

Abstract
Prostate cancer is the fifth most common cancer overall in the world. Androgen ablation therapy is the primary treatment for metastatic prostate cancer. However, most prostate cancer patients receiving the androgen ablation therapy ultimately develop recurrent castration-resistant tumors within 1–3 years after treatment. The median overall survival time is 1–2 years after tumor relapse. Chemotherapy shows little effect on prolonging survival for patients with metastatic hormone-refractory prostate cancer. More than 80% of prostate tumors acquire mutation or deletion of tumor suppressor phosphatase and tensin homolog (PTEN), a negative regulator of PI3K/Akt signaling, indicating that inhibition of PI3K/Akt might be a potential therapy for advanced prostate tumors. Caffeic acid phenethyl ester (CAPE) is a strong antioxidant extracted from honeybee hive propolis. CAPE is a well-known NF-κB inhibitor. CAPE has been used in folk medicine as a potent anti-inflammatory agent. Recent studies indicate that CAPE treatment suppresses tumor growth and Akt signaling in human prostate cancer cells. We discuss the potential of using CAPE as a treatment for patients with advanced prostate cancer targeting Akt signaling pathway in this review article.
Keywords: prostate cancer, caffeic acid phenethyl ester, Akt, LNCaP, PC-3
1. Introduction
Prostate is a gland in the male reproductive system. It secretes a milky or white slightly acidic fluid constituting 50%–75% of semen along with spermatozoa and seminal vesicle fluid. Prostate cancer is the cancer develops in the prostate. Prostate cancer is the second most frequently diagnosed cancer of men and the fifth most common cancer overall in the world. The majority of patients having prostate cancer are over 65 years old and the 5 years survival rate for prostate cancer patients is more than 80%. Nearly 900,000 new cases have been diagnosed in 2008 (GLOBOCAN 2008 database, version 1.2). According to the statistics of Surveillance Epidemiology and End Results (SEER) of National Cancer Institute, more than 24,000 men have been diagnosed with and more than 28,000 men have died of prostate cancer in 2012 in United States. Many factors, including genetics and diet, have been implicated in the development of prostate cancer [1]. Examinations of prostate cancer include physical examination and serum prostate-specific antigen (PSA) test. PSA, also known as kallikrein-3 (KLK3), is a glycoprotein enzyme encoded in humans by the KLK3 gene. PSA level elevates when prostate develop cancer or other diseases [2,3]. PSA is a target gene of androgen receptor (AR) [4,5]. AR plays essential roles in the development of male sex organs and prostate tissues. AR also plays important roles in the development, progression, and metastasis of prostate cancer [6–11]. In prostate cancer cells, AR modulates the expression of proteins regulating cell cycle, survival, and growth [8–14]. AR stimulates the expression of TMPRSS2: ERG, a common gene fusion associated with prostate cancer [15–17]. Elevation of AR mRNA and protein expression has been observed in hormone-refractory prostate tumors compared to the primary androgen-dependent prostate tumors [18–26]. Amplification of the AR locus is reported in nearly one-third of patients developing hormone-refractory prostate cancers [20,23,27–29].
Surgery is often successful for organ-confined prostate cancer. Approximately 20%–40% of patients being treated with radical prostatectomy have tumor recurrence and elevation of serum prostate-specific antigen (PSA) [30]. More than 80% of prostate cancer patients die from bone metastases [31–33]. Bones and lymph nodes are the most common metastatic sites for prostate cancer, and the bone metastases cause severe pain. In 1941, Dr. Charles Huggins discovered that deprivation of androgen caused regression of hormone-responsive metastatic prostate cancer [34]. Since then, androgen ablation therapy has become the primary treatment for metastatic prostate cancer. Current androgen ablation therapy uses luteinizing hormone-releasing hormone agonists (LH-RH) (also known as gonadotropin-releasing hormone, GnRH) [35]. However, the majority of prostate cancer patients receiving androgen ablation therapy develop recurrent castration-resistant tumors within 1–3 years after treatment. The median overall survival time is 1–2 years after cancer relapse [8,36]. Chemotherapy is often used to treat metastatic hormone-refractory prostate cancer [37]. Commonly used chemotherapeutic drugs for prostate cancers include etoposide, paclitaxel, vinblastine, mitoxantrone, and estramustine. Etoposide and mitoxantrone are type II topoisomerase inhibitor [37,38]. Estramustine is a derivative of estrogen with a nitrogen mustard-carbamate ester moiety [37]. Vinblastine binds tubulin and inhibits assembly of microtubules [37]. Paclitaxel disrupts mitotic spindle assembly, chromosome segregation, and cell division. Paclitaxel also stabilizes the microtubule polymer and protects it from disassembly [37]. Treatments with these chemotherapeutic drugs result in decrease of PSA, radiographic response, and improvement of pain and urinary symptoms in a sub-group of patients. However, chemotherapies show little effect on prolonging survival [37]. Undesired side effects of these chemotherapeutic agents include toxic deaths, strokes, thrombosis, neutropenia, edema, dyspnea, malaise, and fatigue [37]. Therefore, alternative treatments for advanced prostate cancers are needed. The aim of this review article is to investigate the possibility of using caffeic acid phenethyl ester (CAPE), a pure natural compound extracted from honeybee hive propolis, for treatment of patients with advanced prostate cancer via inhibition of Akt signaling.
2. Akt Signaling and Prostate Cancer
Akt is a serine/threonine protein kinase regulating a variety of cellular responses, including inhibition of apoptosis and stimulation of cell proliferation. There are three mammalian isoforms of this enzyme, Akt1, Akt2, and Akt3 [39,40]. Phosphatase and tensin homolog (PTEN) protein acts as a phosphatase to dephosphorylate phosphatidylinositol (3,4,5)-trisphosphate. PTEN negatively controls the phosphoinositide 3-kinase/Akt signaling pathway [41]. PTEN is one of the most commonly mutated tumor suppressor genes in human cancers. PTEN is frequently deleted or mutated in prostatic intraepithelial neoplasia (PIN) and prostate cancer, resulting in activation of PI3K/Akt signaling [42,43]. Proteins of the ETS family are group of transcription factors regulating cell proliferation, differentiation, and carcinogenesis. In prostate cancer, fusion of ETS-related genes (in most cases, the ERG) with AR-regulated gene promoter of TMPRSS2 is present in approximately 50% of prostate tumors [44]. Recent studies indicate that gene fusion of TMPRSS2-ERG promotes prostate cancer when PTEN is concurrently lost [44–46]. There are two phosphorylation sites on Akt, threonine 308 and serine 473. Phosphorylation of Thr308 on Akt is activated by PDK1 [47]. Phosphorylation of serine 473 is activated by mTOR kinase, its associated protein rector, and SIN1/MIP1 [48,49].
PI3K/Akt signaling plays important roles in survival and progression of prostate cancer cells [42]. Immunoreactivity assay indicates that level of phospho-Akt correlates with higher Gleason score and immunoreactivity for Ki67 and phospho-epidermal growth factor receptor (EGFR) [50]. EGFR is a member of the ErbB family of receptor tyrosine kinase (RTK) which plays essential role in regulating cell proliferation and signaling transduction [51,52]. Expression levels of phospho-Akt and phospho-mTOR correlate with Gleason score [53]. Up-regulation of PI3K/Akt activity is associated with poor clinical outcome of prostate cancer [43,54–59]. Growth factor signaling pathways have been reported to cross-talk with AR-signaling in prostate cancer cells. Growth factor signaling pathways promote androgen-independent proliferation of prostate cancer cells and activate AR by modifying the phosphorylation status of AR or by altering the expression of coactivators or inhibitors [57,60]. The cross-talks between AR and growth factor signaling pathways sensitize AR to suboptimal level of androgenic stimulation [57,60]. AR transcriptional activity and expression have been reported to be regulated by Akt [61]. Androgens regulate Akt pathway by both genomic and non-genomic effects [61]. Different modes of interaction between the AR and Akt pathways include direct interaction or regulation via downstream Wnt/GSK-3β/β-catenin pathway, NF-κB, and FOXO family members [61]. Activation of PI3 kinase/Akt pathway enhances AR activation in response to low level of androgens [62,63]. Therefore, small molecule inhibitors that can suppress PI3K/Akt signaling with minimal side effects are potential candidates for prostate cancer treatment. There are currently a few drugs targeting Akt signaling under clinical trials for prostate cancer treatments, including agents such as celecoxib, perifosine and genistein [64].
3. Caffeic Acid Phenethyl Ester (CAPE)
Caffeic acid phenethyl ester (CAPE) (Figure 1), a bioactive component extracted from honeybee hive propolis, is a strong antioxidant [65,66]. CAPE is a lipophilic derivative of caffeic acid and a phenolic antioxidant structurally related to 3,4-dihydroxycinnamic acid. CAPE has been used in folk medicine as a potent anti-inflammatory agent and is known to exhibit anti-mitogenic, anti-carcinogenic, anti-inflammatory, anti-viral, and immuno-modulatory properties. CAPE is a well-known NF-κB inhibitor. CAPE treatment at concentrations of 50 μM to 80 μM inhibits the activation of NF-κB via preventing the translocation of p65 unit of NF-κB [66] and preventing the binding between NF-κB and DNA [66].
Figure 1.
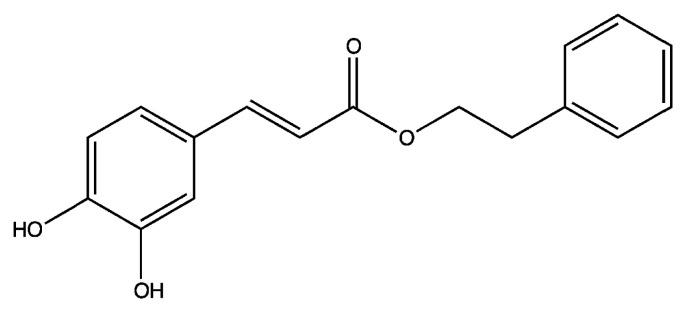
Structure of caffeic acid phenethyl ester (CAPE).
4. Anti-Cancer Effects of CAPE on Human Cancer Cell Lines
CAPE treatment inhibited the transformation of normal cells to cancer cells [67] (Figure 2). CAPE treatment suppressed proliferation of several human cancer cell lines, such as breast [68,69], prostate [70–72], lung [73,74], head and neck [75], cholangio [76], and cervical [77] cancer cells. Non-cancer human cells were much more resistant to CAPE treatment, indicating potential selective cytotoxic effect against cancer cells of CAPE treatment [73,75,78].
Figure 2.

Effects of CAPE treatment on human cancer cells. Treating human cancer cells with CAPE causes suppression of carcinogenesis, cell cycle progression, and cell movement. CAPE also stimulates apoptosis in cancer cells. These changes then give rise to the inhibition of development, growth, and metastasis of tumors.
CAPE treatment caused apoptosis in cancer cells (Figure 2) via stimulation of Bax [69,79–81], Bak [81], p53 [67,77,81], p21cip[77], extracellular signal-regulated kinase (ERKs) [81], c-Jun [77], c-Jun N-terminal kinase (JNK) [69], p38 mitogen-activated protein kinase (p38 MAPK) [69,81], Fas ligand [69], caspase activity [77,79–81]. CAPE treatment decreased expression of Bcl-2 [80,82], cIAP-1, cIAP-2, and XIAP [72,79]. CAPE treatment also reduced release of cytochrome C [80,81] and loss of mitochondrial transmembrane potential [77].
In addition, CAPE treatment caused G1 or G2 cell cycle arrest in several cancer cells (Figure 2) through suppression of protein level of cyclin B1 [73,83], cyclin D1 [70,84,85], cyclin E [85], c-Myc [70,84], S-phase kinase-associated protein 2 (Skp2) [70], phospho-Rb [70,85], and β-catenin [86,87]. CAPE treatment increased protein expression of cyclin dependent kinase inhibitors p21waf1/cip1[70,85], p27Kip1[70,85], and p16INK4A[85].
CAPE treatment hindered motility and invasiveness of cancer cells (Figure 2) via suppression of Akt activity [88], focal adhesion kinase (FAK) activity [89], expression of matrix metalloproteinase MMP-2 and MMP-9, as well as disrupts the arrangement of actin cytoskeleton [89].
5. CAPE Suppresses Tumor Growth and Cancer Metastasis in Animal Models
Oral administration and intraperitoneal (i.p.) injection of CAPE prevented development of colon cancer [90–94] and liver cancer in the mice and the rat models. Intraperitoneal (i.p.) injection of CAPE suppressed tumor growth of melanoma and cholangiocarcinoma xenograft in the mice model [76,95]. Oral administration and i.p. injection of CAPE suppressed metastasis of colon cancer [91,96] and breast cancer [97] in the mice model. Subcutaneous and oral administration of CAPE suppressed liver metastasis of human HepG2 xenografts in the mice model [98].
6. Anticancer Effects of CAPE on Human Prostate Cancer Cells
LNCaP, PC-3, and DU-145 are the most commonly used cell lines for prostate cancer research. LNCaP, PC-3, and DU-145 cell lines are derived from human lymph node, bone, and brain metastatic lesion of prostate adenocarcinoma, respectively [99–102]. LNCaP cells express AR and PSA, while PC-3 and DU-145 cells do not express AR or PSA (Table 1).
Table 1.
Characteristics of commonly used human prostate cancer cell lines. Characteristics, including androgen receptor (AR) level, p53 and phosphatase and tensin homolog (PTEN) status, androgen-dependency, and capability to generate prostate-specific antigen (PSA), of LNCaP 104-S, PC-3, and DU-145 cells are listed.
| LNCaP 104-S | PC-3 | DU-145 | |
|---|---|---|---|
| AR | + | − | − |
| p53 | + | − | mutant |
| PTEN | − | − | + |
| androgen-dependency | dependent | independent | independent |
| androgen effect on cell proliferation | stimulation | no response | no response |
| differentiation | well | poorly | poorly |
| originality | lymph-node metastases | bone-metastases | brain-metastases |
| PSA production | + | − | − |
| EC50 to CAPE treatment (μM) | 0.68 | 18.65 | 9.54 |
Our previous studies suggested that the proliferation of LNCaP 104-S, DU-145, and PC-3 human prostate cancer cells was dosage-dependently suppressed by CAPE treatment (IC50 0.68 μM, 9.54 μM, and 18.65 μM, respectively) [70,71]. LNCaP 104-S is an androgen-dependent subline isolated from LNCaP FGC which is very sensitive to androgen treatment [103–105]. The growth inhibitory effects of CAPE treatment on LNCaP 104-S and PC-3 cells happened within 24 h following CAPE treatment and accumulated over time [70,71]. Treatment with 10 μM CAPE significantly inhibited the formation of LNCaP 104-S and PC-3 colonies in soft agar [70,71]. Flow cytometric analysis revealed that treatment with 3 μM to 20 μM CAPE reduced S phase cell population and caused G1 cell cycle arrest in LNCaP 104-S and PC-3 cells [70,71]. CAPE treatment at high concentrations (88 μM to 176 μM) induced apoptosis in PC-3 cells [72]. Administration of CAPE by gavage (10 mg/kg body weight per day) for six weeks resulted in 50% reduction of LNCaP 104-S xenografts tumor volume in nude mice (p = 0.0008) [70]. Intraperitoneal injection (i.p.) of CAPE (10 mg/kg body weight per day) twice a week for 5 weeks reduced 33% of PC-3 xenografts tumor volume in nude mice (p < 0.05, unpublished data, Figure 3).
Figure 3.

Effects of CAPE treatment on tumor growth of PC-3 xenografts in nude mice. Experiments involving mice were approved by the Institutional Animal Care and Use Committee of National Health Research Institutes. 6–8 week old male Balb/c nu/nu mice were injected subcutaneously in both flanks with 1 × 106 PC-3 cells suspended in 0.5 mL of Matrigel (BD Bioscience, Franklin Lakes, NJ, USA). Tumors were allowed to grow for two weeks until the average tumor volume reached approximately 150 mm3. CAPE (10 mg/kg dissolved in DMSO and diluted in 0.1 mL PBS) or vehicle (DMSO with PBS) was then administered to mice by intraperitoneal injection (i.p.) twice a week for 5 weeks. Tumors were measured weekly using the formula: volume = length × width × height × 0.52. The CAPE treatment group comprised five mice with nine tumors while the vehicle control group comprised five mice with eight tumors. Tumor volume was shown as volume plus standard error (SE).
The enzyme steroid 5α-reductase is responsible for the conversion of testosterone to 5α-dihydrotestosterone (DHT). In men, approximately 5% of testosterone undergoes 5α-reduction to form the more potent androgen, the DHT. 5α-reductase synthesizes DHT in the prostate, testes, hair follicles, and adrenal glands. Therefore, 5α-reductase activity is critical for male sexual differentiation and may be involved in the development of benign prostatic hyperplasia, alopecia, hirsutism, and prostate cancer [106]. CAPE treatment suppresses activity of type 1 and type 2 of 5α-reductase (IC50 8 μM and 7 μM, respectively) [106]. Co-treatment of low concentrations (2.5 μM to 20 μM) of CAPE with chemotherapeutic drugs vinblastine, paclitaxol, or estramustine indicated synergistic suppression effect [71]. These observations suggested that CAPE administration may be useful for the treatment of prostate cancer and androgen-dependent disorders.
7. CAPE Inhibits Akt Signaling in Prostate Cancer Cells
Although CAPE is a well-known NF-κB specific inhibitor, it does not affect NF-κB activity in LNCaP 104-S or PC-3 cells at concentration lower than 20 μM [70–72]. CAPE has previously been shown to suppress Akt signaling in human T cells, coronary smooth muscle cells, and lung cancer cells [88,107,108]. The PTEN in LNCaP cells is mutated, while PC-3 cells acquire a homozygous deletion of PTEN. Therefore, Akt is constantly active in these two cells. Reduction of PDK1 and mTOR activity contribute to the decrease of phosphorylation on Akt. The activities of GSK3α and GSK3β are inhibited by Akt-mediated phosphorylation at Ser21 and Ser9 respectively, limiting their ability to phosphorylate cell cycle regulating proteins, such as cyclin D1 and p21Cip1[109,110]. GSK3β-dependent phosphorylation of cyclin D1 mediates nuclear export and rapid degradation within the cytoplasm of cyclin D1 [111]. Decrease of phosphorylation of GSK3α and GSK3β due to decreased phosphorylation of Akt promotes elevated activity of GSK3α and GSK3β Upregulation of GSK3β activity decreases the abundance of cyclin D1. SGK, like Akt, promotes proliferation through phosphorylation-mediated inactivation of the forkhead transcription factor FoxO3 [112]. Skp2, a member of the F-box protein family, is responsible for ubiquitination and down-regulation of p27Kip cell cycle inhibitors. Skp2 promotes cell cycle progression from G1 phase to S phase. Phosphorylation of Cdk2 on T160 is necessary for its activation [113] and is required for traversing the G1/S checkpoint through phosphorylation of Rb. Phosphorylation of c-Raf on S259 and S621 creates 14-3-3 binding sites which are thought to maintain it in an auto-inhibited state [114]. Treatment of 10 μM CAPE significantly decreased protein abundance of signaling protein involved in Akt signaling and cell cycle regulation, including Akt1, Akt2, Akt3, cyclin A, cyclin D1, cyclin E, SKP2, c-Myc, mTOR, Rb, Bcl-2, phospho-Akt Ser473, phospho-Akt Thr308, phospho-glycogen synthase kinase 3 alpha (Gsk3α Ser21, phospho-Gsk3β Ser9, phospho-mTOR Ser2448, phospho-mTOR Ser2481, phospho-PDK1 Ser241, phospho-Cdk2 Thr160, Ser259, and phospho-serum and glucocorticoid-inducible kinase (SGK) Ser78 in LNCaP 104-S and PC-3 cells [70,71]. CAPE treatment stimulated the protein expression of cell cycle inhibitor p21Cip1 and p27Kip1 as well as phospho-p38 MAPK Thr180/Tyr182 and phospho-p90RSK Ser380 [70,71]. Consistent with inactivation of Cdk2, phospho-Rb Ser807/811 was decreased by 70% under CAPE treatment. CAPE treatment led to increased levels of phospho-c-Raf Ser259. Down-regulation of cyclin A, c-Myc, Skp2, phospho-Rb, and Cdk2, coupled with increased phospho-c-Raf Ser259, p21Cip and p27Kip1 abundance likely contributed to the sustained induction of G1 cell cycle arrest following CAPE treatment in LNCaP 104-S and PC-3 prostate cancer cells (Figure 4).
Figure 4.

Putative model of anti-cancer effect of CAPE in human prostate cancer cells. Protein abundance or activity being stimulated by CAPE treatment are labeled with red upward arrows, while those being suppressed by CAPE treatment are labeled with blue downward arrows.
Insulin or insulin-like growth factor-1 IGF-I receptor signaling pathway activates phosphoinositide 3-kinase (PI3-kinase) and its downstream target Akt [115–117]. The activation of Akt by IGF-1 treatment also promotes protein synthesis via stimulation of mammalian target of rapamycin (mTOR) and p70 S6 kinase, both are downstream targets of Akt, and thereby inhibiting activity of glycogen synthase kinase 3 (GSK3) [118,119]. Stimulating LNCaP 104-S or PC-3 cells with 200 ng/mL IGF-1, the ligand for IGF-1 receptor and an up-stream agonist primarily for the PI3K-Akt signaling pathway, partially rescued the suppressive effect of CAPE (unpublished data, Figure 5A,C). Treating LNCaP 104-S cells with 200 ng/mL epidermal growth factor (EGF), the ligand for EGFR and a potent agonist for the MAPK and Stat pathways, did not block the suppressive effect of CAPE (Figure 5A). Adding PI3K inhibitor LY294002 with CAPE to LNCaP cells caused only moderate additive growth inhibition compared to CAPE treatment alone (unpublished data, Figure 5B). Co-treatment of CAPE with LY294002 did not induce additive growth inhibition compared to LY294002 treatment alone in PC-3 cells (Figure 5C). Taken together, our results suggest that the PI3K-Akt pathway is a major pathway target for CAPE treatment although CAPE treatment alone is not able to completely block PI3K/Akt signaling in human prostate cancer cells.
Figure 5.

CAPE treatment targets PI3K/Akt signaling pathway. (A) LNCaP 104-S cells were treated with 0, 1, 3, or 10 μM CAPE in the presence and absence of 200 ng IGF-1 or 200 ng EGF for 96 h; (B) LNCaP 104-S cells was treated with CAPE (0, 1, 3, or 10 μM) and LY294002 (0, 1, 5, or 10 μM) for 96 h; (C) PC-3 cells was treated with different combinations of CAPE (10 μM), LY294002 (10 μM), and IGF-1 (200 ng/mL) for 72 h. Cell number was determined by 96-well proliferation assay. (*) represents statistically significant difference (p < 0.05) between the groups treated with IGF-1 or LY294002 compared to the control without IGF-1 or LY294002 treatment at each concentration of CAPE.
8. Potential Clinical Application of CAPE
Oral administration of 10 mg/kg CAPE for six weeks caused 50% reduction of tumor growth of LNCaP xenografts while i.p. injection of 10 mg/kg of CAPE for five weeks caused 33% reduction in tumor growth of PC-3 xenografts. The EC50 of 96 h CAPE treatment to cause growth inhibition in advanced human prostate cancer cell lines was approximately 0.7 μM to 18.7 μM [70,71]. The achievable concentration of CAPE in human serum is approximately 17 μM [120]. It is therefore possible to use CAPE as an adjuvant therapeutic agent for advanced prostate cancers.
The pharmacokinetic profile of CAPE was determined in rats after intravenous (i.v.) administration of 5, 10 or 20 mg/kg. The plasma concentration of CAPE was measured with liquid chromatography tandem mass spectrometry and was estimated using non compartmental analysis (NCA) and biexponential fit [121]. Total body clearance values for CAPE ranged from 42.1 to 172 mL/min/kg and decreased with the increasing dose of CAPE. The volume of distribution values for CAPE ranged from 1555 to 5209 mL/kg and decreased with increasing dose. The elimination half-life for CAPE ranged from 21.2 to 26.7 min and was independent of dose [121]. This study suggested that CAPE was distributed extensively into animal tissues and was eliminated rapidly with a short half life. Intraperitoneal injection of CAPE at 10–30 mg/kg for 7 days did not affect mice body weight [95]. Plasma alanine amino transferase (ALT) as well as free thiol content and lipid peroxidation in the liver and kidney tissue were measured to study the in vivo efficacy and toxicology of CAPE treatment in mice [95]. Seven days of i.p. injection of 10 mg/kg of CAPE showed no toxicity while i.p. injection of higher dose (20 and 30 mg/kg) CAPE caused mild dose-dependent liver and kidney toxicity. Therefore, CAPE treatment of dosage lower than 10 mg/kg is plausible for clinical trial in patients.
CAPE treatments have been shown to sensitize cancer cells to chemotherapeutic drugs and radiation treatment by inhibiting pathways that lead to treatment resistance in animal models [122]. CAPE is a protective agent from therapy-associated toxicities in animal models [122]. Doxorubicin is a chemotherapy drug used for hematological malignancies with side effects including acute renal failure [122]. CAPE treatment protected renal, heart, and brain tissues damages caused by doxorubicin treatment in rats [123–125]. Cisplatin is one of the most widely used chemotherapeutic agents for treatment of solid tumors. CAPE treatment protected liver damage caused by cisplatin treatment [126,127]. Methotrexate is an anti-metabolite and anti-folate drug used in treatment of cancer, autoimmune diseases, ectopic pregnancy, and induction of medical abortions. CAPE treatment protected methotrexate-induced renal oxidative impairment in rats [128]. Bleomycin is a glycopeptide antibiotic generated from bacterium Streptomyces verticillus. Bleomycin causes breaks in DNA and is used as a chemotherapy agent for several types of cancers. CAPE treatment inhibited bleomycin-induced lung fibrosis [129]. Tamoxifen is an antagonist of the estrogen receptor (ER) in breast tissue via its active metabolite, hydroxytamoxifen. Tamoxifen is the most common treatment for ER-positive breast cancer in pre-menopausal women. CAPE treatment significantly prevented liver toxicity induced by Tamoxifen treatment [130]. Co-treatment with CAPE and five chemotherapeutic agents commonly used in prostate cancers showed additive suppressive effects in PC-3 cells [71]. CAPE treatment attenuated radiation treatment-induced pulmonary injury in vivo[131]. CAPE treatment sensitized colorectal adenocarcinomas to radiation treatment without affecting bone marrow radio-response in animal model [132]. Therefore, treatment with CAPE not only may suppress tumor growth in patients but also may protect patients from chemotherapy or radiation therapy.
9. Conclusions
According to the above summaries in this review, there are strong evidences that CAPE treatment suppresses tumor growth and Akt signaling in human prostate cancer cells. CAPE treatment reduces the dosage of chemotherapeutic agents required and protects organ damages and toxicity induced by various kinds of cancer chemotherapy drugs or radiation therapies. Therefore, CAPE is a potential treatment for advanced prostate cancer targeting Akt signaling.
Acknowledgments
This work was supported by CS-101-PP-14 and CS-102-PP-14 (National Health Research Institutes), NSC 99-2320-B-400-015-MY3 and NSC 101-2325-B-400-014 (National Science Council) in Taiwan for CPC. CYL is supported by DOH101-TD-C-111-004 (Department of Health). This article is dedicated to our dear mentor Shutsung Liao from Ben May Department for Cancer Research of The University of Chicago for his retirement. He is a member of America Academy of Art & Science (USA) and academician of Academia Sinica (Taiwan).
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Karan D., Thrasher J.B., Lubaroff D. Prostate cancer: Genes, environment, immunity and the use of immunotherapy. Prostate Cancer Prostatic Dis. 2008;11:230–236. doi: 10.1038/pcan.2008.3. [DOI] [PubMed] [Google Scholar]
- 2.Kuriyama M., Wang M.C., Lee C.I., Papsidero L.D., Killian C.S., Inaji H., Slack N.H., Nishiura T., Murphy G.P., Chu T.M. Use of human prostate-specific antigen in monitoring prostate cancer. Cancer Res. 1981;41:3874–3876. [PubMed] [Google Scholar]
- 3.Nadji M., Tabei S.Z., Castro A., Chu T.M., Murphy G.P., Wang M.C., Morales A.R. Prostatic-Specific antigen: An immunohistologic marker for prostatic neoplasms. Cancer. 1981;48:1229–1232. doi: 10.1002/1097-0142(19810901)48:5<1229::aid-cncr2820480529>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 4.Riegman P.H., Vlietstra R.J., van der Korput J.A., Brinkmann A.O., Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol. Endocrinol. 1991;5:1921–1930. doi: 10.1210/mend-5-12-1921. [DOI] [PubMed] [Google Scholar]
- 5.Wolf D.A., Schulz P., Fittler F. Transcriptional regulation of prostate kallikrein-like genes by androgen. Mol. Endocrinol. 1992;6:753–762. doi: 10.1210/mend.6.5.1376410. [DOI] [PubMed] [Google Scholar]
- 6.Feldman B.J., Feldman D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 7.Ricke E.A., Williams K., Lee Y.F., Couto S., Wang Y., Hayward S.W., Cunha G.R., Ricke W.A. Androgen hormone action in prostatic carcinogenesis: Stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis. 2012;33:1391–1398. doi: 10.1093/carcin/bgs153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuu C.P., Kokontis J.M., Hiipakka R.A., Fukuchi J., Lin H.P., Lin C.Y., Huo C., Su L.C. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci. 2011;18:63. doi: 10.1186/1423-0127-18-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuu C.P., Hiipakka R.A., Fukuchi J., Kokontis J.M., Liao S. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res. 2005;65:2082–2084. doi: 10.1158/0008-5472.CAN-04-3992. [DOI] [PubMed] [Google Scholar]
- 10.Chuu C.P., Hiipakka R.A., Kokontis J.M., Fukuchi J., Chen R.Y., Liao S. Inhibition of tumor growth and progression of LNCaP prostate cancer cells in athymic mice by androgen and liver X receptor agonist. Cancer Res. 2006;66:6482–6486. doi: 10.1158/0008-5472.CAN-06-0632. [DOI] [PubMed] [Google Scholar]
- 11.Chuu C.P., Kokontis J.M., Hiipakka R.A., Fukuchi J., Lin H.P., Lin C.Y., Huo C., Su L.C., Liao S. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci. 2011;102:2022–2028. doi: 10.1111/j.1349-7006.2011.02043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q., Li W., Liu X.S., Carroll J.S., Janne O.A., Keeton E.K., Chinnaiyan A.M., Pienta K.J., Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y., Chen S.Y., Ross K.N., Balk S.P. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q., Li W., Zhang Y., Yuan X., Xu K., Yu J., Chen Z., Beroukhim R., Wang H., Lupien M., et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.W., Varambally S., Cao X., Tchinda J., Kuefer R., et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 16.Cerveira N., Ribeiro F.R., Peixoto A., Costa V., Henrique R., Jeronimo C., Teixeira M.R. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–832. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai C., Wang H., Xu Y., Chen S., Balk S.P. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69:6027–6032. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Kwast T.H., Schalken J., Ruizeveld de Winter J.A., van Vroonhoven C.C., Mulder E., Boersma W., Trapman J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer. 1991;48:189–193. doi: 10.1002/ijc.2910480206. [DOI] [PubMed] [Google Scholar]
- 19.Ruizeveld de Winter J.A., Janssen P.J., Sleddens H.M., Verleun-Mooijman M.C., Trapman J., Brinkmann A.O., Santerse A.B., Schroder F.H., van der Kwast T.H. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol. 1994;144:735–746. [PMC free article] [PubMed] [Google Scholar]
- 20.Visakorpi T., Hyytinen E., Koivisto P., Tanner M., Keinanen R., Palmberg C., Palotie A., Tammela T., Isola J., Kallioniemi O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 21.Bubendorf L., Kononen J., Koivisto P., Schraml P., Moch H., Gasser T.C., Willi N., Mihatsch M.J., Sauter G., Kallioniemi O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59:803–806. [PubMed] [Google Scholar]
- 22.Linja M.J., Savinainen K.J., Saramaki O.R., Tammela T.L., Vessella R.L., Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 23.Ford O.H., III, Gregory C.W., Kim D., Smitherman A.B., Mohler J.L. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J. Urol. 2003;170:1817–1821. doi: 10.1097/01.ju.0000091873.09677.f4. [DOI] [PubMed] [Google Scholar]
- 24.Holzbeierlein J., Lal P., LaTulippe E., Smith A., Satagopan J., Zhang L., Ryan C., Smith S., Scher H., Scardino P., et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohler J.L., Gregory C.W., Ford O.H., III, Kim D., Weaver C.M., Petrusz P., Wilson E.M., French F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 26.Stanbrough M., Bubley G.J., Ross K., Golub T.R., Rubin M.A., Penning T.M., Febbo P.G., Balk S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 27.Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T., et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- 28.Brown R.S., Edwards J., Dogan A., Payne H., Harland S.J., Bartlett J.M., Masters J.R. Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J. Pathol. 2002;198:237–244. doi: 10.1002/path.1206. [DOI] [PubMed] [Google Scholar]
- 29.Edwards J., Krishna N.S., Grigor K.M., Bartlett J.M. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br. J. Cancer. 2003;89:552–556. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadar M.D. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011;71:1208–1213. doi: 10.1158/0008-5472.CAN_10-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubendorf L., Schopfer A., Wagner U., Sauter G., Moch H., Willi N., Gasser T.C., Mihatsch M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 32.Ibrahim T., Flamini E., Mercatali L., Sacanna E., Serra P., Amadori D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer. 2010;116:1406–1418. doi: 10.1002/cncr.24896. [DOI] [PubMed] [Google Scholar]
- 33.Keller E.T., Zhang J., Cooper C.R., Smith P.C., McCauley L.K., Pienta K.J., Taichman R.S. Prostate carcinoma skeletal metastases: Cross-Talk between tumor and bone. Cancer Metastasis Rev. 2001;20:333–349. doi: 10.1023/a:1015599831232. [DOI] [PubMed] [Google Scholar]
- 34.Huggins C., Stevens R., Hodges C. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941;43:15. [Google Scholar]
- 35.Seruga B., Tannock I.F. Intermittent androgen blockade should be regarded as standard therapy in prostate cancer. Nat. Clin. Pract. Oncol. 2008;5:574–576. doi: 10.1038/ncponc1180. [DOI] [PubMed] [Google Scholar]
- 36.Hellerstedt B.A., Pienta K.J. The current state of hormonal therapy for prostate cancer. CA Cancer J. Clin. 2002;52:154–179. doi: 10.3322/canjclin.52.3.154. [DOI] [PubMed] [Google Scholar]
- 37.Gilligan T., Kantoff P.W. Chemotherapy for prostate cancer. Urology. 2002;60:94–100. doi: 10.1016/s0090-4295(02)01583-2. [DOI] [PubMed] [Google Scholar]
- 38.Pinto A.C., Moreira J.N., Simoes S. Liposomal imatinib-mitoxantrone combination: Formulation development and therapeutic evaluation in an animal model of prostate cancer. Prostate. 2011;71:81–90. doi: 10.1002/pros.21224. [DOI] [PubMed] [Google Scholar]
- 39.Coffer P.J., Jin J., Woodgett J.R. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez E., McGraw T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell. Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantley L.C., Neel B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarker D., Reid A.H., Yap T.A., de Bono J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 43.Bedolla R., Prihoda T.J., Kreisberg J.I., Malik S.N., Krishnegowda N.K., Troyer D.A., Ghosh P.M. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of PTEN expression and Akt activation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007;13:3860–3867. doi: 10.1158/1078-0432.CCR-07-0091. [DOI] [PubMed] [Google Scholar]
- 44.Squire J.A. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat. Genet. 2009;41:509–510. doi: 10.1038/ng0509-509. [DOI] [PubMed] [Google Scholar]
- 45.King J.C., Xu J., Wongvipat J., Hieronymus H., Carver B.S., Leung D.H., Taylor B.S., Sander C., Cardiff R.D., Couto S.S., et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009;41:524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carver B.S., Tran J., Gopalan A., Chen Z., Shaikh S., Carracedo A., Alimonti A., Nardella C., Varmeh S., Scardino P.T., et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alessi D.R., James S.R., Downes C.P., Holmes A.B., Gaffney P.R., Reese C.B., Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 48.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 49.Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S.Y., Huang Q., Qin J., Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 50.Hammarsten P., Cipriano M., Josefsson A., Stattin P., Egevad L., Granfors T., Fowler C.J. Phospho-Akt immunoreactivity in prostate cancer: Relationship to disease severity and outcome, Ki67 and phosphorylated EGFR expression. PLoS One. 2012;7:e47994. doi: 10.1371/journal.pone.0047994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuu C.P., Chen R.Y., Barkinge J.L., Ciaccio M.F., Jones R.B. Systems-Level analysis of ErbB4 signaling in breast cancer: A laboratory to clinical perspective. Mol. Cancer Res. 2008;6:885–891. doi: 10.1158/1541-7786.MCR-07-0369. [DOI] [PubMed] [Google Scholar]
- 52.Morgan T.M., Koreckij T.D., Corey E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr. Cancer Drug Targets. 2009;9:237–249. doi: 10.2174/156800909787580999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai B., Kong Y.Y., Ye D.W., Ma C.G., Zhou X., Yao X.D. Activation of the mammalian target of rapamycin signalling pathway in prostate cancer and its association with patient clinicopathological characteristics. BJU Int. 2009;104:1009–1016. doi: 10.1111/j.1464-410X.2009.08538.x. [DOI] [PubMed] [Google Scholar]
- 54.Kreisberg J.I., Malik S.N., Prihoda T.J., Bedolla R.G., Troyer D.A., Kreisberg S., Ghosh P.M. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64:5232–5236. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 55.Sircar K., Yoshimoto M., Monzon F.A., Koumakpayi I.H., Katz R.L., Khanna A., Alvarez K., Chen G., Darnel A.D., Aprikian A.G., et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009;218:505–513. doi: 10.1002/path.2559. [DOI] [PubMed] [Google Scholar]
- 56.Wegiel B., Bjartell A., Culig Z., Persson J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer. 2008;122:1521–1529. doi: 10.1002/ijc.23261. [DOI] [PubMed] [Google Scholar]
- 57.McCall P., Gemmell L.K., Mukherjee R., Bartlett J.M., Edwards J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Br. J. Cancer. 2008;98:1094–1101. doi: 10.1038/sj.bjc.6604152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimizu Y., Segawa T., Inoue T., Shiraishi T., Yoshida T., Toda Y., Yamada T., Kinukawa N., Terada N., Kobayashi T., et al. Increased Akt and phosphorylated Akt expression are associated with malignant biological features of prostate cancer in Japanese men. BJU Int. 2007;100:685–690. doi: 10.1111/j.1464-410X.2007.07014.x. [DOI] [PubMed] [Google Scholar]
- 59.Ayala G., Thompson T., Yang G., Frolov A., Li R., Scardino P., Ohori M., Wheeler T., Harper W. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004;10:6572–6578. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- 60.Orio F., Jr, Terouanne B., Georget V., Lumbroso S., Avances C., Siatka C., Sultan C. Potential action of IGF-1 and EGF on androgen receptor nuclear transfer and transactivation in normal and cancer human prostate cell lines. Mol. Cell. Endocrinol. 2002;198:105–114. doi: 10.1016/s0303-7207(02)00374-x. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y., Kreisberg J.I., Ghosh P.M. Cross-Talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets. 2007;7:591–604. doi: 10.2174/156800907781662248. [DOI] [PubMed] [Google Scholar]
- 62.Culig Z., Hobisch A., Cronauer M.V., Radmayr C., Trapman J., Hittmair A., Bartsch G., Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 63.Fan W., Yanase T., Morinaga H., Okabe T., Nomura M., Daitoku H., Fukamizu A., Kato S., Takayanagi R., Nawata H. Insulin-Like growth factor 1/insulin signaling activates androgen signaling through direct interactions of Foxo1 with androgen receptor. J. Biol. Chem. 2007;282:7329–7338. doi: 10.1074/jbc.M610447200. [DOI] [PubMed] [Google Scholar]
- 64.Nelson E.C., Evans C.P., Mack P.C., Devere-White R.W., Lara P.N., Jr Inhibition of Akt pathways in the treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:331–339. doi: 10.1038/sj.pcan.4500974. [DOI] [PubMed] [Google Scholar]
- 65.Bhimani R.S., Troll W., Grunberger D., Frenkel K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res. 1993;53:4528–4533. [PubMed] [Google Scholar]
- 66.Natarajan K., Singh S., Burke T.R., Jr, Grunberger D., Aggarwal B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nomura M., Kaji A., Ma W., Miyamoto K., Dong Z. Suppression of cell transformation and induction of apoptosis by caffeic acid phenethyl ester. Mol. Carcinog. 2001;31:83–89. doi: 10.1002/mc.1043. [DOI] [PubMed] [Google Scholar]
- 68.Wu J., Omene C., Karkoszka J., Bosland M., Eckard J., Klein C.B., Frenkel K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011;308:43–53. doi: 10.1016/j.canlet.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watabe M., Hishikawa K., Takayanagi A., Shimizu N., Nakaki T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J. Biol. Chem. 2004;279:6017–6026. doi: 10.1074/jbc.M306040200. [DOI] [PubMed] [Google Scholar]
- 70.Chuu C.P., Lin H.P., Ciaccio M.F., Kokontis J.M., Hause R.J., Jr, Hiipakka R.A., Liao S., Jones R.B. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prev. Res. (Phila) 2012;5:788–797. doi: 10.1158/1940-6207.CAPR-12-0004-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin H.P., Jiang S.S., Chuu C.P. Caffeic acid phenethyl ester causes p21 induction, Akt Signaling reduction, and growth inhibition in PC-3 human prostate cancer cells. PLoS One. 2012;7:e31286. doi: 10.1371/journal.pone.0031286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McEleny K., Coffey R., Morrissey C., Fitzpatrick J.M., Watson R.W. Caffeic acid phenethyl ester-induced PC-3 cell apoptosis is caspase-dependent and mediated through the loss of inhibitors of apoptosis proteins. BJU Int. 2004;94:402–406. doi: 10.1111/j.1464-410X.2004.04936.x. [DOI] [PubMed] [Google Scholar]
- 73.Chen M.F., Wu C.T., Chen Y.J., Keng P.C., Chen W.C. Cell killing and radiosensitization by caffeic acid phenethyl ester (CAPE) in lung cancer cells. J. Radiat. Res. (Tokyo) 2004;45:253–260. doi: 10.1269/jrr.45.253. [DOI] [PubMed] [Google Scholar]
- 74.Lin H.P., Kuo L.K., Chuu C.P. Combined treatment of curcumin and small molecule inhibitors suppresses proliferation of A549 and H1299 human non-small-cell lung cancer cells. Phytother. Res. 2011;26:122–126. doi: 10.1002/ptr.3523. [DOI] [PubMed] [Google Scholar]
- 75.Lee Y.T., Don M.J., Hung P.S., Shen Y.C., Lo Y.S., Chang K.W., Chen C.F., Ho L.K. Cytotoxicity of phenolic acid phenethyl esters on oral cancer cells. Cancer Lett. 2005;223:19–25. doi: 10.1016/j.canlet.2004.09.048. [DOI] [PubMed] [Google Scholar]
- 76.Onori P., DeMorrow S., Gaudio E., Franchitto A., Mancinelli R., Venter J., Kopriva S., Ueno Y., Alvaro D., Savage J., et al. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF-kappaB and induction of apoptosis. Int. J. Cancer J. Int. Cancer. 2009;125:565–576. doi: 10.1002/ijc.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hung M.W., Shiao M.S., Tsai L.C., Chang G.G., Chang T.C. Apoptotic effect of caffeic acid phenethyl ester and its ester and amide analogues in human cervical cancer ME180 cells. Anticancer Res. 2003;23:4773–4780. [PubMed] [Google Scholar]
- 78.Usia T., Banskota A.H., Tezuka Y., Midorikawa K., Matsushige K., Kadota S. Constituents of Chinese propolis and their antiproliferative activities. J. Nat. Prod. 2002;65:673–676. doi: 10.1021/np010486c. [DOI] [PubMed] [Google Scholar]
- 79.Chen Y.J., Shiao M.S., Hsu M.L., Tsai T.H., Wang S.Y. Effect of caffeic acid phenethyl ester, an antioxidant from propolis, on inducing apoptosis in human leukemic HL-60 cells. J. Agric. Food Chem. 2001;49:5615–5619. doi: 10.1021/jf0107252. [DOI] [PubMed] [Google Scholar]
- 80.Jin U.H., Song K.H., Motomura M., Suzuki I., Gu Y.H., Kang Y.J., Moon T.C., Kim C.H. Caffeic acid phenethyl ester induces mitochondria-mediated apoptosis in human myeloid leukemia U937 cells. Mol. Cell Biochem. 2008;310:43–48. doi: 10.1007/s11010-007-9663-7. [DOI] [PubMed] [Google Scholar]
- 81.Lee Y.J., Kuo H.C., Chu C.Y., Wang C.J., Lin W.C., Tseng T.H. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem. Pharmacol. 2003;66:2281–2289. doi: 10.1016/j.bcp.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 82.Su Z.Z., Lin J., Grunberger D., Fisher P.B. Growth suppression and toxicity induced by caffeic acid phenethyl ester (CAPE) in type 5 adenovirus-transformed rat embryo cells correlate directly with transformation progression. Cancer Res. 1994;54:1865–1870. [PubMed] [Google Scholar]
- 83.Lin Y.H., Chiu J.H., Tseng W.S., Wong T.T., Chiou S.H., Yen S.H. Antiproliferation and radiosensitization of caffeic acid phenethyl ester on human medulloblastoma cells. Cancer Chemother Pharmacol. 2006;57:525–532. doi: 10.1007/s00280-005-0066-8. [DOI] [PubMed] [Google Scholar]
- 84.He Y.J., Liu B.H., Xiang D.B., Qiao Z.Y., Fu T., He Y.H. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J. Gastroenterol. 2006;12:4981–4985. doi: 10.3748/wjg.v12.i31.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuo H.C., Kuo W.H., Lee Y.J., Lin W.L., Chou F.P., Tseng T.H. Inhibitory effect of caffeic acid phenethyl ester on the growth of C6 glioma cells in vitro and in vivo. Cancer Lett. 2006;234:199–208. doi: 10.1016/j.canlet.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 86.Wang D., Xiang D.B., He Y.J., Li Z.P., Wu X.H., Mou J.H., Xiao H.L., Zhang Q.H. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of colorectal cancer cells in vitro. World J. Gastroenterol. 2005;11:4008–4012. doi: 10.3748/wjg.v11.i26.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiang D., Wang D., He Y., Xie J., Zhong Z., Li Z. Caffeic acid phenethyl ester induces growth arrest and apoptosis of colon cancer cells via the beta-catenin/T-cell factor signaling. Anticancer Drugs. 2006;17:753–762. doi: 10.1097/01.cad.0000224441.01082.bb. [DOI] [PubMed] [Google Scholar]
- 88.Shigeoka Y., Igishi T., Matsumoto S., Nakanishi H., Kodani M., Yasuda K., Hitsuda Y., Shimizu E. Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition. J. Cancer Res. Clin. Oncol. 2004;130:146–152. doi: 10.1007/s00432-003-0520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weyant M.J., Carothers A.M., Bertagnolli M.E., Bertagnolli M.M. Colon cancer chemopreventive drugs modulate integrin-mediated signaling pathways. Clin. Cancer Res. 2000;6:949–956. [PubMed] [Google Scholar]
- 90.Mahmoud N.N., Carothers A.M., Grunberger D., Bilinski R.T., Churchill M.R., Martucci C., Newmark H.L., Bertagnolli M.M. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis. 2000;21:921–927. doi: 10.1093/carcin/21.5.921. [DOI] [PubMed] [Google Scholar]
- 91.Nagaoka T., Banskota A.H., Tezuka Y., Harimaya Y., Koizumi K., Saiki I., Kadota S. Inhibitory effects of caffeic acid phenethyl ester analogues on experimental lung metastasis of murine colon 26-L5 carcinoma cells. Biol. Pharm. Bull. 2003;26:638–641. doi: 10.1248/bpb.26.638. [DOI] [PubMed] [Google Scholar]
- 92.Borrelli F., Izzo A.A., Di Carlo G., Maffia P., Russo A., Maiello F.M., Capasso F., Mascolo N. Effect of a propolis extract and caffeic acid phenethyl ester on formation of aberrant crypt foci and tumors in the rat colon. Fitoterapia. 2002;73:S38–S43. doi: 10.1016/s0367-326x(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 93.Carrasco-Legleu C.E., Sanchez-Perez Y., Marquez-Rosado L., Fattel-Fazenda S., Arce-Popoca E., Hernandez-Garcia S., Villa-Trevino S. A single dose of caffeic acid phenethyl ester prevents initiation in a medium-term rat hepatocarcinogenesis model. World J. Gastroenterol. 2006;12:6779–6785. doi: 10.3748/wjg.v12.i42.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carrasco-Legleu C.E., Marquez-Rosado L., Fattel-Fazenda S., Arce-Popoca E., Perez-Carreon J.I., Villa-Trevino S. Chemoprotective effect of caffeic acid phenethyl ester on promotion in a medium-term rat hepatocarcinogenesis assay. Int. J. Cancer. 2004;108:488–492. doi: 10.1002/ijc.11595. [DOI] [PubMed] [Google Scholar]
- 95.Kudugunti S.K., Vad N.M., Ekogbo E., Moridani M.Y. Efficacy of caffeic acid phenethyl ester (CAPE) in skin B16-F0 melanoma tumor bearing C57BL/6 mice. Investig. New Drugs. 2011;29:52–62. doi: 10.1007/s10637-009-9334-5. [DOI] [PubMed] [Google Scholar]
- 96.Liao H.F., Chen Y.Y., Liu J.J., Hsu M.L., Shieh H.J., Liao H.J., Shieh C.J., Shiao M.S., Chen Y.J. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem. 2003;51:7907–7912. doi: 10.1021/jf034729d. [DOI] [PubMed] [Google Scholar]
- 97.Orsolic N., Knezevic A.H., Sver L., Terzic S., Basic I. Immunomodulatory and antimetastatic action of propolis and related polyphenolic compounds. J. Ethnopharmacol. 2004;94:307–315. doi: 10.1016/j.jep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 98.Chung T.W., Moon S.K., Chang Y.C., Ko J.H., Lee Y.C., Cho G., Kim S.H., Kim J.G., Kim C.H. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: Complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004;18:1670–1681. doi: 10.1096/fj.04-2126com. [DOI] [PubMed] [Google Scholar]
- 99.Horoszewicz J.S., Leong S.S., Chu T.M., Wajsman Z.L., Friedman M., Papsidero L., Kim U., Chai L.S., Kakati S., Arya S.K., et al. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980;37:115–132. [PubMed] [Google Scholar]
- 100.Chuu C.P., Kokontis J.M., Hiipakka R.A., Liao S. Modulation of liver X receptor signaling as novel therapy for prostate cancer. J. Biomed. Sci. 2007;14:543–553. doi: 10.1007/s11373-007-9160-8. [DOI] [PubMed] [Google Scholar]
- 101.Stone K.R., Mickey D.D., Wunderli H., Mickey G.H., Paulson D.F. Isolation of a human prostate carcinoma cell line (DU 145) Int. J. Cancer. 1978;21:274–281. doi: 10.1002/ijc.2910210305. [DOI] [PubMed] [Google Scholar]
- 102.Kaighn M.E., Narayan K.S., Ohnuki Y., Lechner J.F., Jones L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest. Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- 103.Kokontis J., Takakura K., Hay N., Liao S. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 1994;54:1566–1573. [PubMed] [Google Scholar]
- 104.Kokontis J.M., Hay N., Liao S. Progression of LNCaP prostate tumor cells during androgen deprivation: Hormone-Independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol. 1998;12:941–953. doi: 10.1210/mend.12.7.0136. [DOI] [PubMed] [Google Scholar]
- 105.Kokontis J.M., Hsu S., Chuu C.P., Dang M., Fukuchi J., Hiipakka R.A., Liao S. Role of androgen receptor in the progression of human prostate tumor cells to androgen independence and insensitivity. Prostate. 2005;65:287–298. doi: 10.1002/pros.20285. [DOI] [PubMed] [Google Scholar]
- 106.Hiipakka R.A., Zhang H.Z., Dai W., Dai Q., Liao S. Structure-Activity relationships for inhibition of human 5alpha-reductases by polyphenols. Biochem. Pharmacol. 2002;63:1165–1176. doi: 10.1016/s0006-2952(02)00848-1. [DOI] [PubMed] [Google Scholar]
- 107.Wang L.C., Chu K.H., Liang Y.C., Lin Y.L., Chiang B.L. Caffeic acid phenethyl ester inhibits nuclear factor-kappaB and protein kinase B signalling pathways and induces caspase-3 expression in primary human CD4+ T cells. Clin. Exp. Immunol. 2010;160:223–232. doi: 10.1111/j.1365-2249.2009.04067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ho H.C., Hsu S.L., Ting C.T., Kuo C.Y., Yang V.C. Caffeic acid phenethyl ester inhibits arterial smooth muscle cell proliferation and migration in vitro and in vivo using a local delivery system. Cell. Mol. Biol. (Noisy-le-grand) 2009;55:OL1161–1167. [PubMed] [Google Scholar]
- 109.Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 110.Liang J., Slingerland J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 111.Alao J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brunet A., Park J., Tran H., Hu L.S., Hemmings B.A., Greenberg M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol. Cell. Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gu Y., Rosenblatt J., Morgan D.O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11:3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morrison D.K., Heidecker G., Rapp U.R., Copeland T.D. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 1993;268:17309–17316. [PubMed] [Google Scholar]
- 115.Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell. Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 116.Dudek H., Datta S.R., Franke T.F., Birnbaum M.J., Yao R., Cooper G.M., Segal R.A., Kaplan D.R., Greenberg M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 117.Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 118.Rommel C., Bodine S.C., Clarke B.A., Rossman R., Nunez L., Stitt T.N., Yancopoulos G.D., Glass D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 119.Duan C., Liimatta M.B., Bottum O.L. Insulin-Like growth factor (IGF)-I regulates IGF-binding protein-5 gene expression through the phosphatidylinositol 3-kinase, protein kinase B/Akt, and p70 S6 kinase signaling pathway. J. Biol. Chem. 1999;274:37147–37153. doi: 10.1074/jbc.274.52.37147. [DOI] [PubMed] [Google Scholar]
- 120.Celli N., Dragani L.K., Murzilli S., Pagliani T., Poggi A. In vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J. Agric. Food Chem. 2007;55:3398–3407. doi: 10.1021/jf063477o. [DOI] [PubMed] [Google Scholar]
- 121.Wang X., Pang J., Maffucci J.A., Pade D.S., Newman R.A., Kerwin S.M., Bowman P.D., Stavchansky S. Pharmacokinetics of caffeic acid phenethyl ester and its catechol-ring fluorinated derivative following intravenous administration to rats. Biopharm. Drug Dispos. 2009;30:221–228. doi: 10.1002/bdd.657. [DOI] [PubMed] [Google Scholar]
- 122.Akyol S., Ginis Z., Armutcu F., Ozturk G., Yigitoglu M.R., Akyol O. The potential usage of caffeic acid phenethyl ester (CAPE) against chemotherapy-induced and radiotherapy-induced toxicity. Cell. Biochem. Funct. 2012;30:438–443. doi: 10.1002/cbf.2817. [DOI] [PubMed] [Google Scholar]
- 123.Yagmurca M., Erdogan H., Iraz M., Songur A., Ucar M., Fadillioglu E. Caffeic acid phenethyl ester as a protective agent against doxorubicin nephrotoxicity in rats. Clin. Chim. Acta. 2004;348:27–34. doi: 10.1016/j.cccn.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 124.Fadillioglu E., Oztas E., Erdogan H., Yagmurca M., Sogut S., Ucar M., Irmak M.K. Protective effects of caffeic acid phenethyl ester on doxorubicin-induced cardiotoxicity in rats. J. Appl. Toxicol. 2004;24:47–52. doi: 10.1002/jat.945. [DOI] [PubMed] [Google Scholar]
- 125.Irmak M.K., Fadillioglu E., Sogut S., Erdogan H., Gulec M., Ozer M., Yagmurca M., Gozukara M.E. Effects of caffeic acid phenethyl ester and alpha-tocopherol on reperfusion injury in rat brain. Cell. Biochem. Funct. 2003;21:283–289. doi: 10.1002/cbf.1024. [DOI] [PubMed] [Google Scholar]
- 126.Iraz M., Ozerol E., Gulec M., Tasdemir S., Idiz N., Fadillioglu E., Naziroglu M., Akyol O. Protective effect of caffeic acid phenethyl ester (CAPE) administration on cisplatin-induced oxidative damage to liver in rat. Cell. Biochem. Funct. 2006;24:357–361. doi: 10.1002/cbf.1232. [DOI] [PubMed] [Google Scholar]
- 127.Yilmaz H.R., Sogut S., Ozyurt B., Ozugurlu F., Sahin S., Isik B., Uz E., Ozyurt H. The activities of liver adenosine deaminase, xanthine oxidase, catalase, superoxide dismutase enzymes and the levels of malondialdehyde and nitric oxide after cisplatin toxicity in rats: protective effect of caffeic acid phenethyl ester. Toxicol. Ind. Health. 2005;21:67–73. doi: 10.1191/0748233705th216oa. [DOI] [PubMed] [Google Scholar]
- 128.Oktem F., Yilmaz H.R., Ozguner F., Olgar S., Ayata A., Uzare E., Uz E. Methotrexate-induced renal oxidative stress in rats: The role of a novel antioxidant caffeic acid phenethyl ester. Toxicol. Ind. Health. 2006;22:241–247. doi: 10.1191/0748233706th265oa. [DOI] [PubMed] [Google Scholar]
- 129.Ozyurt H., Sogut S., Yildirim Z., Kart L., Iraz M., Armutcu F., Temel I., Ozen S., Uzun A., Akyol O. Inhibitory effect of caffeic acid phenethyl ester on bleomycine-induced lung fibrosis in rats. Clin. Chim. Acta. 2004;339:65–75. doi: 10.1016/j.cccn.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 130.Albukhari A.A., Gashlan H.M., El-Beshbishy H.A., Nagy A.A., Abdel-Naim A.B. Caffeic acid phenethyl ester protects against tamoxifen-induced hepatotoxicity in rats. Food Chem. Toxicol. 2009;47:1689–1695. doi: 10.1016/j.fct.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 131.Yildiz O.G., Soyuer S., Saraymen R., Eroglu C. Protective effects of caffeic acid phenethyl ester on radiation induced lung injury in rats. Clin. Invest. Med. 2008;31:E242–E247. doi: 10.25011/cim.v31i5.4870. [DOI] [PubMed] [Google Scholar]
- 132.Chen Y.J., Liao H.F., Tsai T.H., Wang S.Y., Shiao M.S. Caffeic acid phenethyl ester preferentially sensitizes CT26 colorectal adenocarcinoma to ionizing radiation without affecting bone marrow radioresponse. Int. J. Radiat. Oncol. Biol. Phys. 2005;63:1252–1261. doi: 10.1016/j.ijrobp.2005.08.001. [DOI] [PubMed] [Google Scholar]