

SUMMARY
PTEN is one of the most frequently mutated tumor suppressor genes in human cancers. The role of PTEN in carcinogenesis has been validated by knockout mouse models. PTEN heterozygous mice develop neoplasms in multiple organs. Unfortunately, the embryonic lethality of biallelic excision of PTEN has inhibited the study of complete PTEN deletion in the development and progression of cancer. By crossing PTEN conditional knockout mice with transgenic mice expressing a tamoxifen-inducible Cre-ERT under the control of a chicken actin promoter, we have generated a tamoxifen-inducible mouse model that allows temporal control of PTEN deletion. Interestingly, administration of a single dose of tamoxifen resulted in PTEN deletion mainly in epithelial cells, but not in stromal, mesenchymal or hematopoietic cells. Using the mT/mG double-fluorescent Cre reporter mice, we demonstrate that epithelial-specific PTEN excision was caused by differential Cre activity among tissues and cells types. Tamoxifen-induced deletion of PTEN resulted in extremely rapid and consistent formation of endometrial in situ adenocarcinoma, prostate intraepithelial neoplasia and thyroid hyperplasia. We also analyzed the role of PTEN ablation in other epithelial cells, such as the tubular cells of the kidney, hepatocytes, colonic epithelial cells or bronchiolar epithelium, but those tissues did not exhibit neoplastic growth. Finally, to validate this model as a tool to assay the efficacy of anti-tumor drugs in PTEN deficiency, we administered the mTOR inhibitor everolimus to mice with induced PTEN deletion. Everolimus dramatically reduced the progression of endometrial proliferations and significantly reduced thyroid hyperplasia. This model could be a valuable tool to study the cell-autonomous mechanisms involved in PTEN-loss-induced carcinogenesis and provides a good platform to study the effect of anti-neoplastic drugs on PTEN-negative tumors.
INTRODUCTION
PTEN (phosphatase and tensin homolog deleted on chromosome 10) encodes a dual lipid and protein phosphatase that plays a crucial role in the phosphatidylinositol-3-kinase–Akt–mammalian-target-of-rapamycin (PI3K-AKT-mTOR) signaling pathway. PTEN function antagonizes PI3K by catalyzing the dephosphorylation of phosphatidylinositol (3,4,5)-trisphosphate [PIP3; also known as PtdIns(3,4,5)P3] to phosphatidylinositol (4,5)-bisphosphate [PIP2; also known as PtdIns(4,5)P2] (Maehama and Dixon, 1998; Myers et al., 1997). Loss of PTEN function leads to increased levels of PIP3 and a potent activation of 3-phosphoinositide-dependent kinase (PDK) and Akt that stimulates cell growth and survival (Song et al., 2012; Stambolic et al., 1998; Sun et al., 1999).
PTEN is one of the most frequently mutated tumor suppressor genes in human cancers. The tumor suppressive function of PTEN is attributed to chromosome region 10q23, which is partially or completely deleted in multiple neoplasias (Li et al., 1997; Steck et al., 1997). Soon after, germline mutations of the PTEN gene were identified in patients with Cowden disease (Liaw et al., 1997). Since then, loss-of-function mutations of PTEN have been reported in many human sporadic cancers, including glioblastoma and thyroid, breast, colon, prostate or endometrial carcinomas, as well as in familiar cancer predisposition syndromes known as PTEN tumor hamartoma syndromes (PTHS) (Hollander et al., 2011).
PTEN-knockout (KO) mouse models have provided evidence for the role of PTEN in carcinogenesis. Mice with monoallelic deletion of PTEN (PTEN+/−) develop neoplasms in multiple organs, including the endometrium, breast, prostate, gastrointestinal tract, thyroid and lymphoid tissues (Di Cristofano et al., 1998; Podsypanina et al., 1999; Suzuki et al., 1998). PTEN+/− provides a good tool to model the neoplastic phenotype observed in individuals with PTHS (Stambolic et al., 2000). Unfortunately, the embryonic lethality of mice with biallelic excision of PTEN has limited the study of complete PTEN ablation in the development of cancer. Such limitation has been solved by the generation of PTEN conditional-KO mice, which carry both PTEN alleles flanked by loxP sites (Lesche et al., 2002). To generate spatial and temporal control of PTEN deletion, conditional-KO mice have been bred with mouse strains expressing Cre recombinase under tissue-specific or inducible promoters. Such a strategy allowed biallelic ablation of PTEN in different cell types or organs (Knobbe et al., 2008; Suzuki et al., 2008), such as the adrenal gland (Korpershoek et al., 2009), breast (Li et al., 2002), thyroid (Yeager et al., 2007), prostate (Ma et al., 2005; Wang et al., 2003), astrocytes (Fraser et al., 2004) or hepatocytes (Horie et al., 2004). Temporal control of PTEN deletion has been achieved by breeding PTEN conditional-KO mice with mice expressing tamoxifen-inducible Cre recombinase (Cre-ERT) (Feil et al., 1996; Metzger et al., 1995) under the control of the Rosa26 promoter (R26Cre-ERT) (Lu et al., 2007). In this mouse model, tamoxifen causes PTEN excision in a broad spectrum of cells, leading to the development of multiple malignancies. Despite many mouse models for tissue-specific deletion of PTEN being available, the number of mouse models for both inducible and cell-type-specific deletion of PTEN is limited. So far, tamoxifen-inducible and polyinosine-polycytidine PTEN deletion has been achieved in adult prostatic epithelium and hematopoietic stem cells, respectively (Ratnacaram et al., 2008; Yilmaz et al., 2006). In both models, induction of PTEN recombination led to the development of prostatic neoplasias and myeloproliferative disorders, respectively. For other tissues, specific and inducible deletion of PTEN in epithelial cells as a model of carcinogenesis has never been achieved. In the uterus, biallelic deletion of PTEN has been achieved by crossing conditional PTEN-KO mice with a mice expressing Cre under the progesterone receptor promoter (PR-Cre) (Daikoku et al., 2008). PR-Cre mice led to PTEN deletion in all progesterone-receptor-expressing cells, which includes both epithelial and stromal cells. Similarly, tamoxifen-induced activation of Cre in R26Cre-ERT mice results in the deletion of PTEN both in epithelial and stromal compartments (Lu et al., 2007).
TRANSLATIONAL IMPACT.
Clinical issue
PTEN, a lipid and protein phosphatase, is encoded by one of the most frequently mutated tumor suppressor genes in human cancer. Loss of PTEN function is associated with neoplastic transformation in multiple tissue types, including breast, thyroid, prostrate and endometrial tissue. Heterozygous PTEN+/– mice have been used successfully to investigate the role of PTEN in carcinogenesis. However, the consequences of complete PTEN deletion remain unclear, because studies using homozygotes have been limited owing to embryonic lethality. The development of appropriate conditional models is crucial to advance our understanding of the molecular mechanisms underlying PTEN-associated carcinogenesis. Furthermore, such models would enable us to investigate and assay new anti-neoplastic therapies.
Results
Here, the authors present a novel knockout mouse model that carries a tamoxifen-inducible deletion of PTEN. They demonstrate that a single dose of tamoxifen results in complete PTEN ablation, mainly in epithelial cells. Loss of PTEN leads to extremely rapid, reproducible and efficient development of endometrial hyperplasias and in situ carcinomas, prostate neoplasias and thyroid hyperplasias. The team then administered the mTOR inhibitor everolimus to the knockout mice. A dramatic decrease in the progression of endometrial and thyroid carcinomas was observed in the presence of everolimus, validating the model as a tool to investigate drug efficacy.
Implications and future directions
The specificity of PTEN deletion in epithelial cells, together with the high incidence and speed of tumor formation, makes this model an attractive tool to investigate new targeted therapies to treat cancers that are induced by PTEN deficiency. In addition to its use as a platform to test anti-tumor drugs, this model might be useful for the study of molecular mechanisms involved in neoplastic growth caused by PTEN loss, particularly in endometrial, prostate and thyroid tissue. Furthermore, these investigations will be possible within a single mouse strain, facilitating experimental ease and efficiency.
Here, we describe a new tamoxifen-inducible mouse model for inducible and conditional deletion of PTEN. Cre expression driven by the chicken actin promoter (CAG) led to PTEN deletion mainly in epithelial cells, but not in stromal, or hematopoietic, cells. Such deletion caused development hyperplasias and neoplasias in some tissues such as endometrium, prostate or thyroid, but not in other epithelial tissues. We think that this model could be a valuable tool to study the molecular mechanisms underlying PTEN-loss-induced carcinogenesis and is suitable for the rapid assessment of anti-tumor drug effectiveness.
RESULTS
Tamoxifen induces dose-dependent deletion of conditional PTEN alleles in endometrial epithelial cells but not in stromal cells
Our group has experience in the study of the molecular mechanisms that trigger endometrial carcinogenesis. To generate a mouse model for endometrial carcinogenesis, we bred PTEN conditional-KO (PTENfl/fl) mice (Lesche et al., 2002) with mice expressing Cre-ERT (CAG-Cre-ERT) recombinase driven by the chicken β-actin promoter (Hayashi and McMahon, 2002). After two generations, we obtained mice carrying both PTEN floxed alleles and one copy of the Cre-ERT transgene (CAG-Cre-ERT+/−PTENfl/fl). At 3 weeks after birth, mice were weaned and, after 1 or 2 additional weeks, they were intraperitoneally injected with 0.5, 1 or 2 mg of tamoxifen (see Fig. 1A). After 7 days, mice were sacrificed and PTEN expression in the uterus was analyzed by immunohistochemistry (IHC) (Fig. 1B). Increasing doses of tamoxifen caused an increase in the number of PTEN-negative cells in endometrial epithelium (Fig. 1C). Interestingly, loss of PTEN resulted in mosaics of epithelial cells retaining PTEN expression mixed with cells with a complete loss of PTEN labeling. Surprisingly, stromal cells retained PTEN immunostaining, which was comparable to that observed in non-injected mice. To avoid non-desired or possible collateral effects of tamoxifen, we chose the lowest dose (0.5 mg) for all future experiments.
Fig. 1.

Differential Cre-ERT activity and PTEN deletion in endometrial epithelial and stromal compartments. (A) Schematic showing the experimental setup for tamoxifen (TAM) treatment. Briefly, mice were weaned 3 weeks after birth and genotyped. At 4–5 weeks old, animals were given a single intraperitoneal dose of tamoxifen, or corn oil as a vehicle (con). Mice were sacrificed 7 days later. (B) Tamoxifen induces a dose-dependent deletion of PTEN. PTEN immunohistochemistry corresponding to CAG-Cre-ERT+/−PTENfl/fl 7 days after tamoxifen injection. Note that stromal cells remained positive for PTEN staining. (C) Quantification of the percentage of endometrial epithelial cells displaying negative for PTEN immunostaining after injection of the indicated doses of tamoxifen. (D) Analysis of tamoxifen-induced recombination in CAG-Cre-ERT mT/mG reporter mice. Representative images corresponding to CAG-Cre-ERT+/−mT/mG+/− uterus 1 week after tamoxifen injection are shown. Note that, upon tamoxifen treatment, only epithelial cells showed a switch from red to green fluorescence.
These results suggest that tamoxifen treatment causes PTEN deletion preferentially in endometrial epithelial cells. To ascertain whether the differences in PTEN ablation were caused by differential Cre-ERT activity among cell types, we bred a CAG-Cre-ERT mouse with a double-fluorescent Cre reporter mouse that expresses membrane-targeted tandem dimer Tomato (mT) prior to Cre-mediated excision and membrane-targeted green fluorescent protein (mG) after excision (mT/mG mouse) (Muzumdar et al., 2007). The resulting offspring was injected with tamoxifen and 7 days later the expression of green or red fluorescence in the uterus was monitored under confocal microscope. Only epithelial cells of the endometrium switched from red to green fluorescence, indicating that Cre-ERT was only active in epithelial cells (Fig. 1D).
Tamoxifen-induced PTEN deletion led to rapid development of endometrial hyperplasia and adenocarcinoma in situ
Next, we analyzed whether such PTEN deficiency would result in the formation of endometrial proliferations. For this purpose, mice were injected with tamoxifen and analyzed in three intervals of time after tamoxifen injection: 0–2 weeks, 3–5 weeks and 6–8 weeks. For each single animal sacrificed, uterus was dissected and analyzed by routine hematoxylin and eosin (H&E) staining and PTEN IHC. In animals analyzed in the first period of time (0–2 weeks), PTEN was deleted in endometrial epithelial cells, but we did not observe any increase in the number of epithelial glands or gross morphological alteration (Fig. 2A,D). However, morphological alteration became evident between 3 and 5 weeks after tamoxifen administration. Around 40% of mice displayed complex endometrial hyperplasia and an additional 50% of them already developed complex atypical hyperplasia and endometrial in situ adenocarcinoma (Fig. 2B,D), showing architectural complexity, cribriform pattern and solid arrangement. All neoplastic growths were restricted to the endometrium. Between 6 and 8 weeks, 100% of mice showed endometrial in situ adenocarcinoma (Fig. 2C,D). Strikingly, PTEN immunostaining revealed that tumoral tissue was strictly composed of PTEN-negative glands, whereas glands retaining PTEN expression were completely normal. Moreover, stromal cells from all tamoxifen-injected mice displayed a positive PTEN immunostaining that was indistinguishable from that observed in non-injected mice. We want to mention that we did not extend the study for longer times because, by 8 weeks after tamoxifen injection, most mice showed breathing difficulties, lethargy, ruffled fur and hunched posture. To avoid unnecessary animal suffering, mice were euthanized. Such aggravation of health was attributed to the appearance of endometrial and other neoplastic processes (see following parts of Results for explanation). For that reason, mice were not allowed to survive for long enough for the tumors to invade the myometrium and to metastasize to other organs.
Fig. 2.

Tamoxifen-induced PTEN deletion causes rapid and reproducible development of endometrial hyperplasia and in situ adenocarcinoma of the endometrium. (A–D) Comparison of histopathology and PTEN expression of Cre-ERT+/−PTENfl/fl mice injected with tamoxifen (TAM) or corn oil (CON) at different time points. (A–C) Representative images showing PTEN immunohistochemistry (bottom panels) and H&E staining (top panels) of uterus from mice sacrificed in weeks 0–2 (A), weeks 3–5 (B) or weeks 6–8 (C) after tamoxifen injection. (D) Graph showing the percentages of endometrial lesions distributed by periods of time.
Because chronic tamoxifen exposure is a risk factor for the development of endometrial lesions, we injected control mice carrying both PTEN floxed alleles but lacking Cre-ERT expression. At 8 weeks after tamoxifen injection, Cre-ERT−/−PTENfl/fl mice did not show any morphological alteration nor changes in PTEN immunoreactivity (supplementary material Fig. S1).
Tamoxifen-induced Cre-ERT recombination occurs mainly in epithelial tissues
The results obtained with endometrial tissue enabled us to investigate whether Cre-ERT expression would also be restricted to epithelial cells of other tissues or organs. To this end, we analyzed different tissues from CAG-Cre-ERT mT/mG mice, such as lung, colon, thyroid, liver and kidney. mT/mG mice lacking Cre-ERT expression showed only red fluorescence, indicating no recombination. In contrast, CAG-Cre-ERT mT/mG mice injected with tamoxifen displayed a switch from red to green fluorescence (Fig. 3A). Interestingly, most of the tissues that were analyzed displayed green fluorescence in epithelial cells but not in stromal or other cell types. In the lung, green fluorescence appeared in bronchiolar epithelial cells. In colon, a red-to-green switch was observed also in epithelial cells. In the kidneys, green cells appeared in epithelial cells of renal tubules and nephrons. In the thyroid, green fluorescence was observed in follicular cells. These results indicate that Cre-ERT is mainly expressed in epithelial cells.
Fig. 3.
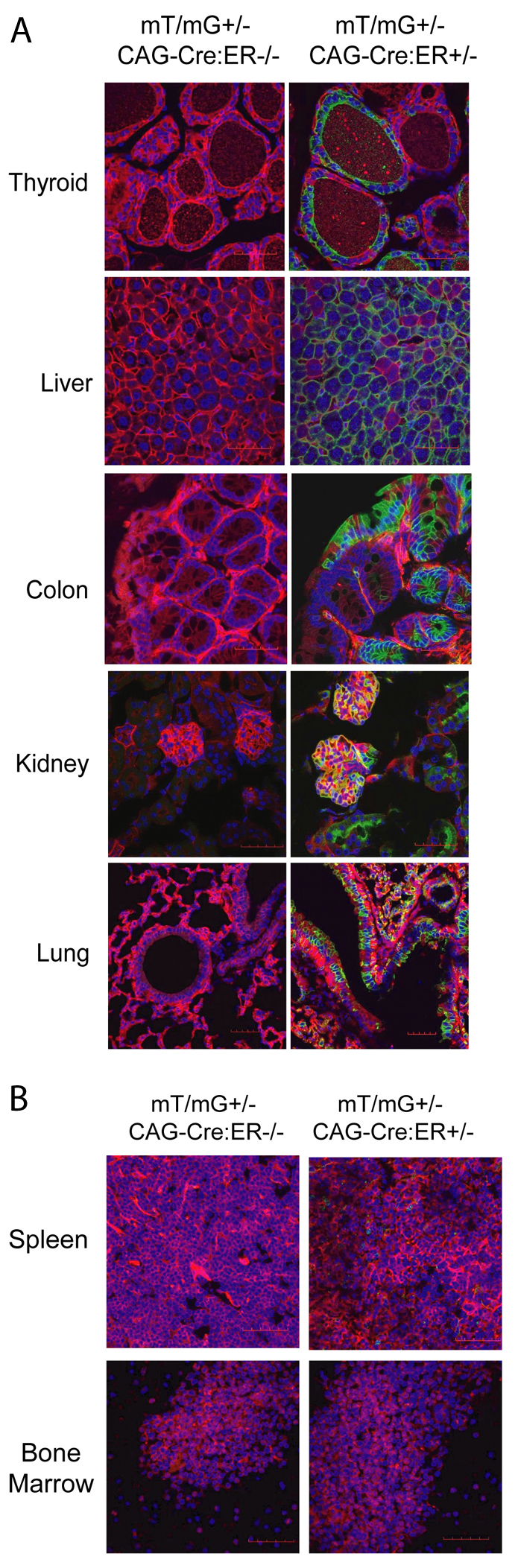
Evaluation of inducible and tissue-specific Cre-ERT recombinase activity in the double-fluorescent mT/mG reporter mice. CAG-Cre-ER−/−mT/mG+/− and CAG-Cre-ERT+/− mT/mG+/− mice were given a single dose of tamoxifen as described in Fig. 1. After 7 days, different tissues were fixed and analyzed for the presence of green (indicative of Cre-ER recombination) or red (indicative of lack of Cre-ER recombination) fluorescence. (A) Representative confocal images corresponding to organs containing epithelial tissues (thyroid, liver, colon, kidney and lung) showing green fluorescence after tamoxifen injection. (B) Representative images of hematopoietic and lymphoid organs (bone marrow and spleen). Note absence of green fluorescence.
We also analyzed bone marrow and spleen from mT/mG mice injected with tamoxifen. None of the lymphoid organs displayed a switch from red to green fluorescence, suggesting the absence of Cre activity in those organs (Fig. 3B).
Tamoxifen-induced PTEN deletion results in a rapid development of prostate intraepithelial neoplasias and thyroid hyperplasia
Tamoxifen-induced Cre-ERT activity was not restricted to endometrial epithelial cells, but was also observed in other epithelial tissues. Because loss of PTEN has been observed in different carcinomas, we extended our investigations to tissues in which PTEN deletion has been associated with carcinogenesis. Among them, PTEN is frequently mutated in thyroid and prostate malignancies. Moreover, conditional deletion of PTEN in the thyroid or the prostate leads to the formation of neoplastic processes.
At 8 weeks after tamoxifen injection, we dissected prostates and thyroids from CAG-Cre-ERT+/−PTENfl/fl mice and analyzed their histopathological features and PTEN expression. As was found for the endometrium, induction of Cre-ERT activity caused a complete loss of PTEN immunoreactivity in epithelial prostatic cells but not in stromal cells. More importantly, PTEN loss resulted in the development of prostate intraepithelial neoplasias (PIN) (Fig. 4A). Mice injected with vehicle (corn oil) or tamoxifen-injected mice carrying conditional PTEN alleles but lacking Cre expression (CAG-Cre-ERT−/−PTENfl/fl) displayed normal morphology and no decrease of PTEN expression. Histopathological analysis and cytokeratin immunostaining revealed high-grade neoplastic growth (PIN) (Fig. 4B). All mice that were studied developed high-grade PIN 8 weeks after tamoxifen injection (Fig. 4C). Neoplastic growth correlated with an increase in Ki-67 immunostaining, suggesting increased proliferation of prostatic tumoral cells.
Fig. 4.
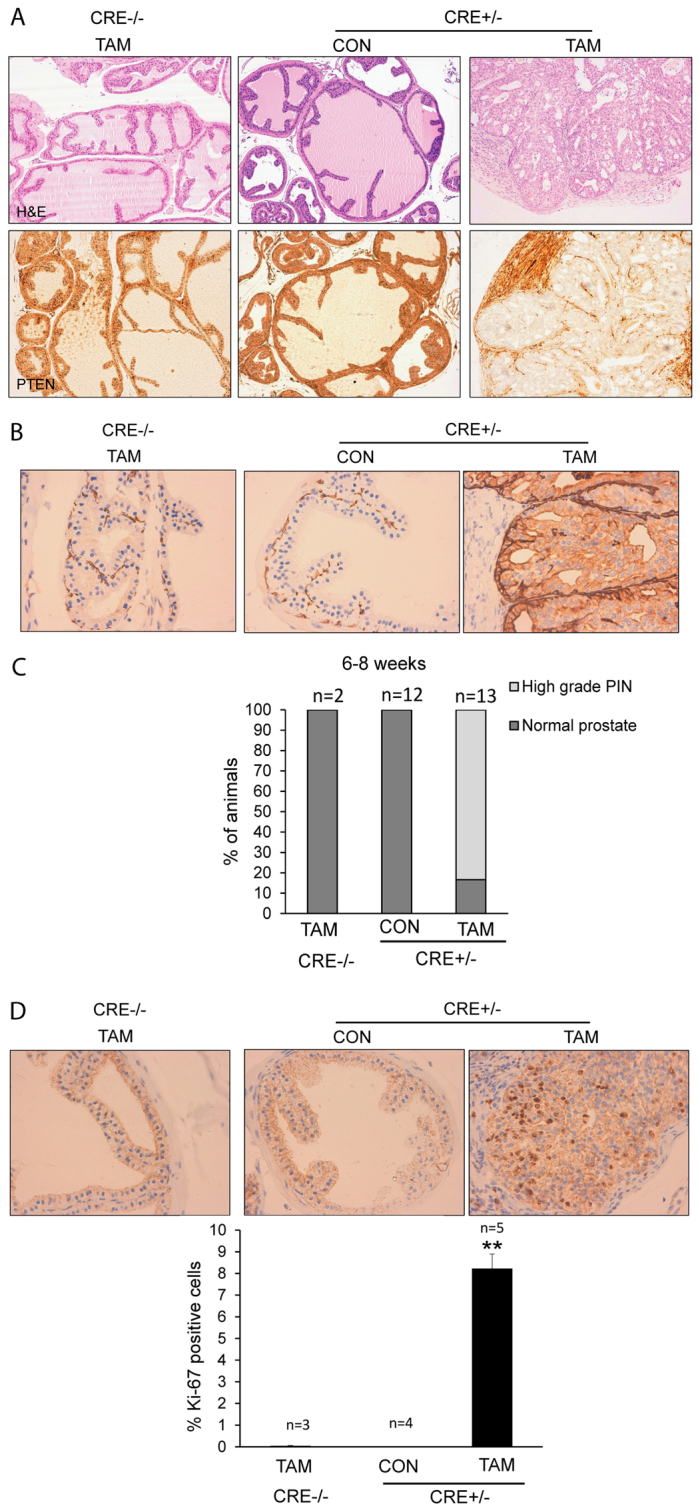
Tamoxifen-induced PTEN deletion causes the rapid development of prostate intraepithelial neoplasia (PIN). Prostates from CAG-Cre-ERT−/−PTENfl/fl or CAG-Cre-ERT+/−PTENfl/fl were analyzed 6–8 weeks after tamoxifen (TAM) or corn oil (CON) injection. (A) Representative images of CAG-Cre-ERT+/−PTENfl/fl tamoxifen-treated males showing high-grade PIN lesions and PTEN-negative immunostaining. Control mice displayed normal histology and retained PTEN immunostaining. (B) Representative images of cytokeratin staining, showing increased immunostaining in high-grade PIN epithelial cells. (C) Quantification of the percentage of mice displaying prostatic lesions 6–8 weeks after tamoxifen injection. (D) Representative images (top) and quantification (bottom) of Ki-67 immunostaining in prostates of CAG-Cre-ERT–/–PTENfl/fl mice injected with tamoxifen, and CAG-Cre-ERT+/−PTENfl/fl mice injected with corn oil (CON) or tamoxifen (TAM). **P≤0.001.
Thyroids from CAG-Cre-ERT+/−PTENfl/fl mice injected with tamoxifen were substantially bigger than those dissected from non-injected mice (Fig. 5A). Pathological analysis of tamoxifen-injected mice revealed the presence of follicular nodular hyperplasia but not invasive thyroid carcinomas (Fig. 5B). Surprisingly, control mice also displayed some focal nodular thyroid hyperplasia. Therefore, we hypothesized that, in the thyroid, Cre-ERT activity was induced in the absence of tamoxifen, resulting in the deletion of PTEN in some cells. To test this hypothesis, we analyzed PTEN expression by immunofluorescence in mice injected or not with tamoxifen. PTEN expression was completely lost in thyroid follicles in CAG-Cre-ERT+/−PTENfl/fl mice injected with tamoxifen (Fig. 5C). In control CAG-Cre-ERT+/−PTENfl/fl mice, injected with corn oil, PTEN expression was retained in zones of normal tissue and was undetectable in hyperplastic areas. This result suggests that spontaneous (tamoxifen-independent) Cre-ERT activity in the thyroid leads to PTEN ablation even in the absence of tamoxifen. Despite such leaking of Cre activity, injection of tamoxifen resulted in a more severe thyroid hyperplasia and increased thyroid size. It is worth noting that, by 8 weeks after tamoxifen treatment, such an increase in thyroid size caused severe breathing difficulties, causing a dramatic worsening of animal health. For this reason, animals required euthanization.
Fig. 5.
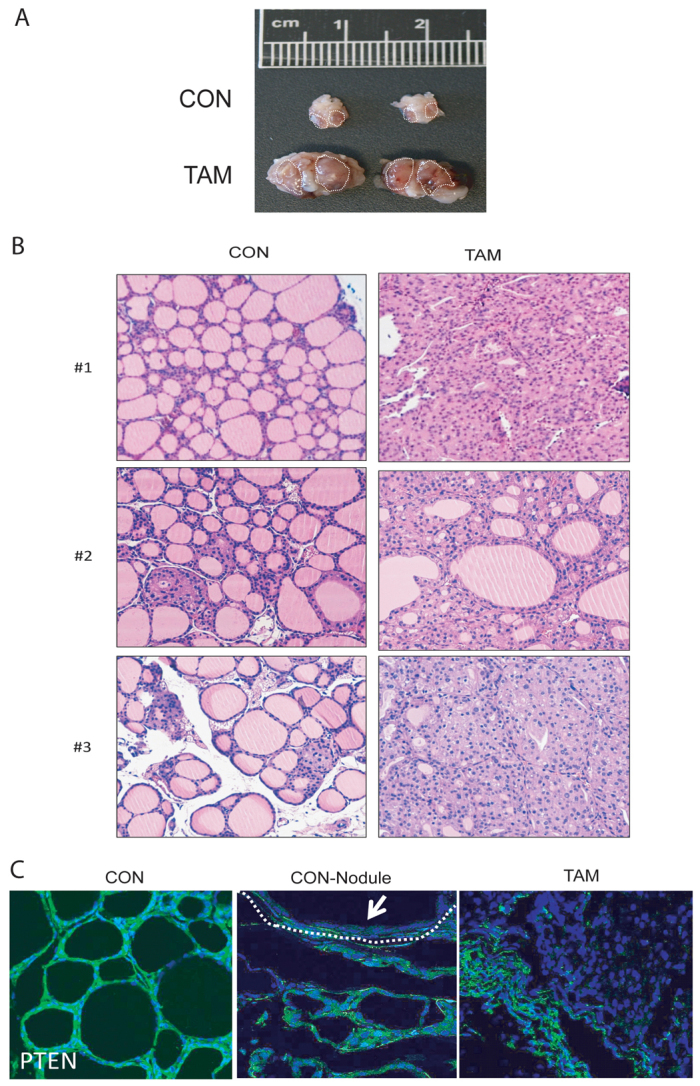
PTEN inactivation leads to the development of thyroid hyperplasia. (A) Comparison of representative thyroids from CAG-Cre-ERT+/−PTENfl/fl mice 6–8 weeks after being injected with corn oil (CON) or tamoxifen (TAM). (B) Representative images of hyperplasia in thyroids dissected from three corn-oil-injected (CON) and three tamoxifen-injected (TAM) mice. (C) Immunofluorescence staining showing PTEN loss in CAG-Cre-ERT+/−PTENfl/fl thyroids. Representative images showing PTEN immunofluorescence of mice injected with corn oil displaying no hyperplasic nodules (CON), mice injected with corn oil displaying hyperplasic nodules (CON-Nodule) or mice injected with tamoxifen (TAM). Arrow indicates PTEN-negative immunofluorescence in hyperplasic module.
PTEN is efficiently deleted in other epithelial organs but does not trigger neoplastic growth
As we describe above, 100% of tamoxifen-treated CAG-Cre-ERT+/−PTENfl/fl mice displayed endometrial hyperplasia/adenocarcinoma in situ or PIN. However, mT/mG reporter mice revealed Cre-ERT activity in other epithelial tissues. For this reason, we decided to analyze the formation of neoplastic growth in other tissues from tamoxifen-injected mice, such as the lungs, kidneys, liver or colon. IHC revealed loss of PTEN immunoreactivity in epithelial cells of the four organs. In the kidney, loss of PTEN immunostaining was observed in epithelial tubular cells (Fig. 6A). In the liver, hepatocytes showed loss of PTEN expression (Fig. 6B). In the lung, loss of PTEN was restricted to bronchiolar columnar epithelial cells (Fig. 6C). Similarly, in colonic tissue, PTEN labeling was also lost in epithelial cells (Fig. 6D). Despite the loss of PTEN observed in all those tissues, none of them displayed morphological alterations or neoplastic growth. It is noteworthy that specific deletion of PTEN in all four tissues correlated with Cre-ERT recombinase activity, evidenced by experiments carried out with mT/mG reporter mice (Fig. 2A).
Fig. 6.
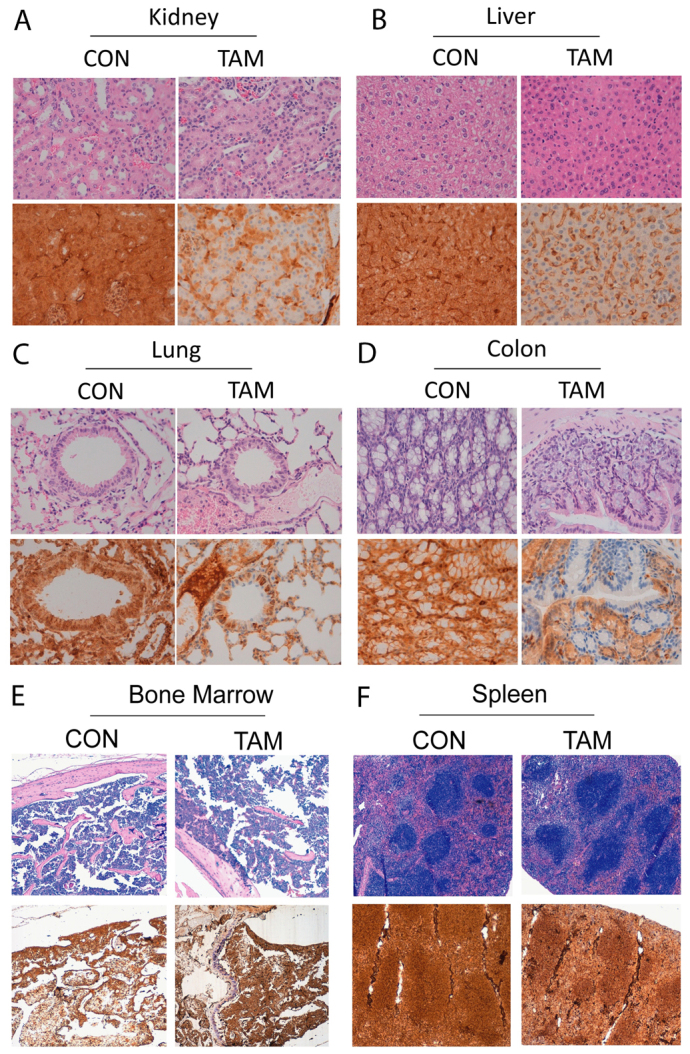
PTEN loss does not cause rapid neoplastic growth in other epithelia or in hematopoietic or lymphoid tissues. Representative images of H&E staining and PTEN immunohistochemistry corresponding to (A) kidney, (B) liver, (C) lung, (D) colon, (E) bone marrow and (F) spleen from CAG-Cre-ERT+/−PTENfl/fl mice 8 weeks after injection with corn oil (CON) or tamoxifen (TAM).
Finally, we analyzed the expression of PTEN in lymphoid tissues. In PTEN+/− mice, tissue conditional PTEN deletion in lymphoid tissues or the inducible ablation of PTEN under the control of R26Cre-ERT led to the development of hematological malignancies such as leukemia and lymphoma. Such malignancies were often the cause of death, thereby limiting the study of PTEN deficiency in other organs in which PTEN alterations are important. Immunohistochemical analysis showed that PTEN expression was similar in mice injected with tamoxifen and control animals in both bone marrow (Fig. 6E) and spleen (Fig. 6F). Concordantly, pathological analysis of lymphoid organs demonstrated the absence of malignancies. Again, the absence of PTEN deletion correlated with the lack of Cre activity in bone marrow and spleen (see Fig. 2B). These results suggest that PTEN is not excised in lymphoid organs because of the lack of Cre-ERT activity in those tissues.
Everolimus reduces the development of endometrial in situ carcinoma and thyroid hyperplasia
Here, we describe a new mouse model that rapidly develops endometrial hyperplasia, intraepithelial carcinoma, PIN and thyroid nodular hyperplasia with 100% incidence. Such features make this mouse an attractive model to test the efficacy of anti-neoplastic drugs targeting the PI3K-Akt pathway. Inhibition of mTOR has been reported as an effective therapy for tumors that have alteration in PI3K-Akt signaling (Engelman, 2009; Hollander et al., 2011). To demonstrate that our model is a suitable tool to test the efficacy of anti-neoplastic drugs, we treated tamoxifen-injected mice with the mTOR inhibitor everolimus (also known as RAD001). For this purpose, 1–2 weeks after weaning, mice were injected with tamoxifen to induce PTEN excision. At 5 weeks after tamoxifen injection, female mice were injected daily with 10 μg everolimus/g body weight for 15 consecutive days and sacrificed (see Fig. 7A). As we mentioned above, tamoxifen-injected mice showed lethargy, ruffled fur, hunched posture and required euthanization. In contrast, everolimus-injected mice remained healthy and active (Fig. 7B). Pathological analysis of the endometrium revealed that everolimus caused a marked reduction in the severity of endometrial lesions (Fig. 7C), decreasing the percentage of mice suffering from endometrial in situ adenocarcinoma from 100% to 25%. The rest of the endometrium from everolimus-treated mice was normal (25%) or hyperplasic (50%). Such a reduction in endometrial lesions correlated with a reduction of Ki-67, suggesting that everolimus caused a reduction of cell proliferation (Fig. 7C). Similarly, everolimus treatment caused a dramatic reduction in thyroid size and decreased the number of Ki-67-positive cells (Fig. 7D).
Fig. 7.

mTOR inhibition reduces endometrial lesions and thyroid hyperplasia. (A) Schematic representation of the protocol used for everolimus administration. Briefly, 5 weeks after tamoxifen injection, animals started to receive a single daily intraperitoneal dose of everolimus for 14 consecutive days and then they were sacrificed. (B) At 7 weeks after tamoxifen injection (TAM), mice exhibited lethargy, ruffled fur and hunched posture. Animals treated with everolimus (TAM+EVE) remained healthy and active, similar to controls (CON). (C) H&E staining and PTEN immunohistochemistry images corresponding to endometria from mice treated with corn oil (CON), tamoxifen (TAM) or tamoxifen plus everolimus (TAM+EVE). Everolimus-treated females showed reduced lesions and PTEN-negative healthy areas. Percentage of animals developing endometrial hyperplasia (EH) and endometrial in situ adenocarcinomas (EC) after injection of corn oil (CON), tamoxifen (TAM) or tamoxifen and everolimus (TAM+EVE) are shown in the top graph. Representative images (bottom left) and quantification (bottom right) of Ki-67 immunostaining performed on uterus dissected from mice injected with corn oil (CON), tamoxifen (TAM) or tamoxifen plus everolimus (TAM+EVE). (D) Representative images of thyroids dissected from mice injected with corn oil (CON), tamoxifen (TAM) or tamoxifen plus everolimus (TAM+EVE). Everolimus treatment also reduced thyromegaly in tamoxifen-injected mice. Everolimus caused a marked decrease of the index of proliferation, with levels similar to control animals, as assessed by Ki-67 immunostaining. *P≤0.05; **P≤0.001.
DISCUSSION
The development of appropriate mouse models of human tumors has been crucial to progress the understanding of cancer biology and to identify better therapeutic and diagnostic strategies. Here, we describe a new mouse model that allows tamoxifen-inducible deletion of PTEN, which results in a rapid development of endometrial hyperplasia and intraepithelial carcinoma, PIN and thyroid nodular hyperplasia.
Molecular alterations of PTEN are very frequent in endometrial carcinomas. To date, only two existing models to achieve conditional deletion of PTEN in the endometrium have been reported: conditional deletion of PTEN has been achieved by breeding conditional PTEN KO mice with mice expressing PR-Cre (Daikoku et al., 2008) or inducible R26Cre-ERT (Lu et al., 2007). Both strategies resulted in ablation of PTEN in both epithelial and stromal cells (apart from other non-endometrial tissues). In contrast, our mouse constitutes the first that allows inducible and epithelial-cell-specific PTEN deletion in endometrial epithelial cells. It is widely accepted that endometrial carcinomas, as other types of carcinomas, arise as a result of the accumulation of mutations in epithelial cells. By contrast, it is well-known that stromal cells can affect tumor behavior (Bhowmick et al., 2004). Such an important role has been recently supported by results showing that specific inactivation of PTEN in mammary stromal cells leads to accelerated breast tumorigenesis (Trimboli et al., 2009; Wallace et al., 2011). Therefore, it is likely that deletion of PTEN in endometrial stromal cells can affect the progression of tumoral epithelial cells. To this end, we think that specific deletion of PTEN in the epithelium is important to understand the cell-autonomous mechanisms that are involved in carcinogenesis. So far, specific PTEN deletion in endometrial epithelial cells has only been achieved by tissue recombination experiments, in which PTEN-deficient epithelial cells have been recombined with wild-type stromal cells and implanted under the renal capsule of immunocompromised mice (Memarzadeh et al., 2010). Because PTEN excision is restricted to the epithelial compartment of the endometrium, our model provides a valuable tool to study cell-autonomous mechanisms of carcinogenesis. Another important point is that PTEN is not deleted in epithelial cells in any organ, but only in those of some specific locations. One single endometrial gland is a mosaic of cells lacking PTEN side-by-side with cells retaining PTEN expression, which mimics what happens in normal human endometrium. PTEN-retaining cells show a completely normal phenotype, strongly demonstrating that carcinogenesis arises exclusively from PTEN-deficient cells. Moreover, in our model, induction of PTEN excision is performed adult mice. This is an important point because, in most tissue-specific conditional models, the onset of deletion depends on the activity of the tissue-specific promoter, but it cannot be temporally regulated. Because hyperplastic and neoplastic processes of human endometrial, prostatic or thyroidal tissues are diseases that are predominantly found in adults, induction of PTEN excision in adult mice mimics the onset of those human malignancies.
We have observed that one single injection of tamoxifen results in the ablation of PTEN in many epithelial cell types. However, we only found endometrial hyperplasia, intraepithelial carcinoma, PIN and thyroid nodular hyperplasia, but did not observe the formation of neoplasia in other epithelial tissues such as the kidney, lung or colon. Previous works have demonstrated that conditional inactivation of PTEN in the lung or colon leads to the development of neoplasias. Such differences can be explained by genetic background. It has been demonstrated that genetic background affects the development of PTEN-loss-induced tumors in mice (Freeman et al., 2006). Spatiotemporal expression of Cre can be different depending on genetic background (Hébert and McConnell, 2000; Smith, 2011; Wang, 2009), leading to different patterns of recombination of both the Cre and the floxed mice. To achieve inducible inactivation of PTEN, we used a transgenic mouse line in which Cre-ERT is expressed under the chicken β-actin promoter. Theoretically, Cre should be ubiquitously expressed in most tissues of the mouse at embryonic and adult stages (Hayashi and McMahon, 2002). However, using a fluorescent reporter mouse, we detected Cre activity mainly in epithelial cells throughout the whole mouse body, but there was little or no activity in hematopoietic or mesenchymal cells. It is worth mentioning that Cre expression completely correlated with PTEN deletion. To this regard, the lack of expression in hematopoietic cells is an important issue. Ablation of PTEN in hematopoietic cells leads to a rapid development of lymphoproliferative and myeloproliferative disorders, shortening mouse lifespan and conditioning the development and study of other PTEN-induced malignancies. Our model overtakes this limitation.
Conditional models allow deletion of a gene in one single tissue and the development of one single type of neoplasm. Our mouse model could represent a platform to assay the effects of chemotherapeutic drugs on hyperplasia and carcinoma in three different organs using one single mouse strain. The use of one single mouse strain reduces the number of mice used and the economic costs of investigations. As a proof for such application, we have used our PTEN mouse model to evaluate the effects of mTOR inhibition by everolimus on endometrial hyperplasia, carcinoma and thyroid hyperplasia. The involvement of mTOR and Akt as downstream signaling effectors of PTEN deficiency has been strongly supported by crossing PTEN hemizygous mice with mice carrying genetic mutations in PTEN downstream effectors. Deficiency in either PDK1 (Bayascas et al., 2005) or Akt-1 (Chen et al., 2006) signaling reduces endometrial carcinogenesis in PTEN+/− mice. Inhibition of mTOR has been reported to reduce the growth of thyroid (Yeager et al., 2008), endometrial (Milam et al., 2007; Podsypanina et al., 2001) or prostate (Zhang et al., 2009) carcinomas in PTEN-deficient mouse models. In our model, everolimus is able to reduce neoplastic growth, validating this model as a platform for the rapid assessment of anti-neoplastic drug efficacy.
MATERIALS AND METHODS
Animals
Mice were housed in a barrier facility and pathogen-free procedures were used in all mouse rooms. Animals were kept in a 12-hour light-dark cycle at 22°C with ad libitum access to food and water. All procedures performed in this study followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were compliant with the guidelines of Universitat de Lleida. Floxed homozygous PTEN (C;129S4-Ptentm1Hwu/J; referred to here as PTENfl/fl), Cre-ERT [B6.Cg-Tg(CAG-Cre/Esr1* 5Amc/J] and reporter mT/mG [B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J] mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Cre-ERT+/−PTENfl/fl mice were bred in a mixed background (C57BL6; 129S4) by crossing PTENfl/fl and Cre-ERT+/− mice. To obtain mice carrying both PTEN floxed alleles (PTENfl/fl) and a single Cre-ERT (Cre-ERT+/−), Cre-ERT+/−PTENfl/+ mice were backcrossed with PTENfl/fl mice. At 3 weeks after birth, animals were weaned and genotyped as previously described (Lesche et al., 2002; Hayashi and McMahon, 2002).
Tamoxifen administration
Tamoxifen (Sigma-Aldrich T5648, St Louis, MO) was dissolved in 100% ethanol at 100 mg/ml. Tamoxifen solution was emulsified in corn oil (Sigma-Aldrich C8267) at 10 mg/ml by vortexing. To induce PTEN deletion, adult mice (4–5 weeks old) were given a single intraperitoneal injection of 0.5 mg of tamoxifen emulsion (30–35 μg per mg body weight).
Cre-ERT reporter assays
For reporter assays of Cre activity, Cre-ERT+/− and mT/mG+/− mice were bred. The resulting Cre-ERT−/− mT/mG+/− and Cre-ERT+/−mT/mG+/− animals were weaned and injected with tamoxifen. At 1 week after tamoxifen treatment, animals were sacrificed and organs were collected and fixed in 4% PFA for 4 hours at room temperature. Samples were cryoprotected in PBS with 30% sucrose overnight at 4°C and embedded in OCT (VWR International). 8-μm sections of frozen tissue were cut with cryostat and nuclei were counterstained with Hoescht and samples mounted with PBS:glycerol (1:1). Tissue immunofluorescence was visualized using a confocal microscope (Olympus, Tokyo, Japan). Confocal images were edited using FluoView software (Olympus).
Histopathology and immunohistochemical analysis
After tamoxifen injection, animals were euthanized by cervical dislocation and organs were collected and formalin-fixed overnight at 4°C. One part of the organs was embedded in paraffin for histological examination. Another part was cryoprotected in PBS with 30% sucrose and embedded in OCT (VWR International) for immunofluorescence staining. H&E-stained samples were reviewed and evaluated by two pathologists, following uniform pre-established criteria. Paraffin blocks were sectioned at 3 μm, dried for 1 hour at 65°C before the pre-treatment procedure of deparaffinization, rehydration and epitope retrieval in the Pre-Treatment Module, PT-LINK (DAKO), at 95°C for 20 minutes in 50× Tris/EDTA buffer, pH 9. The antibodies used were anti-PTEN (Dako, Denmark, clone 6H2.1, 1/100 dilution), Ki-67 (Dako, Denmark, clone TEC-3, 1/50 dilution) and high-molecular-weight cytokeratin (HMW-CK, Dako, Denmark, clone 34βE12, ready to use). The reaction was visualized with the EnVision FLEX detection kit (Dako, Denmark) for PTEN and HMW-CK and polyclonal rabbit anti-rat immunoglobulins/biotinylated and streptavidin (Dako, Denmark) for Ki-67 using diaminobenzidine chromogen as a substrate. Images were taken with a Leica DMD108 microscope.
To optimize PTEN immunostaining with 6H2.1 antibody, we first performed IHC on both wild-type and PTEN-deficient mouse endometrium with 1/100, 1/200 and 1/300 dilutions of the antibody. The staining pattern was similar with 1/100, 1/200 and 1/300 dilutions, but the signal was weaker at higher dilutions. We chose 1/100 dilution as the best signal-to-background ratio for further experiments (supplementary material Fig. S2A). To demonstrate specificity of PTEN immunostaining, normal human endometrial samples containing PTEN-expressing and PTEN-null glands was stained with anti-PTEN antibody (supplementary material Fig. S2B). To further prove the specificity of PTEN immunostaining, PTEN-proficient and PTEN-deficient endometrial carcinoma cell lines were immunostained with PTEN 6H2.1 antibody. For this purpose, the PTEN-deficient RL95 cell line was transfected with a plasmid encoding wild-type PTEN and the PTEN-proficient cell line HEC-1A was transfected with a plasmid encoding PTEN shRNA to downregulate endogenous levels of PTEN expression. Paraffin blocks of RL95 and HEC-1-A were constructed and processed for PTEN immunostaining as described above. The results of PTEN immunostaining on transfected RL95 and HEC-1A cells are shown in supplementary material Fig. S2C.
Immunofluorescence
Frozen tissue was cut in 8-μm slices using a cryostat. Slides were washed in PBS with Tween 0.05% (PBST) for 30 minutes followed by PBS wash (once for 5 minutes) and incubated in blocking solution (PBST with 5% normal goat serum) for 1 hour at room temperature. Sections were incubated overnight at 4°C with rabbit polyclonal anti-PTEN antibody (catalog no. 9552; Cell Signaling, dilution 1/50). Slides were washed four times in PBST for 5 minutes and incubated for 1 hour at room temperature with Alexa-Fluor-488-labeled secondary antibodies. Cell nuclei were counterstained with 5 μg/ml Hoechst 33342 dye. Immunofluorescence was visualized using a confocal microscope (Olympus, Tokyo, Japan).
Everolimus treatment
At 5 weeks after tamoxifen injection, we administered 10 mg/kg body weight/day of Everolimus RAD001 (Sigma-Aldrich) as a microemulsion (2% w/v) diluted in distilled water by intraperitoneal injection. Animals were sacrificed and organs were collected after 14 days of treatment.
Proliferation analysis
Five representative fields of each tissue were photographed for each animal. Proliferation was calculated as the percentage of Ki-67-positive nuclei to the total number of nuclei of each field (1000–2000 total nuclei were evaluated for each animal). Counting was performed using ImageJ software (Wayne Rasband, NIH, Bethesda, MD).
Statistical analysis
Experiments were performed at least three times and statistical significance was determined by Student’s t-test with P≤0.05 (*) or P≤0.001 (**).
Supplementary Material
Acknowledgments
We thank Lidia Mónica Domingo and Lidia Parra for their technical support.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
C.M., N.E., X.M.-G. and X.D. conceived and designed the experiments. C.M., N.E., M.A.D. and L.B. performed the experiments. M.S. and O.M. provided technical support. S.G., J.P. and X.M.-G. analyzed the data. C.M. and X.D. wrote the paper.
FUNDING
Supported by grants FIS PI10/00604, PI10/00922, RD06/0020/1034, 2009SGR794 and 2004XT00090, Grupos estables AECC, Catalunya contra el cáncer and programa de intensificación de la investigación, Instituto Carlos III. L.B. holds an Alicia Cuello de Merigó Fellowship, N.E. and C.M. hold a fellowship from Fondo the Investigaciones Sanitarias (FIS).
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.011445/-/DC1
REFERENCES
- Bayascas J. R., Leslie N. R., Parsons R., Fleming S., Alessi D. R. (2005). Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr. Biol. 15, 1839–1846 [DOI] [PubMed] [Google Scholar]
- Bhowmick N. A., Neilson E. G., Moses H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. L., Xu P. Z., Peng X. D., Chen W. S., Guzman G., Yang X., Di Cristofano A., Pandolfi P. P., Hay N. (2006). The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 20, 1569–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T., Hirota Y., Tranguch S., Joshi A. R., DeMayo F. J., Lydon J. P., Ellenson L. H., Dey S. K. (2008). Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 68, 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 [DOI] [PubMed] [Google Scholar]
- Engelman J. A. (2009). Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562 [DOI] [PubMed] [Google Scholar]
- Feil R., Brocard J., Mascrez B., LeMeur M., Metzger D., Chambon P. (1996). Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 93, 10887–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser M. M., Zhu X., Kwon C.-H., Uhlmann E. J., Gutmann D. H., Baker S. J. (2004). Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 64, 7773–7779 [DOI] [PubMed] [Google Scholar]
- Freeman D., Lesche R., Kertesz N., Wang S., Li G., Gao J., Groszer M., Martinez-Diaz H., Rozengurt N., Thomas G., et al. (2006). Genetic background controls tumor development in PTEN-deficient mice. Cancer Res. 66, 6492–6496 [DOI] [PubMed] [Google Scholar]
- Hayashi S., McMahon A. P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 [DOI] [PubMed] [Google Scholar]
- Hébert J. M., McConnell S. K. (2000). Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 222, 296–306 [DOI] [PubMed] [Google Scholar]
- Hollander M. C., Blumenthal G. M., Dennis P. A. (2011). PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 11, 289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie Y., Suzuki A., Kataoka E., Sasaki T., Hamada K., Sasaki J., Mizuno K., Hasegawa G., Kishimoto H., Iizuka M., et al. (2004). Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 113, 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobbe C. B., Lapin V., Suzuki A., Mak T. W. (2008). The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene 27, 5398–5415 [DOI] [PubMed] [Google Scholar]
- Korpershoek E., Loonen A. J., Corvers S., van Nederveen F. H., Jonkers J., Ma X., Ziel-van der Made A., Korsten H., Trapman J., Dinjens W. N., et al. (2009). Conditional Pten knock-out mice: a model for metastatic phaeochromocytoma. J. Pathol. 217, 597–604 [DOI] [PubMed] [Google Scholar]
- Lesche R., Groszer M., Gao J., Wang Y., Messing A., Sun H., Liu X., Wu H. (2002). Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32, 148–149 [DOI] [PubMed] [Google Scholar]
- Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., Puc J., Miliaresis C., Rodgers L., McCombie R., et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 [DOI] [PubMed] [Google Scholar]
- Li G., Robinson G. W., Lesche R., Martinez-Diaz H., Jiang Z., Rozengurt N., Wagner K. U., Wu D. C., Lane T. F., Liu X., et al. (2002). Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 129, 4159–4170 [DOI] [PubMed] [Google Scholar]
- Liaw D., Marsh D. J., Li J., Dahia P. L., Wang S. I., Zheng Z., Bose S., Call K. M., Tsou H. C., Peacocke M., et al. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 16, 64–67 [DOI] [PubMed] [Google Scholar]
- Lu T. L., Chang J. L., Liang C. C., You L. R., Chen C. M. (2007). Tumor spectrum, tumor latency and tumor incidence of the Pten-deficient mice. PLoS ONE 2, e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Ziel-van der Made A. C., Autar B., van der Korput H. A., Vermeij M., van Duijn P., Cleutjens K. B., de Krijger R., Krimpenfort P., Berns A., et al. (2005). Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 65, 5730–5739 [DOI] [PubMed] [Google Scholar]
- Maehama T., Dixon J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 [DOI] [PubMed] [Google Scholar]
- Memarzadeh S., Zong Y., Janzen D. M., Goldstein A. S., Cheng D., Kurita T., Schafenacker A. M., Huang J., Witte O. N. (2010). Cell-autonomous activation of the PI3-kinase pathway initiates endometrial cancer from adult uterine epithelium. Proc. Natl. Acad. Sci. USA 107, 17298–17303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger D., Clifford J., Chiba H., Chambon P. (1995). Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc. Natl. Acad. Sci. USA 92, 6991–6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milam M. R., Celestino J., Wu W., Broaddus R. R., Schmeler K. M., Slomovitz B. M., Soliman P. T., Gershenson D. M., Wang H., Ellenson L. H., et al. (2007). Reduced progression of endometrial hyperplasia with oral mTOR inhibition in the Pten heterozygote murine model. Am. J. Obstet. Gynecol. 196, 247.e1–247.e5 [DOI] [PubMed] [Google Scholar]
- Muzumdar M. D., Tasic B., Miyamichi K., Li L., Luo L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 [DOI] [PubMed] [Google Scholar]
- Myers M. P., Stolarov J. P., Eng C., Li J., Wang S. I., Wigler M. H., Parsons R., Tonks N. K. (1997). P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 94, 9052–9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K., Ellenson L. H., Nemes A., Gu J., Tamura M., Yamada K. M., Cordon-Cardo C., Catoretti G., Fisher P. E., Parsons R. (1999). Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 96, 1563–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K., Lee R. T., Politis C., Hennessy I., Crane A., Puc J., Neshat M., Wang H., Yang L., Gibbons J., et al. (2001). An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/− mice. Proc. Natl. Acad. Sci. USA 98, 10320–10325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnacaram C. K., Teletin M., Jiang M., Meng X., Chambon P., Metzger D. (2008). Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 105, 2521–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. (2011). Good planning and serendipity: exploiting the Cre/Lox system in the testis. Reproduction 141, 151–161 [DOI] [PubMed] [Google Scholar]
- Song M. S., Salmena L., Pandolfi P. P. (2012). The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 [DOI] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J. L., Brothers G. M., Mirtsos C., Sasaki T., Ruland J., Penninger J. M., Siderovski D. P., Mak T. W. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 [DOI] [PubMed] [Google Scholar]
- Stambolic V., Tsao M. S., Macpherson D., Suzuki A., Chapman W. B., Mak T. W. (2000). High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 60, 3605–3611 [PubMed] [Google Scholar]
- Steck P. A., Pershouse M. A., Jasser S. A., Yung W. K., Lin H., Ligon A. H., Langford L. A., Baumgard M. L., Hattier T., Davis T., et al. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 [DOI] [PubMed] [Google Scholar]
- Sun H., Lesche R., Li D. M., Liliental J., Zhang H., Gao J., Gavrilova N., Mueller B., Liu X., Wu H. (1999). PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 96, 6199–6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., de la Pompa J. L., Stambolic V., Elia A. J., Sasaki T., del Barco Barrantes I., Ho A., Wakeham A., Itie A., Khoo W., et al. (1998). High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 8, 1169–1178 [DOI] [PubMed] [Google Scholar]
- Suzuki A., Nakano T., Mak T. W., Sasaki T. (2008). Portrait of PTEN: messages from mutant mice. Cancer Sci. 99, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimboli A. J., Cantemir-Stone C. Z., Li F., Wallace J. A., Merchant A., Creasap N., Thompson J. C., Caserta E., Wang H., Chong J. L., et al. (2009). Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461, 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J. A., Li F., Leone G., Ostrowski M. C. (2011). Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer Res. 71, 1203–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. (2009). Cre transgenic mouse lines. Methods Mol. Biol. 561, 265–273 [DOI] [PubMed] [Google Scholar]
- Wang S., Gao J., Lei Q., Rozengurt N., Pritchard C., Jiao J., Thomas G. V., Li G., Roy-Burman P., Nelson P. S., et al. (2003). Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221 [DOI] [PubMed] [Google Scholar]
- Yeager N., Klein-Szanto A., Kimura S., Di Cristofano A. (2007). Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 67, 959–966 [DOI] [PubMed] [Google Scholar]
- Yeager N., Brewer C., Cai K. Q., Xu X. X., Di Cristofano A. (2008). Mammalian target of rapamycin is the key effector of phosphatidylinositol-3-OH-initiated proliferative signals in the thyroid follicular epithelium. Cancer Res. 68, 444–449 [DOI] [PubMed] [Google Scholar]
- Yilmaz O. H., Valdez R., Theisen B. K., Guo W., Ferguson D. O., Wu H., Morrison S. J. (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 [DOI] [PubMed] [Google Scholar]
- Zhang W., Zhu J., Efferson C. L., Ware C., Tammam J., Angagaw M., Laskey J., Bettano K. A., Kasibhatla S., Reilly J. F., et al. (2009). Inhibition of tumor growth progression by antiandrogens and mTOR inhibitor in a Pten-deficient mouse model of prostate cancer. Cancer Res. 69, 7466–7472 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.