

Abstract
Motivation: In modern sequencing studies, one can improve the confidence of genotype calls by phasing haplotypes using information from an external reference panel of fully typed unrelated individuals. However, the computational demands are so high that they prohibit researchers with limited computational resources from haplotyping large-scale sequence data.
Results: Our graphics processing unit based software delivers haplotyping and imputation accuracies comparable to competing programs at a fraction of the computational cost and peak memory demand.
Availability: Mendel-GPU, our OpenCL software, runs on Linux platforms and is portable across AMD and nVidia GPUs. Users can download both code and documentation at http://code.google.com/p/mendel-gpu/.
Contact: gary.k.chen@usc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Imputation of untyped genotypes is a critical preliminary in modern genetic association studies. The intuition behind haplotype-based imputation is that typed markers situated on haplotypes in key individuals can be leveraged probabilistically to impute untyped markers at the same position in other individuals of similar ethnicity. If an imputed single-nucleotide polymorphism (SNP) correlates better to a causal variant than neighboring typed SNPs, then imputation can improve statistical power to detect association.
In haplotyping and imputation, computational demands escalate quadratically as the number of reference haplotypes increases. The computational burdens of current datasets are so onerous that most genotyping centers divide their data into small subsets and process each subset on a different node of a computer cluster. For instance, IMPUTE2 documentation recommends this strategy (Howie et al., 2009). Genotyping by second-generation sequencing is quickly becoming cost-competitive with traditional SNP genotyping. Unfortunately, the massive amounts of data generated by sequencing will exacerbate problems and stress traditional computing clusters to the breaking point. Graphics processing units (GPUs) offer one solution to this dilemma. GPUs have been used in recent years to solve several high-dimensional problems in computational biology (Chen, 2012; Zhou et al., 2010) amenable to fine-grained parallelization. In this article, we introduce Mendel-GPU, a software application that addresses the practical need to efficiently impute genotypes on large-scale datasets. Further details on implementation and additional analyses can be found in the Supplementary Information.
2 SOFTWARE
The algorithms behind our imputation method exploit rapid estimation of haplotype frequencies in narrow genomic windows. Carefully chosen penalties eliminate haplotypes with low explanatory power. Penalized estimation is accomplished by harnessing a variant of the standard Expectation Maximization (EM) algorithm known as the Minorization Maximization (MM) algorithm (Lange, 2004). The MM algorithm for haplotype frequency estimation converges in fewer iterations with no loss in imputation accuracy (Ayers and Lange, 2008).
Mendel-GPU is implemented in C++ and OpenCL. If a GPU device is available, users can activate a flag to enable execution of our OpenCL kernels. We include several utilities that ease the transition from other standard formats such as variant call files (VCF), PLINK binary files (Purcell et al., 2007) and the reference haplotype formats used by HapMap and the 1000 Genomes Project (KGP) (Altshuler et al., 2010). Because memory demands can be especially heavy in imputation of whole chromosomes, Mendel-GPU automatically splits large regions into subregions that safely fit within the confines of GPU memory. Unlike competing methods, which do not operate on a sliding window, Mendel-GPU can generate genotype, dosage and quality metric data on the fly, reducing overall memory burden substantially. Imputed genotypes and haplotypes are output in SNP major order to facilitate integration with databases and SNP association testing.
3 RESULTS
3.1 Comparisons with competing programs
We compared the performance of Mendel-GPU with leading programs for genotype imputation: in particular thunder (Li et al., 2010), IMPUTE2 (Howie et al., 2009) and BEAGLE (Browning and Browning, 2007). In contrast to previous evaluations of haplotyping (Browning and Browning, 2011) and imputation performance (Howie et al., 2009), we base our simulations and comparisons on the KGP rather than microarray derived data.
In our first example, we performed genotype imputation and haplotyping with no reference haplotypes present on simulated 2–4× coverage data generated from a 1 MB region of Chromosome 22 taken from the KGP. Table 1 compares our runtime and accuracy results to those from the three competing programs. The second column of the table indicates haplotype phasing performance, measured as 1 minus the switch error, while the third column indicates imputation performance, measured as concordance of imputed heterozygotes to true heterozygotes. Our results indicate comparable haplotyping and imputation accuracies across all programs. In terms of computational efficiency, Mendel-GPU achieved 6-, 33- and 76-fold speed improvements over BEAGLE, IMPUTE2 and thunder, respectively, while requiring only 56, 13 and 33% as much peak memory.
Table 1.
Haplotype phasing and genotype imputation performance ignoring reference haplotypes
| Program | Phasing accuracy | Hetero. accuracy |
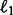 norm of errors norm of errors |
Total runtime | Max memory footprint |
|---|---|---|---|---|---|
| Mendel-GPU | 0.945 | 0.929 | 188 239.305 | 19:44 | 320 MB |
| BEAGLE | 0.968 | 0.948 | 159 772.295 | 2:03:37 | 573 MB |
| IMPUTE2 | 0.925 | 0.820 | 695 491.159 | 10:51:33 | 2.5 GB |
| thunder | 0.964 | 0.952 | 244 652.465 | 25:00:50 | 947 MB |
In our second example, we considered a random 7 MB dataset derived from the KGP where one has ethnically matched reference haplotypes. One half of the KGP was reserved as reference haplotypes, and the other half was used to simulate 2–4× coverage data. In capitalizing on reference haplotypes, Mendel-GPU takes advantage of a computationally efficient middle-thirds algorithm. Because all genotypes in the middle third of a sliding window are imputed simultaneously, the speed and memory improvements for Mendel-GPU are more impressive in Table 2 than Table 1. Mendel-GPU is 104 and 144 times faster than BEAGLE and IMPUTE2 and requires only 8–15% of their peak memory demands. Mendel-GPU’s accuracies fall between those of BEAGLE and IMPUTE2. Note that thunder does not support reference haplotypes.
Table 2.
Genotype imputation in a low pass (2–4×) re-sequencing study using KGP reference haplotypes
| Program | Hetero. accuracy |
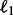 norm of errors norm of errors |
Total runtime | Max memory footprint |
|---|---|---|---|---|
| Mendel-GPU | 0.943 | 767 007.878 | 15:11 | 575 MB |
| BEAGLE | 0.962 | 522 638.033 | 26:21:45 | 7.0 GB |
| IMPUTE2 | 0.903 | 1 604 154.945 | 36:21:12 | 3.7 GB |
4 DISCUSSION
We have described software to meet the challenges of imputation in whole-genome sequencing data. Mendel-GPU supports the use of dense reference haplotypes and genotype penetrances as reported by variant calling pipelines. As the two examples illustrate, Mendel-GPU enjoys similar accuracies to the most highly regarded programs available, while requiring only a fraction of their time and memory demands. The fine-grained parallel algorithms of Mendel-GPU effectively harness the computational efficiency and memory bandwidth of hundreds of GPUs. Although BEAGLE appears to have an edge in accuracy in the scenarios tested, the speed and memory advantages of Mendel-GPU outweigh, in our opinion, its slight losses in accuracy. As GPU devices increase in sophistication and we further tune the code of Mendel-GPU, we expect to see greater gains.
Even as things now stand, Mendel-GPU will prove helpful. For researchers interested in testing rare variants coordinated with the KGP, our simulations highlight potential gains for study data consisting of a few hundred subjects sequenced at modest coverage. For example, whole-genome imputation of low-pass sequencing data on 545 study subjects would complete in ~6.8 days on a machine equipped with a single nVidia Tesla C2050 GPU. The same analysis using IMPUTE2 would require ~2.7 years on a single CPU machine. This difference puts small laboratories back in contention with the sequencing factories. Enabling small projects will ripple productively through the entire fabric of genomics research.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the USC Epigenome Center for GPU computing resources.
Funding: R01 ES019876 and R01 HG006465 (to G.K.C. and K.W., in part); U01 HG004726-01 (to A.H.S.); R01 HG006139 (to E.M.S.) and RO1 GM53275 (to K.L.).
Conflict of Interest: none declared.
REFERENCES
- Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers KL, Lange K. Penalized estimation of haplotype frequencies. Bioinformatics. 2008;24:1596–1602. doi: 10.1093/bioinformatics/btn236. [DOI] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat. Rev. Genet. 2011;12:703–714. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GK. A scalable and portable framework for massively parallel variable selection in genetic association studies. Bioinformatics. 2012;28:719–720. doi: 10.1093/bioinformatics/bts015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, et al. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K. Optimization. Springer Texts in Statistics. New York: Springer; 2004. [Google Scholar]
- Li Y, et al. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, et al. Graphics processing units and high-dimensional optimization. Stat. Sci. 2010;25:311–324. doi: 10.1214/10-STS336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.