

Chromatin modifications contribute to the regulation of gene expression. The genome-wide distribution of a specific chromatin modification, trimethylation of Lys-27 of histone H3, was profiled in five tissues of maize. There is evidence that this chromatin modification plays an important role in regulating tissue-specific expression for a number of maize genes, including many transcription factors and imprinted genes.
Abstract
Trimethylation of histone H3 Lys-27 (H3K27me3) plays a critical role in regulating gene expression during plant and animal development. We characterized the genome-wide distribution of H3K27me3 in five developmentally distinct tissues in maize (Zea mays) plants of two genetic backgrounds, B73 and Mo17. There were more substantial differences in the genome-wide profile of H3K27me3 between different tissues than between the two genotypes. The tissue-specific patterns of H3K27me3 were often associated with differences in gene expression among the tissues and most of the imprinted genes that are expressed solely from the paternal allele in endosperm are targets of H3K27me3. A comparison of the H3K27me3 targets in rice (Oryza sativa), maize, and Arabidopsis thaliana provided evidence for conservation of the H3K27me3 targets among plant species. However, there was limited evidence for conserved targeting of H3K27me3 in the two maize subgenomes derived from whole-genome duplication, suggesting the potential for subfunctionalization of chromatin regulation of paralogs. Genomic profiling of H3K27me3 in loss-of-function mutant lines for Maize Enhancer of zeste-like2 (Mez2) and Mez3, two of the three putative H3K27me3 methyltransferases present in the maize genome, suggested partial redundancy of this gene family for maintaining H3K27me3 patterns. Only a portion of the targets of H3K27me3 required Mez2 and/or Mez3, and there was limited evidence for functional consequences of H3K27me3 at these targets.
INTRODUCTION
Packaging of DNA into chromatin is essential for the regulation of genome function in eukaryotes. Nucleosomes, the basic units of chromatin, are composed of ∼147 bp of DNA that is wrapped around an octomer built by four types of histones. Covalent modifications of histones, along with other factors, such as DNA methylation, chromatin-remodeling proteins, and small RNAs, contribute to the regulation of gene expression (Berger, 2007; Kouzarides, 2007; Berr et al., 2011). Heterochromatin is often used to refer to the parts of the genome that contain chromatin modifications associated with repression of gene expression. Constitutive heterochromatin describes regions of the genome that are stably maintained as heterochromatin throughout development and is often associated with 5-methylcytosine and H3K9me2 in plants (Bernatavichute et al., 2008). By contrast, facultative heterochromatin describes regions of the genome that exist as heterochromatin in some cell types and as euchromatin in other cells. Facultative heterochromatin is often associated with developmental regulation of gene expression and involves a set of chromatin modifications, including trimethylation of histone H3 Lys-27 (H3K27me3) (Schuettengruber et al., 2007; Zheng and Chen, 2011).
In plants, H3K27me3 modifications have been implicated in regulation of seed development, flowering time, vernalization, and organ identity (reviewed in Kohler and Villar, 2008; Schatlowski et al., 2008; Feng et al., 2010; Zheng and Chen, 2011). In both plants and animals, a remarkable number of transcription factors are targeted by H3K27me3 (Boyer et al., 2006; Pasini et al., 2007; Zhang et al., 2007; Kharchenko et al., 2011), further supporting the role of H3K27me3 in regulation of gene activity and development. H3K27me3 broadly marks repressed genes (Turck et al., 2007; Zhang et al., 2007) and is distinct from other epigenetic marks (Roudier et al., 2011). The expression levels of the H3K27me3-modified genes are very low and often exhibit a high degree of tissue specificity (with the majority of them expressed only in one or a few tissues), which suggests that repression of these genes by H3K27me3 is alleviated only in the place(s) where their expression is needed (Kohler and Villar, 2008; Feng et al., 2010). Recently, the levels of H3K27me3 were profiled in several plant tissues, including meristem tissues (Lafos et al., 2011) and roots and shoots (Wang et al., 2009). These studies revealed large tissue-specific variation in H3K27me3 levels. Moreover, hundreds of genes were shown to gain or lose H3K27me3 during cell differentiation, demonstrating dynamic regulation of an epigenetic modification in plants. H3K27me3 was shown to correlate with gene repression, and its release correlated with tissue-specific gene activation, both during differentiation and in mutants for Polycomb group (PcG) proteins (Lafos et al., 2011).
H3K27me3 is catalyzed by PcG proteins and is thought to regulate developmental processes in both plants and animals (reviewed in Schuettengruber et al., 2007; Berr et al., 2011; Zheng and Chen, 2011). The PcG genes were initially identified through the characterization of mutants that failed to properly maintain a repressed state of gene expression for homeotic genes in Drosophila melanogaster (Simon, 1995). A subset of the PcG proteins, including E(z), esc, Su(z)12, and Nurf55 in Drosophila (Schwartz and Pirrotta, 2007), form the PRC2 (for Polycomb-repressive complex) that is involved in catalyzing the methylation of Lys-27 of histone H3. Many plant species have orthologs for the PRC2 genes (reviewed in Hennig and Derkacheva, 2009; Kohler and Hennig, 2010; Zheng and Chen, 2011). Homologs of the E(z)-like gene have been shown to encode H3K27 methyltransferases in animal species and likely encode the same function in plants (Schubert et al., 2006). Arabidopsis thaliana contains three genes, CURLY LEAF (CLF), SWINGER (SWN), and MEDEA (MEA) that are similar to Drosophila E(Z) (Goodrich et al., 1997; Grossniklaus et al., 1998; Chanvivattana et al., 2004). MEA appears to function primarily in gametophyte and seed development, whereas CLF and SWN are broadly expressed and partially redundant in vegetative and reproductive development (Chanvivattana et al., 2004; Hennig and Derkacheva, 2009). The clf swn double mutants in Arabidopsis show severe developmental abnormalities (Chanvivattana et al., 2004) and have no H3K27me3 as shown by immunoblot analysis (Lafos et al., 2011), suggesting that the majority of H3K27me3 in vegetative tissues of Arabidopsis is catalyzed by PRC2 proteins and requires at least one copy of clf or swn. The unique contributions of CLF and SWN to H3K27me3 profiles have not been elucidated at a genome-wide level in Arabidopsis. The maize (Zea mays) genome encodes three E(z) homologs: Mez1, Mez2, and Mez3 (Springer et al., 2002). Mez1 is an imprinted gene that is most closely related to the CLF gene from Arabidopsis (Haun et al., 2007). Mez2 and Mez3 are highly similar to each other (92% nucleotide identity), are located in colinear regions of the maize genome, and are likely paralogs resulting from the ancient allopolyploid event in maize (Springer et al., 2002). These two genes are more closely related to the SWN/MEA genes in Arabidopsis (Haun et al., 2007).
Most studies of H3K27me3 in plants have focused on rice (He et al., 2010) and Arabidopsis (for example, Turck et al., 2007; Zhang et al., 2007; Ha et al., 2011; Lafos et al., 2011), plants with relatively small genomes. The relatively large maize genome with interspersed genes and repetitive sequences (Schnable et al., 2009) provides an opportunity to further understand the profile of H3K27me3 in plant genomes. One study assessed the genome-wide distribution of H3K27me3 in maize roots and shoots using chromatin immunoprecipitation (ChIP) sequencing (Wang et al., 2009). They found a general enrichment for H3K27me3 in genes relative to transposons and documented differences in the distribution of H3K27me3 relative to DNA methylation. However, detailed characterization of the maize genes targeted by H3K27me3 has not been previously reported. We profiled the H3K27me3 distribution in five developmentally distinct tissues in two genetic backgrounds, B73 and Mo17, and in mez2/3 mutants. We found that H3K27me3 targets vary greatly between different tissues and show limited variation between genetic backgrounds.
RESULTS
Profiling H3K27me3 Levels throughout the Maize Genome
We profiled the distribution of H3K27me3 in several tissues and genotypes of maize through Chromatin immunoprecipitation (ChIP) with H3K27me3 antibodies followed by hybridization to a microarray (ChIP-chip). Three independent biological replicates of ChIP-chip were performed for five B73 tissues: deetiolated maize seedlings 12 d after sowing, 3- to 4-cm immature ears, 15- to 20-cm tassels, embryos 14 d after pollination, and endosperm 14 d after pollination (see Methods for details on plant growth and tissue collection). H3K27me3 distribution was also profiled in Mo17 for the same tissues, except for tassels. The microarray platform used for this experiment contains ∼1.4 million probes that are restricted to single-copy sequences in the B73 reference genome (Eichten et al., 2011). Seven sequences that exhibited significant enrichment for H3K27me3 in at least one tissue and four sequences that did not exhibit evidence for H3K27me3 were selected for validation by quantitative PCR (qPCR) (see Supplemental Data Set 1 and Supplemental Figure 1 online). The presence and tissue-specific patterns of H3K27me3 found in ChIP-chip experiments were confirmed by the qPCR assays. There was also significant overlap in the profile of H3K27me3 in our seedling tissue and that reported by Wang et al. (2009) (see Supplemental Figure 2 online).
H3K27me3 was found more frequently in the gene-dense chromosome arms and was less prevalent in the pericentromeric regions (Figure 1A). This is distinct from the abundance of the marks associated with constitutive heterochromatin, such as 5-methylcytosine and H3K9me2 (Eichten et al., 2011, 2012), which are enriched in the gene-poor pericentromeric regions (Figure 1A). This genomic distribution of H3K27me3 and the other marks is quite similar to observations in Arabidopsis (Zhang et al., 2007). Whereas H3K27me3 was more prevalent in gene-rich portions of the maize genome, it was not necessarily enriched over transcribed sequences (Figure 1B; see Supplemental Table 1 online). H3K27me3 was most abundant in sequences that are immediately 5′ or 3′ of genic sequences and was enriched in exons relative to introns (see Supplemental Table 1 online).
Figure 1.

The Genomic Distribution of H3K27me3 and DNA Methylation in Different Tissues of B73 and Mo17.
ChIP-chip analysis was used to profile the genomic distribution of chromatin modifications in maize.
(A) Enrichment for H3K27me3 (red), H3K9me2 (blue) (Eichten et al., 2012), and DNA methylation (blue) (Eichten et al., 2011) was visualized along each of the 10 maize chromosomes. The relative gene density is shown in black.
(B) A more detailed view of the H3K27me3 profile is shown for 400 kb of chromosome 9.
Whereas the analysis of individual probes can provide information on the distribution of H3K27me3, it is often more informative to identify regions of enrichment for a particular modification. DNAcopy (Venkatraman and Olshen, 2007) followed by expectation maximization analysis (Dempster et al., 1977) was used to identify segments within the genome that include multiple adjacent probes with enrichment for H3K27me3 (Table 1; see full details in Supplemental Data Sets 2A to 2E online). This analysis identified over 1000 regions of H3K27me3 in each of the profiled tissues. In total, in B73, the H3K27me3-enriched fraction accounted for 0.13% (ear) to 1.91% (seedling) of the genome. In general, the tissues with higher portions of undifferentiated meristematic cells had fewer regions with H3K27me3, while more differentiated tissues had more prevalent H3K27me3. The median segment length for H3K27me3-enriched regions was 1 to 2 kb in each of the tissues, which implies modification of multiple adjacent nucleosomes. A more detailed analysis of the higher resolution (due to higher density of probes) data for chromosome 9 did not reveal a large number of small regions, which would be expected if the modification were limited to a single nucleosome. A substantial portion of the H3K27me3 segments (24%) did not contain any genes and contained only single-copy intergenic sequences, and 84% of the remaining segments were restricted to a single gene (see Supplemental Figure 3 online). We did not find any examples in which two adjacent genes marked by H3K27me3 exhibited identical patterns of H3K27me3 in all tissues profiled.
Table 1. Characteristics of H3K27me3 Marked Segments in Maize Epigenome.
| Tissue | No. of Segmentsa | Total Length (Mb) | Average Length | Median Length | Proportion of Genome |
|---|---|---|---|---|---|
| Seedlings, all | 4374 | 37.15 | 6890 | 1800 | 1.91% |
| Seedlings, Mez2/3 dependent | 742 | 1.97 | 2661 | 600 | 0.10% |
| Seedlings, Mez2/3 independent | 3530 | 34.96 | 9903 | 3600 | 1.79% |
| Immature Ear | 1183 | 2.49 | 2107 | 1200 | 0.13% |
| Tassel | 3960 | 18.39 | 4645 | 2000 | 0.92% |
| Endosperm | 3392 | 19.37 | 5710 | 1860 | 0.96% |
| Embryo | 4307 | 20.95 | 4863 | 1400 | 1.07% |
The number of segments with a segment mean [log2(H3K27me3 IP/input)] > 1.
Developmental Variation for H3K27me3 Marks
We proceeded to identify the genes that were marked with H3K27me3 in at least one tissue. In total 12,266 (11%) genes in the working gene set (WGS; n = 110,028; maize genome annotation 5b.60) were marked by H3K27me3 in at least one of the tissues. These included 6337 genes (16%) of the filtered gene set (FGS; n = 39,585; Figure 2; see Supplemental Data Set 3 online), which represents a significant (P < 0.01) enrichment for H3K27me3 in the FGS relative to the WGS. The WGS included all putative genes in maize, while the FGS was filtered to remove gene fragments, pseudogenes, and transposon-related sequences (Schnable et al., 2009). The genes that were modified with H3K27me3 exhibited substantial variation among the five profiled tissues (Figure 2). A large portion of the H3K27me3-marked genes were marked by H3K27me3 only in one (40.6%) or two (21%) of the five tissues. Only 8.2% of the H3K27me3 target genes contained the chromatin modification in all five tissues. Clustering of H3K27me3 enrichment levels for all segments in these five tissues revealed that the triploid endosperm was most different from the other tissues, exhibiting many uniquely marked genes (Figure 2A). When we looked at the variation among the diploid vegetative tissues, we found that 30.4% of the genes with H3K27me3 were marked by H3K27me3 only in one of the four tissues (Figure 2B) and 17.9% were marked in all four tissues. The level of tissue-specific gene expression for the genes marked by H3K27me3 was assessed using data from a maize gene expression atlas (Sekhon et al., 2011). High levels of Shannon entropy (Schug et al., 2005; Zhang et al., 2007) indicate genes that have constitutive expression whereas low levels of entropy reflect tissue-specific expression. The genes that were marked with H3K27me3 in one or two of the tissues were enriched for those with lower levels of entropy compared with all maize genes, reflecting some enrichment for genes with tissue-specific expression. The genes that were marked by H3K27me3 in four or five tissues were even more biased toward higher levels of tissue-specific expression (Figure 2C). There were some examples of differences in H3K27me3 levels between B73 and Mo17 (Figure 2D). However, a comparison of H3K27me3 levels in multiple tissues of B73 and Mo17 suggests that there are more differences between tissues than between genotypes (Figure 2D). Our analyses use probes designed from the reference B73 genome sequence and have omitted probes that show high levels of CGH variation, which may result in an underestimation of the level of H3K27me3 variation among genotypes.
Figure 2.

Developmental Variation in H3K27me3 Marks in Five Maize Tissues in B73 and Mo17.
(A) Hierarchical clustering (Ward’s method) of 6337 filtered genes with H3K27me3 marks in at least one of five tissues of B73.
(B) Distribution of 5690 filtered genes marked with H3K27me3 in at least one of four diploid tissues of B73.
(C) Tissue specificity of gene expression in B73 for H3K27me3 target genes marked in one to two tissues (black dotted line), four to five tissues (orange dashed line), and non-H3K27me3 target genes (blue line), measured by Shannon entropy values (low entropy values = high tissue specificity).
(D) Hierarchical clustering of segments marked by H3K27me3 in at least one of the tissues of B73 or Mo17. The labels indicate the tissue and genotype (blue, B73; red, Mo17). The color intensity reflects the level of H3K27me3 enrichment.
Relationship between Gene Expression and H3K27me3
H3K27me3 can play a role in contributing to tissue-specific regulation of gene expression. To understand the effects of H3K27me3 on maize gene expression, we performed RNA sequencing for tissues that were similar (seedling, embryo, and endosperm were from different biological samples but were similarly harvested) or identical (immature ear and tassel) to the tissues used for H3K27me3 profiling. The genes that were marked with H3K27me3 were significantly (P < 0.01) enriched for genes with low or no expression (<1 gene-specific reads per kilobase gene length per million reads [RPKM]) in all tissues (Table 2; see Supplemental Table 2 online). Analysis of the genes with at least 1 RPKM revealed that the genes marked by H3K27me3 were enriched for those with lower expression levels (Figure 3A; see Supplemental Figure 4 online). However, there were also some genes marked with H3K27me3 that were expressed at moderate to high levels. Each of these tissues represents a complex mixture of cell types that might include some cell types with the H3K27me3 modification and others that lack the modification. This complexity of cell types may interfere with the ability to correlate H3K27me3 and gene expression in these tissues. These analyses suggest that in many cases the presence of H3K27me3 is associated with reduced levels of gene expression but that there are some cases in which the presence of H3K27me3 is not associated with low expression.
Table 2. Syntenic Analysis of Genes Targeted by H3K27me3 in B73 Tissues.
| Features | Genome | Subgenome 1 | Subgenome 2 | Proportion Subgenome 1 | Proportion Subgenome 2 |
|---|---|---|---|---|---|
| All genes | 110,029 | 61,541 | 38,923 | 55.9% | 35.4% |
| All methylated genes | 12,266 | 7,649 | 4,078 | 62.4% | 33.2% |
| Proportion of methylated genes | 11.1% | 12.4% | 10.5% | NA | NA |
| Pairs of local duplicates | 9,669 | 5,431 | 3,358 | 56.2% | 34.7% |
| Pairs of methylated local duplicates | 808 | 508 | 268 | 62.9% | 33.2% |
| Only one gene of a local duplicate pair methylated | 2,071 | 1,398 | 593 | 67.5% | 28.6% |
| Proportion of local duplicates with methylation marks | 29.8% | 35.1% | 25.6% | NA | NA |
| Proportion of the pairs with preserved methylation | 28.1% | 26.7% | 31.1% | NA | NA |
| Pairs of syntenic genes | 3,795 | 3,795 | 3,795 | NA | NA |
| Pairs of methylated syntenic genes | 297 | 297 | 297 | NA | NA |
| Only one gene of a syntenic pair methylated | 773 | 405 | 368 | 52.4% | 47.6% |
| Proportion of syntenic duplicates with methylation marks | 28.2% | 18.5% | 17.5% | NA | NA |
| Proportion of the syntenic pairs with preserved methylation | 27.8% | 42.3% | 44.7% | NA | NA |
NA, not applicable.
Figure 3.

H3K27me3 Negatively Correlates with Gene Expression Levels.
(A) Expression level of H3K27me3 target genes (blue and purple) compared with non-H3K27me3 target genes (red and black) in tassel and immature ear tissue. Only FGS genes with expression of at least 1 RPKM were used in this comparison. The numbers in the legend indicate the number of expressed genes with (target) or without (nontarget) H3K27me3 in the indicated tissue.
(B) H3K27me3 (red indicates positive values, while blue indicates negative values) and RNA sequencing profiles (height of black bars reflects number of reads) are shown for two examples of genes that exhibit a negative correlation between H3K27me3 and expression levels. Black bars at the bottom indicate 1-kb scale.
The effects of H3K27me3 on developmental gene regulation were investigated by comparing the presence of the modification and expression in each of the five tissues. One-quarter (25.1%) of the 6337 genes exhibited a negative correlation (r ≤ 0.5) between expression level and H3K27me3 presence among these five tissues. In many of these examples, the loss of H3K27me3 in one or several tissues was associated with activation of expression (Figure 3B). This suggests that H3K27me3 is associated with transcription in maize and that the developmental variation in H3K27me3 levels can be associated with tissue-specific expression. These genes exhibiting tissue-specific expression that was associated with H3K27me3 were observed to have similar patterns of chromatin modification and transcript abundance in both B73 and Mo17 (Figure 3B). There were genes with tissue-specific expression that did not contain H3K27me3 in any of the tissues that were studied, and there were examples of tissue-specific H3K27me3 patterns that were not well correlated with tissue-specific expression levels. It is likely that H3K27me3 provides a component of tissue-specific gene regulation but is certainly not the only factor regulating tissue-specific expression.
Characterization of H3K27me3 Targets
The genes that are targeted by H3K27me3 were further characterized to understand potential functions and conservation among species. Putative Gene Ontology (GO) annotations were assigned for maize genes based on orthology to Arabidopsis. Enrichments for functional GO annotations were assessed in a variety of gene lists using BinGO (Maere et al., 2005). The biological process and molecular function GO annotations were assessed for all 6337 H3K27me3 targets as well as several subsets that exhibited different tissue-specific patterns (see Supplemental Figures 5 and 6 online). There was significant (P = 6*10−92) enrichment for genes annotated as having transcription factor activity in most of the lists of H3K27me3 targets (see Supplemental Figure 5 online). The H3K27me3 targets were also significantly enriched for biological process annotations of “developmental process” and “transcription, DNA-dependent” (see Supplemental Figures 5 and 6 online). The level of enrichment for specific GO terms varies for genes that were marked by H3K27me3 in one to two tissues relative to those marked in four to five tissues. Genes that were constitutively marked by H3K27me3 (targets in four or five tissues) showed very strong overrepresentation of transcription factors and genes involved in developmental processes, while the genes that were targets for H3K27me3 in only one or two tissues did not show such overrepresentation (see Supplemental Figure 5 online). There is evidence that some families of maize transcription factors are enriched for H3K27me3 targets relative to other families (see Supplemental Figure 6B online). Certain families of transcription factors, including YABBY, MADS, G2-like, Homeobox, NAC, and C2C2-DOF, were significantly enriched (P < 0.01) for genes marked by H3K27me3. The genes marked by H3K27me3 were also significantly enriched for classical maize genes (Schnable and Freeling, 2011) that were defined by forward genetics (see Supplemental Figure 7 online).
The ancient tetraploid nature of maize results in two subgenomes that are biased in retention and expression level (Schnable et al., 2011). The proportion of genes with H3K27me3 was slightly higher than expected for subgenome 1 (the more retained and expressed subgenome), but this was not statistically significant. There are many examples of paralog retention following whole-genome duplication, and we assessed whether these paralogs that are located in syntenic positions have conserved patterns of H3K27me3. Among 3795 genes with retained syntenic gene duplications (Schnable et al., 2012), we identified 1070 (28.2%) pairs of genes with at least one gene marked by H3K27me3, which is very similar to the expected proportion. Out of these 1070 pairs, 297 pairs (27.8%) had both copies marked with H3K27me3, while in the remaining 773 pairs, only one of the genes was marked with H3K27me3 (Table 2). The tissue-specific pattern of H3K27me3 was often different in instances in which both paralogs were marked with H3K27me3. Only ∼10% of the 297 pairs of paralogs in which both were marked by H3K27me3 exhibited the same tissue-specific pattern of modification. This suggests that paralogs often have diverged in their regulation by H3K27me3.
The targets of H3K27me3 in maize were compared with the targets of H3K27me3 in Arabidopsis (Lafos et al., 2011) and rice (Oryza sativa) (He et al., 2010). Many (34%) of the maize targets of H3K27me3 that had orthologs in Arabidopsis were marked by H3K27me3 in both species. The conservation of H3K27me3 targets between maize and rice was even higher (64% for maize genes marked with H3K27me3 in seedlings). This conservation was substantially higher for genes marked by H3K27me3 in multiple maize tissues. For example, rice orthologs for 74% of maize genes marked by H3K27me3 in all five profiled tissues were also marked by H3K27me3 in rice seedlings, while only 28 to 38% of the maize genes marked by H3K27me3 in only one of the tissues had rice orthologs marked by H3K27me3. Given the fact that different tissues were sampled in the different species, we would expect some variation in the targets. However, there was evidence for conservation of the targets of H3K27me3 among plant species.
The Majority of Paternally Expressed Imprinted Genes Are Marked by H3K27me3
There is evidence that H3K27me3 plays a role in the regulation of imprinted gene expression in endosperm tissue of Arabidopsis (Wolff et al., 2011) and maize (Haun and Springer, 2008). A recent RNA sequencing analysis of maize endosperm tissue identified 100 imprinted maize genes, including 46 paternally expressed genes (PEGs) and 54 maternally expressed genes (Waters et al., 2011). Nearly all of the PEGs (41/46; 89%) were marked by H3K27me3 in endosperm tissue, whereas only four of 54 maternally expressed genes were marked with H3K27me3. The allele specificity of H3K27me3 was tested for several of the PEGs. ChIP of B73xMo17 hybrid endosperm revealed that the H3K27me3 found in endosperm is restricted to the silenced maternal allele (see Supplemental Figure 8 online). The comparison of expression patterns and H3K27me3 patterns among tissues revealed two subgroups of the H3K27me3-marked PEGs (see Supplemental Figure 8 online). One group (21/41) of genes was expressed in many plant tissues and contained H3K27me3 only in endosperm tissue. The genes from the other group (13/41) were expressed only in endosperm tissue and marked by H3K27me3 in all tissues studied.
Mutations in Mez2 and Mez3 Reduce H3K27me3 in Some Regions but Not Others
Evidence from Arabidopsis suggests that the majority of H3K27me3 in plants is provided by E(z) homologs (Lafos et al., 2011). The three maize genes with homology to E(z), namely, Mez1, Mez2, and Mez3, were expressed in the five tissues that were analyzed (Figure 4A). Several mu-insertion alleles were obtained via reverse-genetic screening, including mez3-m1, mez3-m4, and mez2-m4 (Figure 4B). Although Mu insertions were identified in sequences upstream of Mez1 (Haun et al., 2009), we did not find any Mez1 insertions that resulted in loss-of-function alleles. Mez2 and Mez3 mutant alleles were backcrossed into B73 for at least seven generations. The effect of these insertions upon the transcript was assessed through quantitative RT-PCR (qRT-PCR) and RNA sequencing experiments. qRT-PCR experiments amplifying the regions located 3′ of the insertion detected severely reduced transcript levels in homozygous mutant individuals (Figure 4C) and RNA sequencing data from the mutant lines revealed dramatically reduced expression through the whole transcript (for mez2-m4 and mez3-m1 alleles) and frequent mis-splicing of the affected exons (for mez2-m4 and mez3-m4 alleles). There were no striking phenotypic differences in homozygous mutant plants for each of the three alleles or a mez2-m4 mez3-m4 double mutant relative to wild-type siblings. These mutant lines tended to be somewhat smaller and may have minor quantitative phenotypes but were fully fertile and had a fairly normal morphology.
Figure 4.

Distribution of H3K27me3 in Mez2 and Mez3 Mutants.
(A) Expression levels of three mez genes in five tissues profiled.
(B) Mutant alleles of mez3 and mez2 genes used in the study. Mu insertions in mez genes are shown by black triangles. Exons are shown in blue rectangles, while introns are shown as blue lines. One of the introns in each of the two genes is very long and is not shown completely.
(C) Expression of mez2 and mez3 in mutants used in the study (the level of expression was analyzed by qRT-PCR and normalized to expression levels of mez2 and mez3 genes in B73; GAPC was used as a control gene for normalizations). Error bars show sd between the technical replicates.
(D) Hierarchical clustering of mez2/3-dependent and mez2/3-independent segments. Each row represents a segment marked by H3K27me3 in B73 seedlings and each column represents a different genotype. Red or black indicate tissues in which a particular segment exhibited high or low levels of H3K27me3, respectively.
(E) Examples of mez2/3-dependent and mez2/3-independent genes targeted by H3K27me3 in B73 seedlings. Black bars indicate 1-kb scale.
Despite the lack of a striking morphological phenotype, we were interested in monitoring the molecular phenotypes in these mutant lines. H3K27me3 profiling was performed using ChIP-chip on a pool of homozygous mutant plants for each of the three alleles and for homozygous mez2-m4 mez3-m4 double mutants along with a B73 control to assess the role of these genes in the genome-wide distribution of H3K27me3 in maize (see Supplemental Data Sets 2A and 3 online). RNA sequencing was performed on these same tissues to assess transcript abundance in these mutant genotypes. A simple profile of H3K27me3 abundance over genes revealed that the levels of H3K27me3 in the promoter regions were reduced in each of the single mutants and were most strongly affected in the double mutant (see Supplemental Figure 9A online). A set of 4374 segments enriched for H3K27me3 was detected in the B73 control seedlings that were grown and sampled at the same time as the mutant stocks (see Supplemental Table 1 online). The level of H3K27me3 for these segments in each of the genotypes was used to perform hierarchical clustering (Figure 4D). There was clear evidence that a subset of regions with H3K27me3 was affected in the mez2 and/or mez3 mutants but that many regions with H3K27me3 were not affected by these mutations. The regions that were affected in the mez2/3 mutants were referred to as Mez2/3 dependent, and the segments not affected in mez2/3 mutants were termed Mez2/3 independent. The 742 Mez2/3-dependent segments (17% of the total number of H3K27me3-marked segments) include 21 that were only mez2 dependent, 315 that were only Mez3 dependent, 90 that were affected only in the double mutant, and 316 that were dependent on either of the mutations (Figure 4E, Table 1; see Supplemental Data Set 2A online). The effects of the two mez3 mutant alleles were quite similar. At least one genomic region showing each of these patterns of mez2/3 dependence or independence was selected and used for qPCR validation (see Supplemental Figures 9B and 9C and Supplemental Data Set 1 online). The patterns observed in ChIP-chip were confirmed by qPCR. There was no evidence for genomic clustering of the Mez2/3-dependent regions.
While there was a set of genomic regions that required Mez2/3 for modification by H3K27me3, we did not find evidence that this modification was functionally important at these regions. There were 659 FGS genes located in Mez2/3-dependent regions, and these were enriched for nonsyntenic genes or for instances of H3K27me3 that was not conserved among species. These genes did not show GO enrichments or tissue-specific expression patterns similar to those observed for the mez2/3-independent regions (see Supplemental Figures 10 and 11A online). RNA sequencing was performed to assess expression changes in the Mez2/3 mutants relative to wild-type seedlings. Although there were a number of genes (738) with twofold differences in expression levels in mez2-m4 mez3-m4 double mutants relative to the wild type, these genes were not enriched for targets of Mez2/3-dependent H3K27me3. Instead, the majority of the genes with differential expression was not targets of H3K27me3 or had Mez2/3-independent H3K27me3 (see Supplemental Figure 11B online). Analysis of the expression levels of all genes with mez-dependent H3K27me3 did not reveal evidence for altered expression levels in the mutants relative to the wild type (see Supplemental Figure 11C online), suggesting that the Mez2/3-dependent H3K27me3 does not play a significant role in transcriptional regulation in seedling tissue.
DISCUSSION
In Arabidopsis, H3K27me3 has been shown to play an important role in the regulation of gene expression by providing facultative heterochromatin that can reinforce silencing of gene expression in some tissues (Kohler and Villar, 2008; Schatlowski et al., 2008; Feng et al., 2010; Zheng and Chen, 2011). Relatively little is known about the role of H3K27me3 in other plant species. An analysis of several chromatin modifications in rice revealed that H3K27me3 is present in many genes and is often associated with lower gene expression (He et al., 2010). Analysis of H3K27me3 in maize roots and shoots demonstrated that low expressed genes are enriched for H3K27me3 and that very few genomic regions are enriched for both DNA methylation and H3K27me3 (Wang et al., 2009). Here, we profiled the genomic distribution of H3K27me3 in several tissues of two maize genotypes and compared the variation in H3K27me3 distribution among tissues and genotypes to understand the role that H3K27me3 might play in regulation of tissue-specific gene expression and in gene expression variation due to different genetic backgrounds. This study also developed a catalog of H3K27me3 targets that can be exploited to identify the role of H3K27me3 in developmental regulation of gene expression. Finally, the profiling of H3K27me3 and gene expression in mutants for several maize H3K27 methyltransferases revealed relatively minor contributions of these genes to developmental regulation of gene expression.
Conservation of H3K27me3 Profiles among Tissues, Genotypes, and Species
Epigenetic regulation can contribute to developmental regulation of gene expression or to (epi-)allelic variation among different individuals. We profiled several distinct maize tissues from two genotypes, B73 and Mo17, in order to assess the level of variation for H3K27me3 distribution. There were many examples of tissue-specific differences in the H3K27me3 patterns. This is similar to recent finding in Arabidopsis (Lafos et al., 2011). H3K27me3 is often associated with repression of regulatory gene expression. We found relatively few examples of genotypic differences in H3K27me3. It is possible that the repetitive regions of the genome or regions with structural variation between B73 and Mo17 exhibit H3K27me3 variation among genotypes, but these regions were not included in our analyses. Few studies have compared the H3K27me3 profiles for multiple different genotypes of the same species. Some differences were found between Arabidopsis ecotypes, and those primarily exhibit additive inheritance in F1 offspring (Moghaddam et al., 2011). When variation in H3K27me3 was assessed in two different rice subspecies, a small number of examples of genes with differences in H3K27me3 levels were found, while variation in DNA methylation levels was much more common between the two subspecies (He et al., 2010). Our results suggest that H3K27me3 likely plays an important role in regulating gene expression during development and that these H3K27me3 patterns are mostly conserved among maize inbreds (see Supplemental Table 3 online). This is not to suggest that there is no variation in the patterns of H3K27me3 between genotypes as we do see examples of differences. However, the majority of genes that exhibit differential expression between B73 and Mo17 do not show differences in H3K27me3 abundance.
We also characterized the conservation of H3K27me3 between maize and other species and between the subgenomes derived from the allotetraploid ancestry of maize (Gaut and Doebley, 1997). It is somewhat difficult to compare the H3K27me3 patterns among different species, as collecting similar tissues from species with substantial morphological differences is complicated. However, there was clear evidence for conservation of H3K27me3 targets among species. In particular, when we looked at genes that were marked with H3K27me3 in multiple tissues, we found that a large proportion of maize H3K27me3 targets were marked by H3K27me3 in both rice (He et al., 2010) and Arabidopsis (Lafos et al., 2011). Given the conservation of H3K27me3 targets between species, we expected to find significant similarities in H3K27me3 targets between the two subgenomes of maize. The paralogs that were retained following the whole-genome duplication might be expected to exhibit similar patterns of H3K27me3. However, the majority (72.2%) of pairs of syntenic genes had H3K27me3 at only one of the two paralogs (Table 2). Even when H3K27me3 was found at both paralogs, it frequently exhibited differences in tissue-specific patterns. This divergence in tissue-specific H3K27me3 patterns for retained paralogs may reflect subfunctionalization in gene expression patterns that has arisen through differences in chromatin-based regulation of these paralogs. A recent analysis suggests that H3K27me3 may play a role in functional divergence of Arabidopsis paralogs through contributing to expression differences between paralogs (Berke et al., 2012).
Targets of H3K27me3
In Arabidopsis, H3K27me3 is known to regulate genes that play important roles in development, including AGAMOUS (AG) (Schubert et al., 2006), SHOOT MERISTEMLESS (STM) (Schubert et al., 2006), KNOX genes (Xu and Shen, 2008), and FLOWERING LOCUS C (Bastow et al., 2004). A recent genome-wide profiling of H3K27me3 in several tissues of Arabidopsis provides evidence that H3K27me3 is present at genes coding for many transcription factor families and at many of the genes involved in auxin biosynthesis, transport, and perception (Lafos et al., 2011). We found that many developmentally important maize genes are targets of H3K27me3 as well. The classical maize gene list (Schnable and Freeling, 2011) includes genes that were identified by visible mutant phenotypes. Many (138 of 468) of these genes were targeted by H3K27me3. This list includes a number of genes involved in specific pathways (such as anthocyanin or starch synthesis) as well as genes that contribute to development (such as kn1 and tb1). H3K27me3 marks were present at many of the genes known to be important for proper development. Maize transcription factors were also enriched for H3K27me3 targets. A family-specific analysis showed that certain families of maize transcription factors, including YABBY, MADS, G2-like, homeobox, and NAC domain genes, were particularly likely to be marked by H3K27me3. These include the maize orthologs of AG, STM, and KNOX genes, which are all targets of H3K27me3 in Arabidopsis. This study thus demonstrates that many genes with important regulatory roles are targets of H3K27me3 and provides a catalog of maize genes that are marked by H3K27me3.
Role of H3K27me3 in Imprinting
There is evidence that H3K27me3 is critical for regulating imprinted gene expression in the endosperm of plants (Berger and Chaudhury, 2009; Raissig et al., 2011). MEDEA, the first well-characterized imprinted gene in Arabidopsis (Grossniklaus et al., 1998), encodes a putative H3K27memethyltransferase that is required for endosperm development and is part of a specific FIS-PRC2 complex that functions in endosperm development and includes FIE and FIS2 (Kohler et al., 2003). This FIS-PRC2 complex is necessary for proper imprinting at MEA (Gehring et al., 2006; Jullien et al., 2006), at Pheres1 (Makarevich et al., 2006), and at several other imprinted Arabidopsis genes (Wolff et al., 2011).
We found a striking enrichment for H3K27me3 at maize PEGs. Nearly all of the PEGs (41/46) identified in maize (Waters et al., 2011) had enrichment for H3K27me3 in endosperm tissue and, in the three instances tested, we found that this H3K27me3 was restricted to the silent maternal allele. A combined analysis of the patterns of expression and H3K27me3 through development revealed two different types of pattern. Some of these PEGs were expressed in numerous vegetative tissues. H3K27me3 was observed only in endosperm tissue for these constitutively expressed PEGs. These might require specific recruitment of a FIS-PRC2 complex to mark the maternal allele with H3K27me3 during gametogenesis. The other group of PEGs had endosperm-specific expression. These PEGs were marked by H3K27me3 throughout development and might specifically lose H3K27me3 at the paternal allele. The remodeling of H3K27me3 during gametogenesis could include specific recruitment of H3K27 methyltransferase activity to some loci and recruitment of H3K27 demethylase activities to other loci. The H3K27me3 histone methyltransferase activity is likely supplied by MEA or related genes (Kohler et al., 2005), while the H3K27 demethylase activity could be provided by REF6 or a related gene (Lu et al., 2011). It is worth noting that while endosperm does have unique H3K27me3 patterns, there were also many genomic regions that were marked similarly in endosperm and vegetative tissues. This suggests that the remodeling of H3K27me3 is limited to specific genomic regions.
Mez2 and Mez3 Play Minimal Independent Roles in Gene Regulation through H3K27me3
Most plant species have multiple genes encoding each of the components of the PRC2 complex (reviewed in Hennig and Derkacheva, 2009; Kohler and Hennig, 2010). The Arabidopsis genome contains three orthologs to the Drosophila E(z) gene, MEA, CLF, and SWN (Chanvivattana et al., 2004; Hennig and Derkacheva, 2009; Zheng and Chen, 2011). MEA appears to mainly function in gametes and endosperm development and does not play a major role in vegetative development (Zheng and Chen, 2011). The mutant phenotype of CLF (Goodrich et al., 1997) provides evidence for a unique role of CLF in vegetative tissues. No morphological abnormalities were observed in SWN single mutants (Chanvivattana et al., 2004). However, swn clf double mutants display more severe developmental abnormalities (Chanvivattana et al., 2004) and further reduction in genomic levels of H3K27me3 (Lafos et al., 2011), providing evidence for substantial redundant function of CLF and SWN. There is also biochemical evidence that either CLF or SWN can participate in PRC2 complexes found in vegetative tissues (Chanvivattana et al., 2004; Makarevich et al., 2006).
Maize contains three E(z) orthologs (Springer et al., 2002). The potential for an endosperm-specific function analogous to MEA is less clear in maize. All three of the Mez genes were expressed in all of the tissues that we studied, including endosperm. The imprinted expression of only Mez1 (Haun et al., 2007) may suggest that Mez1 is critical for endosperm functions, but both Mez2 and Mez3 were also detected in this tissue. Although no line with a Mutator transposon inserted into an exon was identified for Mez1 (Haun and Springer, 2008), we were able to identify exon insertion alleles for both of the maize SWN orthologs, Mez2 and Mez3. Profiling the distribution of H3K27me3 in the single and double mutants revealed a subset of H3K27me3-enriched loci that were dependent upon these genes and provided evidence for specific contributions of each of these genes as well as of both of the genes together in genomic distribution of H3K27me3 marks. However, the H3K27me3 that was Mez2/3 dependent often occurred in small regions that were not conserved among species and did not appear to be correlated with gene expression levels or patterns. RNA sequencing analysis of the mutant lines did not reveal evidence for widespread gene expression differences. These results, together with the lack of developmental abnormalities in the mez mutants, suggest that the H3K27me3 that is controlled by Mez2/3 is largely dispensable for proper developmental and gene regulation. It is possible that these mez2/3-dependent H3K27me3 marks are important for precise regulation of gene expression and that loss of these marks might relate to variation in a population of cells. The conserved presence of SWN-like orthologs in many plants suggests that this gene must play an important role, but there is no clear evidence for a specific unique function provided by SWN-like genes in maize or Arabidopsis.
METHODS
Plant Growth and Tissue Collection
The whole aboveground tissue was collected from 12-d-old deetiolated seedlings that were grown in the dark at 30°C in 1:1 mix of autoclaved field soil and MetroMix. For embryo, immature ear (∼5 to 7 cm in length), tassel (green soft tassels prior to shedding, ∼15 to 20 cm in length), and endosperm collection, plants were grown to maturity in the field of the University of Minnesota Saint Paul Agricultural Research station during the summer of 2011. Endosperm and embryos were harvested from multiple B73 and Mo17 ears 14 d after the self-pollination. The seeds for each replicate came from a unique, single source (ear). Each biological replicate was planted at a different time and represents the pooled aboveground tissue from eight to 10 plants. Three replicate tissue collections were performed for all B73 and Mo17 tissues. For each replicate/tissue/genotype combination, 1 g of plant material was harvested on ice, rinsed with water, and cross-linked with 1% formaldehyde for 10 min under vacuum infiltration. Cross-linking was quenched by adding Gly solution to a final concentration of 0.125 M under vacuum infiltration for 5 min. Treated tissue was frozen in liquid nitrogen and stored at −80°C until chromatin extraction.
Isolation and Characterization of mez Mutations
The Pioneer Hi-Bred trait utility system population (Bensen et al., 1995) was screened to identify Mutator transposon insertions in Mez1, Mez2, and Mez3. Several putative loss-of-function mu insertion alleles were obtained for mez2 (-m4) and mez3 (-m1 and -m4). No loss-of-function alleles were identified for Mez1 (Haun et al., 2009). The mez2-m4, mez3-m1, and mez3-m4 alleles were backcrossed into B73 for at least seven generations. The double mutant strain was created by crossing the backcross derived stocks to each other and selecting homozygous double mutant progeny.
Immunoprecipitation of H3K27me3-Linked DNA, Labeling, and Hybridization
Chromatin extractions were performed using the EpiQuik Plant ChIP kit (Epigentek) according to the manufacturer’s recommendations. Extracted chromatin was sheared in 600 μL EpiQuick buffer CP3F with five 10-s pulses on a sonicator. To test and optimize sonication conditions, cross-linking was reversed in a sample of sheared chromatin, and the resulting products were analyzed on agarose gels. Sonication conditions were optimized to yield predominantly 200- to 500-bp DNA samples. ChIP, reverse cross-linking, and DNA cleanup were performed using the EpiQuik Plant ChIP kit according to the manufacturer’s recommendations. Antibodies specific for H3K27me3 (#07-449) were purchased from Millipore. For each genotype, antibody, and replicate, 50 to 100 ng of input and immunoprecipitated DNA was amplified with a whole-genome amplification kit (WGA2; Sigma-Aldrich). The amplification of the no-antibody control (negative control) was always fivefold to 10-fold less efficient, confirming the specificity of immunoprecipitation. For each amplified immunoprecipitated and input sample, 3 μg amplified DNA was labeled using the Dual-Color Labeling Kit (Roche NimbleGen) according to the array manufacturer’s protocol (Roche NimbleGen Methylation User Guide v7.0). Each immunoprecipitated sample was labeled with Cy5, and each input/control sonicated DNA was labeled with Cy3. Samples were hybridized to the custom 1.4 or 2.1 M probe array (Gene Expression Omnibus [GEO] platforms GPL15621 and GPL13499) for 16 to 20 h at 42°C. Slides were washed and scanned according to NimbleGen’s protocol for the Gene-Pix4000B scanner. Images were aligned and quantified using NimbleScan software (Roche NimbleGen) producing raw data reports for each probe on the array.
Array Design and Annotation
Two array platforms were used in this experiment. All seedling tissues were assayed using a NimbleGen 2.1M feature oligonucleotide array (GEO platform GPL13499) as described (Eichten et al., 2011). All other tissues were assayed using a NimbleGen 3 × 1.4M feature oligonucleotide array (GEO platform GPL15621) containing a subset of 1.4 million probes from the larger array design that are single copy in the B73 genome. Only the subset of probes that were present on both microarray platforms were used in the analyses. Adjacent probes were spaced every 56 bp for chromosome 9 and every 200 bp for the other maize (Zea mays) chromosomes.
Normalization and Linear Modeling
XYS files exported from NimbleScan were imported into the Bioconductor statistical environment (http://bioconductor.org/). Sample-dependent H3K27me3 enrichments were estimated for each probe by fitting a fixed linear model accounting for array, dye, and sample effects to the data using the Limma package (Wettenhall and Smyth, 2004). Moderated t-statistics and the log-odds score for differential MeDIP enrichment was computed by empirical Bayes shrinkage of the standard errors with the false discovery rate controlled to 0.05. For the analysis of H3K27me3 levels in Mo17, the probes with substantial variation in CGH signal between B73 and Mo17 were omitted. In addition, a linear model approach was used to control for minor variation in hybridization due to genotype as determined from the CGH data. Results were visualized using Integrated Genomics Viewer (IGV) (Robinson et al., 2011). Microarray results were deposited with the National Center for Biotechnology Information (NCBI) GEO under accession number GSE39456, and data tracks formatted for IGV are available at http://genomics.tacc.utexas.edu/data/h3k27_epigenetic_variation/.
Analysis of H3K27me3 Distribution
Normalized H3K27me3 data, represented as log2(H3K27me3 IP/input), for all B73 tissue samples were segmented into discrete regions using the DNAcopy algorithm (Venkatraman and Olshen, 2007). The resulting segments were filtered to identify H3K27me3-enriched or -depleted segments using the expectation maximization algorithm (Dempster et al., 1977) assuming two subdistributions. Filtered segments required a 95% probability of falling into the higher subdistribution and also required an H3K27me3 segment average [log2(H3K27me3 IP/input)] of >0.8 to be identified as H3K27me3 enriched. The relative levels of H3K27me3 in Mo17 or mez mutant tissues was determined calculating average log2(H3K27me3 IP/input) values in Mo17 or mez mutant samples for the filtered segments defined in their respective tissue in B73. Expectation maximization was applied to the relative levels of H3K27me3 in mez mutant samples compared with the wild type to identify significant differences in H3K27me3 levels in mez mutants compared with the wild type.
Tissue Specificity of Gene Expression
We estimated tissue specificity by calculating Shannon entropy (Schug et al., 2005; Zhang et al., 2007) for genes in a maize gene expression atlas (Sekhon et al., 2011). The data were averaged across biological and technical replicates. Genes that had 0 expression level in all tissues were excluded from the analysis. For the remaining 31,764 genes, entropy was calculated as 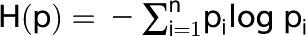 , where pi is a relative abundance of the gene’s transcript in tissue i. We also tried two additional data filters. First, we ignored genes if the sum of their expression levels across all tissues was below a certain threshold (g ∈ [2, 4, 6, 8, 10]). Second, we set expression level in a tissue to 0 whenever it was below a certain cutoff (t ∈ [1, 2, 4, 6, 8]). However, those thresholds didn’t produce any significant impact on the value distribution (data not shown.)
, where pi is a relative abundance of the gene’s transcript in tissue i. We also tried two additional data filters. First, we ignored genes if the sum of their expression levels across all tissues was below a certain threshold (g ∈ [2, 4, 6, 8, 10]). Second, we set expression level in a tissue to 0 whenever it was below a certain cutoff (t ∈ [1, 2, 4, 6, 8]). However, those thresholds didn’t produce any significant impact on the value distribution (data not shown.)
RNA Sequencing
RNA sequencing analysis of all tissues described above was performed as described (Eichten et al., 2012). Three biological replicates of five B73 and Mo17 tissues and one biological replicate of B73, mez3-m1, mez3-m4, mez2-m4, and mez3-m4 mez2-m4 seedlings were prepared, with eight plants pooled for each of the replicates. All RNA samples were prepared by the University of Minnesota BioMedical Genomics Center in accordance with the TruSeq library creation protocol (Illumina). Samples were sequenced on the HiSequation 2000 developing six to 17 million reads per replicate. Raw reads were filtered to eliminate poor quality reads using CASAVA (Illumina). Transcript abundance was calculated by mapping reads to the maize reference genome (AGPv2) using TopHat (Trapnell et al., 2009). A high degree of correlation between replicates was observed (r > 0.98). RPKM values were developed using “BAM to Counts” across the exon space of the maize genome reference WGS (ZmB73_5a) within the iPlant Discovery Environment (www.iplantcollaborative.org).
cDNA Synthesis and ChIP-qPCR Validations of H3K27me3 Levels
cDNA synthesis and qRT-PCR analysis for mez mutants were performed as described (Makarevitch et al., 2012) with the following primers: mez3forward (5′-CCCTATCCCGTTTGCCAGGTCTGACC-3′) and mez3reverse (5′-CCAAGGGGGCCGTCGCAGAAC-3′); mez2forward (5′-AAACTTTCCATTCCGGAGATTCGTG-3′) and mez2reverse (5′-GTTTGCCACTTCTATGCAGGTCTTAAG-3′). H3K27me3 levels were validated for 11 genes in two biological replicates of four B73 tissues and for five genes in seedlings of B73 and in mez2/3 mutants (see Supplemental Data Set 1 online for details). For all samples, three technical replicates were performed. The cycle threshold results were averaged for technical replicates, and percentage of input DNA was calculated for H3K27me3-immunoprecipitated samples as well as for immunoglobulin control samples according to Haring et al. (2007).
Accession Numbers
H3K27me3 profile data were deposited with NCBI GEO under accession number GSE39456 and data tracks formatted for IGV are available at http://genomics.tacc.utexas.edu/data/h3k27_epigenetic_variation/. RNA sequencing data are available at NCBI under numbers SRP013432 and SRP009313.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Validation of Altered H3K27me3 Patterns Using qPCR.
Supplemental Figure 2. Comparison of ChIP-chip H3K27me3 Profiles with ChIP-seq Profiles from Wang et al. (2009).
Supplemental Figure 3. Distribution of Segments Relative to the Number of FGS Genes per Segment.
Supplemental Figure 4. H3K27me3 Negatively Correlates with Gene Expression Levels.
Supplemental Figure 5. GO Annotation of Genes Targeted by H3K27me3.
Supplemental Figure 6. H3K27me3 Modifications Are Enriched at Some Families of Maize Transcription Factors.
Supplemental Figure 7. Correlation between H3K27me3 Levels and Expression Levels for Several Classical Genes.
Supplemental Figure 8. Allele-Specific H3K27me3 Modifications in Maize Endosperm for Imprinted Genes with Parental Expression.
Supplemental Figure 9. Altered Levels of H3K27me3 in mez Mutants.
Supplemental Figure 10. GO Annotation of Gene Targets for H3K27me3.
Supplemental Figure 11. Mez2/3-Dependent H3K27me3 Plays a Minimal Role in Regulation of Gene Expression.
Supplemental Table 1. Localization of H3K27me3 Marks in the Genome of Maize B73 Seedlings.
Supplemental Table 2. Relationships between Gene Expression and H3K27me3 in B73 Tissues.
Supplemental Table 3. Variation in H3K27me3 between B73 and Mo17 Seedlings.
Supplemental Data Set 1. Results of ChIP-qPCR Validations of H3K27me3 Levels.
Supplemental Data Set 2A. Segments Targeted by H3K27me3 in B73 Seedlings.
Supplemental Data Set 2B. Segments Targeted by H3K27me3 in B73 Tassel.
Supplemental Data Set 2C. Segments Targeted by H3K27me3 in B73 Ear.
Supplemental Data Set 2D. Segments Targeted by H3K27me3 in B73 Embryo.
Supplemental Data Set 2E. Segments Targeted by H3K27me3 in B73 Endosperm.
Supplemental Data Set 3. Genes Targeted by H3K27me3 in B73 in at Least One of Five Tissues Profiled.
Supplementary Material
Acknowledgments
We thank Peter Hermanson for assistance in growing and crossing plants and performing ChIP hybridizations and Jennifer Rundquist for performing qPCR validations. The Texas Advanced Computing Center at the University of Texas at Austin provided high performance computing and storage resources. The Minnesota Supercomputing Institute provided access to software and user support for data analyses. The research was supported by a grant from the National Science Foundation (IOS-0922095) to M.V.W. and N.M.S. and by a grant from the National Science Foundation (0957312) to I.M.
AUTHOR CONTRIBUTIONS
I.M., M.W.V., and N.M.S. designed the research. I.M., S.R.E., R.B., and A.J.W. performed research. O.N.D., R.B.M., and C.L.M. contributed new analytic/computational/etc. tools. I.M., S.R.E., R.B., C.L.M., and M.W.V. analyzed data. I.M. and N.M.S. wrote the article.
Glossary
- H3K27me3
trimethylation of histone H3 Lys-27
- PcG
Polycomb group
- ChIP
chromatin immunoprecipitation
- qPCR
quantitative PCR
- WGS
working gene set
- FGS
filtered gene set
- RPKM
gene-specific reads per kilobase gene length per million reads
- GO
Gene Ontology
- PEG
paternally expressed gene
- qRT-PCR
quantitative RT-PCR
- GEO
Gene Expression Omnibus
- NCBI
National Center for Biotechnology Information
References
- Bastow R., Mylne J.S., Lister C., Lippman Z., Martienssen R.A., Dean C. (2004). Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427: 164–167 [DOI] [PubMed] [Google Scholar]
- Bensen R.J., Johal G.S., Crane V.C., Tossberg J.T., Schnable P.S., Meeley R.B., Briggs S.P. (1995). Cloning and characterization of the maize An1 gene. Plant Cell 7: 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F., Chaudhury A. (2009). Parental memories shape seeds. Trends Plant Sci. 14: 550–556 [DOI] [PubMed] [Google Scholar]
- Berger S.L. (2007). The complex language of chromatin regulation during transcription. Nature 447: 407–412 [DOI] [PubMed] [Google Scholar]
- Berke L., Sanchez-Perez G.F., Snel B. (2012). Contribution of the epigenetic mark H3K27me3 to functional divergence after whole genome duplication in Arabidopsis. Genome Biol. 13: R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatavichute Y.V., Zhang X., Cokus S., Pellegrini M., Jacobsen S.E. (2008). Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE 3: e3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berr A., Shafiq S., Shen W.H. (2011). Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta 1809: 567–576 [DOI] [PubMed] [Google Scholar]
- Boyer L.A., et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353 [DOI] [PubMed] [Google Scholar]
- Chanvivattana Y., Bishopp A., Schubert D., Stock C., Moon Y.H., Sung Z.R., Goodrich J. (2004). Interaction of Polycomb-group proteins controlling flowering in Arabidopsis. Development 131: 5263–5276 [DOI] [PubMed] [Google Scholar]
- Dempster A.P., Laird N.M., Rubin D.B. (1977). Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc., B 39: 1–38 [Google Scholar]
- Eichten S.R., Ellis N.A., Makarevitch I., Yeh C.T., Gent J.I., Guo L., McGinnis K.M., Zhang X., Schnable P.S., Vaughn M.W., Dawe K.D., Springer N.M. (2012). Spreading of heterochromatin Is limited to specific families of maize retrotransposons. PLoS Genet. 8: e1003127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichten S.R., et al. (2011). Heritable epigenetic variation among maize inbreds. PLoS Genet. 7: e1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S., Jacobsen S.E., Reik W. (2010). Epigenetic reprogramming in plant and animal development. Science 330: 622–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut B.S., Doebley J.F. (1997). DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. USA 94: 6809–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M., Huh J.H., Hsieh T.F., Penterman J., Choi Y., Harada J.J., Goldberg R.B., Fischer R.L. (2006). DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124: 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich J., Puangsomlee P., Martin M., Long D., Meyerowitz E.M., Coupland G. (1997). A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386: 44–51 [DOI] [PubMed] [Google Scholar]
- Grossniklaus U., Vielle-Calzada J.P., Hoeppner M.A., Gagliano W.B. (1998). Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 280: 446–450 [DOI] [PubMed] [Google Scholar]
- Ha M., Ng D.W., Li W.H., Chen Z.J. (2011). Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 21: 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring M., Offermann S., Danker T., Horst I., Peterhansel C., Stam M. (2007). Chromatin immunoprecipitation: Optimization, quantitative analysis and data normalization. Plant Methods 3: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun W.J., Danilevskaya O.N., Meeley R.B., Springer N.M. (2009). Disruption of imprinting by mutator transposon insertions in the 5′ proximal regions of the Zea mays Mez1 locus. Genetics 181: 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun W.J., Laoueillé-Duprat S., O'Connell M.J., Spillane, C., Grossniklaus, U., Phillips, A.R., Kaeppler, S.M., and Springer, N.M. (2007). Genomic imprinting, methylation and molecular evolution of maize Enhancer of zeste (Mez) homologs. Plant J. 49: 325–337 [DOI] [PubMed] [Google Scholar]
- Haun W.J., Springer N.M. (2008). Maternal and paternal alleles exhibit differential histone methylation and acetylation at maize imprinted genes. Plant J. 56: 903–912 [DOI] [PubMed] [Google Scholar]
- He G., et al. (2010). Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 22: 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig L., Derkacheva M. (2009). Diversity of Polycomb group complexes in plants: Same rules, different players? Trends Genet. 25: 414–423 [DOI] [PubMed] [Google Scholar]
- Jullien P.E., Katz A., Oliva M., Ohad N., Berger F. (2006). Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 16: 486–492 [DOI] [PubMed] [Google Scholar]
- Kharchenko P.V., et al. (2011). Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C., Hennig L. (2010). Regulation of cell identity by plant Polycomb and trithorax group proteins. Curr. Opin. Genet. Dev. 20: 541–547 [DOI] [PubMed] [Google Scholar]
- Kohler C., Hennig L., Bouveret R., Gheyselinck J., Grossniklaus U., Gruissem W. (2003). Arabidopsis MSI1 is a component of the MEA/FIE Polycomb group complex and required for seed development. EMBO J. 22: 4804–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C., Page D.R., Gagliardini V., Grossniklaus U. (2005). The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat. Genet. 37: 28–30 [DOI] [PubMed] [Google Scholar]
- Kohler C., Villar C.B. (2008). Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 18: 236–243 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2007). Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- Lafos M., Kroll P., Hohenstatt M.L., Thorpe F.L., Clarenz O., Schubert D. (2011). Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 7: e1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F., Cui X., Zhang S., Jenuwein T., Cao X. (2011). Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat. Genet. 43: 715–719 [DOI] [PubMed] [Google Scholar]
- Maere S., Heymans K., Kuiper M. (2005). BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449 [DOI] [PubMed] [Google Scholar]
- Makarevich G., Leroy O., Akinci U., Schubert D., Clarenz O., Goodrich J., Grossniklaus U., Kohler C. (2006). Different Polycomb group complexes regulate common target genes in Arabidopsis. EMBO Rep. 7: 947–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarevitch I., Thompson A., Muehlbauer G.J., Springer N.M. (2012). Brd1 gene in maize encodes a brassinosteroid C-6 oxidase. PLoS ONE 7: e30798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam A.M., Roudier F., Seifert M., Berard C., Magniette M.L., Ashtiyani R.K., Houben A., Colot V., Mette M.F. (2011). Additive inheritance of histone modifications in Arabidopsis thaliana intra-specific hybrids. Plant J. 67: 691–700 [DOI] [PubMed] [Google Scholar]
- Pasini D., Bracken A.P., Hansen J.B., Capillo M., Helin K. (2007). The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 27: 3769–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raissig M.T., Baroux C., Grossniklaus U. (2011). Regulation and flexibility of genomic imprinting during seed development. Plant Cell 23: 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. (2011). Integrative genomics viewer. Nat. Biotechnol. 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F., et al. (2011). Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 30: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatlowski N., Creasey K., Goodrich J., Schubert D. (2008). Keeping plants in shape: Polycomb-group genes and histone methylation. Semin. Cell Dev. Biol. 19: 547–553 [DOI] [PubMed] [Google Scholar]
- Schnable J.C., Freeling M. (2011). Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6: e17855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable J.C., Freeling M., Lyons E. (2012). Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol. Evol. 4: 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable J.C., Springer N.M., Freeling M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA 108: 4069–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable P.S., et al. (2009). The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Schubert D., Primavesi L., Bishopp A., Roberts G., Doonan J., Jenuwein T., Goodrich J. (2006). Silencing by plant Polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J. 25: 4638–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G. (2007). Genome regulation by polycomb and trithorax proteins. Cell 128: 735–745 [DOI] [PubMed] [Google Scholar]
- Schug J., Schuller W.P., Kappen C., Salbaum J.M., Bucan M., Stoeckert C.J., Jr (2005). Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 6: R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Y.B., Pirrotta V. (2007). Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 8: 9–22 [DOI] [PubMed] [Google Scholar]
- Sekhon R.S., Lin H., Childs K.L., Hansey C.N., Buell C.R., de Leon N., Kaeppler S.M. (2011). Genome-wide atlas of transcription during maize development. Plant J. 66: 553–563 [DOI] [PubMed] [Google Scholar]
- Simon J. (1995). Locking in stable states of gene expression: transcriptional control during Drosophila development. Curr. Opin. Cell Biol. 7: 376–385 [DOI] [PubMed] [Google Scholar]
- Springer N.M., Danilevskaya O.N., Hermon P., Helentjaris T.G., Phillips R.L., Kaeppler H.F., Kaeppler S.M. (2002). Sequence relationships, conserved domains, and expression patterns for maize homologs of the polycomb group genes E(z), esc, and E(Pc). Plant Physiol. 128: 1332–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. (2009). TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turck F., Roudier F., Farrona S., Martin-Magniette M.L., Guillaume E., Buisine N., Gagnot S., Martienssen R.A., Coupland G., Colot V. (2007). Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PLoS Genet. 3: e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman E.S., Olshen A.B. (2007). A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23: 657–663 [DOI] [PubMed] [Google Scholar]
- Wang X., Elling A.A., Li X., Li N., Peng Z., He G., Sun H., Qi Y., Liu X.S., Deng X.W. (2009). Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21: 1053–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A.J., Makarevitch I., Eichten S.R., Swanson-Wagner R.A., Yeh C.T., Xu W., Schnable P.S., Vaughn M.W., Gehring M., Springer N.M. (2011). Parent-of-origin effects on gene expression and DNA methylation in the maize endosperm. Plant Cell 23: 4221–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall J.M., Smyth G.K. (2004). limmaGUI: A graphical user interface for linear modeling of microarray data. Bioinformatics 20: 3705–3706 [DOI] [PubMed] [Google Scholar]
- Wolff P., Weinhofer I., Seguin J., Roszak P., Beisel C., Donoghue M.T., Spillane C., Nordborg M., Rehmsmeier M., Kohler C. (2011). High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet. 7: e1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Shen W.H. (2008). Polycomb silencing of KNOX genes confines shoot stem cell niches in Arabidopsis. Curr. Biol. 18: 1966–1971 [DOI] [PubMed] [Google Scholar]
- Zhang X., Clarenz O., Cokus S., Bernatavichute Y.V., Pellegrini M., Goodrich J., Jacobsen S.E. (2007). Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 5: e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B., Chen X. (2011). Dynamics of histone H3 lysine 27 trimethylation in plant development. Curr. Opin. Plant Biol. 14: 123–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.