

Abstract
Glucagon is important for maintaining euglycemia during fasting/starvation, and abnormal glucagon secretion is associated with type 1 and type 2 diabetes; however, the mechanisms of hypoglycemia-induced glucagon secretion are poorly understood. We previously demonstrated that global deletion of mitochondrial uncoupling protein 2 (UCP2−/−) in mice impaired glucagon secretion from isolated islets. Therefore, UCP2 may contribute to the regulation of hypoglycemia-induced glucagon secretion, which is supported by our current finding that UCP2 expression is increased in nutrient-deprived murine and human islets. Further to this, we created α-cell–specific UCP2 knockout (UCP2AKO) mice, which we used to demonstrate that blood glucose recovery in response to hypoglycemia is impaired owing to attenuated glucagon secretion. UCP2-deleted α-cells have higher levels of intracellular reactive oxygen species (ROS) due to enhanced mitochondrial coupling, which translated into defective stimulus/secretion coupling. The effects of UCP2 deletion were mimicked by the UCP2 inhibitor genipin on both murine and human islets and also by application of exogenous ROS, confirming that changes in oxidative status and electrical activity directly reduce glucagon secretion. Therefore, α-cell UCP2 deletion perturbs the fasting/hypoglycemic glucagon response and shows that UCP2 is necessary for normal α-cell glucose sensing and the maintenance of euglycemia.
Elevated basal glucagon levels and reduced hypoglycemia-induced glucagon secretion are underappreciated and poorly understood aspects of type 1 and type 2 diabetes (1–3). Although high plasma glucose normally inhibits glucagon secretion, it remains unclear whether this in vivo response is mediated by glucose sensing, neuronal modulation, paracrine/endocrine control, or a combination thereof (4–10). Uncoupling protein 2 (UCP2), an inner mitochondrial membrane protein, is expressed in pancreatic α-cells (11), and its expression can be induced in adipose tissue by a ketogenic diet (12), suggesting a role in the fasting response. While the precise physiological function of UCP2 in islet cells is still debated, it can mildly dissipate the proton motive force generated during mitochondrial electron transport and limit ATP synthesis under certain conditions (13–15). Additionally, UCP2 can limit mitochondrial reactive oxygen species (ROS) production, which can alter associated signaling pathways and/or protect against oxidative stress (16–18). In β-cells, UCP2 deletion elicits only small changes in mitochondrial membrane potential (ΔΨm) with limited effect on ATP (18,19) but rather increases ROS production, which amplifies insulin secretion (18,20). α-Cells, like β-cells, have glucose-sensing machinery that center on KATP channel activity, cellular depolarization, and calcium influx, triggering exocytosis; however, unlike β-cells, they are electrically active and secretory at low glucose concentrations (5,21–24). UCP2 in α-cells could therefore be an important regulator of glucagon secretion via regulation of ATP production, plasma membrane potential, and ROS levels.
Previously, we showed that islets from mice globally lacking UCP2 (UCP2−/−) displayed higher basal glucagon secretion and impaired low glucose–mediated glucagon secretion (11). Due to UCP2’s wide expression profile in glucose-sensitive tissues, these changes in α-cell function in UCP2−/− mice could be the result of β-cell and/or extra-pancreatic deletion. To decipher the role of UCP2 in α-cells and in the response to fasting, we created an α-cell–specific UCP2 knockout (UCP2AKO) deletion mouse model. These mice displayed reduced fasting plasma glucagon levels and impaired glucagon secretion, due in part to elevated ROS, enhanced glucose-induced hyperpolarization of the ΔΨm, and depolarization of plasma membrane potential. Therefore, we conclude that α-cell UCP2 plays a key role in the hypoglycemic response.
RESEARCH DESIGN AND METHODS
Animals.
LoxUCP2 mice on a 129/SV background (25) (Fig. 1A) were crossed for at least four generations to rat glucagon promoter-driven Cre recombinase (Gcg-cre) mice maintained on a C57BL/6 background (26). Male mice were genotyped using standard PCR (Supplementary Fig. 1A and Supplementary Table 1) and used for experiments at age 6–8 weeks. Gcg-cre mice were used as experimental controls, as Gcg-cre and floxed mice had similar phenotypes (Supplementary Fig. 2). For imaging experiments, loxUCP2 mice were crossed to Gcg-cre-ROSA26EYFP mice (27) for at least four generations, producing mice with both α-cell–specific UCP2 deletion and expression of eYFP (UCP2AKO-YFP). All experiments were approved by the Animal Care Committee (University of Toronto) and animals handled according to the Canadian Council of Animal Care guidelines.
FIG. 1.
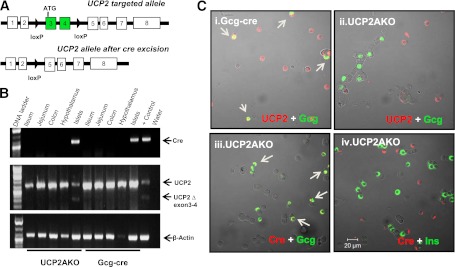
UCP2 is efficiently deleted specifically from islet α-cells of UCP2AKO mice. A: Schematic diagram of the UCP2-targeting construct. B: Cre and UCP2 tissue expression measured by standard PCR. C: Immunostaining for UCP2 protein (red) and glucagon (green) in dispersed islet cells from Gcg-cre and UCP2AKO mice. UCP2AKO islet cells were also stained for Cre (red) and glucagon (green) or insulin (green). Colocalization is indicated by yellow and arrows. N = 3.
Human islets.
Human islets from review board approved healthy donors were isolated using the Edmonton protocol (28,29) and provided by the Clinical Islet Laboratory or IsletCore (University of Alberta, Edmonton, Canada). Islets were picked into RPMI media with 10% FBS and cultured overnight before experimentation.
Antibodies and reagents.
The monoclonal Cre antibody was from Covance (San Diego, CA) and the polyclonal UCP2 antibody from Everest Biotech (Oxfordshire, U.K.). Guinea pig anti-swine insulin (Dako Canada), rabbit anti-human glucagon (Dako Canada), and fluorescein isothiocyanate– and cyanine 5–labeled secondary antibodies (Jackson ImmunoResearch) were also used. Rhodamine 123, H2O2, and RedTaq polymerase were from Sigma-Aldrich. Genipin was from Wako.
Immunofluorescent staining.
Dispersed islet cells plated onto glass coverslips were fixed, stained with antibodies, and fluorescence visualized using a laser-scanning confocal microscope (Zeiss 510 LSM) (11).
Plasma hormone concentration and tolerance tests
Insulin tolerance tests.
After a 4-h fast, insulin (1.75 IU/kg body wt) was injected intraperitoneally and blood glucose levels were measured at 0, 15, 30, 60, and 120 min. Plasma glucagon was measured at 0 and 30 min by RIA (Millipore).
Oral glucose tolerance test.
After a 4-h fast, glucose (3 g/kg body wt) was gavaged and blood glucose levels were measured at 0, 5, 10, 15, 30, 60, and 120 min. Plasma active glucagon-like peptide (GLP)-1 (Meso Scale Discovery, Gaithersburg, MD) was measured at 0 and 10 min.
Fasting/re-feeding hepatic gluconeogenenic gene expression, hepatic glycogen, and plasma metabolites.
Plasma glucose, glucagon, insulin, and corticosterone (MP Biomedicals, Irvine, CA) were measured in fed, 24-h fasted, and 12-h re-fed mice. Mice were killed under fasting or re-fed conditions, and liver RNA was extracted using Trizol (Invitrogen, Canada) for measurement of gluconeogenic and glycolytic enzyme genes (Supplementary Table 1). The glycogen content of livers from 24-h fasted and 12-h re-fed mice was extracted as previously described (30) and measured using a colorimetric kit (Biovision). The relative concentrations of plasma metabolites were measured in 100 μL plasma from fed and 24-h fasted mice by mass spectrometry (Metabolon). Sample preparation, metabolite identification, data normalization, and statistical analysis were performed as previously described (31).
Islet glucose-inhibited glucagon secretion.
Fresh isolated islets (mouse or human) were handpicked into 11 mmol/L glucose RPMI media with 10% FBS and preincubated for 1 h with or without genipin (50 µmol/L) or H2O2 (16 µmol/L), which remained throughout the protocol. Fifteen islets per condition were preincubated for 15 min with 20 mmol/L glucose Krebs-Ringer buffer (KRB) (11) before incubation with either high- (20 mmol/L) or low- (1 mmol/L) glucose KRB for 1 h. Glucagon concentrations were determined by RIA (Millipore) and normalized to DNA content.
Mitochondrial membrane potential (ΔΨm).
α-Cells were identified by YFP fluorescence before rhodamine 123 (25 μg/mL; 10 min) loading in 1 mmol/L glucose KRB on the microscope stage. The cells were then washed and imaged in 1 and 20 mmol/L glucose KRB. NaN3 (5 mmol/L) was added to fully depolarize the ΔΨm (11).
Islet ATP content and intracellular calcium.
Islet ATP content was measured as previously described using 15 islets per condition (18). Cells were preincubated with or without H2O2 (16 µmol/L) or genipin (50 µmol/L) for 1 h in 11 mmol/L glucose RPMI and then loaded with 2 µmol/L Fura-2 AM (Molecular Probes; Invitrogen) in 20 mmol/L glucose KRB for 45 min before imaging of cytosolic calcium in YFP+ α-cells (32).
Electrophysiology.
Electrophysiological recordings of KATP currents in YFP+ α-cells were performed as previously described (33). Plasma membrane potential was measured using a perforated patch technique in whole-cell configuration. The extracellular solution for membrane potential measurements contained (in millimoles per liter) 140 NaCl, 3.6 KCl, 0.5 MgSO4, 1.5 CaCl2, 10 HEPES, 0.5 NaH2PO4, 5 NaHCO3 (pH 7.4 with NaOH), and 20 glucose. The intracellular solution contained (in millimoles per liter unless otherwise indicated) 76 K2SO4, 10 KCl, 10 NaCl, 1 MgCl2, 5 HEPES (pH 7.35 with KOH), and 60 µg/mL gramicidin (34). Cells displaying input resistance <25 MΩ were studied. Average membrane potentials were calculated across 40 s of steady-state recordings.
ROS accumulation in isolated islets and its effect on glucagon secretion.
The levels of superoxide and H2O2 were determined using MitosoxRed and 2′,7′-dichlorodihydro fluorescein diacetate (CM-H2-DCFDA), respectively (Molecular Probes; Invitrogen) (35). Fresh isolated islets were preincubated with or without H2O2 (16 µmol/L) or genipin (50 µmol/L) in 11 mmol/L glucose RPMI for 1 h, followed by 1 mmol/L glucose KRB with or without treatments for 1 h before measurement of superoxide or H2O2 levels. Alternatively, islets were cultured overnight in 11 mmol/L glucose RPMI and then H2O2 levels measured with CM-H2-DCFDA.
Quantitative real-time PCR.
Quantitative PCR was performed as previously described (18,32). Primers are listed in Supplementary Table 1.
Statistics.
Statistical significance was assessed using either the Student t test or a two-way ANOVA for repeated measures followed by a Bonferroni posttest comparison using GraphPad Prism 4. P < 0.05 was considered significant. All data are means ± SEM.
RESULTS
Deletion of α-cell UCP2 impairs glucagon secretion in response to hypoglycemia.
Truncated UCP2, indicative of exon 3 and 4 deletion, was observed only in islets and not in the intestine or hypothalamus of UCP2AKO mice (Fig. 1B). Cre (measured by eYFP staining) was expressed in 72 ± 10% glucagon-positive cells (Supplementary Fig. 1B) suggesting efficient deletion. Immunofluorescent staining of dispersed UCP2AKO islets showed cre protein only in α-cells (Fig. 1C), and specific deletion of UCP2 from UCP2AKO α-cells was confirmed by the absence of UCP2 only in glucagon-positive cells (Fig. 1C). Cre (YFP staining) was not expressed in the nucleus of the solitary tract or the distal ileum, reinforcing the specificity of the glucagon promoter for α-cells (Supplementary Fig. 3).
UCP2AKO and Gcg-cre mice displayed similar insulin sensitivities, as the rate and magnitude of blood glucose reduction during an insulin tolerance test were equal (Fig. 2A); however, the rate of blood glucose recovery was slower in UCP2AKO mice, resulting in significantly lower blood glucose at 120 min (Fig. 2A). This correlated with lower glucagon levels 30 min after insulin injection (just before blood glucose levels increased) (Fig. 2B), suggesting that UCP2AKO mice have impaired hypoglycemia-induced glucagon secretion.
FIG. 2.

α-Cell UCP2 deletion reduces glucagon secretion in vivo. A: Blood glucose during an intraperitoneal insulin tolerance test (ipITT). *P < 0.05. N = 12–15. B: Plasma glucagon levels at 0 and 30 min during the ipITT. N = 6–8 mice/group. *P < 0.05; **P < 0.01.
Mice lacking glucagon receptors are more glucose tolerant compared with wild-type mice (36). Therefore, we tested whether UCP2AKO mice that display impaired glucagon secretion were more glucose tolerant. An oral glucose tolerance test showed that UCP2AKO mice had similar glucose tolerance to Gcg-cre mice (Fig. 3A) and secreted the same amount of active GLP-1 into plasma (Fig. 3B).
FIG. 3.

UCP2AKO mice display normal glucose tolerance and GLP-1 secretion. A and B: Oral glucose tolerance test; mice were gavaged with 3 g/kg glucose after a 4-h fast (N = 7–8 mice/genotype). Blood glucose (A) and plasma active GLP-1 (B) were measured. The inset panel in A shows the incremental area under the glucose curve (iAUC).
α-Cell UCP2 deficiency impairs hepatic gluconeogenic gene expression and causes a switch of fatty acid usage in response to a prolonged fast.
To assess the effect of impaired glucagon secretion in UCP2AKO mice, we examined the effect of prolonged fasting/re-feeding on glucose homeostasis, hepatic gluconeogenesis, and glycogenolysis. Although blood glucose (Fig. 4A) and plasma insulin (Fig. 4C) were similarly decreased upon fasting, the UCP2AKO mice displayed reduced fasting plasma glucagon (Fig. 4B). The difference in plasma glucagon levels was not due to impaired secretion of corticosterone, as the levels were similarly increased upon fasting (Supplementary Fig. 4A).
FIG. 4.

α-Cell UCP2 deletion reduces the gluconeogenic response of the liver and switches fatty acid usage during a prolonged fast. Blood glucose (A), plasma glucagon (B), and plasma insulin (C) measured in fed, 24-h fasted (fasting), and 12-h re-fed mice. *P < 0.05. D–H: Hepatic expression of the enzymes Pepck (D), G6pc (E), Fbp1 (F), Gck (G), and Fasn (H) genes. N = 4–8/group. *P < 0.05 or **P < 0.01 compared with Gcg-cre. I: Hepatic glycogen levels. N = 4–6 mice/condition. *P < 0.05. J: Metabolomic analyses of MCFA and long-chain fatty acid (LCFA) plasma levels. Data are relative to Gcg-cre levels. N = 8 mice/genotype. *P < 0.05 genotype/fed-state interaction.
The liver is the major target organ for glucagon in vivo, and so we explored the effect of prolonged fasting/re-feeding on hepatic glucose metabolism genes and glycogen content. After fasting, UCP2AKO livers displayed impaired induction of the gluconeogenic genes phosphoenolpyruvate carboxykinase (Pepck) (Fig. 4D) and glucose-6-phosphatase (G6pc) (Fig. 4E). Conversely, the glycolytic gene, glucokinase (Gck), was significantly higher (Fig. 4G). The expression of fructose 1,6-bisphosphatase (Fbp1) decreased and fatty acid synthase (Fasn) increased similarly upon re-feeding (Fig. 4F and H), suggesting an intact response for these genes. The reduced gluconeogenic gene expression in the UCP2AKO mice supports their impaired glucagon secretion in response to hypoglycemia. Glycogen levels were 2.5-fold higher in UCP2AKO livers upon fasting (Fig. 4I), also reflecting their lower glucagon levels.
A targeted mass spectrometry approach examining multiple metabolic pathways was used to determine the effect of reduced glucagon secretion and the impaired hepatic gluconeogenic response to fasting on plasma metabolites. Of the 307 identified biochemicals, 157 were significantly changed upon fasting and 63 showed a fed-state, genotype interaction (P < 0.05). Levels of the ketone body 3-hydroxybutarate normally increase upon fasting, and the response of this metabolite was intact in UCP2AKO mice (Supplementary Fig. 4B). Interestingly, several abnormalities in lipid metabolism products were observed in UCP2AKO mice. The levels of medium chain–length fatty acids (MCFA) were similar in fed plasma; however, their concentration decreased in UCP2AKO and increased in Gcg-cre mice upon fasting (Fig. 4J and Supplementary Fig. 4C), suggesting that UCP2AKO mice may preferentially use MCFAs as an energy source under these conditions.
UCP2 expression is increased after nutrient depletion in islets, and α-cell UCP2 deficiency/inhibition impairs glucagon secretion from isolated islets.
Human and mouse islets deprived of nutrients (low serum and low glucose) showed similar induction of glucagon and UCP2 expression, highlighting the potential importance of UCP2 during fasting (Fig. 5A and B). We therefore assessed the ability of fresh-isolated UCP2AKO mouse islets to secrete glucagon under low glucose conditions. When conditions were switched from high to low glucose, glucagon secretion was significantly stimulated in Gcg-cre and UCP2AKO islets (P < 0.001) (Fig. 5D); however, the amount secreted by UCP2AKO islets was 52 and 31% lower under high and low glucose concentrations, respectively. Total glucagon content and islet morphology were similar in the two genotypes, suggesting that the reduced secretion from UCP2AKO islets was not due to reduced glucagon content or α-cell number (data not shown).
FIG. 5.

UCP2 expression is increased after nutrient depletion and glucagon secretion from UCP2AKO islets was impaired. Human (left panel) and mouse (right panel). A: UCP2 (i) and GCG (ii) expression in human islets exposed to low serum (0.01% FBS) and low glucose (2.8 mmol/L) RPMI (low serum and low glucose [LSLG]) for 6 h. N = 6 individual donors. B: Ucp2 (i) and Gcg (ii) expression in control CD1 mouse islets exposed to LSLG for 6 h. N = 4–5/condition. C: Glucose-inhibited glucagon secretion from human islets preincubated with or without genipin (50 µmol/L) during a half-hour static incubation. N = 6–7 donors. D: Glucose-inhibited glucagon secretion was measured from fresh-isolated islets incubated with high glucose (HG) (20 mmol/L), low glucose (LG) (1 mmol/L), or low glucose plus arginine (LG + Arg) (10 mmol/L). N = 8–9 mice/genotype. E: Glucose-inhibited glucagon secretion measured from islets preincubated with or without genipin (50 µmol/L). N = 7–9 mice/genotype. All mouse data are normalized to DNA, but the human data are per islet. *P < 0.05.
Gcg-cre islets exposed to genipin, a compound known to inhibit UCP2 (37), secreted significantly less glucagon in the presence of low glucose, and this level was similar to UCP2AKO islets in the presence of low glucose alone (Fig. 5E). Conversely, preincubation of UCP2AKO islets with genipin did not alter glucagon secretion (Fig. 5E). For assessment of the interspecies effect of UCP2 inhibition on glucagon secretion, human islets were also exposed to genipin. They secreted significantly less glucagon under low glucose conditions, which was similar to high glucose levels alone (Fig. 5C). Therefore, acute inhibition of UCP2 with genipin reduces glucagon secretion under stimulatory low glucose conditions.
α-Cell UCP2 deficiency increases glucose-induced ΔΨm hyperpolarization, which leads to altered oxidative status and impaired glucagon secretion.
To determine the contribution of UCP2 to mitochondrial coupling in α-cells, we measured changes in glucose-induced ΔΨm hyperpolarization (Fig. 6A). Basal (1 mmol/L glucose) ΔΨm was similar, but UCP2AKO-YFP α-cells displayed significantly greater glucose-induced hyperpolarization of ΔΨm (Fig. 6B).
FIG. 6.

UCP2AKO α-cells display enhanced hyperpolarization of ΔΨm and increased superoxide levels. A: Representative traces of ΔΨm (Mmp) relative fluorescent unit (RFU) measurements in YFP+ α-cells. B: ΔΨm (Mmp) was measured in high glucose (20 mmol/L) (Δ1) and NaN3 (5 mmol/L) (Δ2) and normalized to basal fluorescence in the presence of low glucose (1 mmol/L). N = 28–29 cells from 6 mice/genotype. *P < 0.05. C and D: Fresh-isolated islets were cultured with or without H2O2 (16 µmol/L) or genipin (50 µmol/L) before measurement of H2O2 (CM-H2-DCFDA) (C) or superoxide (MitosoxRed) (D). N = 20–28 islets, 6–8 mice/genotype. *P < 0.05, **P < 0.01. E: Islets were cultured overnight, and then intracellular H2O2 (CM-H2-DCFDA) was measured. N = 19–27 islets, 5–7 mice/genotype. *P < 0.05. F: Islet Sod2 expression. N = 4–5 mice/genotype. *P < 0.05. G: Glucose-inhibited glucagon secretion with or without H2O2 (16 µmol/L). Secretion is per microgram of DNA, normalized to Gcg-cre high glucose (HG) conditions. LG, low glucose. N = 3–11 mice/genotype. *P < 0.05, **P < 0.01. H and I: Human islets were incubated with 50 µmol/L genipin for 2 h before measurement of H2O2 (CM-H2-DCFDA) (H) and superoxide (MitosoxRed) (I) levels. N = 4 individual donors. *P < 0.05, **P < 0.01.
Even small increases in ΔΨm can translate into significantly large increases in mitochondrial ROS production, and recently ROS signaling has emerged as an important regulator of insulin secretion (18,20,38). In contrast, chronic oxidative stress can impair secretion (17,35). As such, we compared the levels of intracellular ROS (H2O2 and mitochondrial superoxide) in fresh-isolated islets (to correlate with glucagon secretion studies [Fig. 5D]) and H2O2 in cultured islets. No difference in the amount of H2O2 but significantly more mitochondrial superoxide was observed in fresh-isolated UCP2AKO islets (Fig. 6C and D). Application of genipin increased H2O2 levels 1.37-fold in Gcg-cre islets only (P < 0.01) and increased both H2O2 and superoxide levels in human islets (Fig. 6H and I). Therefore, UCP2 deficiency in mouse α-cells results in superoxide buildup and an inability to convert superoxide to H2O2. However, after overnight culture, UCP2AKO islets displayed ~1.7-fold greater intracellular H2O2 (Fig. 6E). Cultured Gcg-cre islets appeared to recover from the stress of isolation, as H2O2 levels were lower than in fresh-isolated islets (Fig. 6C), suggesting that ROS production is increased in UCP2AKO islets and they experience a chronically altered oxidative status. Whole-islet data were confirmed in dispersed, cultured α-cells, as overall ROS levels were higher in UCP2AKO α-cells (Supplementary Fig. 5). In addition, treatment of Gcg-cre α-cells with genipin increased ROS to a similar level as UCP2AKO α-cells, whereas genipin had no further effect on UCP2AKO α-cells (Supplementary Fig. 5).
Intracellular ROS levels were correlated with expression of several antioxidant-related genes in fresh isolated whole islets. The expression of superoxide dismutase 2 (Sod2) was 88% lower in UCP2AKO islets (Fig. 6F), consistent with their inability to convert superoxide to H2O2, whereas the H2O2-scavenging glutathione peroxidase (Gpx) and catalase (Cat) and the cytoprotective gene, Heme Oxygenase 1 (Ho-1), were unchanged (Supplementary Fig. 6A).
To determine whether increased ROS contribute to impaired glucagon secretion from UCP2AKO islets, we increased intracellular ROS with exogenous H2O2 application (Fig. 6C and D). Gcg-cre islets preincubated with H2O2 secreted significantly more glucagon under high-glucose and less under low glucose conditions (Fig. 6G). The increased glucagon secretion in the presence of high glucose plus H2O2 occurred despite significantly greater insulin secretion (Supplementary Fig. 6C). The addition of H2O2 to UCP2AKO islets did not alter glucagon secretion (Fig. 6G). Similarly, the addition of H2O2 had no effect on glucose-mediated insulin secretion from UCP2AKO islets (Supplementary Fig. 6C). Therefore, elevated ROS levels in control islets impaired glucagon secretion similarly to UCP2AKO islets, which experience endogenously higher ROS levels and altered oxidative status.
Impaired glucagon secretion from UCP2-deficient α-cells correlates with enhanced depolarization of plasma membrane potential and reduced calcium.
Metabolism of glucose and further closure of KATP channels reduce glucagon secretion (24). Therefore, we measured KATP current and plasma membrane potential. KATP currents in Gcg-cre-YFP and UCP2AKO-YFP α-cells could be similarly activated and inhibited by diazoxide and tolbutamide, respectively (Supplementary Fig. 7A). However, despite a similar magnitude of change in plasma membrane potential when switched from high to low glucose, the plasma membrane was more depolarized in UCP2AKO-YFP α-cells under both conditions (Fig. 7A and B). Preincubation of Gcg-cre-YFP α-cells with high glucose plus genipin increased (depolarized) plasma membrane potential but had no further effect in UCP2AKO-YFP α-cells (Fig. 7C). Therefore, the absence of UCP2 in α-cells shifts the operating range of plasma membrane potentials, which might explain the impaired glucagon secretion.
FIG. 7.

UCP2AKO α-cells have more depolarized plasma membranes and reduced intracellular calcium. A: Representative traces of plasma membrane potential in YFP+ α-cells incubated in high (20 mmol/L) and low (1 mmol/L) glucose. B: Average membrane potential in YFP+ α-cells. N = 5–8 cells, 3 mice/genotype. *P < 0.05. C: Average plasma membrane potential measured in YFP+ α-cells (N = 8–13) with or without genipin (50 µmol/L) (N = 5 cells), and at least 3 mice/genotype. *P < 0.05, **P < 0.01. D: Glucose-inhibited glucagon secretion from islets incubated with or without diazoxide (1, 10, and 100 µmol/L) in the presence of high glucose (20 mmol/L) (i) or low glucose (1 mmol/L) (ii). *P < 0.05. N = 3 mice/genotype. E: Representative traces of cytosolic calcium uptake in YFP+ α-cells switched from high glucose (HG) (20 mmol/L) to low glucose (LG) (1 mmol/L) and then in the presence of arginine (Arg) (10 mmol/L), KCl (30 mmol/L), and the l-type calcium blocker nifedipine (Nif) (10 µmol/L). F: Incremental area under the curve (iAUC) for cytosolic calcium, normalized to high glucose plus arginine levels. Representative traces are shown in Supplementary Fig. 8. *P < 0.05. N = 19–40 cells/treatment, 3 mice/genotype.
The more depolarized plasma membrane potential in the presence of high glucose suggests that α-cell UCP2 deficiency may increase ATP levels and close/inactivate KATP channels. There was a trend toward increased ATP content in UCP2AKO islets under high glucose conditions (Supplementary Fig. 7B); however, any effect of α-cell UCP2 deletion may be masked by β-cell ATP content. Therefore, we determined whether the KATP channel opener, diazoxide, could overcome the impaired secretion. Under high glucose conditions, the addition of low-dose diazoxide (1 µmol/L) increased glucagon secretion from UCP2AKO islets to a level similar to that in Gcg-cre islets without diazoxide; however, the UCP2AKO islets were unable to reach a level of secretion as high as that in Gcg-cre islets in the presence of 1 µmol/L diazoxide (Fig. 7D). Conversely, under low glucose conditions, low-dose diazoxide increased glucagon secretion from UCP2AKO islets to a level similar to that in Gcg-cre islets in the absence of diazoxide. The addition of more diazoxide dose-dependently decreased secretion (Fig. 7D), which was similar to previous observations (24). Therefore, under low glucose conditions opening KATP channels can correct the glucagon secretion defect observed in UCP2AKO islets.
Influx of calcium is required for low glucose–mediated exocytosis of glucagon (39), and thus we examined changes in cytosolic calcium. The amount of intracellular calcium under high and low glucose conditions was reduced in YFP-UCP2AKO α-cells (Fig. 7F). Pretreatment of YFP-Gcg-cre cells with genipin also significantly reduced intracellular calcium levels, whereas H2O2 had no effect (Fig. 7F). In addition, intracellular calcium in UCP2AKO α-cells oscillated faster under low glucose conditions (Supplementary Fig. 8E). Reduced cytosolic calcium in UCP2AKO α-cells reflects an impaired ability to secrete glucagon.
DISCUSSION
α-Cells secrete glucagon in response to low blood glucose and are inhibited when blood glucose levels rise (24). We now show that UCP2 expression is increased in islets deprived of nutrients, α-cell UCP2 is necessary for appropriate glucagon secretion, and the absence of UCP2 impairs α-cell function. UCP2AKO mice displayed impaired glucagon secretion in vivo, which inhibited the hepatic response to hypoglycemia (40), including reduced expression of the hepatic gluconeogenic genes, Pepck and G6pc, and higher glycogen levels. Reduced fasting levels of plasma MCFAs in UCP2AKO mice suggest compensation for impaired hepatic gluconeogenesis and increased usage of these fatty acids during hypoglycemia. MCFAs are absorbed directly through the portal vein, making them a direct energy source for the liver (41). MCFAs also produce more ketones per unit of energy than long-chain fatty acids, which could provide crucial energy for the brain during nutrient depletion and when gluconeogenesis is impaired, such as in fasted UCP2AKO mice. Similarly, mice globally lacking glucagon receptors demonstrate impaired activation of hepatic gluconeogenic genes and enhanced lipogenesis, yet have improved glucose tolerance (36,42). Interestingly, UCP2AKO mice, similar to adult mice with 98% α-cell ablation (43), displayed normal glucose tolerance. Therefore, we show that appropriate glucagon secretion is required for normal hepatic function during fasting and that α-cell UCP2 deficiency disrupts this process, solidifying the role of UCP2 in the α-cell response to hypoglycemia.
The amount of glucagon secreted from UCP2AKO islets under stimulatory conditions was the same as that secreted from Gcg-cre islets under inhibitory conditions. Preincubation of Gcg-cre islets with the UCP2 inhibitor genipin reduced low glucose–mediated glucagon secretion by a magnitude similar to that in UCP2 deletion, whereas genipin did not further reduce glucagon secretion from UCP2AKO islets, suggesting its relative specificity of action for UCP2. Interestingly, the glucagon-lowering effect of genipin was not isolated to mouse islets, as it also significantly impaired low glucose–mediated glucagon secretion from human islets. Insulin released from β-cells lowers glucagon secretion via paracrine inhibition (5,7,9); however, the reduced glucagon secretion from UCP2AKO islets was not explained by this mechanism, as insulin secretion was similar from Gcg-cre and UCP2AKO cultured islets (Supplementary Fig. 6B). Therefore, the presence of UCP2 in α-cells is directly required for appropriate glucagon release.
The α-cell secretes glucagon within a narrow range of plasma membrane potentials (24) and is sensitive to small changes in ATP concentration (22), and it is suggested that glucose metabolism does not accelerate ATP synthesis in this cell type (44). We previously showed that UCP2 is expressed at higher levels in α compared with clonal β-cell lines (11). Thus, high expression of UCP2 may normally limit ATP production in α-cells via an uncoupling role. Furthermore, small interfering RNA knockdown of UCP2 in clonal α-cells increased ATP levels (11). Although no difference in ATP concentrations in UCP2AKO versus Gcg-cre islets was detected, the increased glucose-induced hyperpolarization of ΔΨm and more depolarized plasma membrane of UCP2AKO α-cells suggest increased ATP synthesis. Recent studies show that depolarization of α-cells inhibits the activity of Na+ and Ca2+ channels resulting in cessation of glucagon secretion (22,24), and we show reduced intracellular calcium levels in UCP2AKO α-cells under both high and low glucose conditions. Similarly, exposure of Gcg-cre α-cells to genipin depolarized the plasma membrane and reduced calcium entry. As such, we propose that UCP2AKO α-cells have more closed KATP channels, limiting their glucagon secretion capacity (24,45). It was no surprise that the addition of low-dose diazoxide, which opens the KATP channel, corrected this impairment under low glucose conditions. Conversely, in the presence of high glucose, low-dose diazoxide increased glucagon secretion from UCP2AKO islets to a level similar to basal secretion from Gcg-cre islets, yet further addition of diazoxide did not elevate secretion from UCP2AKO islets. These data suggest that altered membrane potential is not the only factor limiting glucagon secretion from UCP2AKO islets under high glucose conditions.
It has been proposed that generation of low-level ROS, likely H2O2, amplifies insulin secretion (20,46), and recently, we showed that β-cell–specific deletion of UCP2 increases insulin secretion in an ROS-dependent manner (18). We now show that the acute addition of exogenous H2O2 to control islets increased glucagon secretion under high glucose and reduced glucagon secretion under low glucose conditions, a situation reminiscent of the disordered glucagon secretion from murine UCP2−/− islets (11) and patients with diabetes (1) and human islets exposed to genipin. It is therefore conceivable that UCP2 regulates the levels of ROS in α-cells and, in turn, glucagon secretion. Acute inhibition of UCP2 in Gcg-cre islets with genipin increased endogenous H2O2 levels, which correlated with reduced low glucose–mediated glucagon secretion. Similarly, UCP2AKO islets had elevated mitochondrial superoxide levels and signs of chronically altered redox-related signaling pathways (i.e., ROS levels remained high after overnight culture and altered antioxidant gene expression [including reduced Sod2 expression] was observed), which also correlated with impaired glucagon secretion. Therefore, elevated intracellular ROS can reduce low glucose–mediated glucagon secretion, although the specific ROS species and its signaling targets are unclear.
We propose that α-cell UCP2 expression is normally increased in response to low nutrient levels and limits ΔΨm hyperpolarization, which, by limiting ATP and/or ROS production, contributes to the cells’ ability to secrete glucagon. Therefore, α-cell–specific deletion of UCP2 results in greater depolarization of the plasma membrane and, although still responsive to changes in glucose, reduces calcium entry and lowers overall secretion capacity (Fig. 8A). Deletion of α-cell UCP2 disrupted the hypoglycemic response in mice, implying that increased UCP2 expression/activity, such as that which occurs after fasting, is required to promote glucagon secretion and is protective in this role (Fig. 8B). Conversely, elevated expression or activity of UCP2 may also contribute to the dysregulated glucagon secretion that is observed in the diabetic state.
FIG. 8.

UCP2 is required for normal glucagon secretion in response to hypoglycemia. A: UCP2AKO islets displayed increased glucose-induced hyperpolarization of ΔΨm, which caused increased ROS and perhaps ATP production. The altered coupling and ROS status resulted in greater depolarization of the plasma membrane, and although still responsive to changes in glucose, the UCP2AKO α-cells showed reduced calcium entry and lower glucagon secretion. B: UCP2 expression is normally increased in islets deprived of nutrients. Therefore, lack of UCP2 in α-cells results in impaired glucagon secretion during hypoglycemia. ITT, insulin tolerance test; TCA, tricarboxylic acid.
Supplementary Material
Acknowledgments
This study was funded by a Canadian Institutes of Health Research (CIHR) grant (MOP 12898) to M.B.W., a Canadian Diabetes Association postdoctoral fellowship award to E.M.A., a CIHR postdoctoral fellowship award to C.A.R.-D., and an Ontario Graduate Scholarship to K.J.P. P.G. is Research Director of the Fonds National de la Recherche Scientifique, Brussels. The human islets used in the study were provided by A.M.J. Shapiro and T. Kin at the Clinical Islet Laboratory and P.E. MacDonald at the IsletCore Laboratory, University of Alberta (Edmonton, Canada). Use of some of the equipment used in the study was supported by the 3D (Diet, Digestive Tract, and Disease) Centre funded by the Canadian Foundation for Innovation and by the Ontario Research Fund, project no. 19442. This study was also funded by a Banting and Best Diabetes Centre Novo Nordisk studentship to S.S.
No potential conflicts of interest relevant to this article were reported.
E.M.A. researched data, contributed to discussion, wrote the manuscript, and reviewed and edited the manuscript. C.A.R.-D., K.J.P., and A.B.H. researched data, contributed to discussion, and reviewed and edited the manuscript. S.S. researched data and contributed to discussion. H.Y.G. contributed to discussion and reviewed and edited the manuscript. D.K. researched data and contributed to discussion. P.G., P.L.H., and B.B.L. contributed to discussion and reviewed and edited the manuscript. M.B.W. designed the study, contributed to discussion, and reviewed and edited the manuscript. M.B.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0981/-/DC1.
REFERENCES
- 1.Burcelin R, Knauf C, Cani PD. Pancreatic alpha-cell dysfunction in diabetes. Diabetes Metab 2008;34(Suppl. 2):S49–S55 [DOI] [PubMed] [Google Scholar]
- 2.Kawamori D, Welters HJ, Kulkarni RN. Molecular pathways underlying the pathogenesis of pancreatic alpha-cell dysfunction. Adv Exp Med Biol 2010;654:421–445 [DOI] [PubMed] [Google Scholar]
- 3.Cryer PE. Hypoglycaemia: the limiting factor in the glycaemic management of Type I and Type II diabetes. Diabetologia 2002;45:937–948 [DOI] [PubMed] [Google Scholar]
- 4.Rorsman P, Berggren PO, Bokvist K, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989;341:233–236 [DOI] [PubMed] [Google Scholar]
- 5.Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol 2003;5:330–335 [DOI] [PubMed] [Google Scholar]
- 6.Gyulkhandanyan AV, Lu H, Lee SC, et al. Investigation of transport mechanisms and regulation of intracellular Zn2+ in pancreatic alpha-cells. J Biol Chem 2008;283:10184–10197 [DOI] [PubMed] [Google Scholar]
- 7.Diao J, Asghar Z, Chan CB, Wheeler MB. Glucose-regulated glucagon secretion requires insulin receptor expression in pancreatic alpha-cells. J Biol Chem 2005;280:33487–33496 [DOI] [PubMed] [Google Scholar]
- 8.Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. J Biol Chem 2010;285:14389–14398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawamori D, Kurpad AJ, Hu J, et al. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 2009;9:350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy AB, Serino AS, Wijesekara N, Chimienti F, Wheeler MB. Regulation of glucagon secretion by zinc: lessons from the β cell-specific Znt8 knockout mouse model. Diabetes Obes Metab 2011;13(Suppl. 1):112–117 [DOI] [PubMed] [Google Scholar]
- 11.Diao J, Allister EM, Koshkin V, et al. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc Natl Acad Sci USA 2008;105:12057–12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millet L, Vidal H, Andreelli F, et al. Increased uncoupling protein-2 and -3 mRNA expression during fasting in obese and lean humans. J Clin Invest 1997;100:2665–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Affourtit C, Jastroch M, Brand MD. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic Biol Med 2011;50:609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Couplan E, del Mar Gonzalez-Barroso M, Alves-Guerra MC, Ricquier D, Goubern M, Bouillaud F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem 2002;277:26268–26275 [DOI] [PubMed] [Google Scholar]
- 15.Bouillaud F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim Biophys Acta 2009;1787:377–383 [DOI] [PubMed] [Google Scholar]
- 16.Arsenijevic D, Onuma H, Pecqueur C, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 2000;26:435–439 [DOI] [PubMed] [Google Scholar]
- 17.Pi J, Bai Y, Daniel KW, et al. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology 2009;150:3040–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robson-Doucette CA, Sultan S, Allister EM, et al. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 2011;60:2710–2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Echtay KS, Roussel D, St-Pierre J, et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002;415:96–99 [DOI] [PubMed] [Google Scholar]
- 20.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007;56:1783–1791 [DOI] [PubMed] [Google Scholar]
- 21.Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PE. K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab 2008;19:277–284 [DOI] [PubMed] [Google Scholar]
- 22.Gromada J, Ma X, Høy M, et al. ATP-sensitive K+ channel-dependent regulation of glucagon release and electrical activity by glucose in wild-type and SUR1-/- mouse alpha-cells. Diabetes 2004;53(Suppl. 3):S181–S189 [DOI] [PubMed] [Google Scholar]
- 23.Braun M, Rorsman P. The glucagon-producing alpha cell: an electrophysiologically exceptional cell. Diabetologia 2010;53:1827–1830 [DOI] [PubMed] [Google Scholar]
- 24.MacDonald PE, De Marinis YZ, Ramracheya R, et al. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 2007;5:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong D, Vong L, Parton LE, et al. Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab 2010;12:545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000;127:2317–2322 [DOI] [PubMed] [Google Scholar]
- 27.Quoix N, Cheng-Xue R, Guiot Y, Herrera PL, Henquin JC, Gilon P. The GluCre-ROSA26EYFP mouse: a new model for easy identification of living pancreatic alpha-cells. FEBS Lett 2007;581:4235–4240 [DOI] [PubMed] [Google Scholar]
- 28.Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000;343:230–238 [DOI] [PubMed] [Google Scholar]
- 29.Kin T, O’Gorman D, Schroeder A, et al. Human islet distribution program for basic research at a single center. Transplant Proc 2011;43:3195–3197 [DOI] [PubMed] [Google Scholar]
- 30.Suzuki Y, Lanner C, Kim JH, et al. Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol 2001;21:2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li LO, Hu YF, Wang L, Mitchell M, Berger A, Coleman RA. Early hepatic insulin resistance in mice: a metabolomics analysis. Mol Endocrinol 2010;24:657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardy AB, Fox JE, Giglou PR, et al. Characterization of Erg K+ channels in alpha- and beta-cells of mouse and human islets. J Biol Chem 2009;284:30441–30452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basford CL, Prentice KJ, Hardy AB, et al. The functional and molecular characterisation of human embryonic stem cell-derived insulin-positive cells compared with adult pancreatic beta cells. Diabetologia 2012;55:358–371 [DOI] [PubMed] [Google Scholar]
- 34.Braun M, Ramracheya R, Bengtsson M, et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010;59:1694–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SC, Robson-Doucette CA, Wheeler MB. Uncoupling protein 2 regulates reactive oxygen species formation in islets and influences susceptibility to diabetogenic action of streptozotocin. J Endocrinol 2009;203:33–43 [DOI] [PubMed] [Google Scholar]
- 36.Conarello SL, Jiang G, Mu J, et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 2007;50:142–150 [DOI] [PubMed] [Google Scholar]
- 37.Zhang CY, Parton LE, Ye CP, et al. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab 2006;3:417–427 [DOI] [PubMed] [Google Scholar]
- 38.Leloup C, Tourrel-Cuzin C, Magnan C, et al. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009;58:673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.González-Vélez V, Dupont G, Gil A, González A, Quesada I. Model for glucagon secretion by pancreatic α-cells. PLoS ONE 2012;7:e32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yabaluri N, Bashyam MD. Hormonal regulation of gluconeogenic gene transcription in the liver. J Biosci 2010;35:473–484 [DOI] [PubMed] [Google Scholar]
- 41.St-Onge MP, Jones PJ. Physiological effects of medium-chain triglycerides: potential agents in the prevention of obesity. J Nutr 2002;132:329–332 [DOI] [PubMed] [Google Scholar]
- 42.Yang J, MacDougall ML, McDowell MT, et al. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: implications on anti-glucagon therapies for diabetes. BMC Genomics 2011;12:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorel F, Damond N, Chera S, et al. Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes 2011;60:2872–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quoix N, Cheng-Xue R, Mattart L, et al. Glucose and pharmacological modulators of ATP-sensitive K+ channels control [Ca2+]c by different mechanisms in isolated mouse alpha-cells. Diabetes 2009;58:412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 2011;13(Suppl. 1):95–105 [DOI] [PubMed] [Google Scholar]
- 46.Martens GA, Cai Y, Hinke S, Stangé G, Van de Casteele M, Pipeleers D. Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J Biol Chem 2005;280:20389–20396 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.