

Abstract
Background: Loss of cardiomyocytes after myocardial infarction (MI) causes heart failure. In this study, we investigate whether the in situ cardiomyocytes can re-enter the cell cycle and to what extent cell division of cardiomyocytes occurs after acute MI (AMI) in rats. Methods: Sprague Dawley (SD) rats were used in this study; the left anterior descending coronary artery was ligated. At time points (3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks) after the operation, five rats were euthanized, respectively. An additional five sham-operated rats serves as a control group and were euthanized at 3 days post-operation. The expressions of cyclin A2, Ki-67, phospho-histone H3 (H3P), and Aurora B in myocardial tissues were detected by Western blot and immunofluorescence. Results: The expression levels of cyclin A2 were significantly higher in all groups with AMI except the 4-week group than those found in the sham-operated group (P < 0.01). The percentage of Ki-67–positive nuclei in the border zones was significantly higher than the percentage in the distant normal myocardium (P < 0.01). Conclusions: our results demonstrate that cardiomyocytes re-enter the cell cycle after AMI and that cyclin A2 is a reliable marker for the detection of cell cycle activity in cardiomyocytes.
Keywords: Acute myocardial infarction, mitosis, cell cycle, cyclin A2
Introduction
Myocardial infarction (MI) is one of the most important ischemic heart diseases. Scarring of the myocardium occurs in the infarction area and the surviving cardiomyocytes become hypertrophic following acute MI (AMI) [1,2]. These processes constitute the cardiac remodeling. The concept that cardiomyocytes cannot divide originated from the difficulty of identifying mitotic figures in vivo and from the inability to induce mitotic division in vitro. Thus, the dogma was introduced that no proliferation of ventricular muscle cells occurs once cell division has ceased, shortly after birth in the mammalian heart. Therefore, many studies focusing on the implantation of stem cells have made some progress for treatment of heart failure; but have still encountered numerous difficulties [3,4]. However, other studies have provided evidence of cardiomyocyte proliferation in the adult heart and especially in the failing human heart [3,4]. It has also been reported that isolated adult cardiomyocytes can re-enter the cell cycle and undergo karyokinesis and cytokinesis under certain conditions [5,6]. If adult cardiomyocytes can really divide effectively, this process may offer a novel way to treat a variety of heart diseases caused by AMI. Cyclin A2 plays a key role in cell cycle regulation that controls both the G1/S transition into DNA synthesis as well as the G2/M entry into mitosis [7,8]. Therapeutic delivery of cyclin A2 induces myocardial regeneration and improves cardiac function following MI [9]. Ki-67 is a vital molecule for cell proliferation that is expressed in proliferating cells during the active cell cycle, but is absent in resting (G0 phase) cells [10]. Both Ki-67 and cyclin A2 are markers of the nuclear synthesis phase of the cell cycle. H3P and Aurora B are mitosis-specific markers that are expressed during karyokinesis and cytokinesis, respectively [5,11]. We test the hypothesis that the in situ cardiomyocytes re-enter the cell cycle and to what extent cell division of cardiomyocytes occurs after AMI in rats by the analysis of these markers.
Materials and methods
Animal model
All animals were housed and handled according to Southeast University Institutional Animal Care and Use Committee guidelines and all animal work was approved by the appropriate committee. The protocol was approved by the local Ethics committee (ethics committee, Southeast University) and all animals received humane care in compliance with “The Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH Publication No. 86-23, revised 1996).
Male Sprague Dawley (SD) rats (n = 25, 8 ± 0.5 weeks old, 210 ± 23 g body weight) were anesthetized with chloral hydrate (320 mg/kg, Sigma-Aldrich, Sheboygan Falls, WI, USA) by intraperitoneal injection, endotracheally intubated with a 14-gauge angiocatheter and mechanically ventilated (tidal volume: 3-4 ml/100 g, frequency: 60 breaths/min). AMI was created by ligation of the left anterior descending coronary artery as described previously [12]. All animals were performed by echocardiography before and after the procedure. Briefly, two-dimensional (2D) guided M-mode echocardiography was conducted in each animal in vivo using a Toshiba PowerVision 6000 ultrasound system (Model SSA-370A, PLM-1204AT 12MHz-transducer) as previously described [13,14], rats were anesthetized by intraperitoneal injection. Chests of the rats were shaved and echocardiography was performed. Diastolic and systolic left ventricle [3] end-diastolic dimension (LVEDD), LV end-systolic dimension (LVESD), and LV Ejection fraction (LVEF) were calculated. AMI was confirmed by echocardiography. Rats were randomized into five groups (each group n = 5) and were euthanized with CO2 according to the time points: 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks post-surgery after echocardiography. An additional sham-operated rat group (n= 5) serves as control group and were euthanized at 3 days following sham-operation. The left LVEF value was measured to assess the severity of the AMI by echocardiography.
Preparation of tissue samples
After 3 day, 1 week, 2 weeks, 3 weeks, and 4 weeks of postoperative echocardiograph, each five rats were euthanized with carbon dioxide (CO2) and heart were resected immediately, respectively. The myocardial samples were obtained and were used for western blot analysis, for histological analysis/immunohistochemistry, and for immunofluorescent staining.
Western blot
Myocardial samples from the border zones and the distant normal myocardium of AMI were harvested at 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks and were frozen immediately in liquid nitrogen. Total protein was isolated from samples using the EpiQuik Nuclear Extraction Kit (Epigentek, Farmingdale, NY, USA), and proteins were separated on a 7.5% gel by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, Life Technologies Corporation, Carlsbad, CA, USA) and transferred to a polyvinylidene difluoride membrane. The membranes were incubated with rabbit anti-cyclin A2 antibody (ab-7956, Abcam, Cambridge, MA, USA) and goat anti-rabbit immunoglobulin G (IgG)-horseradish peroxidase conjugate (GE Healthcare, Life Sciences, Amersham, UK). Targeted immunoreactive proteins were detected by enhanced chemiluminescence (Life Sciences, Amersham, UK) and quantified using ImageJ (Bethesda, MD, National Institutes of Health, USA).
Histological analysis
Hearts were excised with heparin (30 μg/kg, intraperitoneally), weighed and the entire heart from base to apex was cut into 1-mm-thick slices along the short axial plane. The parts of middle capillary muscle were kept in 10% formalin. For histological staining, the slices were further sectioned into 4 μm slides to examine infarct size and its percentile by Masson’s trichrome staining (MTS, Dako North America, Inc., Carpinteria, CA, USA) for assessment of the infarct area. The infarct area was measured on short axial section from the section of slice corresponding to the position of middle papillary muscle of left ventricle [3]. Infarction area percentage was defined as the infarcted area divided by whole LV muscular area using ImageJ (V1.46, NIH Bethesda, MD, USA).
Immunohistochemistry
To detect whether AMI activates the cell cycle, immunohistochemistry analysis was performed for cyclin A2 [7,8]. Paraffin-embedded sections (5µm thickness) were incubated with rabbit anti-cyclin A2 antibody followed by secondary antibody. The sections were then stained with DAB AR1000 (WHIGA, Guangzhou, Guangdong, China). Finally, hematoxylin was used to counterstain the nuclei. The ratio of cyclin A2-positive nuclei to the total number of nuclei was calculated and was averaged over at least five randomly selected fields per specimen.
Immunofluorescence analysis
To determine whether AMI promotes cardiomyocyte proliferation, immunofluorescence analysis was performed for cyclin A2, Ki-67 (Ki-67 antibody, sc-7846-FITC, Santa Cruz Biotech, CA, USA) [10], phospho-histone H3 (phospho-histone H3 antibody, sc-8656-R, Santa Cruz Biotech, CA, USA) and Aurora B (Aurora B antibody, ab2254, Abcam, Cambridge, MA, USA). Frozen sections (5µm thickness) were blocked with 5% normal goat serum and incubated with primary antibodies against one of the above four antigens and α-sarcomeric actin (α-Actin antibody, sc-58670, Santa Cruz Biotech, CA, USA) overnight at 4°C. The sections were then incubated with a secondary antibody mixture containing FITC-conjugated goat anti-rabbit antibody (sc-2012, Santa Cruz Biotech, CA, USA) and PE-conjugated goat anti-mouse antibody (sc-3768, Santa Cruz Biotech, CA, USA) for 1 hr at room temperature. The nuclei were counterstained with DAPI (sc-3598, Santa Cruz Biotech, CA, USA). Subsequently, the specimens were processed for confocal microscopy and examined with a laser scanning confocal microscope (oil lens) (MRC-1000 Confocal Imaging System, Bio-Rad, Hercules CA, USA). The expression data were obtained by counting the number of positive nuclei in five consecutive visual fields (×100, magnification).
Statistical analysis
All data are expressed as mean ± standard deviation (SD). The significance of the differences was determined using Student’s t-test for comparisons of two values and by the Bonferroni method for multiple comparisons. A P value of < 0.05 was considered to be statistically significant, and all analyses were performed with SPSS version 18.0 (SPSS, Chicago, Illinois).
Results
AMI animal model
AMI model was verified by cardiac ultrasound image (Figure 1A and 1B), and re-verified by histopathology at post-operation 1 week (Figure 1C). The LVEF values for these animals was measured at 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks post-surgery (Figure 1D). This LVEF value was significantly lower than that of the sham-operated group, which was 78.31 ± 7.91% (range: 67.13% to 86.27%) (P < 0.001) (Figure 1D). The size of the infarct was measured at 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks post-surgery (Figure 1E).
Figure 1.
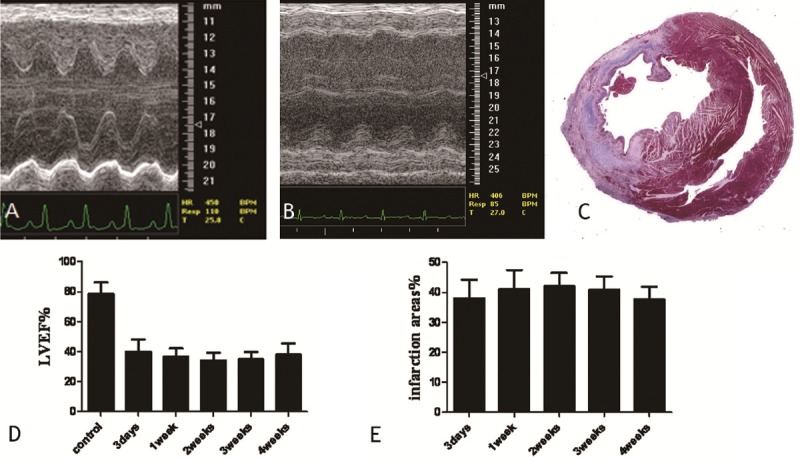
Echocardiography of a normal heart and a heart with AMI. A. a representative echocardiograph from a sham-operated heart; B. a representative echocardiograph from a heart with AMI (1 week after the operation); C. Masson staining to meaure the size of the infarct (1 week after the operation) (red indicates the myofibrils, and blue indicates the collagen fibers); D. The LVEF value at 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks post-surgery. E. The areas of the infarct at 3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks post-surgery.
The expression of cyclin A2
The expression of cyclin A2 was detected in the border zone of AMI with immunohistochemistry and western blot (Figures 2 and 3). The expression levels of cyclin A2 were significantly higher in all groups with AMI except the 4-week group than those found in the sham-operated group (P < 0.01). The expression maximum occurred at 2 weeks after infarction and then declined rapidly. By the fourth week after the operation, there was no significant difference between the operated group and sham-operated group (P > 0.05). At 2 weeks after the operation, the percentage of cyclin A2-positive nuclei in cardiomyocytes from hearts with AMI was 11.46 ± 1.28% in samples from the border zone, which was significantly higher than the percentage observed in the distant normal myocardium. (6.3 ± 0.97%) (P < 0.01). The percentages of cyclin A2-positive nuclei in the border zone and the distant normal myocardium of AMI were significantly higher than those found in the hearts of sham-operated animals (P < 0.001 for both comparisons). Furthermore, an inverse correlation was found between the LVEF value and the number of cyclin A2-positive cell nuclei (Pearson’s product-moment correlation coefficient r = -0.96; P < 0.01) (Figure 4). Subsequently, we detected the expression of cyclin A2 by confocal microscopy at 14 days after AMI and obtained similar results (Figure 5A).
Figure 2.
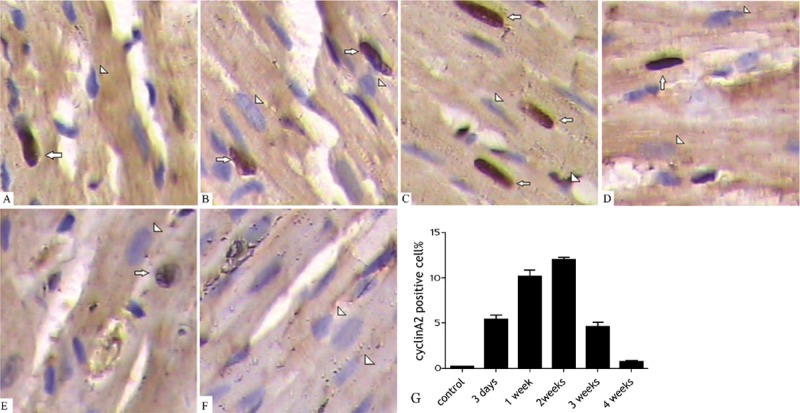
The expression of cyclin A2 in cardiomyocytes (border zones). A. 3 days after operation; B. 1 week after operation; C. 2 weeks after operation; D. 3 weeks after operation; E. 4 weeks after operation; F. The sham-operated group (arrows denote the positive staining, and triangles denote the negative staining; ×40 magnification); G. The percentage of cyclin A2-positive cells (border zone) (mean ± SD).
Figure 3.
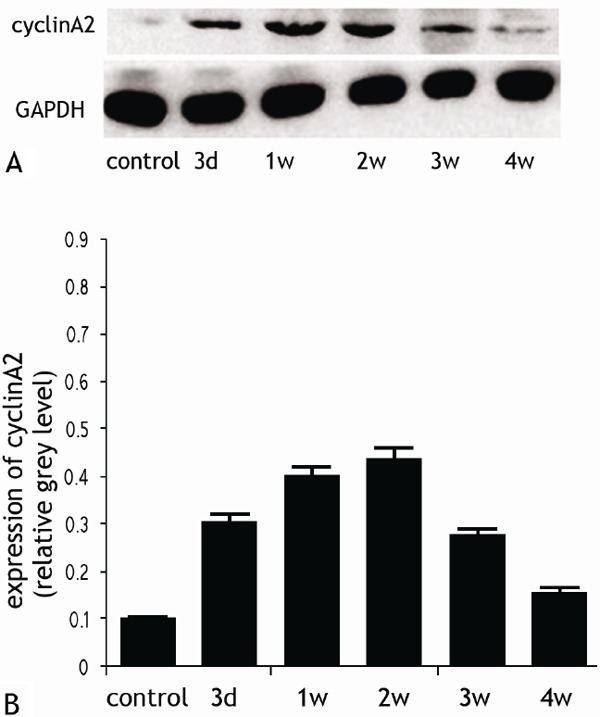
The expression of cyclin A2 in cardiomyocytes by western blotting (A) and quantification (B).
Figure 4.
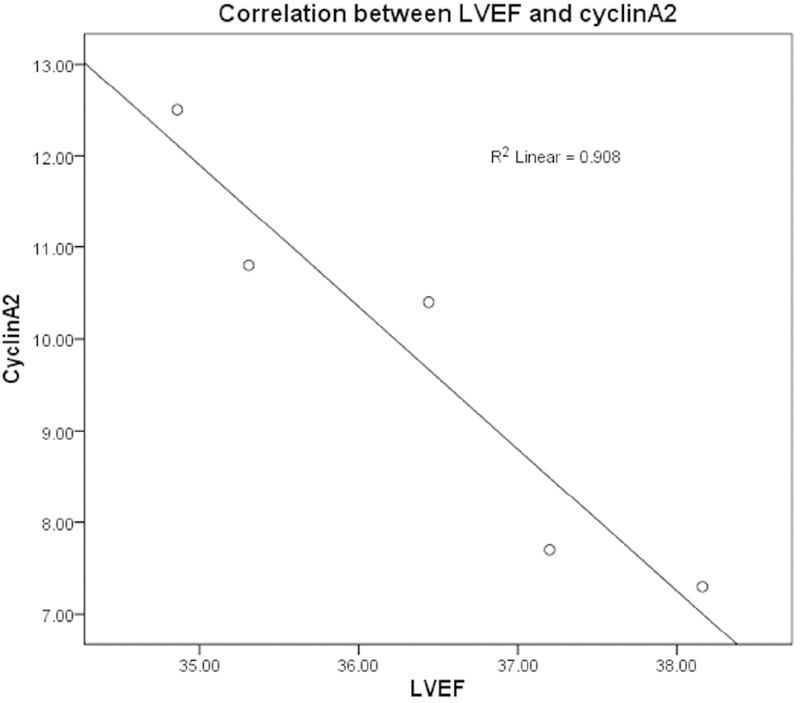
The correlation between LVEF and cyclin A2 at the different time points following AMI.
Figure 5.
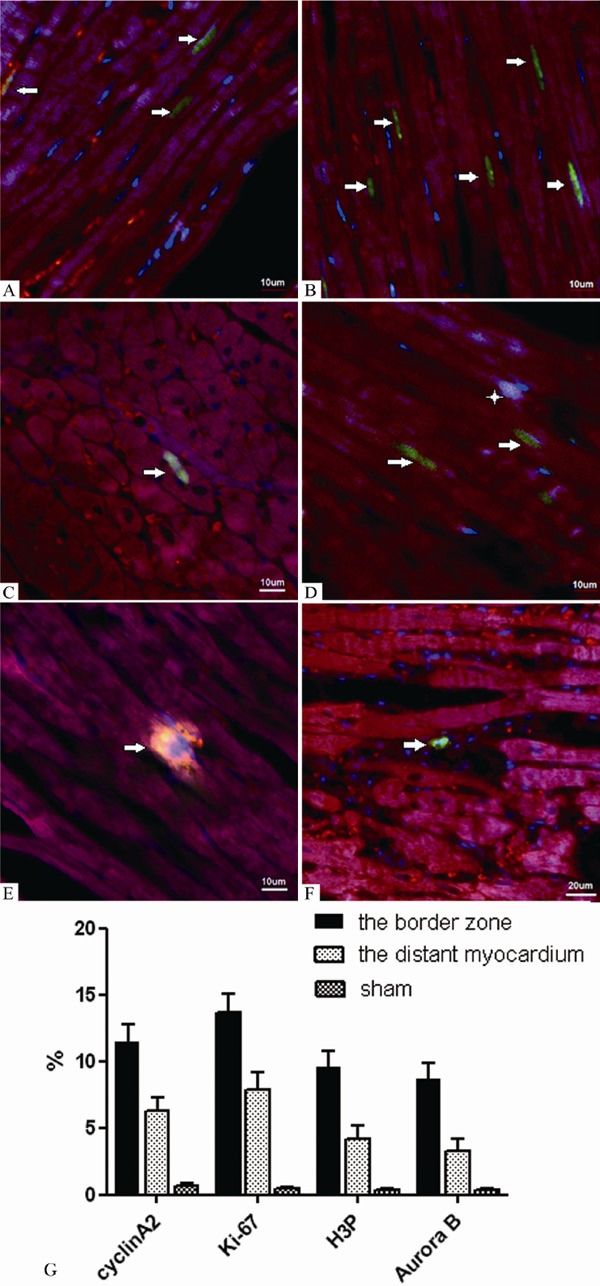
Detection of cyclin A2, Ki-67, H3P and Aurora B expression in cardiomyocytes from infarcted hearts by immunofluorescence analysis (border zone, 2 weeks after infarction) (A-E. ×100 magnification; F. ×60 magnification). Both dark red and red fluorescence show staining of the myocyte cytoplasm by the sarcomeric α-actin antibody, while blue fluorescence shows the nuclei. In panel A, the green fluorescence documents the localization of cyclin A2 in the nuclei (arrows). In panel B, the green fluorescence documents the localization of Ki-67 in the nuclei (arrows). In panels C and D, the green fluorescence documents the localization of H3P (arrows), while the bright fluorescence shows the overlay of the red, blue and green fluorescence (asterisk). In panel E, the bright fluorescence shows a combination of the accumulation of actin and Aurora B by overlay of the dark red and green fluorescence (arrow). In panel F, the green fluorescence documents the localization of Aurora B (arrow). G. The percentage of cyclin A2, Ki-67, H3P, Aurora B-positive cells in the border zone, the distance of cardium, sham group (mean ± SD).
The expression of Ki-67, H3P and aurora B
At 14 days, the percentage of Ki-67–positive nuclei was 13.69 ± 1.40% in samples from the border zone of AMI (Figure 5B). This was significantly higher than the percentage in the distant normal myocardium. (7. 87 ± 1.33%) (P < 0.01), but the percentages of Ki-67-positive nuclei in both regions were significantly higher than those found in the corresponding regions of the sham-operated hearts (0.47 ± 0.10%, both P < 0.001).
Subsequently, we observed the expression of H3P (Figure 5C and 5D) and Aurora B (Figure 5E and 5F) to evaluate the cardiomyocyte mitotic index [5,11]. The expression of these proteins was examined using the same approach as was used to measure the expression of Ki-67. The expression levels of H3B and Aurora B were found to be considerably lower than those of cyclin A2 and Ki-67 when the numbers of positive nuclei were counted in 5 consecutive visual fields (× 100 magnifications). The numbers of H3P-positive and Aurora B-positive cardiomyocytes in the border zone were 9.50 ± 1.26% and 8.66 ± 1.24%, respectively, while in the distant normal myocardium. They were found to be 4.14 ± 1.01% and 3.31± 0.86%, respectively. When compared with the expression levels observed in sham-operated hearts, the expression levels in infarcted hearts were all significantly elevated, and those of the border zone were significantly higher than those of the distant normal myocardium. (Figure 5G) (P < 0.01 for all comparisons).
Discussion
Cyclins are a family of proteins that control the progression of cells through the cell cycle by activating cyclin-dependent kinase (Cdk) enzymes [7,8]. Cyclin A2 complexed with cdk2 is essential for the G1/S transition and cyclin A2/cdk1 promotes entry into mitosis [7,8]. Cyclin A2 is the only cyclin that is completely silenced in quiescent cells. The appearance of cyclin A2 expression occurs at a rate consistent with the rate of entry into the cell cycle [15]. When a cyclin A2-expressing adenoviral vector was delivered to rat hearts after left anterior descending coronary artery ligation-mediated AMI, evidence of cardiomyocyte proliferation, peri-infarct geometric enhancement, and cardiac functional improvement were subsequently observed [9]. This study provides a detailed analysis of the expression of cyclin A2 in cardiomyocytes after AMI. The expression of cyclin A2 increased significantly, and this response peaked 7 to 14 days following AMI. Cyclin A2 expression had declined to levels comparable to those in non-infarcted hearts at 4 weeks following AMI. The data suggest that the number of cardiomyocytes re-entering the cell cycle may follow the same trend. Similar results have been reported previously [9,16]. Furthermore, the number of cycling cardiomyocytes is significantly greater in the border zone of AMI than in the distant normal myocardium. AMI leads to both local and systemic changes and induces the release of various cytokines, which promote cardiomyocyte cell cycle re-entry. Cytokine concentrations also change with time and space. Cytokine concentrations are higher in cases of more extensive infarction and are increased in regions that are in close proximity to the infarcted area, thereby promoting cardiomyocyte proliferation appropriately. However, the specific mechanism of this response needs to be studied further.
Ki-67 is a nuclear antigen that is expressed in all phases of the cell cycle except G0, and it is strongly expressed late in the nuclear synthesis phase (S phase) [10]. Ki-67 is a well-accepted indicator of cellular proliferation, and all types of proliferating cells express cyclin A2 and Ki-67 [17,18]. The expression of cyclin A2 and Ki-67 is required for cells to enter the cell cycle and to undergo cell division [19]. Our study showed that the consistent changes in Ki-67 expression that were observed at 2 weeks after infarction were mirrored by changes in cyclin A2, and this finding suggests that cyclin A2 is also an indicator of nuclear synthesis. Although cyclin A2 and Ki-67 mark cells in S phase and identify cardiomyocytes that have re-entered the cell cycle, positive staining for these factors alone does not demonstrate that the cells are undergoing mitosis.
The incorporation of BrdU or tritiated thymidine into DNA is often used to detect DNA synthesis and cell proliferation but these markers cannot be used directly as unequivocal markers of mitosis because DNA synthesis also occurs in nuclei undergoing DNA repair, hyperplasia and changes in ploidy. There is not a single example of a Ki-67–positive cell that cannot divide [10,17-20], but Ki-67 expression does not directly demonstrate that cardiomyocytes are able to accomplish cell division. Unlike the myocytes of humans [21], some ventricular myocytes in rodents are multinucleated [16,22,23], and it is possible that these arise from a process in which karyokinesis is not followed by cytokinesis. Furthermore, 3 other studies of adenovirus-mediated expression of the indirect cell-cycle regulators E2F [24], E1A [25] and FGF-5 [26] have all demonstrated cell cycle re-entry to some degree, so we subsequently examined the expression of H3P and Aurora B to detect karyokinesis and cytokinesis, respectively, instead of using BrdU and tritiated thymidine. We found that the expression levels of H3P and Aurora B significantly increased to the same degree and that this was especially noticeable in the border zone and in hearts with lower EF values where tissue oxygenation must be largely maintained [27]. This finding is direct proof that cardiomyocytes complete mitotic division, that is, cell proliferation. Both markers were scarcely present in the heart without infarction. Thus, infarction or other heart diseases causing a loss of cardiomyocytes seem to be required for these responses. Injury seems to be required for stem cell migration, multiplication and differentiation into the cell lineages of the damaged heart or other organs [28,29].
In this study, although the expression levels of the 4 markers were significantly increased, it was obvious that the expression levels of Ki-67 and cyclin A2 were significantly higher than those of H3P and Aurora B. The number of cycling cardiomyocytes is much higher, by nearly 50-fold, than that of mitotic cardiomyocytes because karyokinesis and cytokinesis occurring in anaphase are completed in approximately 30 minutes [30] whereas the duration of the myocyte cell cycle in vivo is approximately 25 hrs [31]. Furthermore, as mentioned above, some ventricular myocytes in rodents are multinucleated [16,22,23], which means that some cycling cardiomyocytes do not accomplish mitosis and that karyokinesis in this case is not followed by myocyte cytokinesis.
This study has demonstrated that some cardiomyocytes can divide. In contrast to most adult cardiomyocytes, fetal cardiomyocytes do proliferate [32], so we assume that the dividing cells we observed were immature cardiomyocytes. Subsequently, the key issue is the origin of the immature cells in normal and diseased hearts. To date, two types of stem cells, including bone marrow–derived stem cells that reach the scarred myocardium after infarction [28] and cardiac stem cells [33], have been shown to differentiate into cardiomyocytes. The cyclin A2-positive cardiomyocytes we observed should originate from one or both of these stem cell types. As in the damaged brain [34], repair of the necrotic myocardium may involve interventions that promote the migration of endogenous and/or exogenous types of stem cells to the infarcted region.
Proliferation and differentiation of stem cells [33,35-38] into cardiomyocytes can compensate for the loss of cardiomyocytes, but the number of these stem cells is relatively small and cannot prevent cardiac remodeling, including cardiac hypertrophy and fibrosis of the infarcted area, and the onset and evolution of cardiac failure after AMI. Because restoration of the infarcted myocardium, even in part, might interfere with the progression of the structural and functional alterations of the diseased heart [28] and thereby delay any irreversible ventricular dysfunction, these responses are still very important in determining the prognosis. The proliferative response is enough to compensate for the loss of a few cardiomyocytes and to maintain normal heart function, and this is especially true for the continuous replacement of the aging cardiomyocytes [39]. It may be a theoretically ideal approach to apply specific extracellular factors to induce native cardiomyocyte proliferation with the aim of enhancing cardiac regeneration [40-43].
Based on our present study, we conclude that there is still a subpopulation of cardiomyocytes that can re-enter the cell cycle and undergo cell division soon after infarction and that cyclin A2 is a reliable marker for the detection of cardiomyocytes actively undergoing the cell cycle. The proliferative response of the organism may be a spontaneous compensatory reaction, but it is not enough to compensate for the damage caused by AMI, and follow-up studies should be focused on how to strengthen this compensatory response.
Acknowledgments
The authors are grateful to Hai-Yan Chen (China Pharmaceutical University), Wei-na Zhu (People’s Hospital of Jiangsu Province) and Xin-Gui Pen, Guo-Qing Wang (Zhongda Hospital) for technical assistance. This work was supported by grants from the National Natural Science Foundation (No. 6590000029), the Chinese Medical Association (No. 09010140169) and the Jiangsu Province Key Medical Education Foundation (No. RC2007103).
References
- 1.Kiessling A, Fussel S, Wehner R, Bachmann M, Wirth MP, Rieber EP, Schmitz M. Advances in specific immunotherapy for prostate cancer. Eur Urol. 2008;53:694–708. doi: 10.1016/j.eururo.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 2.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 4.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci U S A. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Amerongen MJ, Diehl F, Novoyatleva T, Patra C, Engel FB. E2F4 is required for cardiomyocyte proliferation. Cardiovasc Res. 2010;86:92–102. doi: 10.1093/cvr/cvp383. [DOI] [PubMed] [Google Scholar]
- 6.Novoyatleva T, Diehl F, van Amerongen MJ, Patra C, Ferrazzi F, Bellazzi R, Engel FB. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc Res. 2010;85:681–690. doi: 10.1093/cvr/cvp360. [DOI] [PubMed] [Google Scholar]
- 7.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 9.Woo YJ, Panlilio CM, Cheng RK, Liao GP, Atluri P, Hsu VM, Cohen JE, Chaudhry HW. Therapeutic delivery of cyclin A2 induces myocardial regeneration and enhances cardiac function in ischemic heart failure. Circulation. 2006;114:I206–213. doi: 10.1161/CIRCULATIONAHA.105.000455. [DOI] [PubMed] [Google Scholar]
- 10.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc Natl Acad Sci U S A. 1998;95:7480–7484. doi: 10.1073/pnas.95.13.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youn TJ, Piao H, Kwon JS, Choi SY, Kim HS, Park DG, Kim DW, Kim YG, Cho MC. Effects of the calcineurin dependent signaling pathway inhibition by cyclosporin A on early and late cardiac remodeling following myocardial infarction. Eur J Heart Fail. 2002;4:713–718. doi: 10.1016/s1388-9842(02)00120-4. [DOI] [PubMed] [Google Scholar]
- 13.Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S, Williams C, Torres RL, Wagle D, Ulrich P, Cerami A, Brines M, Regan TJ. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc Natl Acad Sci U S A. 2000;97:2809–2813. doi: 10.1073/pnas.040558497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez Y, Lutjemeier B, Seshareddy K, Trevino EM, Hageman KS, Musch TI, Borgarelli M, Weiss ML. Wharton’s Jelly or Bone Marrow Mesenchymal Stromal Cells Improve Cardiac Function Following Myocardial Infarction for More Than 32 Weeks in a Rat Model: A Preliminary Report. Curr Stem Cell Res Ther. 2013;8:46–59. doi: 10.2174/1574888x11308010007. [DOI] [PubMed] [Google Scholar]
- 15.Yoshizumi M, Lee WS, Hsieh CM, Tsai JC, Li J, Perrella MA, Patterson C, Endege WO, Schlegel R, Lee ME. Disappearance of cyclin A correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. J Clin Invest. 1995;95:2275–2280. doi: 10.1172/JCI117918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kajstura J, Zhang X, Reiss K, Szoke E, Li P, Lagrasta C, Cheng W, Darzynkiewicz Z, Olivetti G, Anversa P. Myocyte cellular hyperplasia and myocyte cellular hypertrophy contribute to chronic ventricular remodeling in coronary artery narrowing-induced cardiomyopathy in rats. Circ Res. 1994;74:383–400. doi: 10.1161/01.res.74.3.383. [DOI] [PubMed] [Google Scholar]
- 17.Hall PA, McKee PH, Menage HD, Dover R, Lane DP. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 1993;8:203–207. [PubMed] [Google Scholar]
- 18.Hall PA, Woods AL. Immunohistochemical markers of cellular proliferation: achievements, problems and prospects. Cell Tissue Kinet. 1990;23:505–522. doi: 10.1111/j.1365-2184.1990.tb01343.x. [DOI] [PubMed] [Google Scholar]
- 19.MacCallum DE, Hall PA. The location of pKi67 in the outer dense fibrillary compartment of the nucleolus points to a role in ribosome biogenesis during the cell division cycle. J Pathol. 2000;190:537–544. doi: 10.1002/(SICI)1096-9896(200004)190:5<537::AID-PATH577>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 20.MacCallum DE, Hall PA. The biochemical characterization of the DNA binding activity of pKi67. J Pathol. 2000;191:286–298. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH628>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 21.Olivetti G, Cigola E, Maestri R, Corradi D, Lagrasta C, Gambert SR, Anversa P. Aging, cardiac hypertrophy and ischemic cardiomyopathy do not affect the proportion of mononucleated and multinucleated myocytes in the human heart. J Mol Cell Cardiol. 1996;28:1463–1477. doi: 10.1006/jmcc.1996.0137. [DOI] [PubMed] [Google Scholar]
- 22.Kajstura J, Zhang X, Liu Y, Szoke E, Cheng W, Olivetti G, Hintze TH, Anversa P. The cellular basis of pacing-induced dilated cardiomyopathy. Myocyte cell loss and myocyte cellular reactive hypertrophy. Circulation. 1995;92:2306–2317. doi: 10.1161/01.cir.92.8.2306. [DOI] [PubMed] [Google Scholar]
- 23.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, Schneider MD. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997;100:2722–2728. doi: 10.1172/JCI119817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akli S, Zhan S, Abdellatif M, Schneider MD. E1A can provoke G1 exit that is refractory to p21 and independent of activating cdk2. Circ Res. 1999;85:319–328. doi: 10.1161/01.res.85.4.319. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki G, Lee TC, Fallavollita JA, Canty JM Jr. Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle. Circ Res. 2005;96:767–775. doi: 10.1161/01.RES.0000162099.01268.d1. [DOI] [PubMed] [Google Scholar]
- 27.Parodi O, De Maria R, Oltrona L, Testa R, Sambuceti G, Roghi A, Merli M, Belingheri L, Accinni R, Spinelli F, Pellegrini A, Baroldi G. Myocardial blood flow distribution in patients with ischemic heart disease or dilated cardiomyopathy undergoing heart transplantation. Circulation. 1993;88:509–522. doi: 10.1161/01.cir.88.2.509. [DOI] [PubMed] [Google Scholar]
- 28.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 29.Lagasse E, Connors H, Al-Dhalimy M, Reitsma M, Dohse M, Osborne L, Wang X, Finegold M, Weissman IL, Grompe M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–1234. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 30.Rieder CL. The formation, structure, and composition of the mammalian kinetochore and kinetochore fiber. Int Rev Cytol. 1982;79:1–58. doi: 10.1016/s0074-7696(08)61672-1. [DOI] [PubMed] [Google Scholar]
- 31.Antonio C, Ferby I, Wilhelm H, Jones M, Karsenti E, Nebreda AR, Vernos I. Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell. 2000;102:425–435. doi: 10.1016/s0092-8674(00)00048-9. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 33.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 34.Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J, Korc M, Endo TL, Duong A, Nguyen G, Karkehabadhi M, Twardzik D, Patel S, Loughlin S. In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci U S A. 2000;97:14686–14691. doi: 10.1073/pnas.97.26.14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuura K, Nagai T, Nishigaki N, Oyama T, Nishi J, Wada H, Sano M, Toko H, Akazawa H, Sato T, Nakaya H, Kasanuki H, Komuro I. Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem. 2004;279:11384–11391. doi: 10.1074/jbc.M310822200. [DOI] [PubMed] [Google Scholar]
- 37.Martin CM, Meeson AP, Robertson SM, Hawke TJ, Richardson JA, Bates S, Goetsch SC, Gallardo TD, Garry DJ. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev Biol. 2004;265:262–275. doi: 10.1016/j.ydbio.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bicknell KA, Coxon CH, Brooks G. Forced expression of the cyclin B1-CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem J. 2004;382:411–416. doi: 10.1042/BJ20031481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Regula KM, Rzeszutek MJ, Baetz D, Seneviratne C, Kirshenbaum LA. Therapeutic opportunities for cell cycle re-entry and cardiac regeneration. Cardiovasc Res. 2004;64:395–401. doi: 10.1016/j.cardiores.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Regula KM, Kirshenbaum LA. Breaking down cell-cycle barriers in the adult heart. Circ Res. 2004;94:1524–1526. doi: 10.1161/01.RES.0000134761.05562.a6. [DOI] [PubMed] [Google Scholar]
- 43.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]