006;7:769–778. doi: 10.1111/j.1600-0854.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, Marks MS, Rubinstein E, Raposo G. The Tetraspanin CD63 integrates ESCRT-independent and dependent sorting at a common endosome. Dev Cell. 2011;21:708–721. doi: 10.1016/j.devcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennegoor C, Hageman P, van Nouhuijs H, Ruiter DJ, Calafat J, Ringens PJ, Rümke P. A monoclonal antibody specific for cells of the melanocytic lineage. Am J Pathol. 1988;130:179–192. [PMC free article] [PubMed] [Google Scholar]
- Vijayasaradhi S, Doskoch PM, Houghton AN. Biosynthesis and intracellular movement of the melanosomal membrane glycoprotein gp75, the human b (brown) locus product. Exp Cell Res. 1991;196:233–240. doi: 10.1016/0014-4827(91)90256-t. [DOI] [PubMed] [Google Scholar]
- Vijayasaradhi S, Xu YQ, Bouchard B, Houghton AN. Intracellular sorting and targeting of melanosomal membrane proteins: identification of signals for sorting of the human brown locus protein, gp75. J Cell Biol. 1995;130:807–820. doi: 10.1083/jcb.130.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–37746. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- Wang F, Yang F, Hu Y, Wang X, Wang X, Jin C, Ma J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46:7045–7053. doi: 10.1021/bi700299h. [DOI] [PubMed] [Google Scholar]
- Wang N, Hebert DN. Tyrosinase maturation through the mammalian secretory pathway: bringing color to life. Pigment Cell Res. 2006;19:3–18. doi: 10.1111/j.1600-0749.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- Wang R, Tang P, Wang P, Boissy RE, Zheng H. Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer’s disease mutation. Proc Natl Acad Sci USA. 2006;103:353–358. doi: 10.1073/pnas.0509822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B, Raposo G, Marks MS. Pmel17: An amyloid determinant of organelle structure. In: Rigacci S, Bucciantini M, editors. Functional Amyloid Aggregation. Trivandrum, Kerala, India: Research Signpost; 2010. pp. 89–113. [Google Scholar]
- Watt B, Tenza D, Lemmon MA, Kerje S, Raposo G, Andersson L, Marks MS. Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet. 2011;7:e1002286. doi: 10.1371/journal.pgen.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B, van Niel G, Fowler DM, Hurbain I, Luk KC, Stayrook SE, Lemmon MA, Raposo G, Shorter J, Kelly JW, et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J Biol Chem. 2009;284:35543–35555. doi: 10.1074/jbc.M109.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Edskes HK, Roberts BT, Baxa U, Pierce MM, Ross ED, Brachmann A. Prions: proteins as genes and infectious entities. Genes Dev. 2004;18:470–485. doi: 10.1101/gad.1177104. [DOI] [PubMed] [Google Scholar]
- Zhou BK, Kobayashi T, Donatien PD, Bennett DC, Hearing VJ, Orlow SJ. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning”. Proc Natl Acad Sci USA. 1994;91:7076–7080. doi: 10.1073/pnas.91.15.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]