

Abstract
Takayasu arteritis (TA) is a chronic idiopathic and granulomatous vasculitis, manifesting mainly as a panaortitis. Autoimmune cell-mediated immunity is probably responsible for the disease. The inflammation commences from the adventitia and progresses to the intima and leads to, both in adults and children, segmental stenosis, occlusion, dilatation, and/or aneurysm formation. This review focuses briefly on the etiopathogenesis, and describes the pathological and clinical features in adults and children.
Keywords: Large vessel vasculiti, Takayasu arteritis, hypertension
INTRODUCTION
Vasculitides refers to a heterogeneous group of disorders characterized by inflammation of blood vessels resulting in destruction and distortion of the layered components of their walls. The inflammation can result in clinical syndromes ranging from minor self-limiting illness to life-threatening systemic disorder. The categorization of vasculitides hinges on the sizes of the vessels affected, and excludes a direct infective etiology. The diagnosis, however, depends on the site and extent of vascular injury, which in turn is reflected by protean clinical and radiological manifestations, along with altered laboratory parameters. The Chapel Hill Consensus Conference Nomenclature of Vasculitides classification is still widely used and has been recently refined.[1] For children and adolescents, the European League Against Rheumatism (EULAR) and the Pediatric Rheumatology European Society (PRES) endorsed a new classification which particularly addressed the problems of childhood vasculitides, and Takayasu arteritis (TA) is the only large vessel vasculitidis in this age group.[2] The term “large-vessel vasculitis” (LVV) refers to chronic inflammation affecting the elastic arteries, mainly aorta and its major branches. In adults, this group also includes giant cell arteritis (rarely occurring before the age of 50 years) and TA (rarely occurring after the age of 50 years).[3] TA forms the third most frequent vasculitis in childhood in the EULAR/PRES classification of vasculitis.[4] TA is a chronic idiopathic and granulomatous vasculitis, manifesting mainly as a panaortitis. The inflammation commences from the adventitia and progresses to the intima and leads to, both in adults and children, segmental stenosis, occlusion, dilatation, and/or aneurysm formation. This review focuses briefly on the etiopathogenesis, and the pathological features in adults and children.
ETIOPATHOGENESIS
TA is known worldwide with an estimated incidence of 1.2-2.6/million per year in the western population and is seen in all races, but the incidence is higher in Southeast Asia, Central and South America, and Africa.[5] It is generally considered a disease of young adults with a peak onset in second and third decades of life and a striking predilection for females. The predisposing and etiological factors of TA have still to be clearly elucidated. An autoimmune basis, influenced by genetic and environmental factors, is strongly suggested; the resulting inflammation is largely a cell-mediated immune response. There is an association between TA and Mycobacterium tuberculosis, but this connection has not been well substantiated.[6] TA also occurs with a variety of autoimmune disorders such as systemic lupus erythematosus, rheumatoid arthritis, sero-negative spondyloarthropathies, inflammatory bowel disease, anterior uveitis, Wegener's granulomatosis, sarcoidosis, amyloidosis, Sweet syndrome, and even some immunodeficiency syndromes.[7] The general understanding is that an unknown stimulus triggers expression of heat shock protein-65 in the aortic tissue, which is also synthesized by other tissues in response to stress.[8] The protein has its homolog in Mycobacteria and many other bacterial species. This protein induces the Major Histocompatibility Class 1 chain-related A on vascular cells. These are recognized by subsets of T lymphocytes, which along with macrophages, produce pro-inflammatory cytokines. This finally results in acute inflammation, necroses, neo-vascularization, smooth muscle migration, intimal proliferation, and giant cell formation. Further, there is recruitment of B lymphocytes with production of anti-endothelial, anti-cardiolipin, and anti-aorta auto-antibodies, which accentuates the inflammatory process. Further, human leukocyte antigen (HLA)-B52 and B39 are associated with TA in Japan, and HLA-DR B1-1301/1302 in Mexico.[8]
CLINICAL AND PATHOLOGICAL FEATURES OF TAKAYASU ARTERITIS
The disease progression is said to occur in a triphasic pattern.[9] Phase I is the systemic or pre-pulseless period, which is characterized by constitutional symptoms such as low-grade fever, malaise, night sweats, arthralgia, anorexia, and weight loss. Hence, TA should form a differential diagnosis for fever of unknown origin. Phase II is the vasculitic stage where constitutional symptoms are accompanied by features of vascular involvement like tenderness or pain over vessels (angiodynia). The final phase refers to the late, fibrotic, occlusive, quiescent, or “burnt-out” phase, wherein characteristic features of TA (pulseless disease) related to arterial stenosis or occlusion appear. Only a minority of patients show such a temporal progression. The active or acute phases of the disease, which are difficult to diagnose, remit spontaneously in about 3 months or may progress insidiously and relentlessly for months to years into the chronic phase. It is to be noted that inflammatory and fibrotic changes can co-exist.
Diagnosis of TA presents a challenge to the clinician. The disease has non-specific clinical symptoms and has protean manifestations: Ranging from asymptomatic patients with incidental findings such as unequal pulses, to catastrophic presentation such as cerebrovascular accidents. Specific diagnostic criteria have been described by Ishikawa[10] [Table 1] and American College of Rheumatology[11] [Table 2]. Apart from constitutional complaints, nearly all patients with TA sooner or later present with symptoms and signs related to vascular stenosis and/or occlusion. They include hypertension with blood pressure differences in the extremities, pulse deficits, bruit, and upper and/or lower extremity claudications. Neurological, musculoskeletal, and dermatological manifestations are less common. These vascular changes are well delineated through advances in imaging technology.[12]
Table 1.
Ishikawa criteria of Takayasu arteritis[15]

Table 2.
American college of rheumatology classification criteria of Takayasu arteritis[16]

Around 5% of patients with TA are children and adolescents. Most children are diagnosed between ages 8 and 13 years, and like adults, females outnumber males with a ratio of 3:1.[4,10] The largest pediatric series of 70 patients was reported by Hong et al;[13] the youngest reported case was 7 months old.[12] Early clinical diagnosis in children is even more challenging, TA should be considered in the differential diagnosis when they present with fever, vomiting, and weight loss; other symptoms include night sweats, back pain, mylagias, and arthralgias. This also simulates acute rheumatic fever, leading to misdiagnosis.[14] In comparison to limb claudication, poor/absent peripheral pulses, or bruits, hypertension is not only the most common mode of presentation, but also very often the only manifestation of TA in children.[14,15] Hence, modification of the diagnostic criteria for TA has been advocated [Table 3].[2] One-third of children present with inactive disease, in which clinical features represent vascular sequelae. Presentation with heart failure is also common.
Table 3.
EULAR/PRES criteria of Takayasu arteritis[2]

Histopathologic features
In TA, biopsy specimens are seldom available and hence morbid anatomic features are based on autopsy findings[16] or segments excised during bypass surgery.[17] On histology, the lesions can be active, chronic, or healed. Though TA is a panarteritis, the initial site of inflammation is around the vasa vasora and at the medio-adventitial junction. There is edema, and mononuclear cell infiltration (CD4 and CD8 lymphocytes, plasma cells, and macrophages) in the outer thirds of the media and adventitia. Giant cell granulomatous reaction [Figure 1a] and laminar necrosis can also be present. Fragmentation of elastic fibers with “elasticophagia” is prominent. Rapid or more severe inflammation leads to loss of smooth muscle cells, medial weakening, vascular dilatation, and even aneurysm formation. There is reactive fibrosis and increased ground substance in the intima, with overlying layer of thrombus or a band of neo-vascularization at the intimo-medial junction. In the chronic phase [Figures 1b and 1c], there is patchy mononuclear inflammatory infiltrate with medial scarring and vascularization, while the healed phase [Figure 2] shows only fibrosis in all layers; these two phases are often seen.
Figure 1.

(a) Active phase of TA in descending thoracic aorta showing a granulomatous inflammation at the medio-adventitial junction of aorta destroying the structure of an intercostal artery; (b) chronic phase of TA in descending thoracic aorta showing media (M) at the outer third with clusters of mononuclear cells. Note marked fibrosis of the adventitia (A) (Hematoxylin and Eosin, ×250). (c) The cells are lymphocytes, and few plasma cells and histiocytes (Hematoxylin and Eosin, ×250)
Figure 2.

Healed phase of TA in descending thoracic aorta: (a) Loose fibrocellular thickening of the intima (I); (b) junctional area between intima (I) and media (M) showing destruction of the elastic lamellae/smooth muscle with replacement fibrosis; (c) similar features are also seen toward the outer third of the media (M) and adventitia (A); (d) fibrotic adventitia (A) showing endarteritis obliterans of a branch (elastic van Gieson, ×250)
Clinically, though aortic arch and renal arteries are often involved, at autopsy it is rare to find disease restricted to aorta and its branches.[16] One or more patterns of disease are noted: (1) Localized aortic segment involvement for a length of 2–7 cm; (2) multiple short segment disease with normal or “skip” areas in between; or (3) diffuse aortic disease with or without a small stretch of normal segments in between. The disease may start at the aortic annulus, but by and large, the infra-renal aorta, inferior mesenteric artery, and iliac arteries are usually not affected. The disease is classified into five types on the basis of the pattern of anatomical involvement: Type I, aortic arch and its branches; Type II, descending thoracic and abdominal aorta; Type III, aortic arch and thoraco-abdominal aorta; Type IV, pulmonary artery in addition to the aforementioned types; and Type V, coronary artery in addition to the other types.[18] Another classification[19] categorizes TA into six types: Type I, aortic arch and its branches; Type IIa, ascending aorta, arch, and its branches; Type IIb, ascending aorta, arch, and its branches, and descending thoracic aorta; Type III, thoracic aorta and abdominal aorta; Type IV, abdominal aorta; and Type V, combination of Type IIb and Type IV. Involvement of coronary arteries is labeled C (+) and involvement of pulmonary arteries is denoted as P (+). All patterns of vascular involvement are observed in all regions, though a distinct pattern is also seen. Japanese patients have a higher incidence of arch involvement (reverse coarctation), while in Southeast Asia, India, and Africa, it is the “middle aortic syndrome” with affection of thoraco-abdominal aorta and renal arteries; the pattern is often diffuse [Figure 3]. In North America, arch vessels and abdominal aorta are more frequently affected.
Figure 3.

Scanned images of the ascending aorta, arch, left subclavian artery, descending thoracic aorta (elastic van Gieson), and abdominal aorta (Hematoxylin and Eosin, and elastic van Gieson) in a diffuse pattern of TA. All segments show marked adventitial fibrosis, the major cause of wall thickening. Lymph nodes (arrows) are seen around abdominal aorta
The extent of the aortic disease is apparent on gross examination and it is best seen in children. The aorta feels stiff and rigid on palpation. The most striking feature is the extraordinary degree of adventitial and periadventitial fibrosis [Figure 3], which is in excess to that seen in any other inflammatory disorder of the aorta. This may produce adherence to the neighboring structures. In the early stages of the disease, the thickened adventitia may have a gelatinous appearance. Another feature is the presence of enlarged para-aortic lymph nodes, particularly in the vicinity of renal [Figure 3] and subclavian arteries. The nodes may show caseation necrosis or active hyperplasia. The intimal fibrocellular hyperplasia is seen as plaques, and depending on the amounts of acid mucopolysaccharides and collagen, it appears gelatinous or white, respectively. Gelatinous plaques are characteristically seen in children [Figure 4a]. Sometimes the plaques are large or even circumferential or may even be covered by mural thrombi [Figure 4b]. Sometimes the involvement is seen as diffuse intimal thickening with mild affection of the adventitia [Figure 4c]. Localized disease is also often seen in children and there is always a sharp line of demarcation between normal and diseased segments [Figure 4d]. Superimposed calcification and atherosclerosis accentuates the vascular rigidity [Figure 5]; lesions in children are usually bereft of secondary atherosclerosis.
Figure 4.
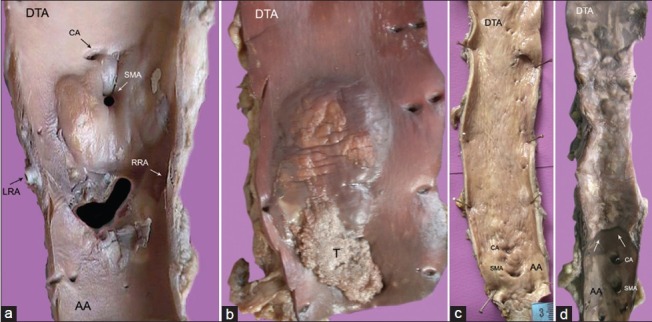
(a) Abdominal aorta with large, glistening intimal plaques seen around the superior mesenteric (SMA) and left (LRA), right (RRA) renal arteries; (b) single large gelatinous plaque with superimposed thrombosis (t) in descending thoracic aorta (DTA); (c) diffuse intimal thickening with superficial scars and furrows of entire descending aorta; (d) the localized disease affecting the descending thoracic aorta (DTA) is abruptly separated from normal abdominal aorta (AA) by a distinct shelf of intimal thickening
Figure 5.
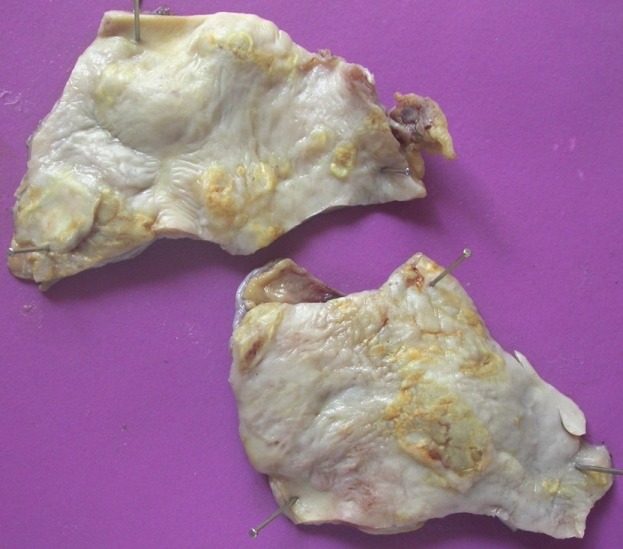
Cobble-stoned intima with atherosclerosis, calcification, and focal ulceration in an excised segment of ectatic descending aorta
When large and/or circumferential, these plaques contribute greatly to luminal stenosis or obliteration. These mainly affect the mid-thoracic level or the region of renal arterial origins [Figure 6]. Depending on the number and sizes of the intimal plaques, the aortic lumen may be irregular or obstructed [Figure 6]. The external diameter of the aorta is, however, the same or is slightly reduced. The exuberant adventitial and intimal fibrosis with consequent aorto-arterial stenosis/occlusion characterizes the pathology of TA, and these features are not seen with other types of LVV. Luminal occlusion results from superimposition of mural thrombus. Medial destruction results in not only vascular ectasia [Figure 5], but also frank aneurysms [Figure 7a]. The aneurysms (saccular or fusiform) occur in 2-6%, but usually co-exist with stenotic lesions; these are also uncommon in children. Aneurysms devoid of stenosis or pseudo-aneurysms are extremely rare, and so are dissections.[20] Dissections may not have acute presentations, are localized, usually affect the abdominal aorta, and at times are not grossly evident [Figures 7b and c].
Figure 6.
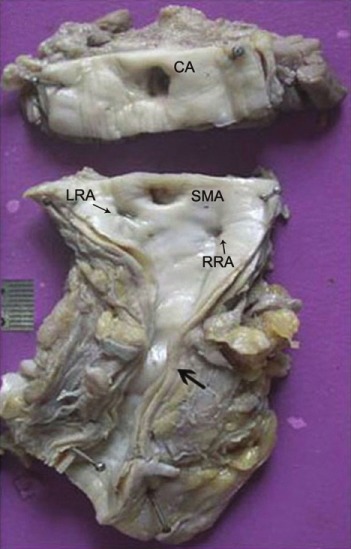
Intimal thickening of the abdominal aorta with extensive adventitial/peri-adventitial fibrosis and adherent adipose tissue. Note the presence of narrowing of the aortic lumen in the infra-renal region, and renal arterial ostia (CA, celiac artery; SMA, superior mesenteric artery; RRA, right renal artery; LRA, left renal artery)
Figure 7.
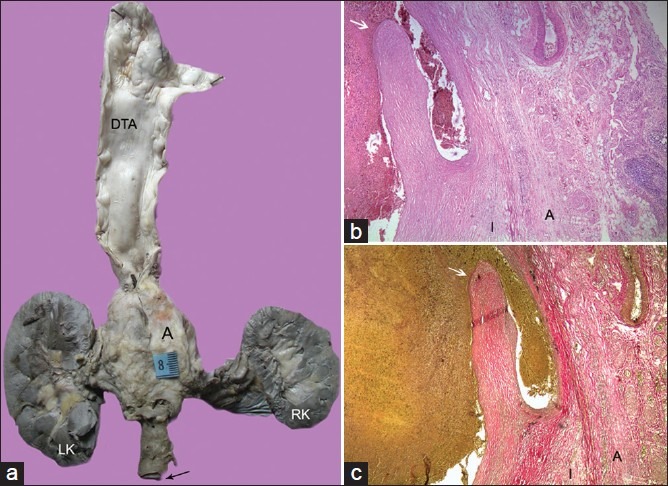
(a) Multi-focal aortic disease with skip areas showing a large aneurysm in the abdominal aorta. The wall of the aneurysm when studied histologically also showed the presence of chronic dissection as seen in (b) (Hematoxylin and Eosin, ×100) and (c) (Elastic van Gieson, ×100). Arrow points to the intimal flap. The wall is represented only by thickened intima (I) and adventitia (A)
With branch involvement, the lesions are noted around the ostia. The subclavian (especially the left) and renal arteries are commonly affected. Rarely, all arch arteries can be involved [Figure 8a]. It was initially hypothesized that the disease began at the subclavian arteries, but a latest study shows that TA lesions mostly develop in a symmetric manner in paired vascular territories and are contiguous in the aorta.[21] Pulmonary arterial involvement is found in about 70% of patients [Figure 8b],[22] is usually mild, and can extend into segmental and sub-segmental branches [Figure 8c]. Therefore, some patients may have features related to pulmonary thromboembolism or hypertension.
Figure 8.
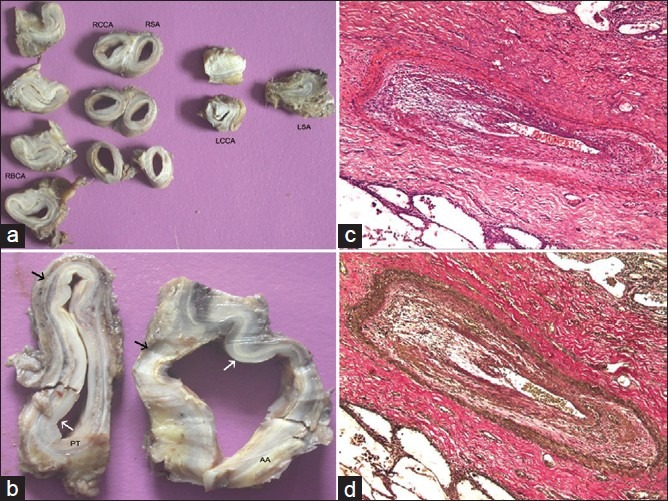
(a) There is involvement of all the arch arteries. Luminal obliteration is present in left common carotid artery (LCCA) and left subclavian artery (RBCA, right brachiocephalic artery; RCCA, right common carotid artery; RSA, right subclaivan artery). (b) The involvement of ascending aorta (AA) and pulmonary trunk (PT) is comparable. There is prominent intimal (white arrow) and adventitial (black arrow) thickening; The case also showed inflammation of the segmental branches seen in (c) (Hematoxylin and Eosin, ×250) and (d) (Elastic van Gieson, ×250)
Coronary arterial involvement is usually rare, ostial, and/or proximal.[23] It must be remembered that ascending aorta [Figure 9], pulmonary trunk and its branches, and coronary arteries can be affected in isolation. About 40% of patients develop cardiac abnormalities, manifesting as cardiac failure, seen often in children. The underlying basis for this in most cases is hypertension or ischemia of the hypertrophied myocardium. But not uncommonly, dilated, poorly contractile hearts with features akin to dilated cardiomyopathy occur, and a misdiagnosis of dilated cardiomyopathy is often made by those not familiar with this entity. Rarely, there can be organic aortic valvular involvement or ischemia/infarction related to stenosing or occlusive coronary arterial lesion. At times, there may be myocarditis or aortic valvular incompetence.
Figure 9.
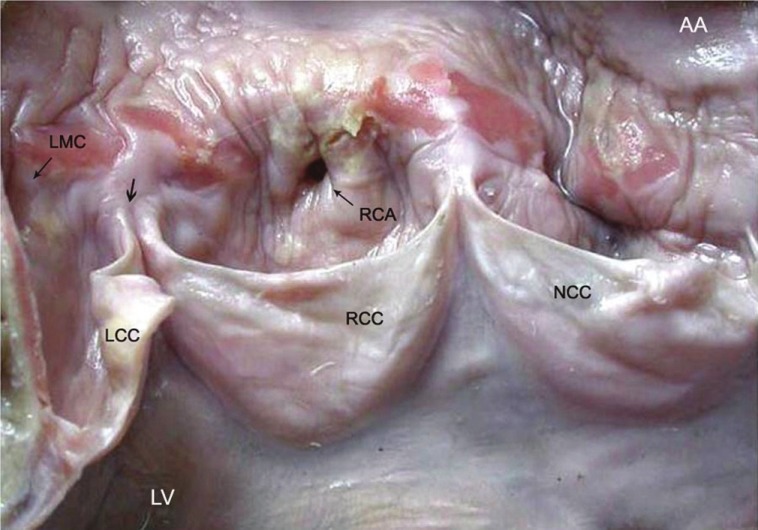
A 21-year-old woman, who presented with sudden cardiac death, showed TA manifesting as a band of aortitis affecting the ascending aorta (AA) with narrowing (thin arrow) of the dominant right coronary arterial (RCA) ostium. There is widening of the commissure (thick arrow) between right RCC and left LCC coronary cusps (LV, left ventricle; NCC, non-coronary cusp, LMC, left main coronary ostium)
ACTIVITY ASSESSMENT AND THERAPY
Assessing disease activity in TA is challenging as there is frequently a lack of clinical and investigational correlation. New onset or worsening of two or more of the set criteria of TA are said to be indicative of active disease,[24] though angiographic disease progression and histological progression can be seen clinically even during inactive phase of the disease. Similar situation is faced with assessment of laboratory markers such as erythrocyte sedimentation rate (ESR) and several other acute phase reactants. Recent investigations have focused on identification of novel and sensitive biomarkers such as pentraxin-3[25] and imaging techniques.[11,12]
In general, there is higher morbidity and mortality in children as compared to adults due to rapid development of hypertension and heart failure. A mortality of 10-30% has been reported on follow-up.[26] With better treatment options now available, the prognosis appears to be improved. The mainstay of treatment is high-dose corticosteroids.[13] Patients who do not respond to steroids or who have a relapse may need to be given methotrexate, azathioprine, or cyclophosphamide.[13] Cyclosporine has been used as an alternative to cyclophosphamide. Tumor necrosis factor inhibition has also been used in relapsing disease or glucocorticoid-resistant cases. There are also interventional or surgical options available.[27,28] Balloon dilatation alone, stenting if required, and bypass grafting may be done, but the management requires a refined judgment because of the range of patho-anatomic and clinical issues involved.
CONCLUSIONS
TA, a large vessel vasculitis in children, remains a fascinating and difficult disease characterized by constitutional features, hypertension, heart failure, and pulse loss. The pathological features vary with the stage of the disease and include granulomatous inflammation, elasophagia, adventitial and medial fibrosis, vessel dilatation, and stenosis or occlusions of the involved arteries. The clinical course of the disease is variable, with the active and quiescent phases, and treatment strategies need individualized management.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 2.Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PRES endorsed consensus criteria for the classification of childhood vasculitis. Ann Rheum Dis. 2006;65:936–41. doi: 10.1136/ard.2005.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luqmani R. Large vessel vasculitides: Update for the cardiologist. Curr Opin Cardiol. 2012;27:578–84. doi: 10.1097/HCO.0b013e32835895ea. [DOI] [PubMed] [Google Scholar]
- 4.Weiss PF. Pediatric vasculitis. Pediatr Clin N Am. 2012;59:407–23. doi: 10.1016/j.pcl.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards BL, March L, Gabriel SE. Epidemiology of large-vessel vasculitides. Best Pract Res Clin Rheumatol. 2010;24:871–3. doi: 10.1016/j.berh.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Kothari SS. Aetiopathogenesis of Takayasu's arteritis and BCG vaccination: The missing link. Med Hypotheses. 1995;45:227–30. doi: 10.1016/0306-9877(95)90109-4. [DOI] [PubMed] [Google Scholar]
- 7.Al abrawi S, Fouillet-Desjonqueras M, David L, Barral X, Cochat P, Cimaz R. Takayasu arteritis in children. Pediatr Rheumatol Online J. 2008;6:17. doi: 10.1186/1546-0096-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z. Pathogenesis of Takayasu's arteritis: A 2011 update. Autoimmun Rev. 2011;11:61–7. doi: 10.1016/j.autrev.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Miller DV, Maleszewski JJ. The pathology of large-vessel vasculitides. Clin Exp Rheumatol. 2011;29:S92–8. [PubMed] [Google Scholar]
- 10.Brunner J, Feldman BM, Tyrrell PN, Kuemmerle-Descher JB, Zimmerhackl LB, Gassner I, et al. Takayasu arteritis in children and adolescents. Rheumatology. 2010;49:1806–14. doi: 10.1093/rheumatology/keq167. [DOI] [PubMed] [Google Scholar]
- 11.Gornik HL, Creager MA. Aortitis. Circulation. 2008;117:3039–51. doi: 10.1161/CIRCULATIONAHA.107.760686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanley P, Roebuck D, Barboza A. Takayusu's arteritis in children. Tech Vasc Interven Radiol. 2003;6:158–68. doi: 10.1053/j.tvir.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Ozen S, Duzova A, Bakkaloglu A, Bilginer Y, Cil BE, Demircin M, et al. Takayasu arteritis in children: Preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr. 2007;150:72–6. doi: 10.1016/j.jpeds.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 14.Muranjan MN, Bavdekar SB, More V, Deshmukh H, Tripathi M, Vaswani R. Study of Takayasu's arteritis in children: Clinical profile and management. J Postgrad Med. 2000;46:3–8. [PubMed] [Google Scholar]
- 15.Jain S, Sharma N, Singh S, Bali HK, Kumar L, Sharma BK. Takayasu arteritis in children and young Indians. Int J Cardiol. 2000;75:S153–7. doi: 10.1016/s0167-5273(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 16.Kinare SG, Gandhi MA, Deshpande JR. Non-specific aortoarteritis (Pathology and Radiology) Mumbai: Quest Publications; 1998. [Google Scholar]
- 17.Walker M, Gallagher PJ. The surgical pathology of large vessel disease. Diagn Histopathol. 2009;16:10–6. [Google Scholar]
- 18.Gulati A, Bagga A. Large vessel vasculitides. Pediatr Nephrol. 2010;25:1037–48. doi: 10.1007/s00467-009-1312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hata A, Noda M, Moriwaki R, Numano F. Angiographic findings in Takayasu arteritis: New classification. Int J Cardiol. 1996;54:S155–63. doi: 10.1016/s0167-5273(96)02813-6. [DOI] [PubMed] [Google Scholar]
- 20.Khalife T, Alsac JM, Lambert M, Messas E, Duong Van Huyen JP, Bruneval P, et al. Diagnosis and surgical treatment of a takayasu disease on an abdominal aortic dissection. Ann Vasc Surg. 2011;25:556.e1–5. doi: 10.1016/j.avsg.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Arnaud L, Haroche J, Toledano D, Cacoub P, Mathian A, Costedoat-Chalumeau N, et al. Cluster analysis of arterial involvement in Takayasu arteritis reveals symmetric extension of the lesions in paired arterial beds. Arthritis Rheumat. 2011;63:1136–40. doi: 10.1002/art.30240. [DOI] [PubMed] [Google Scholar]
- 22.Toledano K, Guralnik L, Lorber A, Ofer A, Yigla M, Rozin A, et al. Pulmonary arteries involvement in Takayasu's arteritis: Two cases and literature review. Semin Arthritis Rheum. 2011;41:461–70. doi: 10.1016/j.semarthrit.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Lanjewar C, Kerkar P, Vaideeswar P, Pandit S. Isolated bilateral coronary ostial stenosis--an uncommon presentation of aortoarteritis. Int J Cardiol. 2007;114:e126–8. doi: 10.1016/j.ijcard.2006.07.220. [DOI] [PubMed] [Google Scholar]
- 24.Kerr GS, Hallahan CW, Giordan J, Leavitt RY, Fauci AS, Rottem M, et al. Takayasu arteritis. Ann Intern Med. 1994;120:919–29. doi: 10.7326/0003-4819-120-11-199406010-00004. [DOI] [PubMed] [Google Scholar]
- 25.Dagna L, Salvo F, Tiraboschi M, Bozzolo EP, Franchini S, Doglioni C, et al. Pentraxin-3 as a marker of disease activity in Takayasu's arteritis. Ann Intern Med. 2011;155:425–33. doi: 10.7326/0003-4819-155-7-201110040-00005. [DOI] [PubMed] [Google Scholar]
- 26.Kothari SS. Takaysau's arteritis in children – A review. Images Paediatr Cardiol. 2001;3:4–23. [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy E, Robbs JV. Surgical management of Takayasu's arteritis in children and adolescents. Cardiovasc J Afr. 2007;18:393–6. [PubMed] [Google Scholar]
- 28.Saxena A, Kothari SS, Sharma S, Juneja R, Srivastava S. Percutaneous transluminal angioplasty of the aorta in children with nonspecific aortoarteritis: Acute and follow-up results with special emphasis on left ventricular function. Catheter Cardiovasc Interv. 2000;49:419–24. doi: 10.1002/(sici)1522-726x(200004)49:4<419::aid-ccd15>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]