

Abstract
Protein Phosphatase 2A (PP2A) consists of a collection of heterotrimeric serine/threonine phosphatase holoenzymes that play multiple roles in cell signaling via dephosphorylation of numerous substrates of a large family of serine/threonine kinases. PP2A substrate specificity is mediated by B regulatory subunits of four different families, which selectively recognize diverse substrates by mechanisms that are not well understood. Among the many signaling pathways with critical PP2A functions are several deregulated in cancer cells, and PP2A is a know tumor suppressor. However, the precise composition of the heterotrimeric PP2A complexes with tumor supressor activity is not well understood. This review is centered on the emerging role of the B regulatory subunit B55α and related subfamilly members in the modulation of the phosphorylation state of pocket proteins and mitotic CDK substrates, as well as the implications of PP2A function disruption in cancer in the context of these activities.
Keywords: cyclins, p107, p130, PPP2R2A, B55α, tumor suppressor, PP2A inhibitors
Introduction
Protein Phosphatase 2A (PP2A) is an abundant serine/threonine phosphatase, which attains substrate specificity via the assembly of a large pool of holoenzymes consisting at the minimum of three subunits. A catalytic subunit (PP2A/C) and a scaffold subunit (PP2A/A), each encoded by two different genes, form core dimers, which are assembled with a collection of PP2A/B regulatory subunits forming trimeric holoenzymes (Figure 1). B subunits belong to four separate families, designated B, B′, B′′ and B′′′, each with multiple members encoded by different genes. The crystal structure of two separate holoenzymes assembled with regulatory subunits of the B (B55α) or the B′ (B56g) families, has provided insight on how the B subunit forms a pocket with the catalytic subunit that may confer substrate specificity1-3. PP2A plays a major role in signaling by counteracting the action of Ser/Thr kinases in multiple pathways and is implicated in a large number of cellular processes including those that are often defective in cancer. Major targets of relevance to cancer include the MAP kinase and AKT pathways, the tumor suppressors pRB and p53, and CDK1 substrates among others3-5. This review is mainly focused on the emerging role of B55α and perhaps other members of the B family in the modulation of the phosphorylation state of pocket proteins and mitotic CDK substrates, as well as the implications of PP2A function disruption in cancer in the context of these activities. Recent reviews focused on the role of serine/threonine phosphatases in the regulation of pocket protein function have been published recently3,5.
Figure 1.
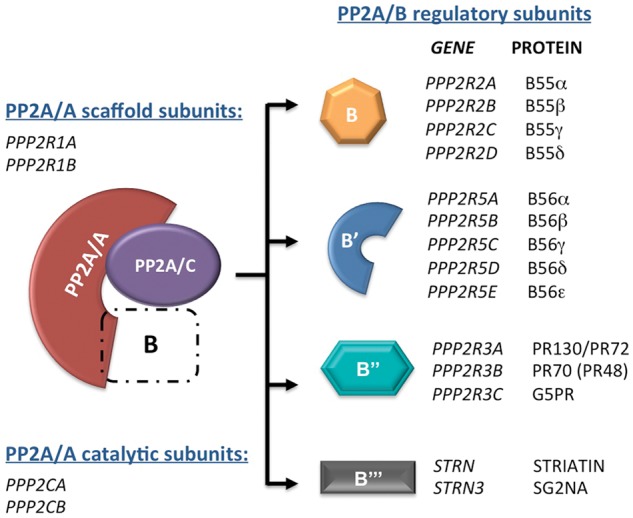
Heterotrimeric PP2A holoenzymes consist of a catalytic, a scaffold, and a regulatory subunit. The B regulatory subunit is responsible for substrate specificity and/or subcellular localization and can be any member from 4 unrelated families (B, B’, B’’, and B’’’). Scaffold and catalytic subunits are encoded each by 2 separate genes. Gene symbols and common protein names are listed.
Evidence That PP2A Is a Tumor Suppressor
Early evidence suggesting that PP2A could function as a tumor suppressor comes from studies showing that okadaic acid (OA) and other toxins in the same family that inhibit PP2A promote tumors in mice and the finding that SV-40 small t antigen, which inhibits PP2A, can transform human cells in cooperation with other oncogenes and facilitates transformation of rodent cells.
Toxins that inhibit PP2A are potent tumor promoters
OA is a toxin extracted from marine sponges and is one of the causative agents of diarrheic shellfish poisoning.6 OA was shown to inhibit PP2A, PP2B, and PP1 in vitro, with ID50s in the nanomolar and micromolar ranges for PP2A and PP1, respectively.7 OA was also found to induce skin irritation in mice in a screen for tumor promoters8 using a 2-stage carcinogenesis protocol.9 In this carcinogenesis experimental system, in which mice were treated with DMBA (a tumor initiator) followed by OA (tumor promoter), 93% of the mice displayed tumors by week 16 in a 30-week treatment. Of note, this was not mediated via activation of PKC, indicating that its mechanism of action was different from that of TPA-tumor promoters.9 Calyculin A, another potent PP1/PP2A inhibitor isolated from marine sponges, was found to have tumor-promoting activity in mice comparable to that of OA10; however, Calyculin A appeared to inhibit PP1 and PP2A with comparable potency.11 Other inhibitors of PP1/PP2A, microcystin-LR and tautomycin, were subsequently identified that were equally potent toward both types of serine/threonine protein phosphatases.12
The transforming activities of the small t antigens (st) of SV40 and related polyomaviruses are mediated via PP2A inhibition
SV40 and related polyomaviruses encode tumor antigens that target and inhibit tumor suppressors in mammalian cells, and this results in their immortalization and/or transformation.13 The small t antigen (st) of SV40 and the small and middle T antigens of polyomaviruses were found to interact with 2 cellular proteins, 36 and 63 kD in size,14-16 which appeared important for transformation, as a middle T transformation-defective mutant was unable to form a complex with the 63 kD protein.17 Peptide microsequencing, co-migration in 2D gels, limited proteolysis, and Western blot analyses were used to identify these proteins as the scaffold and catalytic subunits of PP2A,18,19 hence implicating PP2A in oncogenic transformation of mammalian cells. Importantly, no B subunits were identified associated with these tumor antigens. It was later shown that st interacts directly with the amino terminal HEAT domains present in the PP2A/A scaffold subunit. This association does not interfere with binding of PP2A/C, which interacts with the C-terminal HEAT domains of PP2A/A, but appears to compete with B regulatory subunits, as these also interact with the amino-terminal HEAT domains of PP2A/A.20 This competition was elegantly observed in vitro, as purified ERK and MEK treated with PP2A/B55α heterotrimers preincubated with an excess of st were as potently inactivated as if the PP2A/B55α treatment had been done in the presence of OA.21 In contrast, st did not affect the purified active free PP2A/C catalytic subunit and affected PP2A A-C dimers to a lower extent in the same assay. Moreover, the ability of st to compete with B subunits in cells for binding to PP2A A-C dimers has been shown in cells and may be specific to particular heterotrimers,21-23 suggesting a mechanism by which st may inhibit dephosphorylation of a subset of PP2A substrates. It has also been shown that the binding of st to the core dimer inhibits PP2A′s enzymatic activity toward multiple substrates.20,24 More recently, a crystal structure of st in complex with the PP2A core dimer showed that st has 2 C-terminal zinc binding motifs. One of these motifs and the N-terminal J domain of st overlap the site where the B56γ subunit binds the scaffold subunit. In contrast, the other zinc binding motif is positioned in a manner that may inhibit the catalytic activity of the C subunit,25 suggesting that both B subunit displacement and direct inhibition of PP2A/C may mediate the effects of st on PP2A. In contrast, another study showed that in vitro, st does not efficiently displace B subunits from heterotrimeric complexes unless st is in excess. However, as pointed out by the authors of the study, the J domain of st has been shown to interact with HSC70 increasing its activity, which hypothetically might help disassemble heterotrimers in cells.26
It is also important to underscore the critical role of the pathways inactivated by st in the transformation of human cells. Human cells are more difficult to both immortalize and transform than rodent cells, which often spontaneously immortalize and even transform as passaged extensively in culture. Transformation of normal human fibroblasts, mammary epithelial cells, and kidney epithelial (HEK) cells can be accomplished via expression of the early region of SV40, oncogenic H-RAS, and expression of catalytic subunit of human telomerase (hTERT).27 The early region of SV40 encodes the large antigen (LT), which disables both the pRB and p53 pathways, and st, which inhibits PP2A.28 In these cells, st promotes proliferation, even with nutrient deprivation, and permits anchorage independent growth and tumor formation in immunocompromised mice.29 Subsequently, it was shown that in HEK cells immortalized with LT, hTERT, and H-RAS, knockdown of B56γ3, a regulatory subunit of PP2A from the B′ family, substitutes for st in the anchorage independent and tumorigenicity assays described above.22 Reciprocally, ectopic expression of B56γ3 in HEK cells expressing LT, hTERT, H-RAS, and st inhibited tumorigenicity. Importantly, reintroduction of B56γ3 in lung cancer cell lines that expressed no detectable levels of this B subunit reduced cell proliferation and anchorage independent growth. Thus, this study identified PP2A/B56γ3 holoenzymes as one of the key targets of st in HEK cells, as HEK cells with reduced B55α do not behave in the same manner. A later study using a panel of shRNAs targeting various B subunits demonstrated that the loss of B56α, B56γ, PR72/PR130, and PTPA in immortalized HEK cells results in increased anchorage-independent colony formation as well as the activation of the PI3K/AKT, c-MYC, and WNT pathways.30
Adenovirus E4ORF4 modulates the activity of B55α trimeric PP2A holoenzymes
Another viral protein known to target PP2A is the adenoviral protein E4ORF4, which directly binds to the B55 subunit,31 and this interaction is required for E4ORF4-mediated apoptosis, which is independent of p53.32 As opposed to SV40 st, E4ORF4 forms complexes with PP2A heterotrimers via direct interaction with the B subunit and may direct PP2A to substrates, a mechanism that is significantly different than that described above for st. Members of the B′ regulatory subunit family also bind E4ORF4, but E4ORF4-mediated apoptosis is dependent on E4ORF4 binding to B55α.33-35 E4ORF4 also downregulates MYC transcription by targeting B55α.36 Moreover, E4ORF4 has been shown to induce G2/M arrest and mitotic catastrophe, and this may result from a decrease in the global activity of B55α PP2A heterotrimers.37 This is consistent with a critical role for B55α in mitotic exit (see below). Although it appears counteractive that a viral protein might be that toxic to cells, it has been proposed that this facet of E4orf4 may facilitate release of the viral progeny.38
PP2A activity can also be inhibited by endogenous inhibitors that are deregulated in cancer
PP2A activity can also be deregulated in cancer via endogenous inhibitors such as Cancerous Inhibitor of PP2A (CIP2A) and I2PP2A/SET. CIP2A was first identified as a 90 kD protein (p90) recognized by auto-antibodies detected in the sera of hepatocellular carcinoma and gastric cancer patients but not in normal human serum.39 The cellular function of p90 and relation to malignancy were unveiled when it was identified as a PP2A-interacting protein in a tandem mass spectrometric analysis and renamed CIP2A. As MYC protein stability had been previously linked to PP2A destabilizing dephosphorylation of serine 62 (S62) on MYC,40,41 the effect of CIP2A knockdown was investigated. CIP2A siRNA reduced MYC-S62 phosphorylation and MYC expression and increased PP2A association with MYC, suggesting that CIP2A facilitates MYC stability by inhibiting PP2A.42 Consistent with its MYC-stabilizing function, CIP2A was found to be overexpressed in head and neck squamous cell carcinomas (HNSCC); to be able to substitute for st in transformation of HEK cells expressing hTERT, LT, and oncogenic RAS; and to promote tumorigenesis in HNSCC cells. Moreover, expression of CIP2A was associated with reduced survival in gastric cancer patients and with breast cancer aggressivity, suggesting a prognostic value for CIP2A levels in these cancers.43-45
Another endogenous inhibitor of PP2A, I2PP2A, was first purified from bovine kidney along with I1PP2A, and both were shown to inhibit PP2A activity in vitro.46 Subsequently, I2PP2A was found to be a truncated form of a protein designated SET.47 Importantly, in acute myeloid leukemia (AML), the SET gene is fused to the NUP214 gene resulting in a chimeric SET-NUP214 protein,48 implicating I2PP2A/SET in deregulation of PP2A activity in leukemia. Next, SET was found to be induced by BCR-ABL in chronic myelogenous leukemia (CML), which results in PP2A inhibition. This in turn results in stabilization of BCR-ABL and stimulation of its downstream signaling pathways. In addition, BCR-ABL mediated upregulation of SET may inhibit attenuation of MAP kinase pathways by PP2A.44,49 Consequently, forced upregulation of PP2A activity suppresses BCR-ABL oncogenicity in vitro and impairs leukemogenesis in SCID mice injected with 32D cells expressing BCR-ABL. Interestingly, pharmacological activation of PP2A with forskolin in these mice increased survival significantly, suggesting a potential new therapeutic avenue (see below).
PP2A Is a Haploinsufficient Tumor Suppressor
Mutations have also been found in the 2 genes (PPP2R1A and PPP2R1B) encoding the scaffolding PP2A/A subunit in several types of cancers (Table 1). In one study, the PPP2R1B locus was analyzed in primary lung and colon samples as well as lung cancer derived cell lines. Various point and frameshift mutations were identified, including a mutant PP2A/Aβ scaffold subunit in a small cell lung cancer line that was unable to bind PP2A/C.50 Another study analyzed breast, lung, and melanoma tumors and cell lines and found mutations in both PPP2R1A and PPP2R1B.51 The most frequent mutation found in this study, the deletion of exon 9 in PPP2R1B, results in the deletion of helix 9, which is needed for the binding of the B subunit.52 Recapitulation of mutations of the Aα53 and Aβ54 scaffold subunits described by Calin et al.51 via site-directed mutagenesis and investigation of their effects on the integrity of various PP2A heterotrimers revealed defects in the binding of various B family member subunits as well as the catalytic subunit. Interestingly, the E64D and E64G mutations of the PP2A/Aα subunit are unable to bind members of the B′ family, whereas binding to B and B′′ members as well as the C subunit is relatively unaffected. Mutations in the C-terminal end are deficient in binding both B and C subunits.53 Subsequently, it was shown that although PP2A/Aα mutants ectopically expressed in HEK-TER cells exhibited defects in binding other PP2A subunits, they were not oncogenic in transformation assays in which inactivation of PP2A by other means is transforming.55 In contrast, knockdown of the PP2A/Aα subunit in these cells promotes survival via activation of the AKT pathway and induced tumorigenesis in immunocompromised mice.55 This suggests that mutations affecting PP2A/A act by reducing the amount of functional heterotrimers in the cells. More recently, transgenic mice expressing the E64D and E64G mutations and a Δ5-Δ6 mutant with exons 5 and 6 of PPP2R1A deleted have been generated.56 PP2A/Aα extracted from the tissues of the E64D and E64G mice was defective in binding the B′ subunit, as previously reported.53 These mice were around 60% more susceptible to lung tumors than their wild-type counterparts when treated with benzopyrene.56,57 Together, these studies strongly suggest that the 2 genes (PPP2R1A and PPP2R1B) encoding PP2A/A scaffolding subunits are haploinsufficient tumor suppressors.
Table 1.
Alterations in PP2A Subunits in Cancer
| Mutation | Cancer Type | Ref. | Effect on Binding | Ref. |
|---|---|---|---|---|
| PP2A/Aα | ||||
| E64D | Lung | 51 | B’ binding deficient | 54 |
| E64G | Breast | 51 | B’ binding deficient | 54 |
| R418W | Melanoma | 51 | No B or C binding | 54 |
| D171-589 | Breast | 51 | No B or C binding | 54 |
| P179L | Endometrial | 95 | Unknown | |
| R182W | Endometrial | 95 | Unknown | |
| S256Y | Endometrial | 95 | Unknown | |
| R183Q | Endometrial | 95 | Unknown | |
| R249H | Endometrial | 95 | Unknown | |
| R182W | Endometrial | 95 | Unknown | |
| E216K | Endometrial | 96 | Unknown | |
| R258Y | Endometrial | 96 | Unknown | |
| R258H | Endometrial | 96 | Unknown | |
| R258C | Ovarian | 95 | Unknown | |
| W257C | Endometrial, ovarian | 95, 96 | Unknown | |
| P179R | Endometrial, ovarian | 95 | Unknown | |
| R183W | Endometrial, ovarian | 95 | Unknown | |
| S256F | Endometrial, ovarian | 95 | Unknown | |
| W257G | Endometrial, ovarian | 95 | Unknown | |
| PP2A/Aβ | ||||
| D504G | Lung | 50 | Increased B’’ and C binding | 53 |
| G8R | Lung | 50 | Normal B’’ and C binding | 53 |
| G90D | Lung | 50 | Normal B’’ and C binding | 53 |
| Breast | 97 | Reduced B’ binding | 97 | |
| K343E | Lung | 50 | Normal B’’ and C binding | 53 |
| D344-388 | Lung | 50 | Reduced B’’ binding | 53 |
| P65S | Lung | 50 | Reduced B’’ binding | 53 |
| L101P | Colon | 50 | Reduced B’’ binding | 53 |
| V545A | Colon | 50 | Reduced B’’ and C binding | 53 |
| V448A | Colon | 50 | Reduced B’’ and C binding | 53 |
| L101P/V448A | Colon | 50 | Reduced B’’ and C binding | 53 |
| D230-518 | Lung | 50 | Unknown | |
| G15A | Colorectal | 98 | Unknown | |
| S365P | Colorectal | 98 | Unknown | |
| V498E | Colorectal | 98 | Unknown | |
| L499I | Colorectal | 98 | Unknown | |
| V500G | Colorectal | 98 | Unknown | |
| Deletions | Liver | 99 | Unknown | |
| PP2A/B55α | ||||
| Deletions | Breast | 64 | ||
| Deletions | Prostate | 58 | ||
| Deletions | Ovarian | 65 | ||
| PP2A/B55β | ||||
| Hypermethylation | Colorectal | 66 | ||
| Hypermethylation | Breast | 67 | ||
Alterations on PPP2R2A and PPP2R2B in Cancer
Although mutations on the scaffold subunit of PP2A are common in a variety of cancers, mutations in the regulatory subunits of PP2A are rare. Only 4 point mutations in PPP2R2A, which encodes the B55α B subunit of PP2A, have been reported in the COSMIC database from 652 sequenced samples, and mutations in other B subunits are uncommon. However, recent studies have shown that deletions of PPP2R2A, which maps to chromosome 8p21.2, are extremely common in prostate cancer (PCa) and are also frequent in breast cancer (Table 1). Using Affymetrix 500k/6.0 SNP array analysis of 141 paired normal/cancer prostate samples, investigators found that 84 tumors (59.6%) presented hemizygous deletion of the PPP2R2A gene and that 3 tumors (2.1%) had a homozygous deletion. However, there was no apparent correlation between PPP2R2A deletion and PCa-specific death, Gleason score, prostate specific antigen levels, or stage. These deletions were all somatic, and no germline deletions were found in 500 nonaggressive and 448 aggressive prostate tumors.58 An independent study conducted to identify recurrent breakpoints in prostate cancer also using the Affymetrix array 6.0 and 500K SNP identified 5 genomic breakpoints in the PPP2R2A gene in 78 prostate tumor samples, ranking PPP2R2A as the fourth most common genomic breakpoint, ahead of PTEN, BRCA1, and BRCA2. In addition, qRT-PCR analysis revealed that 8 of 9 paired normal/tumor samples exhibited decreased PPP2R2A mRNA expression in the tumors.59 Finally, analysis of the gene copy number alteration (CNA) data from another recent study shows that PPP2R2A was homozygously deleted in 7 of 128 (5.5%) and hemizygously deleted in 47 of 128 (36.7%) prostate tumors (Taylor et al.,60 supplementary data). Matched mRNA expression Z-scores versus controls showed a correlation with CNA, where median expression values decreased with copy number. Of note, in all these studies the homozygous deletions affecting PPP2R2A also eliminate BNIP3L, another potential tumor suppressor gene58,60,61; therefore, the contribution of disruption of each of these 2 genes in tumorigenesis remains to be elucidated. Moreover, hemizygous deletions are larger and variable and may affect the expression of other genes. NKX3.1, a gene that has been previously implicated in prostate gland development and as a potential haploinsufficient PCa tumor suppressor,62,63 was found to be homozygously deleted in 4 of 81 prostate tumors (4.9%); however, no co- correlation between mRNA expression and CNA was found, suggesting the alternative presence of other tumor suppressors in this region.60 Importantly, deletion of PPP2R2A is not restricted to PCa, as a recently published study of the genomic and transcriptomic architecture of about 2,000 breast tumors has revealed that PPP2R2A is 1 of 3 novel deletions that is associated with outlier underexpression of its product. Loss of PPP2R2A expression in breast cancer is enriched in mitotic ER-positive cancers (luminal B), allowing for new molecular stratification of breast cancers.64 Moreover, homozygous deletions of PPP2R2A are also detected in serous ovarian cancer (4.5%), all indicating that alterations of this gene are not restricted to PCa.65
Promoter silencing via methylation of PPP2R2B, which encodes B55β, a regulatory subunit in the same family as B55α, has been found in colorectal cancer (CRC) (Table 1). Downregulation of PPP2R2B in CRC-derived patient samples was observed when compared with matched normal colon-derived mucosa in gene expression array analysis, as was hypermethylation of the PPP2R2B promoter. Reintroduction of B55β reduced the ability of DLD1 and HCT116 cells to form anchorage-independent colonies in soft agar, and the ability of DLD1 cells to form tumors in nude mice was reduced when B55β was expressed. In this study, it was shown that B55β targets PDK1 to regulate MYC signaling, and in CRC, loss of B55β results in a PDK1-dependent, but PI3K-independent induction of MYC phosphorylation in response to rapamycin, which is reversed upon readdition of B55β.66 Aberrant methylation of PPP2R2B has also been detected in ductal carcinoma in situ when compared with normal breast tissue.67
Taken together, the observation that PP2A inactivation occurs by different mechanisms affecting distinct trimeric PP2A holoenzymes in different types of cancer strongly suggests that the targets of PP2A inactivation are likely to be specific in subsets of tumors. In other words, the tumor relevant pathways deregulated as result of loss of particular PP2A activities are likely tumor specific.
Alterations on B55α May Be Linked to Inactivation of the Pocket Protein Pathway
The 3 members of the pocket protein family—pRB, p107, and p130—are inactivated by phosphorylation in a cell cycle dependent manner.3,5,68,69 Pocket proteins are hypophosphorylated in quiescent cells or in the G1 phase of the cell cycle preceding the restriction point. Mitogenic stimulation of D-type cyclins′ expression and downregulation and/or sequestration of p27 result in the sequential activation of D-type cyclin/CDK4/6 and cyclin E/CDK2 complexes. These G1/S cyclin/CDK complexes cooperate to hyperphosphorylate and inactivate pocket proteins in mid- to late G1, resulting in disruption of E2F/pocket protein transcriptional repressor complexes and the initiation of a wave of E2F dependent gene transcription required for progression through S phase and mitosis.69,70 Pocket proteins remain hyperphosphorylated through S, G2, and most M phase, as other cyclin/ CDK complexes are active through these phases. Coinciding with CDK inactivation in mitosis, pocket proteins are abruptly dephosphorylated and reset to their active state, as cells exit mitosis and enter the next G1 phase. The finding that protein phosphatase 1 (PP1) associates with pRB in late mitosis and early G1 and subsequent studies led to a model in which pocket proteins were switched off by inducible CDKs from mid- to late G1 though the point in mitosis where CDKs were inactivated, which in turn was associated with activation of PP1 and the reactivation of pocket proteins.71-73 However, our laboratory found that treatment of exponentially growing cells with small pharmacological compounds that rapidly inhibit expression of D-type cyclin or CDK activity resulted in immediate dephosphorylation of pocket proteins.74 This dephosphorylation could be mediated by a phosphatase that is constitutively active throughout the cell cycle or, alternatively, the recruitment of a phosphatase to pocket proteins following CDK inhibition. This dephosphorylation could be prevented pharmacologically with concentrations of OA that inhibit PP2A but not PP1 and via expression of SV40 st, implicating PP2A as the phosphatase responsible. In agreement with these results, PP2A/C was found to co-immunoprecipitate with p107 and p130 in both quiescent and exponentially growing cells. Altogether this suggested that a heterotrimeric PP2A holoenzyme(s) acts in a dynamic equilibrium with inducible CDKs to maintain the phosphorylation state of pocket proteins through the cell cycle and in quiescent cells74 (Figure 2). The data also showed that reduction of particular cyclin/CDK activities had an intermediate effect, which could be explained by a shift in the PP2A/CDK equilibrium or by dephosphorylation of sites specific to the cyclin/CDK complexes being downregulated.74 The notion that the phosphorylation state of pRB could be determined by the ratio of pRB kinases and phosphatases has also been proposed.75
Figure 2.
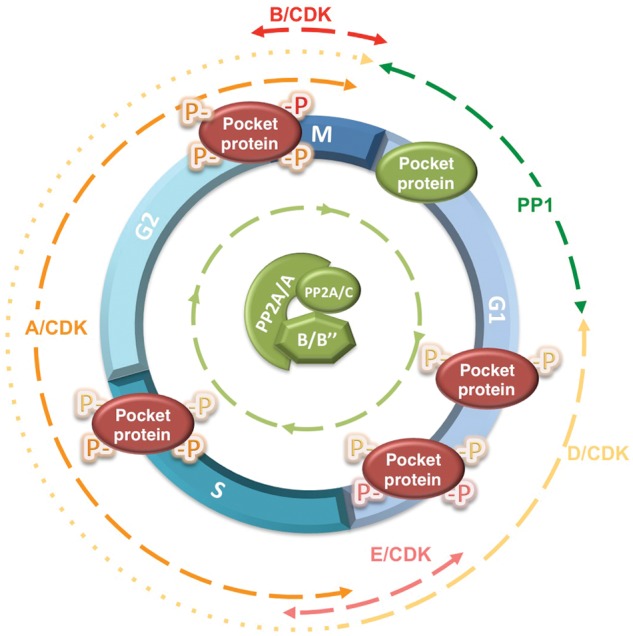
The phosphorylation state of pocket proteins is regulated by an equilibrium between inducible cyclin/CDK complexes and 2 major types of Ser/Thr phosphatases. The active, hypophosphorylated form of pocket proteins is shown in green, and the inactive, hyperphosphorylated pocket protein is shown in red. Although PP1 has been implicated in dephosphorylation of pRB from late mitosis to G1, when CDK activity is low, PP2A appears to restrict pocket protein phosphorylation throughout the cell cycle and in quiescent cells (not shown in this figure). Two heterotrimeric complexes have been directly implicated in dephosphorylation of pocket proteins, PP2A/B55α and PP2A/PR70. PP2A/B55α was shown to preferentially target p107 and p130, but our unpublished observations indicate that pRB is also a target. PP2A/PR70 has been implicated in pRB dephosphorylation upon oxidative stress. However, its contribution to the phosphorylation state of pocket proteins in the absence of inducible signals has not been investigated. The approximate portion of the cell cycle in which each participating CDK and Ser/Thr phosphatase is active toward pocket proteins is indicated by dashed lines. Of note, the expression of D-type cyclins during the cell cycle, which is stimulated by mitogens, varies in different cell types. In some cell types, D-type cyclins are only expressed from mid-G1 through the G1/S transition (this is denoted by dashes changing to dots).
More recently, it has been shown that PP2A/PR70 holoenzymes target pRB for dephosphorylation upon oxidative stress,76 and our laboratory identified PP2A/B55α holoenzymes as being responsible for the dephosphorylation of p107 and, likely, p130.23 Of note, expression of SV40 st in cells dissociated the PP2A/A-C core dimer from B55α, but p107 remained bound to B55α. In agreement with this result, we also found that a B55α point mutant (D197K), which lacks a residue in a surface predicted to interact with substrates,77 was unable to interact with p107 yet retained, albeit reduced, an interaction with the PP2A/ A-C core dimer.23 Strikingly, both loss and gain of function assays suggested that the steady state phosphorylation of p107 is highly sensitive to variations in B55α expression in a variety of cells tested thus far, suggesting that regulation of B55α activity in cells is likely to have an impact on the activation state of pocket proteins. Obviously, this has implications in cancer, as genetic/epigenetic alterations leading to downregulation of pocket protein phosphatases could function as an alternative mechanism to inactivate the pRB pathway (Figure 3). One conceivable example is downregulation of B55α as a result of deletions on chromosome 8p21 that eliminated PPP2R2A in a variety of cancers, but most prominently in PCa.58,60 More work is needed to test this idea.
Figure 3.
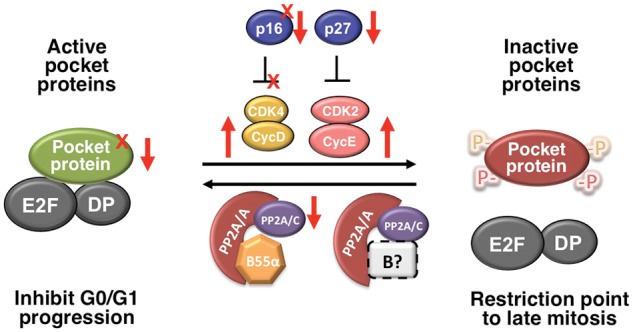
The pocket protein regulatory network is disrupted by multiple mechanisms in cancer. Tumor suppressors in the network are inactivated via point mutations or deletions (X) or via epigenetic silencing or increased proteolytic degradation (↓). Mutations and or deletions affecting pRB, p130, and p16 have been described (reviewed in Malumbres & Barbacid94). Amplification of cyclins or defects in their degradation pathways is also common (↑). The pathway can be disrupted by a mutation on CDK4 that makes it resistant to CDK inhibitors (X). Deletions affecting expression of B55α have been described in a variety of cancers (see text) and may affect this equilibrium.
Alterations on PPP2R2A May Be Linked to Mitotic Defects
PP2A/B55 complexes play a critical role in promoting mitotic exit in higher eukaryotes by dephosphorylating many substrates phosphorylated by CDK1 in mitosis.78 Of note, as opposed to the universal involvement of CDK1 in triggering mitotic events in all eukaryotes, dephosphorylation of CDK1 substrates seems to be accomplished by different phosphatases in yeast and animal cells. An RNAi-based live-cell imaging screen identified B55α and the nuclear transport factor importin-β as key regulators in postmitotic spindle breakdown, nuclear envelop reassembly, chromatin decondensation, and Golgi assembly.79 In this study, mass spectrometry was used to identify regulated phosphorylation sites of the subunits of the heterotrimeric complex. Phosphorylation on Ser 167 of the B55α subunit was by far the most abundant and was enriched more than 5-fold in mitotic cells.79 Consistently, a B55α phosphorylation mimic mutant bound the PP2AC A-C dimer less efficiently than wild-type B55α in in vitro pulldown assays. Although these results suggested that the heterotrimeric PP2A/B55α complex may be negatively regulated via phosphorylation, further work is needed to clarify the kinetics of phosphorylation, its effects on the holoenzyme activity, and the upstream regulators.
Previous studies showed that Drosophila B55 mutants with reduced activity exhibited mitotic defects and reduced ability to dephosphorylate CDK1 substrates in vitro.80 In Xenopus extracts, PP2A/B55δ holoenzymes also appear to be responsible for dephosphorylation of many CDK substrates in interphase, and their activity is negatively regulated by the Greatwall (GW) kinase in mitosis, which is activated by CDK1. In the absence of GW, Xenopus extracts cannot enter M phase, but this is corrected by depletion of B55δ,81 and GW depletion promotes mitotic exit even with high CDK1 activity.82 Conversely, depletion of B55δ accelerates entry into mitosis.83 Two substrates of GW, ENSA and ARPP19, have been identified as novel inhibitors of PP2A during mitosis. In one study, ENSA and ARPP19 were both shown to bind PP2A; however, only ARPP19 was required to promote mitotic entry in Xenopus egg extracts.84 In another study, it was shown that phosphorylated ENSA specifically targets and inhibits PP2A/B55δ holoenzymes in Xenopus, ultimately leading to CDK1 activation.85 Moreover, in mammals GW is designated MASTL (MASTL/GW), and at least 2 B55 subunits, B55α (mentioned above) and B55δ, appear to work downstream. In MEFs, targeted inactivation of CDC20, the activating subunit of the anaphase promoting complex (APC), results in metaphase arrest. In these cells, mitotic exit can be induced by pharmacological inhibition of both CDK1 (with Roscovitine) and MASTL/GW (siRNA). Under these conditions, depletion of B55α and/or B55δ with specific siRNAs blocks mitotic exit.86 Thus, the requirement of members of the B55 family of heterotrimeric complexes for mitotic exit and their negative regulation by MASTL/GW is conserved in vertebrates. Of note, B55β and B55γ are not expressed at high levels in MEFs, as their expression is tissue specific, so it is conceivable that they would play a similar role in those cell types were they are expressed at significant levels.
In closing, the critical importance of B55α in mitotic exit, so far analyzed in HeLa cells and MEFs, also has implications in cancer given the high frequency of deletions affecting PPP2R2A, at least in prostate, ovarian and breast cancer. Hemizygous deletions on PPP2R2A may result in limiting levels of B55α for effective mitotic exit, which may facilitate mitotic defects and potentially genetic instability. However, the role of B55α in modulating the activation state of p107/p130 and perhaps also pRB suggests that elimination of PPP2R2A may be pleiotropic, especially if one also considers that B55α has been implicated in the negative regulation of mitogenic and survival pathways. The consequences of B55α deregulation will most likely vary in a cell type specific manner, reflecting the relative importance of this B regulatory subunit in each cell type.
In contrast, the high frequency of hemizygous deletions affecting the PPP2R2A locus, without alteration in the other allele, provides a potential strategy for therapeutics using small pharmacologic molecules with the ability to boost the activity of the product of the remaining intact allele. This idea has been tested in preclinical studies in leukemia where PP2A activity is reduced by deregulated endogenous inhibitors of PP2A (see sections above). FTY720 is a pharmacological activator of PP2A, which the FDA approved for use in the treatment of multiple sclerosis in humans, and is well tolerated.57,87-89 FTY720 has been shown to have antitumoral activity in mouse xenografts of human tumors with no toxicity to mice. In addition to the established effect of FTY720 on leukemias, it has been shown that prostate cancer cell lines are more sensitive to FTY720 than prostate stromal cells.90 FTY720 also induced cytoskeletal changes in murine breast cancer cells and decreased their ability to adhere and migrate to extracellular matrix components,91 and it inhibited metastasis of hepatocellular carcinoma cells in nude mice via inhibition of Rac-mediated cell motility and VEGF-mediated angiogenesis.92 Because FTY720 is known to activate PP2A enzymatic activity independently of trimeric holoenzyme composition,93 it is conceivable that it might also alleviate the effects of reduction on B55α holoenzymes in mitosis and/or the activation of pocket proteins. Future studies should address these issues.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in the XG laboratory was supported by the National Institutes of Health Grant MH083585 and by a grant from the Pennsylvania Department of Health to X.G.
References
- 1. Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell 2009;139:468-484 [DOI] [PubMed] [Google Scholar]
- 2. Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537-545 [DOI] [PubMed] [Google Scholar]
- 3. Kurimchak A, Graña X. PP2A holoenzymes negatively and positively regulate cell cycle progression by dephosphorylating pocket proteins and multiple CDK substrates. Gene. 2012;499:1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795:1-15 [DOI] [PubMed] [Google Scholar]
- 5. Kolupaeva V, Janssens V. PP1 and PP2A phosphatases—cooperating partners in modulating retinoblastoma protein activation. FEBS J. Epub 2012. February 2 [DOI] [PubMed] [Google Scholar]
- 6. Cohen P, Holmes CF, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15:98-102 [DOI] [PubMed] [Google Scholar]
- 7. Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases:xpecificity and kinetics. Biochem J. 1988;256:283-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujiki H, Suganuma M, Suguri H, et al. Induction of ornithine decarboxylase activity in mouse skin by a possible tumor promoter, okadaic acid. Proc Jpn Acad Ser B Phys Biol Sci. 1987;63:51-53 [Google Scholar]
- 9. Suganuma M, Fujiki H, Suguri H, et al. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci U S A. 1988;85:1768-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suganuma M, Fujiki H, Furuya-Suguri H, et al. Calyculin A, an inhibitor of protein phosphatases, a potent tumor promoter on CD-1 mouse skin. Cancer Res. 1990;50:3521-3525 [PubMed] [Google Scholar]
- 11. Ishihara H, Martin B. L, Brautigan DL, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871-877 [DOI] [PubMed] [Google Scholar]
- 12. Suganuma M, Fujiki H, Okabe S, et al. Structurally different members of the okadaic acid class selectively inhibit protein serine/threonine but not tyrosine phosphatase activity. Toxicon. 1992;30:873-878 [DOI] [PubMed] [Google Scholar]
- 13. Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol. 2001;11:15-23 [DOI] [PubMed] [Google Scholar]
- 14. Pallas DC, Cherington V, Morgan W, et al. Cellular proteins that associate with the middle and small T antigens of polyomavirus. J Virol. 1988;62:3934-3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walter G, Carbone-Wiley A, Joshi B, Rundell K. Homologous cellular proteins associated with simian virus 40 small T antigen and polyomavirus medium T antigen. J Virol. 1988;62:4760-4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang YC, Hearing P, Rundell K. Cellular proteins associated with simian virus 40 early gene products in newly infected cells. J Virol. 1979;32:147-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grussenmeyer T, Scheidtmann KH, Hutchinson MA, Eckhart W, Walter G. Complexes of polyoma virus medium T antigen and cellular proteins. Proc Natl Acad Sci U S A. 1985;82:7952-7954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pallas DC, Shahrik LK, Martin BL, et al. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60:167-176 [DOI] [PubMed] [Google Scholar]
- 19. Walter G, Ruediger R, Slaughter C, Mumby M. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc Natl Acad Sci U S A. 1990;87:2521-2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746-7755 [DOI] [PubMed] [Google Scholar]
- 21. Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell. 1993;75:887-897 [DOI] [PubMed] [Google Scholar]
- 22. Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127-136 [DOI] [PubMed] [Google Scholar]
- 23. Jayadeva G, Kurimchak A, Garriga J, et al. B55alpha PP2A holoenzymes modulate the phosphorylation status of the retinoblastoma-related protein p107 and its activation. J Biol Chem. 2010;285:29863-29873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11:1988-1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 2007;5:e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Xu Y, Bao Q, et al. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat Struct Mol Biol. 2007;14:527-534 [DOI] [PubMed] [Google Scholar]
- 27. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464-468 [DOI] [PubMed] [Google Scholar]
- 28. Chen W, Hahn WC. SV40 early region oncoproteins and human cell transformation. Histol Histopathol. 2003;18:541-550 [DOI] [PubMed] [Google Scholar]
- 29. Hahn WC, Dessain SK, Brooks MW, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111-2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sablina AA, Hector M, Colpaert N, Hahn WC. Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res. 2010;70:10474-10484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kleinberger T, Shenk T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J Virol. 1993;67:7556-7560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shtrichman R, Sharf R, Barr H, Dobner T, Kleinberger T. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc Natl Acad Sci U S A. 1999;96:10080-10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shtrichman R, Sharf R, Kleinberger T. Adenovirus E4orf4 protein interacts with both Balpha and B′ subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by the interaction with Balpha. Oncogene. 2000;19:3757-3765 [DOI] [PubMed] [Google Scholar]
- 34. Van Hoof C, Goris J. Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97-104 [DOI] [PubMed] [Google Scholar]
- 35. Marcellus RC, Chan H, Paquette D, Thirlwell S, Boivin D, Branton PE. Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the Balpha subunit of protein phosphatase 2A. J Virol. 2000;74:7869-7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ben-Israel H, Sharf R, Rechavi G, Kleinberger T. Adenovirus E4orf4 protein downregulates MYC expression through interaction with the PP2A-B55 subunit. J Virol. 2008;82:9381-9388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li S, Brignole C, Marcellus R, et al. The adenovirus E4orf4 protein induces G2/M arrest and cell death by blocking protein phosphatase 2A activity regulated by the B55 subunit. J Virol. 2009;83:8340-8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Branton PE, Roopchand DE. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene. 2001;20:7855-7865 [DOI] [PubMed] [Google Scholar]
- 39. Soo Hoo L, Zhang JY, Chan EK. Cloning and characterization of a novel 90 kDa “companion” auto-antigen of p62 overexpressed in cancer. Oncogene. 2002;21:5006-5015 [DOI] [PubMed] [Google Scholar]
- 40. Yeh E, Cunningham M, Arnold H, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308-318 [DOI] [PubMed] [Google Scholar]
- 41. Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol. 2006;26:2832-2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Junttila MR, Puustinen P, Niemela M, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51-62 [DOI] [PubMed] [Google Scholar]
- 43. Khanna A, Bockelman C, Hemmes A, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst. 2009;101:793-805 [DOI] [PubMed] [Google Scholar]
- 44. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152-160 [DOI] [PubMed] [Google Scholar]
- 45. Come C, Laine A, Chanrion M, et al. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res. 2009;15:5092-5100 [DOI] [PubMed] [Google Scholar]
- 46. Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry. 1995;34:1988-1996 [DOI] [PubMed] [Google Scholar]
- 47. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059-11062 [DOI] [PubMed] [Google Scholar]
- 48. von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12:3346-3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355-368 [DOI] [PubMed] [Google Scholar]
- 50. Wang SS, Esplin ED, Li JL, et al. Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284-287 [DOI] [PubMed] [Google Scholar]
- 51. Calin GA, di Iasio MG, Caprini E, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19:1191-1195 [DOI] [PubMed] [Google Scholar]
- 52. Ruediger R, Hentz M, Fait J, Mumby M, Walter G. Molecular model of the A subunit of protein phosphatase 2A: interaction with other subunits and tumor antigens. J Virol. 1994;68:123-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruediger R, Pham HT, Walter G. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene. 2001;20:10-15 [DOI] [PubMed] [Google Scholar]
- 54. Ruediger R, Pham HT, Walter G. Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the A beta subunit gene. Oncogene. 2001;20:1892-1899 [DOI] [PubMed] [Google Scholar]
- 55. Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65:8183-8192 [DOI] [PubMed] [Google Scholar]
- 56. Ruediger R, Ruiz J, Walter G. Human cancer-associated mutations in the Aalpha subunit of protein phosphatase 2A increase lung cancer incidence in Aalpha knock-in and knockout mice. Mol Cell Biol. 2011;31:3832-3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walter G, Ruediger R. Mouse model for probing tumor suppressor activity of protein phosphatase 2A in diverse signaling pathways. Cell Cycle. 2012;11:451-459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cheng Y, Liu W, Kim ST, et al. Evaluation of PPP2R2A as a prostate cancer susceptibility gene: a comprehensive germline and somatic study. Cancer Genet. 2011;204:375-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mao X, Boyd LK, Yanez-Munoz RJ, et al. Chromosome rearrangement associated inactivation of tumour suppressor genes in prostate cancer. Am J Cancer Res. 2011;1:604-617 [PMC free article] [PubMed] [Google Scholar]
- 60. Taylor BS, Schultz N, Hieronymus H, et al. L. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu W, Xie CC, Zhu Y, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008;10:897-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. He WW, Sciavolino PJ, Wing J, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43:69-77 [DOI] [PubMed] [Google Scholar]
- 63. Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966-977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Network TCGAR Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tan J, Lee PL, Li Z, et al. B55beta-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010;18:459-471 [DOI] [PubMed] [Google Scholar]
- 67. Muggerud AA, Ronneberg JA, Warnberg F, et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. 2010;12:R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein—is the whole a sum of its parts? Cell Div. 2007;2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sotillo E, Graña X. Escape from cellular quiescence. In: Cell cycle deregulation in cancer, contemporary cancer research New York, NY: Springer Publishing; 2010. p. 3-22 [Google Scholar]
- 70. Rowland BD, Bernards R. Re-evaluating cell-cycle regulation by E2Fs. Cell. 2006;127:871-874 [DOI] [PubMed] [Google Scholar]
- 71. Durfee T, Becherer K, Chen PL, et al. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 1993;7:555-569 [DOI] [PubMed] [Google Scholar]
- 72. Nelson DA, Krucher NA, Ludlow JW. High molecular weight protein phosphatase type 1 dephosphorylates the retinoblastoma protein. J Biol Chem. 1997;272:4528-4535 [DOI] [PubMed] [Google Scholar]
- 73. Nelson DA, Ludlow JW. Characterization of the mitotic phase pRb-directed protein phosphatase activity. Oncogene. 1997;14:2407-2415 [DOI] [PubMed] [Google Scholar]
- 74. Garriga J, Jayaraman AL, Limon A, et al. A dynamic equilibrium between CDKs and PP2A modulates phosphorylation of pRB, p107 and p130. Cell Cycle. 2004;3:1320-30 [DOI] [PubMed] [Google Scholar]
- 75. An B, Dineley K, Zhang L, Termin T, Meijer L, Dou Q. Involvement of RB kinases and phosphatases in life and death decisions. Oncol Rep. 1997;4:1129-1134 [DOI] [PubMed] [Google Scholar]
- 76. Magenta A, Fasanaro P, Romani S, et al. Protein phosphatase 2A subunit PR70 interacts with pRb and mediates its dephosphorylation. Mol Cell Biol. 2008;28:873-882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell. 2008;31:873-885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469-482 [DOI] [PubMed] [Google Scholar]
- 79. Schmitz MH, Held M, Janssens V, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol. 2009;12:886-893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mayer-Jaekel RE, Ohkura H, Ferrigno P, et al. Drosophila mutants in the 55 kDa regulatory subunit of protein phosphatase 2A show strongly reduced ability to dephosphorylate substrates of p34cdc2. J Cell Sci. 1994;107(Pt 9):2609-2616 [DOI] [PubMed] [Google Scholar]
- 81. Castilho PV, Williams BC, Mochida S, Zhao Y, Goldberg ML. The M phase kinase Greatwall (Gwl) promotes inactivation of PP2A/B55delta, a phosphatase directed against CDK phosphosites. Mol Biol Cell. 2009;20:4777-4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vigneron S, Brioudes E, Burgess A, Labbe JC, Lorca T, Castro A. Greatwall maintains mitosis through regulation of PP2A. EMBO J. 2009;28:2786-2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mochida S, Ikeo S, Gannon J, Hunt T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009;28:2777-2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gharbi-Ayachi A, Labbe JC, Burgess A, et al. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science. 2010;330:1673-1677 [DOI] [PubMed] [Google Scholar]
- 85. Mochida S, Maslen SL, Skehel M, Hunt T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science. 2010;330:1670-1673 [DOI] [PubMed] [Google Scholar]
- 86. Manchado E, Guillamot M, de Carcer G, et al. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell. 2010;18:641-654 [DOI] [PubMed] [Google Scholar]
- 87. Wallington-Beddoe CT, Don AS, Hewson J, et al. Disparate in vivo efficacy of FTY720 in xenograft models of Philadelphia positive and negative B-lineage acute lymphoblastic leukemia. PLoS One. 2012;7:e36429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu Q, Zhao X, Frissora F, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111:275-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408-2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang JD, Takahara S, Nonomura N, et al. Early induction of apoptosis in androgen-independent prostate cancer cell line by FTY720 requires caspase-3 activation. Prostate. 1999;40:50-55 [DOI] [PubMed] [Google Scholar]
- 91. Azuma H, Takahara S, Ichimaru N, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410-1419 [PubMed] [Google Scholar]
- 92. Lee TK, Man K, Ho JW, et al. FTY720; a promising agent for treatment of metastatic hepatocellular carcinoma. Clin Cancer Res. 2005;11:8458-8466 [DOI] [PubMed] [Google Scholar]
- 93. Matsuoka Y, Nagahara Y, Ikekita M, Shinomiya T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br J Pharmacol. 2003;138:1303-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222-231 [DOI] [PubMed] [Google Scholar]
- 95. McConechy MK, Anglesio MS, Kalloger SE, et al. Subtype-specific mutation of PPP2R1A in endometrial and ovarian carcinomas. J Pathol. 2011;223:567-573 [DOI] [PubMed] [Google Scholar]
- 96. Nagendra DC, Burke J, Maxwell GL, Risinger JI. PPP2R1A mutations are common in the serous type of endometrial cancer. Mol Carcinog. 2012;51:826-831 [DOI] [PubMed] [Google Scholar]
- 97. Esplin ED, Ramos P, Martinez B, Tomlinson GE, Mumby MC, Evans G. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer. 2006;45:182-190 [DOI] [PubMed] [Google Scholar]
- 98. Takagi Y, Futamura M, Yamaguchi K, Aoki S, Takahashi T, Saji S. Alterations of the PPP2R1B gene located at 11q23 in human colorectal cancers. Gut. 2000;47:268-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chou HC, Chen CH, Lee HS, et al. Alterations of tumour suppressor gene PPP2R1B in hepatocellular carcinoma. Cancer Lett. 2007;253:138-143 [DOI] [PubMed] [Google Scholar]