

Abstract
The cell cycle ensures genome maintenance by coordinating the processes of DNA replication and chromosome segregation. Of particular importance is the irreversible transition from the G1 phase of the cell cycle to S phase. This transition marks the switch from preparing chromosomes for replication (“origin licensing”) to active DNA synthesis (“origin firing”). Ubiquitin-mediated proteolysis is essential for restricting DNA replication to only once per cell cycle and is the major mechanism regulating the G1 to S phase transition. Although some changes in protein levels are attributable to regulated mRNA abundance, protein degradation elicits very rapid changes in protein abundance and is critical for the sharp and irreversible transition from one cell cycle stage to the next. Not surprisingly, regulation of the G1-to-S phase transition is perturbed in most cancer cells, and deregulation of key molecular events in G1 and S phase drives not only cell proliferation but also genome instability. In this review we focus on the mechanisms by which E3 ubiquitin ligases control the irreversible transition from G1 to S phase in mammalian cells.
Keywords: cell cycle, E3 ubiquitin ligase, DNA replication, origin licensing, Cdk, cyclin
E3 Ubiquitin Ligases Control Cell Cycle Transitions
Changes in protein abundance are critical for transitions in the cell division cycle. Cell cycle changes at both the protein and transcript levels have been analyzed globally and reveal that the proteome is even more dynamic than the transcriptome.1 Such findings highlight the importance of ubiquitin-mediated protein degradation for properly-regulated cell cycle progression. Ubiquitination control is lost or perturbed in many cancers through the amplification or inactivation of key E3 ubiquitin ligases.2-5 These perturbations often impact the critical transition from G1 phase to S phase of the cell cycle. Replication licensing occurs during the G1 phase of the cell cycle at thousands of sites throughout the human genome known as origins of replication. These origins are the genomic loci where DNA replication is initiated during S phase. Replication licensing is accomplished by the assembly of prereplication complexes or “preRCs.” Licensing is complete once MCM complexes have been loaded onto origin DNA by ORC, Cdc6, and Cdt1 (Figure 1). Replication origins are first recognized and bound by the origin recognition complex (ORC) that remains associated with origins throughout the cell cycle. ORC is composed of constitutively expressed Orc2-6 and cell cycle regulated Orc1 (reviewed in DePamphilis6). Cdc6 is then recruited to origins and interacts directly with ORC. Cdt1 interacts with the MCM complex in the nucleus during G1 through direct binding to the Mcm6 subunit.7-10 Cdt1 and MCM complexes are then recruited to origins through direct interaction of Cdt1 with both Cdc69 and Orc6.11 Cdt1 is absolutely required for MCM recruitment but lacks any enzymatic activity. It is through the ATPase activity of ORC and Cdc6 that MCM helicases are finally loaded onto origin DNA.
Figure 1.

DNA replication origin licensing control in G1 and S phase. Origins are licensed by the DNA loading of MCM complexes in late G1. During early G1, MCM loading is blocked through the APCCdh1-mediated destruction of Cdc6. Cdc6 binding to APCCdh1 is blocked in late G1 by Cyclin E/Cdk2-mediated phosphorylation (Ser54), whereas the licensing inhibitor geminin is ubiquitinated by APCCdh1 throughout G1 phase. In late G1, MCM complexes are loaded at origins by the combined action of ORC/ORCA, Cdc6, and Cdt1 with contributions from the PR-Set7 and Hbo1 enzymes that may function through modification of histones at origins and/or nonhistone proteins. Once cells enter S phase, Cdc6 is phosphorylated at Ser106 phosphorylation, which promotes Cdc6 nuclear export. Three overlapping mechanisms suppress Cdt1 activity during S phase: (1) geminin accumulates and prevents Cdt1 from binding MCM. (2) Cdt1 is phosphorylated by Cdk2, and this phosphorylation induces Cdt1 ubiquitination by SCFSkp2. (3) Cdt1 and PR-Set7 both associate with DNA-loaded PCNA at replication forks, and PCNA interaction triggers ubiquitination by CRL4Cdt2.
Once MCM complexes have been loaded, the origins are licensed for replication, although MCM is not yet active as a DNA helicase in G1 because activation requires phosphorylation events mediated by S phase protein kinases. After MCM loading, however, ORC, Cdc6, and Cdt1 are no longer required for DNA synthesis in S phase.12,13 Origin licensing is tightly regulated to prevent MCM loading outside of G1 phase because new licensing during S and G2 phases can lead to rereplication that ultimately promotes genome instability. The consequences of rereplication include gene amplification and aneuploidy that drive oncogenesis.14 Therefore, it is imperative that cells prevent rereplication. Origin licensing is restricted to G1 primarily through ubiquitin-mediated proteolysis of licensing components and cell cycle regulators.
Ubiquitin-mediated proteolysis by the 26S proteasome is initiated through polyubiquitination of substrate proteins. Polyubiquitination is accomplished through the activities of E1, E2, and E3 enzymes, and substrate specificity is determined by the E3 ubiquitin ligases (reviewed in Wilkinson15 and Pickart16). The 3 most important E3 ubiquitin ligase complexes that regulate the G1/S transition in mammalian cells are the anaphase promoting complex/cyclosome (APC/C), Skp1-Cul1-F-box protein complex (SCF), and Cul4-RING-E3 Ligase 4 (CRL4) (Figure 2). These complexes are members of the largest single class of RING E3 ubiquitin ligases, the cullin-RING ligase (CRL) superfamily (reviewed in Jackson & Xiong17 and Zimmerman et al.18). The basic components of the CRL E3 complexes are a cullin protein, a small RING-containing protein (such as Roc1, Rbx1, etc.), and an adaptor protein that confers substrate specificity. The human genome encodes 9 cullins (Cul1-3, 4A/B, 5-7, PARC, and Apc2) that function as scaffolds to assemble ubiquitin ligase complexes. The RING-containing proteins are required to recruit the activated E2 enzyme to the E3 ligase complex. There are a very large number of adaptor proteins that function with specific CRL E3 ligases and provide additional layers of regulation.
Figure 2.

Three E3 ubiquitin ligases, APCCdh1, SCFSkp2, and CRL4Cdt2, at the G1/S transition. (A) APCCdh1 is active until late G1 but inactivated in late G1 and S phase by a combination of CDK-mediated phosphorylation of the Cdh1 subunit, degradation of the E2 enzyme UbcH10, and inhibition by the pseudosubstrate Emi1 protein that accumulates after S phase onset. (B) SCFSkp2 is inactive during early G1 because APCCdh1 targets Skp2 for degradation. CDK-mediated phosphorylation of Skp2 protects it from APCCdh1 in late G1. (C) CRL4Cdt2 is inactive during G1 because its substrates cannot be recognized by the Cdt2 substrate receptor unless they are first bound to DNA-loaded PCNA at replication forks during S phase.
The APC/C complex is active not only during the second half of mitosis (from anaphase on) but also during G1. APC/C contains the cullin protein Apc2, the Apc11 RING protein, either the Cdh1 (during G1) or Cdc20 (during G2/M) adaptor protein, and 11 other proteins whose exact functions remain unknown (reviewed in Nakayama & Nakayama19). The adaptor proteins mediate substrate interactions although some core APC/C proteins (Apc10 and Apc5) can also interact with substrates.20-23 Nearly all APC/C targets contain at least 1 peptide sequence known as a “destruction motif” or “degron” that mediates interaction with the adapter proteins.24-27 The 2 most common motifs are the destruction box (D-box) and the KEN-box, although there are several others that have been more recently identified (reviewed in McLean et al.28). Protein-protein interactions between substrates and the APC/C can be disrupted upon substrate phosphorylation, thereby protecting substrates from proteasomal degradation (reviewed in Song & Rape29).
Our focus here is on APCCdh1 since it functions primarily during telophase and the G1 phase of the cell cycle as opposed to APCCdc20 that functions in anaphase. Cdh1 protein levels remain constant throughout the cell cycle, but Cdh1 activity is regulated by phosphorylation.30-33 During early to mid-G1, Cdh1 is dephosphorylated and APCCdh1 is active, but as cells reach the G1/S transition, Cdh1 is phosphorylated by Cyclin-dependent kinases (CDKs) and this phosphorylation disrupts its interaction with the APC/C. Additionally, E2F transcription factor activity during late G1 induces accumulation of the APC/C pseudosubstrate, Emi1, which binds Cdh1 in S phase and thereby competitively inhibits APCCdh1 activity.34-36 Moreover, the two E2 enzymes that interact with the APC/C, UbcH10 and UbcH5, are degraded during S phase, and this downregulation further suppresses APC/C activity.37-39 Although loss of Cdh1 does not prevent cell cycle progression, it does result in a shorter G1 phase with impaired origin licensing followed by a prolonged and defective S phase with accumulation of DNA damage.40-42
In contrast to APC/C, the SCFSkp2 ubiquitin ligase complex becomes active later at the G1/S transition and remains active throughout S phase. The SCF complex is composed of only 4 subunits: Cul1 (cullin scaffold), Roc1 (RING-domain protein), Skp1 (adaptor protein), and one of many F-box protein substrate adaptors. Skp1 associates with many different F-box proteins that confer substrate specificity. During G1, APCCdh1 inhibits SCFSkp2 activity through ubiquitination and degradation of Skp2; this degradation is dependent on the N-terminal D-box of Skp2.43,44 Later in G1, however, Skp2 is phosphorylated at Ser64/72 by Cyclin E-Cdk2, and Skp2 continues to be phosphorylated, but by Cyclin A-Cdk2 in S phase. Phosphorylation at Ser64, and to a lesser extent at Ser72, prevents Skp2 ubiquitination by APCCdh1.43-45 Once active, SCFSkp2 promotes S phase progression and contributes to the inhibition of origin licensing during S phase (described below).
There are 69 F-box proteins encoded in the human genome, but substrates have been elucidated for fewer than 20%.46 For substrates that have been identified, substrate phosphorylation is typically essential for direct interaction with the F-box protein and thus the SCF enzyme. Phosphorylation creates a negatively charged site on the substrate that docks with a positively charged surface on the F-box protein (e.g., Skp2).47,48 A notable exception to this requirement is p21, which does not require phosphorylation for interaction with Skp2. For efficient ubiquitination of the p21 and p27 CDK inhibitors, the SCFSkp2 complex instead requires the cofactor Cks1.49-51 Cks1 performs a docking function to facilitate the interaction between Cyclin-CDKs and their substrates. In addition, Cks1 promotes interactions between Cdk2-bound, phosphorylated CKIs and Skp2.47 There are two CKS (Cyclin-dependent kinase subunit) paralogs in mammals, Cks1 and Cks2, which tightly associate with Cyclin-CDK complexes, compete for Skp2 binding, and are essential for cell viability.52,53 Only Cks1 can promote substrate interaction with Skp2.52 It is clear that SCFSkp2 requires Cks1 association for a subset of its substrates, but the p21 and p27 CDK inhibitors are the only ones identified in this group thus far; others may yet be discovered (reviewed in Krishnan et al.54). It has been suggested that Cks1 is also a target of APCCdh1 during G1.44 Cks1 possesses a C-terminal D-box that is required for its degradation, although direct ubiquitination by APCChd1 in vitro has not been reported.
The most recently characterized E3 ubiquitin ligase required for proper cell cycle progression is CRL4Cdt2. This complex is composed of Cul4, Roc1/2, an adaptor protein DDB1, and a substrate receptor or DCAF (DDB1- and Cul4-associated factor) that interacts directly with DDB1. There are at least 20 known DCAFs encoded in the human genome (reviewed in Havens & Walter55). We focus primarily on the CRL4Cdt2 complex in this review because it has the highest relevance for the G1/S transition. The DCAF Cdt2 (Cdc10-dependent transcript 2) is unique among DCAFs in that its interaction with substrates is dependent on the substrate first interacting with DNA-loaded PCNA. PCNA is a processivity factor for DNA polymerase δ that is loaded at replication forks during S phase and during the DNA synthesis steps of DNA repair. Proteins that interact with PCNA contain a motif known as a PCNA-interacting protein box (PIP box). CRL4Cdt2 substrates contain a specialized PIP box known as the PIP degron. The PIP degron differs from the canonical PIP box in 2 ways. First, a TD motif is present within the PIP box that strengthens the interaction with PCNA.56-59 Second, the PIP degron contains a conserved basic residue 4 amino acids downstream from the PIP box that is required for interaction with Cdt2.56,59,60 All bona fide substrates of CRL4Cdt2 contain this PIP degron. Interestingly, PCNA must be loaded onto DNA by the RFC complex to promote interaction between the substrate and Cdt2, although the basis for substrates to distinguish soluble PCNA from DNA-loaded PCNA is not yet clear.59,61 Through the requirement for substrate interaction with PCNA, CRL4Cdt2-mediated proteolysis is coupled to DNA replication during S phase.
DNA Replication Origin Licensing Regulation before the G1/S Transition
Geminin and Cdt1
During S phase and G2, Cdt1 is bound by its inhibitor, geminin.62,63 Structural studies of mouse geminin and Cdt1 found that a dimer of geminin binds to Cdt1.64 The human geminin-Cdt1 complex was also crystallized, and interestingly this study identified not only the trimeric form but also that the complex could dimerize to form a heterohexamer. Further analysis suggested that the trimeric geminin/Cdt1 complex may be permissive for origin licensing, whereas the heterohexameric complex is inhibitory for origin licensing due to the burial of Cdt1 residues required for licensing.65,66 More recently, Mcm9 was shown to interact with Cdt1, and this interaction prevents formation of the inhibitory geminin/Cdt1 heterohexamer to promote origin licensing.67 The molecular mechanism controlling the apparent switch between licensing-competent and licensing-incompetent Cdt1 is still not fully understood (reviewed in Caillat & Perrakis68). During G1, APCCdh1 targets geminin for degradation, allowing Cdt1 to recruit MCM complexes to origins. Geminin interacts with the Cdh1 adaptor protein through a D-box at its N-terminus.69 Overexpression of a D-box mutant, nondegradable form of geminin in primary fibroblasts blocks chromatin-association of MCM complexes and inhibits origin licensing.70 Thus, low levels of geminin are required for efficient licensing during late G1.
In addition to geminin degradation, another Cdt1 regulatory mechanism in G1 has been suggested. Cdt1 was shown to interact with APCCdh1 in a proteomics study, and cdh1 overexpression decreased Cdt1 levels. However, mutation of the D-boxes that were proposed to mediate this interaction also disrupted interaction of Cdt1 with SCFSkp2 (discussed in a later section), making it difficult to interpret those results.71 Further, in vitro ubiquitination assays demonstrated that geminin was a much stronger APCCdh1 substrate than Cdt1. The authors suggested that Cdt1 degradation by the APCCdh1 is required to maintain cells in quiescence although it could also be important for balancing Cdt1 levels during G1. It seems counterintuitive that APCCdh1 would both inhibit (through Cdt1 degradation) and promote (through geminin degradation) Cdt1 activity during G1. Additional studies will be necessary to determine the importance of the reported APCCdh1-Cdt1 interaction. Another Cdt1 interacting protein, ORCA (also known as LRWD1), is also required for origin licensing and progression through G1 phase.72 ORCA is required at many origins to stabilize the chromatin association of ORC. The ORCA protein is most abundant during G1, and levels decrease as cells transition into S phase.73 ORCA associates with CRL4DDB1, and polyubiquitinated ORCA was detected at the G1/S transition. ORCA binds to Cdt1 and Orc1 during G1 phase and Orc2 throughout the cell cycle.72,73 Only nonubiquitinated ORCA associates specifically with the Orc2 subunit of ORC, and this binding may protect ORCA from ubiquitination.72
Cdc6
APCCdh1 targets not only geminin but also Cdc6 for degradation during early G1.74 Cdh1 binding to Cdc6 is dependent on both a KEN-box and a D-box found at the N-terminus of Cdc6. APCCdh1 prevents Cdc6 accumulation until late in G1, when Cdc6 is phosphorylated at Ser54 by Cyclin E-Cdk2. This phosphorylation protects Cdc6 from APCCdh1-mediated degradation by disrupting the interaction between Cdc6 and Cdh1.75 Additionally, APCCdh1 activity is inhibited late in G1 (as previously described) to allow cell cycle progression into S phase. Importantly, Cdc6 is stabilized, but geminin is not. Thus, in late G1, ORC is already bound to origins, Cdt1 is actively recruiting MCM complexes to origins, and now that Cdc6 is present, MCM loading can be completed. Cdc6, Cdt1, ORC, and MCM complexes are the essential replication licensing proteins and, together with nucleoplasmin, are fully sufficient for licensing Xenopus laevis sperm chromatin in vitro. 76
PR-Set7 (Set8)
The requirement for nucleoplasmin in the reconstituted licensing reaction with purified proteins has been attributed to a need to decondense chromatin. This notion underscores the importance of chromatin structure near origins in controlling the efficiency of both origin licensing in G1 and origin firing in S phase. Recently, the methyltransferase PR-Set7 (also known as Set8) has emerged as a new critical player in origin licensing. PR-Set7 is the sole enzyme responsible for monomethylation of histone H4 at lysine 20 (H4K20me1), and both PR-Set7 and H4K20me1 are tightly cell cycle- regulated. Importantly, H4K20me1 is enriched at origins of replication in Drosophila. 77 Several lines of evidence support a role for PR-Set7 in origin licensing. First, PR-Set7 knockout in mice results in embryonic lethality.78 Second, artificially tethering PR-Set7 to a genomic locus is sufficient to promote the chromatin-association of ORC and MCM.79 Third, knockdown of PR-Set7 results in decreased chromatin association of ORC, Cdc6, and MCM complexes.79 Additionally, L3MBTL1, a human homolog of the Drosophila polycomb tumor suppressor, binds H4K20me180 and interacts with components of the replication machinery (Cdc45, PCNA, and MCM), and such interactions may facilitate replication protein recruitment to chromatin.81
As with many origin licensing factors, PR-Set7 abundance is regulated both transcriptionally and posttranslationally. pr-set7 gene expression fluctuates throughout the cell cycle, peaking in mitosis. PR-Set7 protein levels drop during S phase even when pr-set7 is expressed from a constitutive promoter,82 and ubiquitin-mediated proteolysis by both APCCdh1 and CRL4Cdt2 is largely responsible for regulating PR-Set7 protein abundance.83 During mitosis, PR-Set7 is phosphorylated at Ser29 by Cyclin B-Cdk1 preventing PR-Set7 ubiquitination by APCCdh1. During early anaphase, however, PR-Set7 is dephosphorylated by the Cdc14 phosphatases resulting in APCCdh1-mediated proteolysis and reduced protein levels during early G1.84 As a result, H4K20me1 levels peak in G2/M and are dramatically reduced during mid- to late G1 and remain low during S phase.
The absence of the PR-Set7 methyltransferase can only result in corresponding changes in histone methylation if histone H4K20 monomethylation is simultaneously converted either to unmethylated K20 or to di- or trimethylated H4K20 (or both). H4K20me1 could promote origin licensing in G1 by serving as the template for di- and trimethylation by both Suv4-20h1 and 2 methyltransferases.85-87 Interestingly, the ORCA protein can specifically bind to trimethylated H4K20.73,88 The decrease in H4K20me1 during S phase could thus be due in part to the transition to these higher methylation states, although these marks have not yet been specifically examined at replication origins. Loss of both Suv4-20h1 and Suv4-20h2 causes defects in S phase entry, but the effects are less severe than loss of PR-Set7, suggesting additional roles for PR-Set7 in the G1/S transition.78,86 One such role could be the methylation of an as yet unidentified nonhistone protein required for progression from G1 to S phase. Currently, the only nonhistone substrate of PR-Set7 that has been identified is p53.89 However, it is unlikely that p53 methylation is fully responsible for the replication defects caused by PR-Set7 depletion because these phenotypes are not entirely p53 dependent, and p53 null cells are viable.77,90,91 The mechanism by which PR-Set7 promotes cell cycle progression is therefore still unclear.
E2F and the mixed-lineage leukemia methyltransferase
The proteins responsible for origin licensing are clearly regulated by ubiquitin-mediated proteolysis, but they are also the products of transcriptionally regulated genes. The transcription factors controlling these genes are themselves also regulated by ubiquitin-mediated proteolysis. The most important class of transcription factors involved in progression from G1 to S phase is the E2F family. E2F proteins play key roles in cell cycle progression. They are often categorized as activator (E2F1-3) or repressor (E2F4-8) E2Fs, although members of either group can both activate and repress gene transcription depending on the circumstances (reviewed in Wong et al.92). The activator E2Fs are functionally redundant but differ in regulatory sequences that control their spatiotemporal expression.93 In early G1 cells, repressor E2Fs are exchanged with activator E2Fs on target gene promoters, but transcription remains inhibited through the E2F-Rb interaction until later in G1.94,95 In this manner, E2F-dependent genes are negatively regulated through interaction of activator E2Fs with hypophosphorylated Rb (and Rb-related proteins), but this interaction is disrupted upon Rb phosphorylation first by Cyclin D-Cdk4 and later by Cyclin E-Cdk2 (reviewed in Henley & Dick,96 Nevins,97 and Lundberg & Weinberg98). The activator E2Fs promote G1 progression by inducing the expression of genes encoding replication licensing factors and cell cycle regulators such as Cdc6, geminin, PCNA, Skp2, and Cyclins E and A as well as the activator E2Fs themselves.96,99-101 Once transcriptional targets of E2Fs have been activated, cells are committed to completing the cell cycle, and this commitment reflects passage through what is known as the “restriction point.”
Tight control over E2F activity is required to prevent aberrant DNA replication. Specifically, overexpression of e2f1 or inactivation of Rb (as seen in nearly all human cancers) promotes uncontrolled DNA replication, resulting in genome instability in later cell cycles.102,103 E2Fs are regulated at the transcriptional, translational, and posttranslational levels (reviewed in Wong et al.92). Recently, several E2Fs were identified as targets of APCCdh1. The activating E2Fs, as well as the repressor E2F4, have been shown to interact with Cdh1 in vitro and were destabilized upon overexpression of cdh1. 104,105 The interaction between Cdh1 and E2F1 was dependent on the C-terminal 79 amino acid residues of E2F1, which are highly conserved among all the E2F proteins. Previous work identifying E2F1 as a target of APCCdc20 in mitosis also confirmed the interaction between E2F1 and Cdh1.106 Interestingly, this interaction was not dependent on the two D-box motifs present in E2F1 or the four present in E2F3.105,106 E2F1 does, however, interact with the core APC protein Apc5 that, together with Cdh1, could constitute a bipartite degron. Further work is needed to determine precisely how these E2Fs are recognized by APCCdh1. Nevertheless, it is clear that E2F degradation by APCCdh1 is an important mechanism regulating E2F activity during G1.
Recent studies have described an MLL-E2F axis that promotes transcription of E2F target genes, particularly cyclin D and cyclin E. 107 The mixed-lineage leukemia (MLL) family of methyltransferases is responsible for histone H3 lysine 4 (H3K4) methylation, and this mark promotes active gene transcription.108,109 E2F1 recruits MLL to E2F target gene promoters during G1, leading to a corresponding increase in H3K4 trimethylation.110 Knockdown of Mll impaired proliferation nearly 2-fold and in synchronized HeLa cells delayed S phase entry, indicating that MLL is required for proper G1 progression.111 Together, the E2F transcription factors and MLL stimulate progression through G1 phase by activating transcription of genes encoding the replication machinery and the cyclins required to promote cell cycle progression. Another chromatin-modifier, the acetyltransferase Hbo1, is also required for cell cycle progression. Hbo1 interacts with Cdt1 at origins of replication and, like Cdt1, is inhibited by geminin. Hbo1 acetylates histone H4 and this acetyltransferase activity is required for MCM loading in G1.112-114 The importance of chromatin modification at origins to promote DNA replication is becoming increasingly clear, yet we still lack a complete understanding of how enzymes like MLL, PR-Set7, and Hbo1 function together to regulate the G1/S transition.
Cyclin-CDK Regulation before the G1/S Transition
Cyclin D
Cyclin D1 is the first cyclin expressed during G1 in response to mitogenic signals and peaks immediately before the G1-S transition. It interacts with both Cdk4 and Cdk6 to form active Cyclin-CDK complexes that phosphorylate Rb to relieve repression of E2F-dependent genes, including cyclin E, cyclin A, cdc6, orc1, and pola1 (DNA polymerase alpha), to promote progression through G1 (reviewed in Wells et al.,94 Takahashi et al.,95 Henley & Dick,96 Sherr & Roberts,115 and Johnson & Schneider-Broussard116). Tob1, a member of the BTG/TOB family of antiproliferative proteins, represses cyclin D1 expression. Tob1 is phosphorylated by Erk1/Erk2 kinases after growth factor stimulation, and this phosphorylation relieves the transcriptional repression of cyclin D1. 117 In combination with a host of other growth factor regulated transcription factors, including SRF and AP-1, the cyclin D1 gene is upregulated in early G1. More recently, Tob1 was shown to be a substrate of SCFSkp2 although, like p21, phosphorylation of Tob1 was not required for and had no effect on its interaction with Skp2.118 Inactivation and degradation of Tob1 ensure proper expression of cyclin D1 mRNA during G1. Spontaneous tumors develop in Tob1–/– mice, and decreased tob1 expression has been documented in various cancer types.119 Tob1 reduction correlates well with the high level of Cyclin D1 seen in many cancers that contributes to enhanced cell proliferation through E2F activation.120-124
Cyclin E
Cyclin E transcription is activated by E2F, and Cyclin E-Cdk2 activity in turn promotes E2F activity in a positive feedback loop. Cyclin E-Cdk2 phosphorylates Rb, causing its dissociation from E2F and allowing transcription of E2F target genes (reviewed in Wong et al.92 and Henley & Dick96). During early G1, however, the CDK inhibitors p21 and p27 are present and inhibit Cyclin E-Cdk2 activity. The switch from the inactive, CKI-bound state to an active state later in G1 is accomplished through several mechanisms. First, increased transcription of cyclin E generates more Cyclin E protein, making it more difficult for the existing p27 to inhibit Cyclin E/Cdk2 complexes. Second, p27 phosphorylation by low levels of active CDK can promote its cytoplasmic relocalization or disrupt its association with Cyclin E-Cdk2. Finally, p21 and p27 can be targeted for proteasomal degradation by both SCFSkp2 and CRL4 E3 ubiquitin ligases (discussed in the next section). Eliminating both p21 and p27 allows the full activation of Cdk2 complexes in late G1. As mentioned previously, Cyclin E-Cdk2 (as well as Cyclin A-Cdk2) phosphorylates Cdc6 at Ser54 to prevent its degradation by APCCdh1.75 This Cdc6 stabilization allows a window of opportunity in which origin licensing can take place (Figure 1).
CKIs: p21 and p27
High CDK activity is required for progression from G1 to S phase, and one critical CDK-mediated event is the recruitment of MCM activating subunits to stimulate origin unwinding. Destruction of the CKIs p21 and p27 prior to the G1/S transition is required for full CDK activity (Figure 3). P21 and p27 are regulated posttranslationally, in part, through ubiquitination by the SCFSkp2 E3 ubiquitin ligase complex. This SCFSkp2-mediated ubiquitination requires that p21 and p27 be in a complex with Cyclin-CDK.125,126 Phosphorylation of p27 by Lyn or BCR/ABL kinases at Tyr88 ejects the inhibitory domain of p27 from the Cdk2 active site.127,128 The now-active Cyclin E-Cdk2 can then phosphorylate p27 at Thr187 to stimulate its interaction with the SCFSkp2-Cks1 E3 ubiquitin ligase.47,126 Cyclin E-Cdk2 also phosphorylates p21 at Ser130 to stimulate its interaction with SCFSkp2-Cks1.51,129 Although p21 and p27 both require association with Cks1 and Cyclin A/E-Cdk2 for proper SCFSkp2-mediated degradation, they differ in that phosphorylation of p21 is not strictly required for proteolytic degradation as it is for p27. In fact, Ser130 phosphorylation of p21 by mitogen-activated protein kinases (MAPKs) has been shown to stabilize p21.130 Although it has not been tested, it is possible that Ser130 phosphorylation strengthens the interaction between p21 and Cyclin A/E-Cdk2 that is absolutely required for proper degradation of p21 by SCFSkp2-Cks1.
Figure 3.
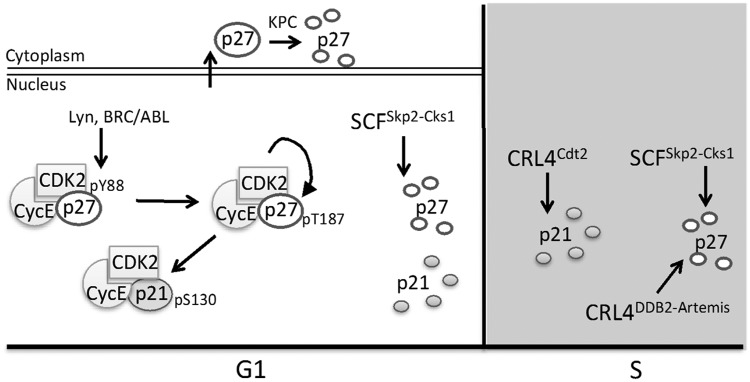
CDK inhibitor control in G1 and S phase. During G1, CDK activity is lowest due to high levels of p21 and p27. Lyn and BRC/ABL kinases phosphorylate p27 in late G1, relieving its inhibition of Cyclin E-Cdk2 without disrupting CDK association. The active Cyclin E-Cdk2 then phosphorylates both p27 and p21, promoting their interaction with and ubiquitination by SCFSkp2-Cks1. High CDK activity is maintained during S phase through ubiquitination of p27 and p21 by SCFSkp2-Cks1, p27 ubiquitination by CRL4DDB2-Artemis, and p21 ubiquitination by CRL4Cdt2.
As previously mentioned, Cks1 is required for degradation of both p21 and p27 by SCFSkp2. Cks1 promotes the stable interaction between Skp2 and phosphorylated p27.49,50,131 Cks1 also promotes the phosphorylation-independent interaction between p21 and Skp2.51 Knockout of Cks1 prevented p27 degradation and resulted in its accumulation and impaired S phase entry.132 The Cks1 paralog Cks2 lacks the Skp2 interaction domain and competes with Cks1 for p27 binding. In contrast to Cks1 knockout, Cks2–/– MEF cells showed increased p27 degradation and consequently high Cyclin A-Cdk2 activity, increased origin firing, and faster cell cycle progression.52 These differences suggest that a balance between Cks1 and Cks2 is required for proper regulation of p27.
In addition to changes in stability, free p27 (not bound to Cyclin-Cdk2) is exported from the nucleus during G1 through interaction with Cyclin D2-Cdk4/ 6. Additionally, p27 phosphorylation by PI3K/Akt and Ras signaling pathways (induced by growth factors) promotes its cytoplasmic relocalization.133-135 Cytoplasmic p27 can be targeted by the Kip1 ubiquitylation-promoting complex (KPC).136-138 Although KPC inactivation has little effect on cell cycle progression, when combined with Skp2 downregulation cells cannot enter S phase from quiescence.136
Summary of G1 events
By the end of G1, origins have been licensed and are ready for replication initiation during S phase. Once MCM complexes have been loaded at origins, the MCM loading factors Cdc6, Cdt1, and ORC are no longer required for cell cycle progression. APCCdh1 is inactive and has allowed Skp2 to accumulate and interact with the SCF E3 ubiquitin ligase. Another consequence of inactive APCCdh1 is accumulation of geminin that prevents additional MCM loading at S phase onset through direct Cdt1 inhibition. Finally, Cyclin E-Cdk2 is active and promotes the transition into S phase. At this point, the business of G1 phase is complete and cells are poised to initiate DNA replication through phosphorylation-mediated recruitment and activation of MCM helicase activators, DNA polymerases (α, δ), and processivity factors (PCNA, RFC, etc.).
DNA Replication Origin Licensing Regulation after the G1/S Transition
As cells enter S phase, it is imperative that origin licensing is inhibited. Once a given origin has fired and bidirectional replication forks have already been established, relicensing of that same origin could be followed by a refiring or “rereplication” event.139 Rereplication is an aberrant phenomenon (unlike endoreduplication), and rereplication ultimately results in double-strand DNA breaks.140,141 Most recently, rereplication has also been established as a direct cause of gene amplification.14,142 The induction of both DNA damage and gene amplification drives genome instability and may be one underlying cause of oncogenesis. In normal cells, however, several overlapping mechanisms suppress rereplication, and these are discussed below.
Geminin and Cdt1
Arguably the most important mechanism to prevent rereplication during S phase is the simultaneous degradation and inhibition of Cdt1. Cdt1 is a substrate of both CRL4Cdt2 and SCFSkp2 during S phase.143 CRL4Cdt2 targets Cdt1 for degradation only when Cdt1 is bound to PCNA loaded onto DNA at replication forks. Since PCNA is DNA loaded both in S phase and after DNA damage, Cdt1 ubiquitination by CRL4Cdt2 can be induced outside of S phase by exogenous DNA damage.144-147 Cdt1 contains a well-conserved PIP degron at its N-terminus through which it interacts with both DNA-loaded PCNA and Cdt2.59,148-150 Overproduction of Cdt1 or expression of Cdt1 lacking a PIP degron promotes rereplication in many experimental systems.148,149,151-155 Removal of Cdt1 from chromatin after ubiquitination by CRL4Cdt2 is partially dependent on the activity of the p97 ATPase and its cofactor UFD1 (which possesses a ubiquitin-binding domain). Depletion of p97 in HeLa cells resulted in stabilization of Cdt1 in S phase and after DNA damage.156
During S phase, SCFSkp2 can also ubiquitinate Cdt1 to promote its degradation. Cdt1 interaction with Skp2 is dependent upon a Cy-motif that mediates interaction with and phosphorylation by Cyclin E/A-Cdk2.143 Cdk2 can phosphorylate human Cdt1 at Thr29 to stimulate ubiquitination by SCFSkp2.153 Interestingly, the same site was recently shown to be phosphorylated by JNK after cellular stress, although JNK-mediated phosphorylation had no effect on Skp2 binding or Cdt1 stability.157 It is not yet clear how Skp2 binding and Cdt1 degradation were blocked when Thr29 was phosphorylated by JNK, but several other JNK-mediated phosphorylation sites in Cdt1 that block targeting by CRL4Cdt2 likely have a much stronger effect on overall Cdt1 stability than the T29 site.158 For instance, five phosphorylation sites in the C-terminal one third of human Cdt1 were recently mapped that confer resistance to CRL4Cdt2 despite being quite distant from the PCNA and Cdt2 binding sites at the N-terminus of Cdt1.158 Both the JNK and p38 stress MAP kinases phosphorylate these amino acids not only in response to exogenous stress but also during G2 and M phases. Stabilization of Cdt1 in G2 sets the stage for Cdt1’s second cell cycle function in establishing robust microtubule-kinetochore attachments.159
Degradation of Cdt1 can be prevented not only by MAPK-mediated phosphorylation but also by acetylation at lysines 24 and 29 by p300 and PCAF in asynchronously dividing 293T cells.160 These acetylations were detected during early G1. During S phase, however, HDAC11 associated with and deacetylated Cdt1, promoting Cdt1 degradation.160 It is unclear which E3 ubiquitin ligase Cdt1 is protected from by acetylation, but the proximity of Lys24 to Thr29 suggests that it could interfere with Skp2 binding, although this has not yet been tested, and the PIP degron that directs CRL4Cdt2 binding is also relatively close to Thr29 (amino acids 3-14). Further work is needed to fully understand the importance of Cdt1 acetylation/deacetylation during G1 and S phases.
Finally, Cdt1 inhibition is accomplished through the reaccumulation of geminin after APCCdh1 inactivation as cells transition from G1 to S phase and APC/C is inactivated. Geminin prevents the association of Cdt1 with the MCM complex and with Cdc6.9,10 Additionally, geminin accumulation disrupts the ORCA-Cdt1 interaction at origins.161 Like Cdt1, ORCA is degraded during S phase, so its downregulation may also contribute to origin licensing inhibition. ORCA interacts with CRL4 and is polyubiquitinated during S phase; inhibiting CRL4 and DDB1 had no effect on ORCA stability, however, suggesting that multiple E3s could target ORCA for degradation.161
Cdc6
Although many mechanisms are in place to prevent aberrant Cdt1 accumulation during S phase, there are also safeguards to inhibit other origin licensing factors. As cells exit G1, APCCdh1 is inactivated and no longer targets Cdc6 for proteasomal degradation (see above). As a consequence, Cdc6 protein levels are paradoxically much higher in S phase and G2 when licensing is blocked than during G1 when licensing should occur. Nevertheless, during S phase, Cdc6 is exported from the nucleus through a 2-step mechanism. First, the acetyltransferase Gcn5 acetylates Cdc6 at 3 lysine residues (92, 105, and 109).162 These acetylation events are required for subsequent Cdc6 phosphorylation at Ser106 by Cyclin A-Cdk2, which is active because the p21 and p27 inhibitors are degraded in S phase (see below). Ser106 phosphorylation promotes CRM1-dependent nuclear export of Cdc6, and, of course, cytoplasmic Cdc6 cannot participate in origin licensing.163,164 Cdc6 persists in the cytoplasm and is eventually degraded by APCCdh1 in the next G1 phase.
PR-Set7
PR-Set7 is required for proper S phase progression and origin firing. Similar to Cdt1, PR-Set7 levels sharply decrease during S phase.84 Treatment with proteasome inhibitors to block degradation revealed that PR-Set7 associates with PCNA at replication foci.90 Moreover, PR-Set7 has recently been shown to be a target of the CRL4Cdt2 ubiquitin ligase that relies on PCNA as a cofactor for substrate recognition.82,165,166 PR-Set7 has 2 PIP boxes, but only one functions as the PIP degron.55,82,165 Just as with Cdt1, removal of polyubiquitinated PR-Set7 from chromatin is dependent upon p97/UFD1.156 Inhibition of CRL4Cdt2 or deletion of the PR-Set7 PIP degron results in aberrant PR-Set7 accumulation during S phase and rereplication that is dependent on the catalytic activity of PR-Set7.79,165
E2F and the mixed-lineage leukemia methyltransferase
As cells transition from G1 to S phase, both E2F1 and MLL are targeted for degradation by SCFSkp2 through Skp2 binding to their N-termini.111,167 Degradation of E2F1 is presumed necessary since prolonged E2F1 activity leads to induction of apoptosis.168-170 Additionally, E2F1 degradation prevents aberrant expression of cyclin E and genes encoding licensing proteins that could promote rereplication (reviewed in Wong et al.92). Similarly, transient overexpression of MLL in HEK293T cells inhibits S phase progression.111 The underlying mechanism of S phase inhibition by overexpressing MLL has not yet been determined. A clue to the mechanism comes from the effects of DNA damage. After DNA damage, MLL is protected from Skp2-mediated degradation and delays S phase by trimethylating H3K4 at late replicating origins.171 It may be that high levels of MLL during S phase could have similar effects in normal cell cycles and that this inhibition of origin firing explains the need for MLL downregulation during normal S phase.
Cyclin-CDK Regulation after the G1/S Transition
Cyclin D1
As cells reach the G1/S transition, Cyclin D1-Cdk4/6 is inactivated through proteasome-mediated degradation of Cyclin D1. This degradation is necessary for proper S phase progression in part because overexpressed Cyclin D1 can bind PCNA and prevent DNA replication.172,173 Phosphorylation of Cyclin D1 at Thr286 is required for its nuclear export and subsequent cytoplasmic ubiquitination and proteolysis; mutation of Thr286 to alanine therefore results in nuclear accumulation of Cyclin D1.174,175 ATM and ATR kinases are responsible for Cyclin D1 Thr286 phosphorylation during S phase.176 Interestingly, nuclear accumulation of active Cyclin D1/Cdk4 indirectly promotes Cdt1 stabilization in S phase through transcriptional repression of Cul4 by an unknown mechanism.177
The SCF E3 ubiquitin ligase has been implicated in targeting phosphorylated Cyclin D1 for proteolysis at the G1/S transition. Studies conducted in various human cancer cell lines identified multiple F-box proteins that promote Cyclin D1 ubiquitination by the SCF complex including Fbx4,178,179 Fbxw8,180 and Fbxo31.181 Downregulation of Fbx4 promotes the accumulation of phosphorylated Cyclin D1 in the nucleus, and this accumulation facilitates oncogenic transformation.178 Tumors isolated from Fbx4–/– mice had elevated levels of Cyclin D1, as did Fbx4–/– MEF cells.182 The Fbx4 null allele was constructed by deleting exon 2, resulting in a frameshift and premature termination. In direct contrast to this and other reports, a recent study detected no changes in Cyclin D1 protein stability in a more complete knockout of Fbx4, Fbxw8, or Fbxo31 (either singly or in combination).183 The more complete Fbx4 null allele used in the latter study was constructed by deleting exons 1-4. In addition, SCF or APC/C inhibition failed to promote Cyclin D1 accumulation in these experiments. These findings suggest that there are additional F-box proteins or E3 ubiquitin ligases responsible for destabilizing Cyclin D1 or that the different biological consequences are due to the different strategies used to create the null alleles in these mouse models. Immediately prior to the publication of the above results, NIRF (UHRF2) was identified as an E3 ubiquitin ligase that targets both Cyclin E and Cyclin D1 for proteasome-mediated degradation in COS-7 and HEK293T cells.184 As previously shown by the same group, transient overexpression of NIRF resulted in a G1 arrest consistent with a role for NIRF in the G1/S transition.185 Further investigation is required to determine which E3 ubiquitin ligases are principally responsible for Cyclin D1 regulation.
Cyclin E
As cells progress into late S phase, Cyclin E is targeted for degradation by 2 mechanisms. The first is through SCF-mediated ubiquitination and the second is through CRL4- mediated ubiquitination. Cyclin E- Cdk2 auto-phosphorylates at Thr62 and Thr384, and these phosphorylations promote interaction with the SCF adaptor protein Fbxw7, resulting in Cyclin E ubiquitination and degradation.186-188 Phosphorylation at these 2 residues is maximally detected during S phase and mitosis, respectively.189 Mutation of the homologous residues to alanines in a knock-in mouse model resulted in aberrant proliferation and aneuploidy, presumably due to perturbations from inappropriate Cyclin E-Cdk2 activity during late S phase and G2.190 The second mechanism for Cyclin E degradation is through ubiquitination by CRL42. Knockdown of Cul4B, the scaffolding subunit of CRL4 complexes, resulted in the accumulation and increased half-life of Cyclin E, causing a prolonged S phase in both HeLa and HEK293T cells.191 It is not yet known which substrate receptor links Cul4B to Cyclin E, however. Nevertheless, these Cyclin E degradation mechanisms function together in normal cells to suppress Cyclin E levels in mid- to late S phase.
CKIs: p21 and p27
In addition to SCFSkp2-Cks1 activity, CRL4Cdt2 also promotes destruction of p21 during S phase.192-197 As for the Cdt1 and PR-Set7 targets of CRL4Cdt2, ubiquitination of p21 in S phase requires interaction with DNA-loaded PCNA. P21 contains a PIP degron near its C-terminus through which it interacts with both PCNA and Cdt2 (reviewed in Abbas & Dutta198). Interestingly, like Cdt1, p21 is also a stress MAP kinase substrate,130 and MAP kinase activation blocks the replication-dependent degradation of human p21158 by an as-yet unknown mechanism. In contrast to p21, p27 does not contain a PIP degron or even a minimal PIP-box and therefore cannot interact with CRL4Cdt2. SCFSkp2 constitutes the main p27 degradation mechanism during S phase. P27 is, however, still degraded in a CRL4-dependent manner as evidenced by its accumulation after knockdown of DDB1 or Cul4 and reduced p27 levels upon overexpression of Cul4. 196,197,199-201 Interestingly, CRL4-mediated p27 degradation requires the association of Skp2 with the Cul4-DDB1-p27 complex.199 More recently, Cul4 and DDB1 were found to interact with Artemis to efficiently target p27 for proteasomal degradation.202 The relative contributions of these different mechanisms to p27 regulation are still unknown.
Summary of S-phase events
The regulatory mechanisms discussed above are all in place to prevent inappropriate origin licensing and subsequent rereplication during the S and G2 cell cycle phases. These regulatory mechanisms are disrupted in many human cancers, and in many cases those disruptions not only drive tumor initiation but also likely contribute to the progression to metastatic disease and/or the development of chemotherapy resistance. After the G1/S transition, cells inhibit origin licensing by exporting Cdc6 from the nucleus, inhibiting and degrading Cdt1, and degrading PR-Set7. Simultaneously, origins initiate DNA synthesis as a consequence of high Cyclin-Cdk2 activity. Amplification or overexpression of genes encoding D-type and E-type Cyclins are frequent events in human cancers, and the high CDK activity from these genetic perturbations not only inactivates Rb but also disrupts the normal fluctuations in CDK activity that maintain replication precision and genome stability. It is clear that protein degradation plays an integral role in regulating the G1 to S transition and preventing rereplication.
Transformed cells show a higher propensity to undergo rereplication than normal cells do. For example, HeLa cells undergo low and constitutive rereplication even during unperturbed cell cycles,203 and many tumor-derived cell lines can be readily induced to rereplicate whereas normal cells are relatively resistant to such induction.141,151,204,205 Replication licensing proteins are overexpressed in a number of human cancers, in part due to deregulation of the Rb-E2F pathway and higher expression of genes encoding replication licensing proteins.206-208 Even very modest overproduction of the origin licensing factors Cdt1 or Cd6 promotes tumorigenesis in cultured human and mouse cells as well as in mouse xenograft models, presumably due at least in part to low levels of rereplication and consequent genome instability.151,209-214 Unregulated expression of replication licensing factors is one potential reason for higher rereplication in cancer cell lines,206-208 but other mechanisms such as oncogene signaling (e.g., Ras) may also contribute to this difference.215 In addition to promoting oncogenesis, aberrant regulation of replication licensing also contributes to gene amplification through increased genome instability. Gene amplification promotes resistance of cancer cells to therapeutic agents, increasing the likelihood of developing second cancers.14,142
The Ubiquitin-Proteasome System as a Target for Anticancer Therapies
Given the previously discussed importance of ubiquitin-mediated proteolysis in cell cycle regulation, the ubiquitin-proteasome system (UPS) and the specific proteins involved are promising targets of anticancer therapies. These regulatory pathways are frequently mutated in diverse cancer types. Many drugs have been developed to target the UPS, and several are currently being used in the clinic to successfully treat many different cancers. Initial efforts to develop proteasome inhibitors for cancer therapies were met with skepticism as these drugs were predicted to be highly toxic to normal cells. Surprisingly, cancer cells are particularly sensitive to proteasomal inhibition.216,217 The mechanism for this sensitivity is not fully understood, but in the context of this discussion, cancer cells already suffering from perturbed cell cycle control may not tolerate additional deregulation and still maintain chromosome integrity, whereas normal cells are better able to tolerate such pressure.
The boronic-acid derivative bortezomib was the first proteasome inhibitor designed for cancer treatment.218 It specifically and reversibly inhibits the active sites in the 26S proteasome. Proteasome inhibition promotes the upregulation of p21 and p27 and suppression of proinflammatory response genes resulting in apoptosis in tumor cells.219,220 Combination therapies with bortezomib have increased cancer cell sensitivity to other cancer drugs and led to the development of bortezomib derivatives such as carfilzomib.216,219 For example, a combination of melphalan, a DNA damaging agent, and bortezomib was successful in treating multiple myeloma patients.221 Carfilzomib as well as bortezomib is now being used effectively to treat multiple myeloma and relapsed mantle cell lymphoma.222-225 These proteasome inhibitors sensitize cancer cells to other chemotherapeutic agents by abrogating the DNA damage response, upregulating the CKIs p21 and p27, and promoting apoptosis.219,226
The most recent effort to design a novel proteasome inhibitor specifically targets the CRL ubiquitin ligases. All members of the CRL ubiquitin ligase family require the addition of NEDD8, a 9-kD ubiquitin-like protein, for full activity. Neddylation of the cullin proteins produces a conformational change that enhances interaction with the activated E2 enzyme as well as ubiquitin transfer and may prevent association with the negative regulator CAND1.227,228 MLN4924 is the first drug that specifically inhibits the NEDD8-activating enzyme (NAE) to prevent activation of CRL E3 ubiquitin ligases. In the first proof-of-principle study, MLN4924 treatment of many different cancer cell types induced apoptosis and prevented tumor progression in mouse xenograft models.229 This result was later attributed to high levels of rereplication that could be partially suppressed by Cdt1 knockdown, indicating that MLN4924 treatment promoted stabilization of Cdt1 through inhibition of CRL4Cdt2 activation.230 Additionally, MLN4924 treatment was found to increase tumor sensitivity to ionizing radiation in both pancreatic and breast cancers.231,232 Several Phase I clinical trials are currently underway.
Acknowledgments
The authors thank members of the Cook laboratory for helpful comments on the manuscript.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. [DOI] [PubMed] [Google Scholar]
- 2. Wang Q, Moyret-Lalle C, Couzon F, et al. Alterations of anaphase-promoting complex genes in human colon cancer cells. Oncogene. 2003;22(10):1486-90 [DOI] [PubMed] [Google Scholar]
- 3. Yokoi S, Yasui K, Iizasa T, Takahashi T, Fujisawa T, Inazawa J. Down-regulation of SKP2 induces apoptosis in lung-cancer cells. Cancer Sci. 2003;94(4):344-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yokoi S, Yasui K, Mori M, Iizasa T, Fujisawa T, Inazawa J. Amplification and overexpression of SKP2 are associated with metastasis of non-small-cell lung cancers to lymph nodes. Am J Pathol. 2004;165(1):175-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dowen SE, Neutze DM, Pett MR, et al. Amplification of chromosome 5p correlates with increased expression of Skp2 in HPV-immortalized keratinocytes. Oncogene. 2003;22(16):2531-40 [DOI] [PubMed] [Google Scholar]
- 6. DePamphilis ML. The “ORC cycle”: a novel pathway for regulating eukaryotic DNA replication. Gene. 2003;310:1-15 [DOI] [PubMed] [Google Scholar]
- 7. Wei Z, Liu C, Wu X, et al. Characterization and structure determination of the Cdt1 binding domain of human minichromosome maintenance (Mcm) 6. J Biol Chem. 2010;285(17):12469-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J, Yu L, Wu X, et al. The interacting domains of hCdt1 and hMcm6 involved in the chromatin loading of the MCM complex in human cells. Cell Cycle. 2010;9(24):4848-57 [DOI] [PubMed] [Google Scholar]
- 9. Cook J, Chasse D, Nevins J. The regulated association of Cdt1 with minichromosome maintenance proteins and Cdc6 in mammalian cells. J Biol Chem. 2004;279(10):9625-33 [DOI] [PubMed] [Google Scholar]
- 10. Yanagi K-I, Mizuno T, You Z, Hanaoka F. Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J Biol Chem. 2002;277(43):40871-80 [DOI] [PubMed] [Google Scholar]
- 11. Chen S, de Vries MA, Bell SP. Orc6 is required for dynamic recruitment of Cdt1 during repeated Mcm2-7 loading. Genes Dev. 2007;21(22):2897-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donovan S, Harwood J, Drury L, Diffley J. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci U S A. 1997;94(11):5611-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hua XH, Newport J. Identification of a preinitiation step in DNA replication that is independent of origin recognition complex and cdc6, but dependent on cdk2. J Cell Biol. 1998;140(2):271-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen L, Nishioka T, Guo J, Chen C. Geminin functions downstream of p53 in K-ras-induced gene amplification of dihydrofolate reductase. Cancer Res. 2012;72:6153-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkinson KD. Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin Cell Dev Biol. 2000;11(3):141-8 [DOI] [PubMed] [Google Scholar]
- 16. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-33 [DOI] [PubMed] [Google Scholar]
- 17. Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34(11):562-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zimmerman ES, Schulman BA, Zheng N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2010;20(6):714-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369-81 [DOI] [PubMed] [Google Scholar]
- 20. Hayes MJ, Kimata Y, Wattam SL, et al. Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat Cell Biol. 2006;8(6):607-14 [DOI] [PubMed] [Google Scholar]
- 21. Buschhorn BA, Petzold G, Galova M, et al. Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc1. Nat Struct Mol Biol. 2011;18(1):6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. da Fonseca PCA, Kong EH, Zhang Z, et al. Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature. 2011;470(7333):274-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamano H, Gannon J, Mahbubani H, Hunt T. Cell cycle-regulated recognition of the destruction box of cyclin B by the APC/C in Xenopus egg extracts. Mol Cell. 2004;13(1):137-47 [DOI] [PubMed] [Google Scholar]
- 24. Burton JL, Solomon MJ. D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 2001;15(18):2381-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14(6):655-65 [PMC free article] [PubMed] [Google Scholar]
- 26. Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 2002;16(17):2274-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Araki M, Yu H, Asano M. A novel motif governs APC-dependent degradation of Drosophila ORC1 in vivo. Genes Dev. 2005;19(20):2458-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McLean JR, Chaix D, Ohi MD, Gould KL. State of the APC/C: organization, function, and structure. Crit Rev Biochem Mol Biol. 2011;46(2):118-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song L, Rape M. Substrate-specific regulation of ubiquitination by the anaphase-promoting complex. Cell Cycle. 2011;10(1):52-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bar-On O, Shapira M, Skorecki K, Hershko A, Hershko DD. Regulation of APC/C (Cdh1) ubiquitin ligase in differentiation of human embryonic stem cells. Cell Cycle. 2010;9(10):1986-9 [DOI] [PubMed] [Google Scholar]
- 31. Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell. 2000;11(5):1555-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282(5394):1721-4 [DOI] [PubMed] [Google Scholar]
- 33. Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9(5):227-36 [DOI] [PubMed] [Google Scholar]
- 34. Hsu JY, Reimann JDR, Sørensen CS, Lukas J, Jackson PK. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat Cell Biol. 2002;4(5):358-66 [DOI] [PubMed] [Google Scholar]
- 35. Di Fiore B, Pines J. Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J Cell Biol. 2007;177(3):425-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Machida YJ, Dutta A. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 2007;21(2):184-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A. 2009;106(43):18213-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rape M, Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004;432(7017):588-95 [DOI] [PubMed] [Google Scholar]
- 39. Peters J-M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7(9):644-56 [DOI] [PubMed] [Google Scholar]
- 40. Sigl R, Wandke C, Rauch V, Kirk J, Hunt T, Geley S. Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis. J Cell Sci. 2009;122(Pt 22):4208-17 [DOI] [PubMed] [Google Scholar]
- 41. García-Higuera I, Manchado E, Dubus P, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nature. 2008;10(7):802-11 [DOI] [PubMed] [Google Scholar]
- 42. Engelbert D, Schnerch D, Baumgarten A, Wäsch R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene. 2008;27(7):907-17 [DOI] [PubMed] [Google Scholar]
- 43. Rodier G, Coulombe P, Tanguay P-L, Boutonnet C, Meloche S. Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APC(Cdh1) in G1 phase. EMBO J. 2008;27(4):679-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428(6979):190-3 [DOI] [PubMed] [Google Scholar]
- 45. Wei W, Ayad NG, Wan Y, Zhang G-J, Kirschner MW, Kaelin WG. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428(6979):194-8 [DOI] [PubMed] [Google Scholar]
- 46. Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18(21):2573-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ungermannova D, Gao Y, Liu X. Ubiquitination of p27Kip1 requires physical interaction with cyclin E and probable phosphate recognition by SKP2. J Biol Chem. 2005;280(34):30301-9 [DOI] [PubMed] [Google Scholar]
- 48. Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112(2):243-56 [DOI] [PubMed] [Google Scholar]
- 49. Hao B, Zheng N, Schulman BA, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20(1):9-19 [DOI] [PubMed] [Google Scholar]
- 50. Sitry D, Seeliger MA, Ko TK, et al. Three different binding sites of Cks1 are required for p27-ubiquitin ligation. J Biol Chem. 2002;277(44):42233-40 [DOI] [PubMed] [Google Scholar]
- 51. Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278(28):25752-7 [DOI] [PubMed] [Google Scholar]
- 52. Frontini M, Kukalev A, Leo E, et al. The CDK subunit CKS2 counteracts CKS1 to control cyclin A/CDK2 activity in maintaining replicative fidelity and neurodevelopment. Dev Cell. 2012;23(2):356-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martinsson-Ahlzén H-S, Liberal V, Grünenfelder B, Chaves SR, Spruck CH, Reed SI. Cyclin-dependent kinase-associated proteins Cks1 and Cks2 are essential during early embryogenesis and for cell cycle progression in somatic cells. Mol Cell Biol. 2008;28(18):5698-709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krishnan A, Nair SA, Pillai MR. Loss of cks1 homeostasis deregulates cell division cycle. J Cell Mol Med. 2010;14(1-2):154-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25(15):1568-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Michishita M, Morimoto A, Ishii T, et al. Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4(Cdt2) -mediated proteolysis. Genes Cells. 2011;16(1):12-22 [DOI] [PubMed] [Google Scholar]
- 57. Nakanishi M, Robetorye RS, Pereira-Smith OM, Smith JR. The C-terminal region of p21SDI1/WAF1/CIP1 is involved in proliferating cell nuclear antigen binding but does not appear to be required for growth inhibition. J Biol Chem. 1995;270(29):17060-3 [DOI] [PubMed] [Google Scholar]
- 58. Chuang L-C, Zhu X-N, Herrera CR, et al. The C-terminal domain of the Xenopus cyclin-dependent kinase inhibitor, p27Xic1, is both necessary and sufficient for phosphorylation-independent proteolysis. J Biol Chem. 2005;280(42):35290-8 [DOI] [PubMed] [Google Scholar]
- 59. Havens CG, Walter JC. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol Cell. 2009;35(1):93-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Havens CG, Shobnam N, Guarino E, et al. Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J Biol Chem. 2012;287(14):11410-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shiomi Y, Hayashi A, Ishii T, et al. Two different replication factor C proteins, Ctf18 and RFC1, separately control PCNA-CRL4Cdt2-mediated Cdt1 proteolysis during S phase and following UV irradiation. Mol Cell Biol. 2012;32(12):2279-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290(5500):2309-12 [DOI] [PubMed] [Google Scholar]
- 63. Tada S, Li A, Maiorano D, Mechali M, Blow JJ. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol. 2001;3(2):107-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee C, Hong B, Choi JM, et al. Structural basis for inhibition of the replication licensing factor Cdt1 by geminin. Nature. 2004;430(7002):913-7 [DOI] [PubMed] [Google Scholar]
- 65. De Marco V, Gillespie PJ, Li A, et al. Quaternary structure of the human Cdt1-Geminin complex regulates DNA replication licensing. Proc Natl Acad Sci U S A. 2009;106(47):19807-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lutzmann M, Maiorano D, Méchali M. A Cdt1-geminin complex licenses chromatin for DNA replication and prevents rereplication during S phase in Xenopus. EMBO J. 2006;25(24):5764-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lutzmann M, Méchali M. MCM9 binds Cdt1 and is required for the assembly of prereplication complexes. Mol Cell. 2008;31(2):190-200 [DOI] [PubMed] [Google Scholar]
- 68. Caillat C, Perrakis A. Cdt1 and geminin in DNA replication initiation. Subcell Biochem. 2012;62:71-87 [DOI] [PubMed] [Google Scholar]
- 69. McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93(6):1043-53 [DOI] [PubMed] [Google Scholar]
- 70. Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002;21(43):6624-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sugimoto N, Kitabayashi I, Osano S, et al. Identification of novel human Cdt1-binding proteins by a proteomics approach: proteolytic regulation by APC/CCdh1. Mol Biol Cell. 2008;19(3):1007-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shen Z, Chakraborty A, Jain A, et al. Dynamic association of ORCA with prereplicative complex components regulates DNA replication initiation. Mol Cell Biol. 2012;32(15):3107- 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shen Z, Sathyan KM, Geng Y, et al. A WD-repeat protein stabilizes ORC binding to chromatin. Mol Cell. 2010;40(1):99-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petersen BO, Wagener C, Marinoni F. Cell cycle-and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000;14:2330-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mailand N, Diffley JFX. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122(6):915-26 [DOI] [PubMed] [Google Scholar]
- 76. Gillespie PJ, Li A, Blow JJ. Reconstitution of licensed replication origins on Xenopus sperm nuclei using purified proteins. BMC Biochem. 2001;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Houston SI, McManus KJ, Adams MM, et al. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J Biol Chem. 2008;283(28):19478-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Oda H, Okamoto I, Murphy N, et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol. 2009;29(8):2278-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tardat M, Brustel J, Kirsh O, et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12(11):1086-93 [DOI] [PubMed] [Google Scholar]
- 80. Kalakonda N, Fischle W, Boccuni P, et al. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1. Oncogene. 2008;27(31):4293-304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gurvich N, Perna F, Farina A, et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci U S A. 2010;107(52):22552-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Oda H, Hübner MR, Beck DB, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40(3):364-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 2002;16(17):2225- 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu S, Wang W, Kong X, et al. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010;24(22):2531-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol. 2008;28(1):468-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schotta G, Sengupta R, Kubicek S, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22(15):2048-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yang H, Pesavento JJ, Starnes TW, et al. Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J Biol Chem. 2008;283(18):12085-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vermeulen M, Eberl HC, Matarese F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967-80 [DOI] [PubMed] [Google Scholar]
- 89. Shi X, Kachirskaia I, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27(4):636-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol. 2007;179(7):1413-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215-21 [DOI] [PubMed] [Google Scholar]
- 92. Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L. Network calisthenics: control of E2F dynamics in cell cycle entry. Cell Cycle. 2011;10(18):3086-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tsai S-Y, Opavsky R, Sharma N, et al. Mouse development with a single E2F activator. Nature. 2008;454(7208):1137-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20(16):5797-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14(7):804-16 [PMC free article] [PubMed] [Google Scholar]
- 96. Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9(8):585-93 [PubMed] [Google Scholar]
- 98. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18(2):753-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245-62 [DOI] [PubMed] [Google Scholar]
- 100. Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10(7):699-703 [DOI] [PubMed] [Google Scholar]
- 101. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3(1):11-20 [DOI] [PubMed] [Google Scholar]
- 102. Bartkova J, Hořejší Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864-70 [DOI] [PubMed] [Google Scholar]
- 103. Pickering MT, Kowalik TF. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene. 2006;25(5):746-55 [DOI] [PubMed] [Google Scholar]
- 104. Budhavarapu VN, White ED, Mahanic CS. Regulation of E2F1 by APC/CCdh1 via K11 linkage-specific ubiquitin chain formation. Cell cycle (Georgetown, Tex). 2012;11:2030–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ping Z, Lim R, Bashir T, Pagano M, Guardavaccaro D. APC/CCdh1 controls the proteasome-mediated degradation of E2F3 during cell cycle exit. Landes Bioscience. 2012;11(10):1999-2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Peart MJ, Poyurovsky MV, Kass EM, et al. APC/C(Cdc20) targets E2F1 for degradation in prometaphase. Cell Cycle. 2010;9(19):3956-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Takeda S, Chen DY, Westergard TD, et al. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20(17):2397-409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Milne TA, Briggs SD, Brock HW, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10(5):1107-17 [DOI] [PubMed] [Google Scholar]
- 109. Wang P, Lin C, Smith ER, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29(22):6074-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27(1):107-19 [DOI] [PubMed] [Google Scholar]
- 111. Liu H, Cheng EH-Y, Hsieh JJ-D. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007;21(19):2385-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wong PG, Glozak MA, Cao TV, Vaziri C, Seto E, Alexandrow MG. Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle. 2010;9(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Miotto B, Struhl K. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 2008;22(19):2633-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell. 2010;37(1):57-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501-12 [DOI] [PubMed] [Google Scholar]
- 116. Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci. 1998;3:d447-8 [DOI] [PubMed] [Google Scholar]
- 117. Suzuki T, K-Tsuzuku J, Ajima R, Nakamura T, Yoshida Y, Yamamoto T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev. 2002;16(11):1356-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hiramatsu Y, Kitagawa K, Suzuki T, et al. Degradation of Tob1 mediated by SCFSkp2-dependent ubiquitination. Cancer Res. 2006;66(17):8477-83 [DOI] [PubMed] [Google Scholar]
- 119. Yoshida Y, Nakamura T, Komoda M, et al. Mice lacking a transcriptional corepressor Tob are predisposed to cancer. Genes Dev. 2003;17(10):1201-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750-62 [DOI] [PubMed] [Google Scholar]
- 121. Jin M, Inoue S, Umemura T, et al. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer. 2001;34(2):207-18 [DOI] [PubMed] [Google Scholar]
- 122. Barnes DM, Gillett CE. Cyclin D1 in breast cancer. Breast Cancer Res Treat. 1998;52(1-3):1-15 [DOI] [PubMed] [Google Scholar]
- 123. Bartkova J, Lukas J, Müller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55(4):949-56 [PubMed] [Google Scholar]
- 124. Shamma A, Doki Y, Shiozaki H, et al. Cyclin D1 overexpression in esophageal dysplasia: a possible biomarker for carcinogenesis of esophageal squamous cell carcinoma. Int J Oncol. 2000;16(2):261-6 [DOI] [PubMed] [Google Scholar]
- 125. Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 1997;16(17):5334-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Montagnoli A, Fiore F, Eytan E, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13(9):1181-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Grimmler M, Wang Y, Mund T, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128(2):269-80 [DOI] [PubMed] [Google Scholar]
- 128. Chu I, Sun J, Arnaout A, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128(2):281-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhu H, Nie L, Maki CG. Cdk2-dependent inhibition of p21 stability via a C-terminal cyclin-binding motif. J Biol Chem. 2005;280(32):29282-8 [DOI] [PubMed] [Google Scholar]
- 130. Kim G-Y, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem. 2002;277(33):29792-802 [DOI] [PubMed] [Google Scholar]
- 131. Yao Z-P, Zhou M, Kelly SE, Seeliger MA, Robinson CV, Itzhaki LS. Activation of ubiquitin ligase SCF(Skp2) by Cks1: insights from hydrogen exchange mass spectrometry. J Mol Biol. 2006;363(3):673-86 [DOI] [PubMed] [Google Scholar]
- 132. Spruck C, Strohmaier H, Watson M, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7(3):639-50 [DOI] [PubMed] [Google Scholar]
- 133. Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153-60 [DOI] [PubMed] [Google Scholar]
- 134. Kfir S, Ehrlich M, Goldshmid A, Liu X, Kloog Y, Henis YI. Pathway- and expression level-dependent effects of oncogenic N-Ras: p27(Kip1) mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005;25(18):8239-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20(1):47-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kamura T, Hara T, Matsumoto M, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6(12):1229-35 [DOI] [PubMed] [Google Scholar]
- 137. Kotoshiba S, Kamura T, Hara T, Ishida N, Nakayama KI. Molecular dissection of the interaction between p27 and Kip1 ubiquitylation-promoting complex, the ubiquitin ligase that regulates proteolysis of p27 in G1 phase. J Biol Chem. 2005;280(18):17694-700 [DOI] [PubMed] [Google Scholar]
- 138. Hara T, Kamura T, Kotoshiba S, et al. Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol. 2005;25(21):9292-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sclafani R, Holzen T. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lin JJ, Dutta A. ATR pathway is the primary pathway for activating G2/M checkpoint induction after re-replication. J Biol Chem. 2007;282(42):30357-62 [DOI] [PubMed] [Google Scholar]
- 141. Liu E, Lee A, Chiba T, Olson E, Sun P, Wu X. The ATR-mediated S phase checkpoint prevents rereplication in mammalian cells when licensing control is disrupted. J Cell Biol. 2007;179(4):643-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Green BM, Finn KJ, Li JJ. Loss of DNA replication control is a potent inducer of gene amplification. Science. 2010;329(5994):943-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nishitani H, Sugimoto N, Roukos V, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006;25(5):1126-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21(5):497-518 [DOI] [PubMed] [Google Scholar]
- 145. Higa LAA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5(11):1008-15 [DOI] [PubMed] [Google Scholar]
- 146. Machida Y, Hamlin J, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123(1):13-24 [DOI] [PubMed] [Google Scholar]
- 147. Teer JK, Machida YJ, Labit H, et al. Proliferating human cells hypomorphic for origin recognition complex 2 and pre-replicative complex formation have a defect in p53 activation and Cdk2 kinase activation. J Biol Chem. 2006;281(10):6253-60 [DOI] [PubMed] [Google Scholar]
- 148. Arias EE, Walter JC. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005;19(1):114-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol. 2006;8(1):84-90 [DOI] [PubMed] [Google Scholar]
- 150. Guarino E, Shepherd MEA, Salguero I, Hua H, Deegan RS, Kearsey SE. Cdt1 proteolysis is promoted by dual PIP degrons and is modulated by PCNA ubiquitylation. Nucleic Acids Res. 2011;39(14):5978-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Vaziri C, Saxena S, Jeon Y, et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11(4):997-1008 [DOI] [PubMed] [Google Scholar]
- 152. Li A, Blow JJ. Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 2005;24(2):395-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Takeda DY, Parvin JD, Dutta A. Degradation of Cdt1 during S phase is Skp2-independent and is required for efficient progression of mammalian cells through S phase. J Biol Chem. 2005;280(24):23416-23 [DOI] [PubMed] [Google Scholar]
- 154. Yoshida K, Takisawa H, Kubota Y. Intrinsic nuclear import activity of geminin is essential to prevent re-initiation of DNA replication in Xenopus eggs. Genes Cells. 2005;10(1): 63-73 [DOI] [PubMed] [Google Scholar]
- 155. Kerns SL, Torke SJ, Benjamin JM, McGarry TJ. Geminin prevents rereplication during xenopus development. J Biol Chem. 2007;282(8): 5514-21 [DOI] [PubMed] [Google Scholar]
- 156. Raman M, Havens CG, Walter JC, Harper JW. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol Cell. 2011;44(1):72-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Miotto B, Struhl K. JNK1 phosphorylation of Cdt1 inhibits recruitment of HBO1 histone acetylase and blocks replication licensing in response to stress. Mol Cell. 2011;44(1):62-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chandrasekaran S, Tan TX, Hall JR, Cook JG. Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Mol Cell Biol. 2011;31(22):4405-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Varma D, Chandrasekaran S, Sundin LJR, et al. Recruitment of the human Cdt1 replication licensing protein by the loop domain of Hec1 is required for stable kinetochore-microtubule attachment. Nature. 2012;14(6):593-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Glozak MA, Seto E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J Biol Chem. 2009;284(17):11446-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Shen Z, Prasanth SG. Orc2 protects ORCA from ubiquitin-mediated degradation. Cell Cycle. 2012;11(19):3578-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Paolinelli R, Mendoza-Maldonado R, Cereseto A, Giacca M. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat Struct Mol Biol. 2009;16(4):412-20 [DOI] [PubMed] [Google Scholar]
- 163. Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci U S A. 1999;96(11):6193-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Petersen BO, Lukas J, Sørensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999;18(2):396-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40(1):9-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Centore RC, Havens CG, Manning AL, et al. CRL4Cdt2-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40(1):22-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Marti A, Wirbelauer C, Scheffner M, Krek W. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat Cell Biol. 1999;1(1):14-9 [DOI] [PubMed] [Google Scholar]
- 168. Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14(12):8166-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69(4):2491-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sun B, Wingate H, Swisher SG, Keyomarsi K, Hunt KK. Absence of pRb facilitates E2F1-induced apoptosis in breast cancer cells. Cell Cycle. 2010;9(6):1122-30 [DOI] [PubMed] [Google Scholar]
- 171. Liu H, Takeda S, Kumar R, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467(7313):343-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Pagano M, Theodoras AM, Tam SW, Draetta GF. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994;8(14):1627-39 [DOI] [PubMed] [Google Scholar]
- 173. Guo Y, Harwalkar J, Stacey DW, Hitomi M. Destabilization of cyclin D1 message plays a critical role in cell cycle exit upon mitogen withdrawal. Oncogene. 2005;24(6):1032-42 [DOI] [PubMed] [Google Scholar]
- 174. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(8):957-72 [DOI] [PubMed] [Google Scholar]
- 176. Hitomi M, Yang K, Stacey AW, Stacey DW. Phosphorylation of cyclin D1 regulated by ATM or ATR controls cell cycle progression. Mol Cell Biol. 2008;28(17):5478-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Aggarwal P, Lessie MD, Lin DI, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21(22):2908-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Barbash O, Zamfirova P, Lin DI, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008;14(1):68-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Lin DI, Barbash O, Kumar KGS, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24(3):355-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Okabe H, Lee S-H, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS One. 2006;1:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459(7247):722-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Vaites LP, Lee EK, Lian Z, et al. The Fbx4 tumor suppressor regulates cyclin D1 accumulation and prevents neoplastic transformation. Mol Cell Biol. 2011;31(22):4513-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kanie T, Onoyama I, Matsumoto A, et al. Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol. 2012;32(3):590-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Mori T, Ikeda DD, Fukushima T, Takenoshita S, Kochi H. NIRF constitutes a nodal point in the cell cycle network and is a candidate tumor suppressor. Cell Cycle. 2011;10(19):3284-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Li Y, Mori T, Hata H, Homma Y, Kochi H. NIRF induces G1 arrest and associates with Cdk2. Biochem Biophys Res Commun. 2004;319(2):464-8 [DOI] [PubMed] [Google Scholar]
- 186. Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10(16):1979-90 [DOI] [PubMed] [Google Scholar]
- 187. Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173-7 [DOI] [PubMed] [Google Scholar]
- 188. Welcker M, Singer J, Loeb KR, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003;12(2): 381-92 [DOI] [PubMed] [Google Scholar]
- 189. Grim JE, Gustafson MP, Hirata RK, et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol. 2008;181(6):913-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Minella AC, Loeb KR, Knecht A, et al. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev. 2008;22(12):1677-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zou Y, Mi J, Cui J, et al. Characterization of nuclear localization signal in the N terminus of CUL4B and its essential role in cyclin E degradation and cell cycle progression. J Biol Chem. 2009;284(48):33320-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22(18):2496-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283(43):29045-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Stuart SA, Wang JYJ. Ionizing radiation induces ATM-independent degradation of p21Cip1 in transformed cells. J Biol Chem. 2009;284(22):15061-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Li B, Jia N, Kapur R, Chun KT. Cul4A targets p27 for degradation and regulates proliferation, cell cycle exit, and differentiation during erythropoiesis. Blood. 2006;107(11):4291-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Miranda-Carboni GA, Krum SA, Yee K, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008;22(22):3121-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10(2):241-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bondar T, Kalinina A, Khair L, et al. Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol Cell Biol. 2006;26(7):2531-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8(11):1277-83 [DOI] [PubMed] [Google Scholar]
- 201. Cang Y, Zhang J, Nicholas SA, et al. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell. 2006;127(5):929-40 [DOI] [PubMed] [Google Scholar]
- 202. Yan Y, Zhang X, Legerski RJ. Artemis interacts with the Cul4A-DDB1DDB2 ubiquitin E3 ligase and regulates degradation of the CDK inhibitor p27. Cell Cycle. 2011;10(23):4098-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Dorn E, Chastain P, Hall J, Cook J. Analysis of re-replication from deregulated origin licensing by DNA fiber spreading. Nucleic Acids Res. 2009;37(1):60-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hall J, Lee H, Bunker B, et al. Cdt1 and Cdc6 are destabilized by rereplication-induced DNA damage. J Biol Chem. 2008;283(37):25356-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Zhu W, DePamphilis ML. Selective killing of cancer cells by suppression of geminin activity. Cancer Res. 2009;69(11):4870-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Xouri G, Lygerou Z, Nishitani H, Pachnis V, Nurse P, Taraviras S. Cdt1 and geminin are down-regulated upon cell cycle exit and are over-expressed in cancer-derived cell lines. Eur J Biochem. 2004;271(16):3368-78 [DOI] [PubMed] [Google Scholar]
- 207. Blow JJ, Gillespie PJ. Replication licensing and cancer—a fatal entanglement? Nat Rev Cancer. 2008;8(10):799-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Gonzalez MA, Tachibana K-EK, Laskey RA, Coleman N. Control of DNA replication and its potential clinical exploitation. Nat Rev Cancer. 2005;5(2):135-41 [DOI] [PubMed] [Google Scholar]
- 209. Arentson E, Faloon P, Seo J, et al. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene. 2002;21(8):1150-8 [DOI] [PubMed] [Google Scholar]
- 210. Liontos M, Koutsami M, Sideridou M, et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007;67(22):10899-909 [DOI] [PubMed] [Google Scholar]
- 211. Karakaidos P, Taraviras S, Vassiliou LV, et al. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability—evidence of E2F-1 transcriptional control over hCdt1. Am J Pathol. 2004;165(4):1351-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Seo J, Chung YS, Sharma GG, et al. Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene. 2005;24(55):8176-86 [DOI] [PubMed] [Google Scholar]
- 213. Melixetian M, Ballabeni A, Masiero L, et al. Loss of geminin induces rereplication in the presence of functional p53. J Cell Biol. 2004;165(4):473-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zhu W, Chen Y, Dutta A. Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol Cell Biol. 2004;24(16):7140-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319(5868):1352-5 [DOI] [PubMed] [Google Scholar]
- 216. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438-44 [DOI] [PubMed] [Google Scholar]
- 217. Ludwig H, Khayat D, Giaccone G, Facon T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer. 2005;104(9):1794-807 [DOI] [PubMed] [Google Scholar]
- 218. Adams J. Development of the proteasome inhibitor PS-341. Oncologist. 2002;7(1):9-16 [DOI] [PubMed] [Google Scholar]
- 219. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5(5):417-21 [DOI] [PubMed] [Google Scholar]
- 220. Ling Y-H, Liebes L, Jiang J-D, et al. Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines. Clin Cancer Res. 2003;9(3):1145-54 [PubMed] [Google Scholar]
- 221. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906-17 [DOI] [PubMed] [Google Scholar]
- 222. Tobinai K. Proteasome inhibitor, bortezomib, for myeloma and lymphoma. Int J Clin Oncol. 2007;12(5):318-26 [DOI] [PubMed] [Google Scholar]
- 223. McCormack PL. Carfilzomib: in relapsed, or relapsed and refractory, multiple myeloma. Drugs. 2012;72(15):2023-32 [DOI] [PubMed] [Google Scholar]
- 224. Vij R, Wang M, Kaufman JL, et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol. 2012;158(6):739-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101(6):2377-80 [DOI] [PubMed] [Google Scholar]
- 227. Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10(6):1511-8 [DOI] [PubMed] [Google Scholar]
- 228. Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32(1):21-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732-6 [DOI] [PubMed] [Google Scholar]
- 230. Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71(8):3042-51 [DOI] [PubMed] [Google Scholar]
- 231. Wei D, Li H, Yu J, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Yang D, Tan M, Wang G, Sun Y. The p21- dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS One. 2012;7(3):e34079. [DOI] [PMC free article] [PubMed] [Google Scholar]