

Abstract
In humans multiple pathways can induce TH-17 cell differentiation, whereas in mice this process is mostly modulated by IL-6 and TGF-β. IL-17 produced by TH-17 cells has been associated with a number of inflammatory autoimmune diseases including psoriasis, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, and rheumatoid arthritis. In this review, we have primarily focused on the role of TH-17 cells/IL-17 in the pathogenesis of rheumatoid arthritis and experimental arthritis. The potential role of TH-17 cells in rheumatoid arthritis progression has been demonstrated by correlating the percent TH-17 cells or levels of IL-17 with rheumatoid arthritis disease activity score and C-reactive protein levels. Further, previous studies suggest that IL-17 mediated vascularization may lay the foundation for rheumatoid arthritis joint neutrophil and monocyte recruitment as well as cartilage and bone destruction. The profound role of IL-17 in the pathogenesis of experimental arthritis may be due to its synergistic effect with TNF-α and IL-1β. Although the initial clinical trial employing anti-IL-17 antibody has been promising for rheumatoid arthritis, future studies in humans will shed more light on how anti-IL-17 therapy affects rheumatoid arthritis and other autoimmune disease pathogenesis.
Keywords: TH-17, IL-17, rheumatoid arthritis, autoimmune diseases, experimental arthritis, inflammation, angiogenesis
INTRODUCTION
TH-17 cells are a subgroup of CD4+ lymphocytes that function predominantly at mucosal surfaces to protect against extracellular pathogens and are involved in the inflammatory process through the recruitment of neutrophils and in autoimmune diseases [1,2]. Human TH-17 cell development is regulated by transcription factor, RAR-related orphan receptor C (RORC) and these cells express IL-17A, IL-17F, IL-21, IL-22 and IL-26 [3]. Originally TH-17 cells were characterized by expression of IL-17A, commonly referred to as IL-17, though more recently, the lectin receptor, CD161, has been identified as a specific marker for human TH-17 cells [4].
IL-17A was the first member identified of the IL-17 cytokine family, which consists of 6 members IL-17A through IL-17F (Table 1). The IL-17 family members function as either homodimers or heterodimers [5,6]. IL-17A and IL-17F are both produced by TH-17 cells, however IL-17A is significantly more potent in initiating signaling and causing autoimmune responses. IL-17B is produced by cells of the gastrointestinal tract, pancreas and neurons, IL-17C is produced by cells of the prostate and fetal kidney, and IL-17D is secreted by cells of the muscles, brain, heart, lung, pancreas and adipose tissues [7,8]. IL-17E also known as IL-25 is produced by lymphocytes, lung epithelial cells, alveolar macrophages, eosinophils, basophils, NKT cells, TH-2 cells, and mast cells [9,10]. IL-17E is capable of initiating a TH-2 response which suppresses TH-17 cell differentiation. IL-17 was initially believed to be solely expressed by TH-17 cells, however it is now known that IL-17 can be produced by γδ T cells [11], lymphoid tissue inducer (LTi) cells [12], mast cells [13], and neutrophils [14].
Table 1.
The human IL-17 cytokine family members source of expression and pathological role.
| Family member | Receptor | Source | Pathological role |
|---|---|---|---|
| IL-17(A) | IL-17RA and RC | TH-17 cells, CD8+ T cells, γδ T cells, NK cells, NKT cells | Involved in autoimmune pathology, neutrophil and monocyte trafficking and angiogenesis |
| IL-17B | IL-17RB | Cells of the gastrointestinal tract, pancreas and neurons | Not identified |
| IL-17C | IL-17RE | Cells of the prostate and fetal kidney | Not identified |
| IL-17D | unknown | Cells of the muscles, brain, heart, lung, pancreas and adipose tissue | Not identified |
| IL-17E (IL-25) | IL-17RA and RB | lymphocytes, lung epithelial cells, alveolar macrophages, eosinophils, basophils, NKT cells, TH-2 cells, mast cells | Induces TH-2 cell responses and therefore suppresses TH-17 cell polarization |
| IL-17F | IL-17RA and RC | TH-17 cells, CD8+ T cells, γδ T cells, NK cells, NKT cells | Neutrophil recruitment |
The IL-17 receptor family consists of 5 members, IL-17RA through IL-17RE, whose structures are not homologus to other cytokine receptor families [15,16]. IL-17 receptors are type I transmembrane proteins that have conserved structural elements including a cytoplasmic SEF/IL17R domain (SEFIR) and two extracellular fibronectin III-like domains [5]. IL-17RA can bind to either IL-17A, IL-17E, or IL-17F, however it binds to IL-17A with the highest affinity [17]. IL-17RB can bind to either IL-17B or IL-17E and is also known as IL-25 receptor [16]. IL-17RC can bind either IL-17A or IL-17F, while IL-17RE binds only to IL-17C [18,19]. IL-17RD does not have a known ligand. IL-17 receptor family members function as receptor complexes with multiple members binding to ligands, possibly in a 2 step binding process where one IL-17 ligand must bind to a receptor before the second IL-17 ligand can bind a second receptor in the complex [17]. It is thought that these complexes can be either homomeric or heteromeric, for example, IL-17RA can pair with IL-17RC to bind and transmit signals from ligands such as IL-17A and IL-17F [20]. Of the IL-17 receptor family members, IL-17RA is expressed ubiquitously while IL-17RC is expressed mostly on non-hemopoietic cells [17,21].
DIFFERENTIATION OF MICE AND HUMAN TH-17 CELLS
Origin of and factors that modulate human TH-17 cells have been discussed intensely over the past few years. Three independent studies demonstrated that in mice TGF-β and IL-6 are responsible for polarizing naïve CD4+ T cells to TH-17 and the transcription factor RORγt is an essential component in this process [22-24]. However, the role of TGF-β in human TH-17 differentiation is less clear. It was shown that TGF-β can inhibit human TH-17 differentiation while IL-6 and IL-1β were the driving factors for this development [25]. Consistently, others have shown that while IL-1β or IL-23 is necessary for human TH-17 differentiation from naïve human CD4+ T cells, the presence of TGF-β was not required [26]. Interestingly, direct contact of LPS or peptidoglycan activated monocytes with naïve CD4+ T cells was able to polarize naïve CD4+ T cells to TH-17 cells [27]. In contrast to these findings, others have shown that TGF-β, together with other proinflammatory factors such as IL-23, IL-1β and IL-6, was critical for human TH-17 cell development [28,29] and lack of TGF-β induces a shift from TH-17 to a TH-1 profile [29]. It was also demonstrated that IL-21 and TGF-β could uniquely promote human naïve CD4+ T cells into TH-17 cells by activating RORC2 which is the human homologue of mouse RORγt [30]. It was thought that the presence of TGF-β in serum containing media, and therefore purity and/or naivety of cells, may have lead to the discrepancy in these results [31]. Further, while both human and mouse TH-17 cells secrete IL-17, IL-17F, IL-21, IL-22 and CCL20 production of IL-26 is only induced by human TH-17 cells as there is no known murine homolog for this cytokine [32-34]. Collectively, previous studies indicate that while in mice IL-6 and TGF-β can induce robust levels of TH-17 cells (40-80%) this phenomenon may not be as straight forward in humans where multiple factors can differentially modulate TH-17 cell development to modest levels (10%).
TH-17 CELLS IN HUMAN AUTOIMMUNE INFLAMMATORY DISEASE
A number of inflammatory autoimmune diseases including psoriasis, SLE, MS, inflammatory bowel disease and RA have been associated with TH-17 cells. In psoriasis, which is a chronic skin disease characterized by keratinocyte hyperplasia, inflammation, T cell invasion, and angiogenesis, TH-17 cells are predominantly located in the dermis of skin lesions [35]. Further, IL-17 and other TH-17 cytokines such as IL-22 and IL-23 are involved in the pathogenesis of psoriasis [36-38]. In psoriasis skin lesions, IL-17 mRNA levels increase with disease activity [35]. Disease resolution correlated with decreased IL-17 and IL-23 expression levels in psoriasis patients who responded to treatment with the soluble TNFα receptor fusion protein, Etanercept [39]. In vitro experiments with human keratinocytes showed that IL-17 treatment resulted in increased production of ICAM-1, IL-6 and IL-8 [36]. Also IL-17F treatment of kerotinocytes showed greater increase in IL-6 expression compared to IL-17 or TNFα- activated cells suggesting that IL-17F produced by TH-17 cells causes the inflammation in psoriasis partly through induction of IL-6 by keratinocytes [40]. Studies demonstrate that kerotinocytes treated with IL-17 and IL-22 had increased CCL20/MIP-3α expression which may drive CCR6+ TH-17 cell recruitment into the skin lesions perpetuating the disease process [41]. These results suggest that several TH-17 associated cytokines such as IL-17, IL-17F and IL-22 play an important role in pathogenesis of psoriasis by inducing the production of similar down stream targets. Therefore, response to therapy can suppress these common inflammatory pathways.
Another chronic autoimmune disease, SLE, also features elevated levels of IL-17. SLE is a systemic disease in which autoantibodies initiate immune complex formation resulting in chronic inflammation in locations including the skin and kidneys. In patients with SLE, IL-17 is produced by both TH-17 cells and TCRαβ CD4- CD8- T cells [42,43].
Additionally, in these patients IL-17 levels are elevated in the sera [44] and in sites of inflammation such as the skin [45] and kidneys [46,47]. The over production of IL-17 is thought to potentiate the systemic inflammation observed in patients with SLE in a couple of ways. First, IL-17 stimulation of B cells can increase B cell activation and antibody production resulting in greater amounts of autoantibody production and immune complex formation [43,48]. Secondly, IL-17 released at inflammation locations such as the skin can amplify the immune response by increasing the influx of effector cells [49].
MS is an autoimmune central nervous system disease that involves the destruction of myelin sheets resulting in impairment of nerve signal transduction. IL-17 production was elevated in MS cerebral spinal fluid and blood [50] however, in comparison, greater IL-17 levels were detected in the cerebral spinal fluid which correlated with clinical exacerbations and neutrophil infiltration [51]. More recently, histological studies demonstrated that IL-17 expressing cells were more likely found in active rather than inactive areas of MS lesions [52]. It is hypothesized that IL-17 and TH-17 cytokine IL-22 may be involved in the pathogenesis of MS by weakening the blood brain barrier. Endothelial cells of the blood brain barrier express receptors for IL-17 and IL-22 and it was shown that IL-17 and IL-22 could disrupt the blood brain barrier possibly through the production of reactive oxygen species [53,54]. Hence, disruption of the blood brain barrier would allow autoantibodies and inflammatory mediator’s access to the myelin sheets allowing the pathology to occur.
Inflammatory bowel disease includes both Crohn’s disease and ulcerative colitis and is a chronic autoimmune inflammatory condition of the gastrointestinal tract. IL-17 has been found to be elevated in the intestinal mucosa and within the sera of patients with inflammatory bowel disease compared to normal controls [55,56]. IL-17 was also produced from gut TH-17 cells in patients with Crohn’s disease [57]. Further IL-17F mRNA has been found in greater levels in inflamed lesions of Crohn’s disease patients compared to non-inflamed areas [58]. These human studies suggest a role for TH-17 cells and IL-17 in the pathogenesis of inflammatory bowel disease. However, the exact function of IL-17 in inflammatory bowel disease is somewhat controversial as animal studies have shown both protective and pathogenic roles for IL-17 [59,60].
ROLE OF TH-17 CELLS IN RA PATHOGENESIS
Synovial tissue explants from RA but not osteoarthritis (OA) spontaneously produce biologically active IL-17 [61]. Increased levels of IL-17 are also detected in RA synovial fluid and in T cell rich areas of RA synovial tissues compared to OA synovial tissue and fluid [61-63]. We found that TH-17 cells were significantly elevated in RA synovial fluid compared to RA and normal peripheral blood cells [63]. Others have shown that expression of CCR4 and CCR6 on RA TH-17 cells demonstrates selective migration of these cells to the site of inflammation [64]. This group of investigators shows that increased TH-17 presence in peripheral blood of early stage RA patients is suggestive of the contribution of TH-17 cells to disease onset. Further, percentage of TH-17 cells and levels of IL-17 strongly correlate with disease activity score (DAS 28) and C-reactive protein (CRP) suggesting the importance of these cells in disease progression [64]. The potential importance of TH-17 cells/IL-17 in RA is supported by a randomized, placebo-controlled and double blind phase I study where RA patients that received DMARD plus neutralizing monoclonal antibody against IL-17 achieved an ACR 20 more rapidly compared to those receiving DMARD alone [65]. Consistently a two year prospective study analyzing RA synovial tissues demonstrated that IL-17 and TNF-α mRNA levels are synergistic prognostic factors for worse out come [66]. These studies clearly demonstrate that TH-17 cells play an important role in RA pathogenesis.
SYNERGISTIC EFFECT OF IL-17 WITH OTHER PROINFLAMMATORY CYTOKINES
The direct proinflammatory effects of IL-17 may be small when compared to those of IL-1β and TNF-α. However, IL-17 enhances many of the effects of IL-1β and TNF-α. IL-17 stimulates the production of IL-1 and TNF-α from human macrophages [67]. IL-17 also enhances IL-1-mediated IL-6 production by RA synovial tissue fibroblasts [68], as well as TNF-α induced synthesis of IL-1, IL-6 and IL-8 [69]. Many of the IL-17 activated genes contain CCAAT/enhancer binding proteins (C/EBP) in their promoter which cooperates with NF-κB in inducing the transcription of these proinflammatory factors [70]. In RA synovial tissue fibroblasts, IL-17 interacts with IL-1 and TNF-α to amplify the secretion of CCL20 [71]. IL-17, in combination with TNF-α, induces significantly higher levels of nitric oxide and osteoclastic resorption compared to each cytokine alone [72,73]. The mechanism by which IL-17 mediates synergistic effect is through enhancing mRNA stability of AU-rich elements in the 3’ untranslated region (UTR) of many cytokines and chemokines [74,75]. In short, a major role of IL-17 may be amplifying the effects of macrophage derived proinflammatory cytokines and hence be the missing link between T cells in RA joint and the effector phase of RA.
ROLE OF TH-17 IN GRANULOPOIESIS AND NEUTROPHIL MIGRATION
It is shown that neutrophils are critical in the early stage of arthritis and are abundantly present in the RA joint and synovial fluid of patients with active disease [76]. In mice, overexpression of IL-17 can expand both neutrophil progenitors in bone marrow and spleen [77]. This effect is due to IL-17-induced G-CSF production since neutralization of G-CSF, but not deletion of IL-17RA, markedly attenuates this effect [77-79] suggesting that IL-17 plays an indirect role in granulopoiesis. Local expression of IL-17 enhances neutrophil migration in mouse ankle joints as well as the peritoneal cavity [80,81]. In the rat airway, IL-17 mediates neutrophil recruitment via induction of macrophage inflammatory protein protein-2 (rMIP-2) [82]. Like granulopoiesis, neutrophil chemotaxis caused by conditioned media from IL-17-stimulated gastric epithelial cells was inhibited by a neutralizing antibody to IL-8 but not to IL-17, suggesting that IL-17 is unable to directly induce neutrophil chemotaxis [83]. Further, results from our laboratory demonstrates that neutralizing antibody to CXCL5, but not CXCL1, significantly suppresses neutrophil trafficking to IL-17-induced arthritis ankle joints indicating that IL-17 mediated CXCL5 plays a role in this process [84]. Therefore if these studies in rodents translate into human, IL-17 may contribute to RA disease onset by inducing IL-8 and/or CXCL5 production that is involved in recruitment of neutrophils to the RA joint.
EFFECTS OF IL-17 ON CARTILAGE DEGRADATION AND BONE EROSION
Local inflammation is involved in cartilage degradation by suppressing proteoglycan and collagen synthesis as well as causing extracellular matrix breakdown. Earlier studies have demonstrated that IL-17 alone and in synergy with IL-1β inhibits cartilage proteoglycan synthesis in murine explants by inducing production of nitric oxide [85]. IL-17 treated bovine chondrocytes demonstrated increased expression of matrix metalloproteinases (MMP)1, 3 and 13 which are factors involved in degradation of extracellur matrix [86]. IL-17 can also promote cartilage breakdown by synergizing with TNF-α, IL-1β and IL-6 to increase collagen degradation and MMP release [86,87]
IL-17 induces the expression of RANKL in osteoblasts [88] and can further synergize with TNF-α in osteoclastic resorption [73,89]. A naturally occurring decoy protein osteoprotegerin (OPG) can inhibit the interaction of RANK and RANKL, however high dose of OPG could only partially inhibit IL-17/TNF-α mediated bone resorption [89]. In collagen induced arthritis (CIA) local expression of IL-17 in ankle joints enhances bone erosion by mediating an imbalance in RANKL/OPG in favor of RANKL levels [90]. Taken together these observations suggest that IL-17 can directly and in combination with IL-1β and TNF-α result in RA bone and cartilage destruction.
UNIQUE CHARACTERISTICS OF IL-17 IN RA
Angiogenesis is an early and a critical event in the pathogenesis of RA. Hence, neovascularization is involved in leukocyte ingress into the synovium during the development and progression of RA [91,92]. Our recent data demonstrates that IL-17 can intensify inflammation by promoting angiogenesis and subsequently recruiting inflammatory cells to the RA joint [93]. We show that IL-17 is capable of endothelial chemotaxis at concentrations present in RA synovial fluid. Further, ligation to IL-17RC on endothelial cells and activation of PI3K/AKT pathway is responsible for IL-17-induced endothelial migration and tube formation [93]. Expression of IL-17 in RA synovial fluid and IL-17RC on endothelial cells plays an important role in RA synovial fluid mediated migration [93]. In vivo, IL-17 enhances vascularity in experimental arthritis and induces blood vessel development in matrigel plugs [93] (Fig. 1). However, there are also data to suggest that IL-17 can indirectly induce angiogenesis, by promoting production of proangiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and hepatocyte growth factor (HGF) [94,95] from cells in the RA joint. We demonstrate that in addition to the direct effect of IL-17/IL-17R on angiogenesis, joint IL-17-mediated CXCL5, but not CXCL1, plays a key role in IL-17-induced arthritis and vascularization [84]. Angiogenesis mediated by IL-17 may lay the foundation for recruitment of leukocytes and therefore studies were performed to better understand the mechanism by which IL-17 may promote inflammation and monocyte trafficking in RA. It was shown that IL-17 induces monocyte migration at concentrations available in RA joint by ligation to IL-17RA and IL-17RC through activation of p38 MAPK pathway [96]. However, the direct effects of IL-17 does not account for all its chemotactic ability in vivo since neutralization of CCL2 significantly reduces IL-17 induced monocyte migration into the peritoneal cavity [81] (Fig. 1). Production of CCL2 was detected in IL-17 activated macrophages and RA synovial tissue fibroblasts, two cells types important for RA pathogenesis [81]. The potential role of IL-17 in RA angiogenesis and monocyte extravasation was also documented when local expression of IL-27 in CIA ankle joints suppressed TH-17 differentiation as well as IL-17-mediated vascularization and macrophage staining [97]. These studies suggest that IL-17 can perpetuate inflammation by driving angiogenesis which can result in subsequent recruitment of neutrophils (acute phase) and monocytes (chronic phase) thereby amplifying chronic inflammation in RA through multiple pathways.
Figure 1. Schematic overview of the pathological role of IL-17 in RA.
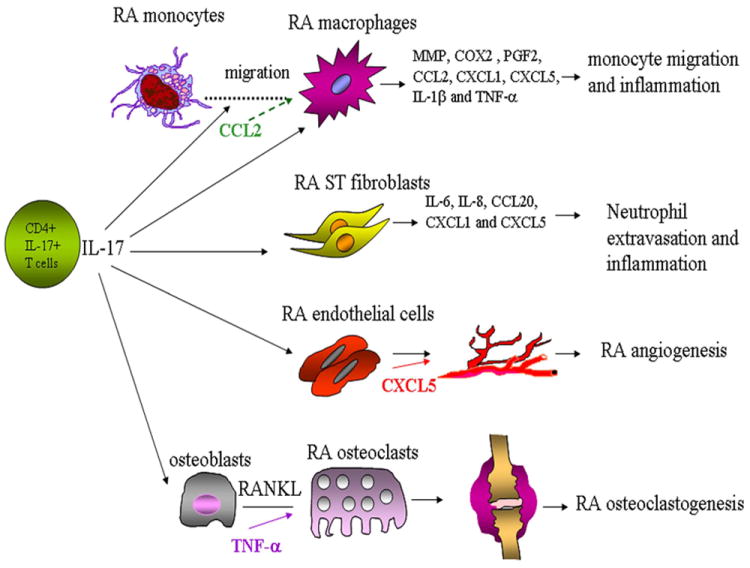
Several cell types are affected by IL-17 in RA joints. IL-17 can induce migration of RA peripheral blood monocytes to the inflamed joints where they differentiate to macrophages. RA synovial tissue macrophages and fibroblasts activated with IL-17 can produce a number of proinflammatory factors that can mediate inflammation and neutrophil recruitment. Angiogenesis mediated by IL-17 can provide the inflamed joint with nutrients and oxygen and thereby perpetuate the vicious inflammatory cycle. IL-17 can also play a role in RA bone destruction by enhancing RANKL production alone or in synergy with TNF-α.
ROLE OF IL-17 IN EXPERIMENTAL ARTHRITIS
It has been shown that IL-17 plays a profound role in experimental arthritis. CIA was markedly reduced in IL-17-/- mice [98]. Early neutralization of IL-17 using an IL-17R IgG Fc fusion protein in CIA suppresses the onset of the disease [99]. Consistently, treatment of CIA after disease onset using anti-IL-17 antibody decreases the severity of inflammation and bone destruction in CIA [100]. Blocking of IL-17 in antigen induced arthritis suppresses both IL-1β and TNF-α indicating that IL-17 is an important upstream inflammatory mediator [101]. Local overexpression of IL-17 using an adenoviral vector results in joint inflammation and cartilage proteoglycan depletion in the knees of naive mice [81,84,102]. Further, IL-17-induced joint inflammation and cartilage erosion was markedly reduced in TNF-α-/- mice but not in IL-1 deficient mice [102]. These data suggest a requirement for TNF-α and not IL-1 for the induction of IL-17-induced arthritis. However, IL-17 may also act independently of TNF-α after disease onset [102]. Interestingly a very recent paper shows that neutralizing both IL-17 and TNF-α ameliorates CIA joint damage to a greater extent compared to each factor alone implicating the synergistic inflammatory effect of IL-17 and TNF-α [87]. IL-23-/- mice were resistant to CIA and this correlated with an absence of IL-17-producing CD4+ T cells, despite normal IFN-γ production by TH1 cells [103]. In contrast to the significant role of TH-17 cells in CIA pathogenesis, proteoglycan-induced arthritis model (PGIA) is dependent on IFN-γ production and hence suppression of TH-17 differentiation by IL-27 or deletion of IL-17 does not affect disease pathogenesis [104,105]. Ectopic expression of IL-27 in CIA mice reduces TH-17 mediated angiogenesis and monocyte trafficking as well as TH-17 differentiation by downregulating IL-1β and IL-6, two important TH-17 cell polarizing factors [97]. Mounting evidence from experimental arthritis models and RA demonstrates that IL-17 is involved in the initial and progression phase of disease which supports IL-17 as a therapeutic target in RA.
CONCLUSION
Identification of TH-17 cells has been a paradigm shifting event and has therefore questioned the importance of TH-1 cell involvement in autoimmune disorders. Strong evidence supports that TH-17 cells are pathologically important in chronic inflammatory and autoimmune disorders, namely psoriasis, SLE, inflammatory bowel disease, MS, and RA. In this review we have specifically focused on the implication of TH-17 cells/IL-17 in RA. Interestingly, IL-17 is one of the few T cell derived cytokines found in RA joints where the majority of inflammatory factors are produced from synovial tissue fibroblasts and macrophages. The role of IL-17 in neutrophil recruitment as well as cartilage and bone erosion is well established and more recently a novel function of IL-17 in RA angiogenesis and monocyte extravasation has been identified. The direct proinflammatory effect of IL-17 is often smaller than TNF-α and IL-1β, however IL-17 synergizes with these cytokines by enhancing their production and action in experimental arthritis and RA joint. Inhibition of TH-17 differentiation or IL-17 function ameliorates pathogenesis of experimental arthritis models and neutralizing anti-IL-17 was able to improve RA symptoms. Since blockade of IL-6 function (an important downstream target of IL-17) has yielded to some success in RA patients, it is tempting to speculate that anti-IL-17 therapy can be employed in anti-TNF-α non responders or in adjunct to anti-TNF-α therapy. Therefore, more human studies are required to respond to these inquiries.
Acknowledgments
This work was supported in part by awards from the National Institutes of Health AR056099, Arthritis National Research Foundation, grants from Within Our Reach from The American College of Rheumatology and funding provided by Department of Defense PR093477.
Abbreviations
- bFGF
basic fibroblast growth factor
- CIA
collagen induced arthritis
- CRP
C-reactive protein
- DAS 28
disease activity score
- HGF
hepatocyte growth factor
- LTi
lymphoid tissue inducer
- MMP
matrix metalloproteinases
- OA
osteoarthritis
- OPG
osteoprotegerin
- PGIA
proteoglycan-induced arthritis model
- RA
rheumatoid arthritis
- RORC
RAR-related orphan receptor C
- SLE
systemic lupus erythematosus; SEF/IL17R domain (SEFIR)
- VEGF
vascular endothelial growth factor C/EBP, CCAAT/enhancer binding proteins
- UTR
3’ untranslated region
References
- 1.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct cd4 t cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 2.Langrish CL, Chen Y, Blumenschein WM, et al. Il-23 drives a pathogenic t cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ t helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 4.Santarlasci V, Maggi L, Capone M, et al. Tgf-beta indirectly favors the development of human th17 cells by inhibiting th1 cells. Eur J Immunol. 2009;39:207–15. doi: 10.1002/eji.200838748. [DOI] [PubMed] [Google Scholar]
- 5.Yao Z, Fanslow WC, Seldin MF, et al. Herpesvirus saimiri encodes a new cytokine, il-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–21. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 6.Chang SH, Dong C. A novel heterodimeric cytokine consisting of il-17 and il-17f regulates inflammatory responses. Cell Res. 2007;17:435–40. doi: 10.1038/cr.2007.35. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Chen J, Huang A, et al. Cloning and characterization of il-17b and il-17c, two new members of the il-17 cytokine family. Proc Natl Acad Sci USA. 2000;97:773–8. doi: 10.1073/pnas.97.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aggarwal S, Gurney AL. Il-17: Prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 9.Fort MM, Cheung J, Yen D, et al. Il-25 induces il-4, il-5, and il-13 and th2-associated pathologies in vivo. Immunity. 2001;15:985–95. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda K, Nakajima H, Suzuki K, et al. Mast cells produce interleukin-25 upon fc epsilon ri-mediated activation. Blood. 2003;101:3594–6. doi: 10.1182/blood-2002-09-2817. [DOI] [PubMed] [Google Scholar]
- 11.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta t cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–30. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 12.Takatori H, Kanno Y, Watford WT, et al. Lymphoid tissue inducer-like cells are an innate source of il-17 and il-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hueber AJ, Asquith DL, Miller AM, et al. Mast cells express il-17a in rheumatoid arthritis synovium. J Immunol. 2010;184:3336–40. doi: 10.4049/jimmunol.0903566. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Huang L, Vergis AL, et al. Il-17 produced by neutrophils regulates ifn-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120:331–42. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Z, Spriggs MK, Derry JM, et al. Molecular characterization of the human interleukin (il)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- 16.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and il-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–74. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 17.Ely LK, Fischer S, Garcia KC. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol. 2009;10:1245–51. doi: 10.1038/ni.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuestner RE, Taft DW, Haran A, et al. Identification of the il-17 receptor related molecule il-17rc as the receptor for il-17f. J Immunol. 2007;179:5462–73. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaffen SL. Structure and signalling in the il-17 receptor family. Nat Rev Immunol. 2009;9:556–67. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toy D, Kugler D, Wolfson M, et al. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–9. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 21.Ishigame H, Kakuta S, Nagai T, et al. Differential roles of interleukin-17a and -17f in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector th17 and regulatory t cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 23.Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the t(h)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 24.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. Tgfbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of il-17-producing t cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human t helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 26.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper t cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 27.Evans HG, Suddason T, Jackson I, Taams LS, Lord GM. Optimal induction of t helper 17 cells in humans requires t cell receptor ligation in the context of toll-like receptor-activated monocytes. Proc Natl Acad Sci USA. 2007;104:17034–9. doi: 10.1073/pnas.0708426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manel N, Unutmaz D, Littman DR. The differentiation of human t(h)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor rorgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human t(h)-17 responses. Nat Immunol. 2008;9:650–7. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Anderson DE, Baecher-Allan C, et al. Il-21 and tgf-beta are required for differentiation of human t(h)17 cells. Nature. 2008 doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Garra A, Stockinger B, Veldhoen M. Differentiation of human t(h)-17 cells does require tgf-beta! Nat Immunol. 2008;9:588–90. doi: 10.1038/ni0608-588. [DOI] [PubMed] [Google Scholar]
- 32.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 t helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol. 2009;5:325–31. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 33.Korn T, Bettelli E, Oukka M, Kuchroo VK. Il-17 and th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 34.Laurence A, O’Shea JJ. T(h)-17 differentiation: Of mice and men. Nat Immunol. 2007;8:903–5. doi: 10.1038/ni0907-903. [DOI] [PubMed] [Google Scholar]
- 35.Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of th1 and th17 t cells. J Invest Dermatol. 2008;128:1207–11. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 36.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 37.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of il-23 by keratinocytes in healthy skin and psoriasis lesions: Enhanced expression in psoriatic skin. J Immunol. 2006;176:1908–15. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- 38.Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a t(h)17 cytokine, mediates il-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 39.Zaba LC, Cardinale I, Gilleaudeau P, et al. Amelioration of epidermal hyperplasia by tnf inhibition is associated with reduced th17 responses. J Exp Med. 2007;204:3183–94. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujishima S, Watanabe H, Kawaguchi M, et al. Involvement of il-17f via the induction of il-6 in psoriasis. Arch Dermatol Res. 2010;302:499–505. doi: 10.1007/s00403-010-1033-8. [DOI] [PubMed] [Google Scholar]
- 41.Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate ccl20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175–83. doi: 10.1038/jid.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of il-23 and il-17 in patients with systemic lupus erythematosus: Implications for th17-mediated inflammation in auto-immunity. Clin Immunol. 2008;127:385–93. doi: 10.1016/j.clim.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 43.Doreau A, Belot A, Bastid J, et al. Interleukin 17 acts in synergy with b cell-activating factor to influence b cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–85. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 44.Wong CK, Ho CY, Li EK, Lam CW. Elevation of proinflammatory cytokine (il-18, il-17, il-12) and th2 cytokine (il-4) concentrations in patients with systemic lupus erythematosus. Lupus. 2000;9:589–93. doi: 10.1191/096120300678828703. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, Chu Y, Yang X, et al. Th17 and natural treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1472–83. doi: 10.1002/art.24499. [DOI] [PubMed] [Google Scholar]
- 46.Crispin JC, Oukka M, Bayliss G, et al. Expanded double negative t cells in patients with systemic lupus erythematosus produce il-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–6. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Ito S, Chino Y, et al. Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin Exp Immunol. 2010;159:1–10. doi: 10.1111/j.1365-2249.2009.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong G, Ye R, Shi W, et al. Il-17 induces autoantibody overproduction and peripheral blood mononuclear cell overexpression of il-6 in lupus nephritis patients. Chin Med J (Engl) 2003;116:543–8. [PubMed] [Google Scholar]
- 49.Apostolidis SA, Crispin JC, Tsokos GC. Il-17-producing t cells in lupus nephritis. Lupus. 2011;20:120–4. doi: 10.1177/0961203310389100. [DOI] [PubMed] [Google Scholar]
- 50.Matusevicius D, Kivisakk P, He B, et al. Interleukin-17 mrna expression in blood and csf mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 51.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 52.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating t cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kebir H, Kreymborg K, Ifergan I, et al. Human th17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huppert J, Closhen D, Croxford A, et al. Cellular mechanisms of il-17-induced blood-brain barrier disruption. Faseb J. 2010;24:1023–34. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- 55.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sugihara T, Kobori A, Imaeda H, et al. The increased mucosal mrna expressions of complement c3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol. 2010;160:386–93. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seiderer J, Elben I, Diegelmann J, et al. Role of the novel th17 cytokine il-17f in inflammatory bowel disease (ibd): Upregulated colonic il-17f expression in active crohn’s disease and analysis of the il17f p.His161arg polymorphism in ibd. Inflamm Bowel Dis. 2008;14:437–45. doi: 10.1002/ibd.20339. [DOI] [PubMed] [Google Scholar]
- 59.Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 60.Yang XO, Chang SH, Park H, et al. Regulation of inflammatory responses by il-17f. J Exp Med. 2008;205:1063–75. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: A t cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 62.Honorati MC, Meliconi R, Pulsatelli L, Cane S, Frizziero L, Facchini A. High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology. 2001;40:522–7. doi: 10.1093/rheumatology/40.5.522. [DOI] [PubMed] [Google Scholar]
- 63.Shahrara S, Huang Q, Mandelin AM, 2nd, Pope RM. Th-17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leipe J, Grunke M, Dechant C, et al. Role of th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010;62:2876–85. doi: 10.1002/art.27622. [DOI] [PubMed] [Google Scholar]
- 65.Genovese MC, Van den Bosch F, Roberson SA, et al. Ly2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase i randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929–39. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]
- 66.Kirkham BW, Lassere MN, Edmonds JP, et al. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: A two-year prospective study (the damage study cohort) Arthritis Rheum. 2006;54:1122–31. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 67.Jovanovic DV, Di Battista JA, Martel-Pelletier J, et al. Il-17 stimulates the production and expression of proinflammatory cytokines, il-beta and tnf-alpha, by human macrophages. J Immunol. 1998;160:3513–21. [PubMed] [Google Scholar]
- 68.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of il-17 on il-1-induced il-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by th2 cytokines. J Immunol. 1998;161:409–14. [PubMed] [Google Scholar]
- 69.Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: A possible role as a “Fine-tuning cytokine” In inflammation processes. Arthritis Rheum. 2001;44:2176–84. doi: 10.1002/1529-0131(200109)44:9<2176::aid-art371>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 70.Yang TT, Chow CW. Transcription cooperation by nfat.C/ebp composite enhancer complex. J Biol Chem. 2003;278:15874–85. doi: 10.1074/jbc.M211560200. [DOI] [PubMed] [Google Scholar]
- 71.Chabaud M, Page G, Miossec P. Enhancing effect of il-1, il-17, and tnf-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: Regulation by soluble receptors and th2 cytokines. J Immunol. 2001;167:6015–20. doi: 10.4049/jimmunol.167.10.6015. [DOI] [PubMed] [Google Scholar]
- 72.LeGrand A, Fermor B, Fink C, et al. Interleukin-1, tumor necrosis factor alpha, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin e2 production in explants of human osteoarthritic knee menisci. Arthritis Rheum. 2001;44:2078–83. doi: 10.1002/1529-0131(200109)44:9<2078::AID-ART358>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 73.Van bezooijen RL, Farih-Sips HC, Papapoulos SE, Lowik CW. Interleukin-17: A new bone acting cytokine in vitro. J Bone Miner Res. 1999;14:1513–21. doi: 10.1359/jbmr.1999.14.9.1513. [DOI] [PubMed] [Google Scholar]
- 74.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger rna stability by a surface-mediated t cell activation pathway. Science. 1989;244:339–43. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 75.Henness S, Johnson CK, Ge Q, Armour CL, Hughes JM, Ammit AJ. Il-17a augments tnf-alpha-induced il-6 expression in airway smooth muscle by enhancing mrna stability. J Allergy Clin Immunol. 2004;114:958–64. doi: 10.1016/j.jaci.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 76.Cornish AL, Campbell IK, McKenzie BS, Chatfield S, Wicks IP. G-csf and gm-csf as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:554–9. doi: 10.1038/nrrheum.2009.178. [DOI] [PubMed] [Google Scholar]
- 77.Schwarzenberger P, La Russa V, Miller A, et al. Il-17 stimulates granulopoiesis in mice: Use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383–9. [PubMed] [Google Scholar]
- 78.Schwarzenberger P, Huang W, Ye P, et al. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for il-17-mediated granulopoiesis. J Immunol. 2000;164:4783–9. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 79.Tan W, Huang W, Zhong Q, Schwarzenberger P. Il-17 receptor knockout mice have enhanced myelotoxicity and impaired hemopoietic recovery following gamma irradiation. J Immunol. 2006;176:6186–93. doi: 10.4049/jimmunol.176.10.6186. [DOI] [PubMed] [Google Scholar]
- 80.Lubberts E, Joosten LA, Oppers B, et al. Il-1-independent role of il-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–13. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- 81.Shahrara S, Pickens SR, Mandelin AM, 2nd, et al. Il-17-mediated monocyte migration occurs partially through cc chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol. 2010;184:4479–87. doi: 10.4049/jimmunol.0901942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human il-17 via c-x-c chemokine release in the airways. J Immunol. 1999;162:2347–52. [PubMed] [Google Scholar]
- 83.Luzza F, Parrello T, Monteleone G, et al. Up-regulation of il-17 is associated with bioactive il-8 expression in helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–7. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 84.Pickens SR, Chamberlain ND, Volin MV, et al. Anti-cxcl5 therapy ameliorates il-17-induced arthritis by decreasing joint vascularization. Angiogen. 2011 doi: 10.1007/s10456-011-9227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lubberts E, Joosten LA, van de Loo FA, van den Gersselaar LA, van den Berg WB. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheum. 2000;43:1300–6. doi: 10.1002/1529-0131(200006)43:6<1300::AID-ANR12>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 86.Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD. Interleukin 17 induces cartilage collagen breakdown: Novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis. 2002;61:704–13. doi: 10.1136/ard.61.8.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koenders MI, Marijnissen RJ, Devesa I, et al. Tumor necrosis factor-interleukin-17 interplay induces s100a8, interleukin-1beta, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: Rationale for combination treatment during arthritis. Arthritis Rheum. 2011;63:2329–39. doi: 10.1002/art.30418. [DOI] [PubMed] [Google Scholar]
- 88.Kotake S, Udagawa N, Takahashi N, et al. Il-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Bezooijen RL, Papapoulos SE, Lowik CW. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: Is there dependency on nuclear factor-kappab and receptor activator of nuclear factor kappab (rank)/rank ligand signaling? Bone. 2001;28:378–86. doi: 10.1016/s8756-3282(00)00457-9. [DOI] [PubMed] [Google Scholar]
- 90.Lubberts E, van den Bersselaar L, Oppers-Walgreen B, et al. Il-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of nf-kappa b ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–62. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- 91.Szekanecz Z, Koch AE. Mechanisms of disease: Angiogenesis in inflammatory diseases. Nat Clin Pract Rheumatol. 2007;3:635–43. doi: 10.1038/ncprheum0647. [DOI] [PubMed] [Google Scholar]
- 92.Szekanecz Z, Koch AE. Angiogenesis and its targeting in rheumatoid arthritis. Vascul Pharmacol. 2009;51:1–7. doi: 10.1016/j.vph.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pickens SR, Volin MV, Mandelin AM, 2nd, Kolls JK, Pope RM, Shahrara S. Il-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol. 2010;184:3233–41. doi: 10.4049/jimmunol.0903271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ryu S, Lee JH, Kim SI. Il-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clin Rheumatol. 2006;25:16–20. doi: 10.1007/s10067-005-1081-1. [DOI] [PubMed] [Google Scholar]
- 95.Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2006;14:345–52. doi: 10.1016/j.joca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 96.Shahrara S, Pickens SR, Dorfleutner A, Pope RM. Il-17 induces monocyte migration in rheumatoid arthritis. J Immunol. 2009;182:3884–91. doi: 10.4049/jimmunol.0802246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pickens SR, Chamberlain ND, Volin MV, et al. Local expression of interleukin-27 ameliorates collagen-induced arthritis. Arthritis Rheum. 2011;63:2289–98. doi: 10.1002/art.30324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in il-17-deficient mice. J Immunol. 2003;171:6173–7. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 99.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor igg1 fc fusion protein. Arthritis Rheum. 2002;46:802–5. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 100.Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–9. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 101.Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing rankl and interleukin-1. Am J Pathol. 2005;167:141–9. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koenders MI, Lubberts E, van de Loo FA, et al. Interleukin-17 acts independently of tnf-alpha under arthritic conditions. J Immunol. 2006;176:6262–9. doi: 10.4049/jimmunol.176.10.6262. [DOI] [PubMed] [Google Scholar]
- 103.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for il-23 and il-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Doodes PD, Cao Y, Hamel KM, et al. Development of proteoglycan-induced arthritis is independent of il-17. J Immunol. 2008;181:329–37. doi: 10.4049/jimmunol.181.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cao Y, Doodes PD, Glant TT, Finnegan A. Il-27 induces a th1 immune response and susceptibility to experimental arthritis. J Immunol. 2008;180:922–30. doi: 10.4049/jimmunol.180.2.922. [DOI] [PubMed] [Google Scholar]