

Abstract
The retinoblastoma tumor suppressor protein (pRB) plays an integral role in G1-S checkpoint control and consequently is a frequent target for inactivation in cancer. The RB protein can function as an adaptor, nucleating components such as E2Fs and chromatin regulating enzymes into the same complex. For this reason, pRB’s regulation by posttranslational modifications is thought to be critical. pRB is phosphorylated by a number of different kinases such as cyclin dependent kinases (Cdks), p38 MAP kinase, Chk1/2, Abl, and Aurora b. Although phosphorylation of pRB by Cdks has been extensively studied, activities regulated through phosphorylation by other kinases are just starting to be understood. As well as being phosphorylated, pRB is acetylated, methylated, ubiquitylated, and SUMOylated. Acetylation, methylation, and SUMOylation play roles in pRB mediated gene silencing. Ubiquitinylation of pRB promotes its degradation and may be used to regulate apoptosis. Recent proteomic data have revealed that pRB is posttranslationally modified to a much greater extent than previously thought. This new information suggests that many unknown pathways affect pRB regulation. This review focuses on posttranslational modifications of pRB and how they influence its function. The final part of the review summarizes new phosphorylation sites from accumulated proteomic data and discusses the possibilities that might arise from this data.
Keywords: phosphorylation, methylation, acetylation, cell cycle, proteomics
Introduction
The decision to enter the cell cycle and initiate DNA replication is an intricately choreographed process by the cell and results from the integration of information regarding the nutritional state, presence or absence of growth factors, and other external signals. Inherent to the cell cycle are checkpoints that monitor the state of the cell and block progression until all the requirements to proceed are met.1 One of these checkpoints, called the restriction point, lies at the G1-S phase boundary and is regulated by the retinoblastoma tumor suppressor protein (pRB; hypophosphor- ylated human retinoblastoma protein)2,3 In higher metazoans, pRB is one of a family of proteins including p107 and p130 collectively called the “pocket proteins.”4 Although these proteins retain a high degree of structural similarity, they perform distinct functions with respect to regulation and protein binding partners.5 The tumor suppressor activity of pRB resides mainly in its ability to restrict growth by suppressing the activity of members of the E2F family of transcription factors both through direct binding and in the recruitment of co-repressors such as chromatin remodeling factors.3 The importance of the E2Fs lies in their function as regulators of cell cycle genes and, in particular, genes necessary to traverse S phase.6,7
The underlying etiology of cancer involves a loss of proliferative control, and because regulation of G1 progression is highly sensitive to oncogenic processes,2,8 disruption of the pRB pathway appears to be universal in almost all cancers.9 The human retinoblastoma gene RB1 was originally identified as the susceptibility locus for the childhood cancer syndrome retinoblastoma. In addition to developing retinal malignancies, retinoblastoma survivors also show a predisposition to osteosarcoma.10,11 However, among other cancers, only small cell lung cancer is strongly associated (90%) with direct mutations in RB1. 12,13 Indeed, tumors harboring mutations in RB1 generally occur infrequently.14,15 Rather, in most cancers pRB function is lost due to dysregulation of its upstream regulators, which renders it constitutively inactive with respect to cell cycle control.9,12,16
Much of the function of pRB can be understood within the context of posttranslational modifications and in particular phosphorylation. Protein posttranslational modification is one of the fundamental mechanisms underlying cellular metabolic regulation.17 In almost all instances, biochemical information is transduced through molecular changes, such as phosphorylation/dephosphorylation, made to existing proteins or by the targeted degradation of specific proteins through ubiquitinylation. In cells resting in G0, or in early G1, pRB is largely dephosphorylated and is often described as hypophosphorylated (Fig. 1). Under these conditions, pRB binds to E2Fs to form a repressor complex.3 However, as the cell progresses through G1 to S phase, pRB is sequentially phosphorylated at specific sites by cyclin D/Cdk4/6 in early G1 and cyclin E/Cdk2 in late G1.3,18 Phosphorylation leads to conformational changes and reduced affinity for pRB’s interacting partners.19,20 Complete dissociation occurs at the restriction point toward the end of G1 and allows for the E2F dependent transcription of S-phase specific genes.2,3,8,21
Figure 1.

A model for cell cycle regulation by pRB. In G1, pRB is mostly dephosphorylated (hypophosphorylated) and binds to E2F/DP1 heterodimers to repress transcription. As part of the repressor complex, pRB also recruits chromatin modifying proteins that contribute in the repression of E2F dependent transcription. As the cell progresses through G1, hyperphosphorylation of pRB (ppRB) by cyclin D/Cdk4/6 and cyclin E/Cdk2 compromises the integrity of the repressor complex, resulting in the dissociation of E2F and allowing for the transcription of S-phase genes.
In addition to its role in G1 checkpoint control, pRB performs many other functions. Included among these are regulation of apoptosis, differentiation, chromosomal stability, and senescence.15 Many if not all of these functions are governed by posttranslational modifications. For example, within the context of G1 progression, hyperphosphorylation is equated with loss of E2F binding, whereas under conditions where DNA is damaged, hyperphosphorylated ppRB retains the ability to bind E2F1.22-24 Although these interactions remain poorly characterized, current data suggest that phosphorylation of pRB under these circumstances may be distinct from that observed in normal S-phase.22,24 This implies the involvement of kinases other than Cdks, and indeed pRB has been observed to be a substrate for kinases such as p38 MAPK.25-28 In addition to p38 MAPK, pRB has been shown to be a target of other serine/threonine kinases and also the tyrosine kinase c-Abl.18 Although the functional aspects of these phosphorylation events are just beginning to be appreciated, they point to an emerging level of complexity inherent to pRB regulation.
Another layer of transcriptional control, in addition to direct regulation of E2F transcription factors, is pRB’s ability to recruit chromatin modifying proteins such as histone deacetylases and methyltransferases.29 The RB protein is acetylated and methylated in response to a number of external stimuli,18 and these modifications are important in dictating specific functions of pRB within a given context. In addition, acetylation and methylation of pRB are necessary prerequisites for facilitating interactions with proteins involved in differentiation and transcriptional repression.30-34 pRB is also a target for ubiquitinylation and SUMOylation. The Mdm2 oncoprotein binds to pRB and mediates its ubiquitinylation and subsequent degradation.35,36 The role of SUMOylation in pRB function is less clear. Given that viral proteins such as adenovirus E1A inhibit pRB function and also antagonize SUMOylation,37 this modification is likely part of an activation signal. Since SUMOylation is required for senescence,38,39 this cell cycle exit paradigm may use this modification of pRB as part of this process.
In a recent review, Munro et al.18 summarized many of the posttranslational modifications inherent to pRB function, in particular phosphorylation by Cdks as it relates to the function of pRB during the G1-S phase transition. However, due to the advent of proteomics, it has become apparent that phosphorylation of pRB is far more extensive than has thus far been realized. Many of these “undocumented” phosphorylation sites occur within the context of specific physiological paradigms and suggest additional regulatory functions for pRB. The purpose of this review is to provide a comprehensive summary of the posttranslational modifications that influence pRB function particularly as it is defined by protein-protein interactions.
Phosphorylation
Serine/Threonine Phosphorylation
Regulation of pRB-E2F interactions during the G1 phase by Cyclin-Cdk phosphorylation
The initial observations linking pRB phosphorylation and the cell cycle were based on altered electrophoretic migration rates that correlated with different stages of the cell cycle.40 In noncycling, quiescent cells (G0 phase) or cells in early G1, pRB is phosphorylated at only a few Cdk sites41,42 and is bound to E2F transcription factors.3 As cells progress through G1 toward S phase, pRB is increasingly phosphorylated, resulting in the dissociation of E2F and facilitating the transcription of S phase genes (Fig. 1). Phosphorylation of pRB by Cdks has been extensively studied, and consequently much is known about the process. Analysis of human pRB reveals 12 sequences matching the consensus phosphorylation motif of S/T-P-X-K/R for Cdks.43 Using the minimum sequence requirement for Cdk of S/T-P, a total of 16 sites are found in 3 main areas within pRB (Fig. 2). Seven sites are clustered between S780 and T826 at the carboxyl-terminus and a further 6 in the amino terminus between T5 and T373.44-46 The remaining sites (S567, S608, S612) are found within the small pocket.44,45 Murine pRb is missing T5 but contains an additional site at S364, corresponding to P370 in human.44 Although the minimum consensus sequence S/T-P applies for all Cdks, upstream or downstream amino acid sequence may contribute to specificity among the Cdk family.46,47 By a combination of site specific methods and mass spectrometry, phosphorylation of all 16 consensus Cdk phosphorylation sites in pRB has been observed.44-46,48-52 In actively cycling cells, the initial phosphorylation events in early G1 phase are catalyzed by Cdk4 or 6 activated by cyclin D41,46 and can be inferred from in vitro phosphorylation data to occur on S249, T252, T356, S608, S788, S807, S811, and S826.46 Thereafter, phosphorylation by cyclin D/Cdk4/6 and/or cyclin E/Cdk2 phosphorylates T5, T373, and S795. Finally, in late G1, phosphorylation of S612 and T821 occurs mediated by cyclin E Cdk2.46 Much of the data described above were obtained by measuring in vitro kinase specificities followed by phosphopeptide mapping or by site directed mutagenesis of consensus Cdk sites followed by reporter or in vitro kinase assays. Together these studies reveal a mechanistic framework for understanding the sequential nature of pRB phosphorylation.
Figure 2.

Schematic representation of the Cdk phosphorylation sites in pRB. Position of the consensus Cdk phosphorylation sites in relation to the pRB protein is indicated. Phosphorylation of S230 has not been confirmed by either site directed methods or mass spectrometry in human pRB, but phosphorylation of the corresponding serine in mouse pRb (S224) has been observed by site directed methods (see text). Unless specified, amino acid numbering throughout the text will refer to human pRB. The A and B domains of the small pocket and large pocket and the carboxyl-terminus are indicated.
Phosphoprotein analysis by mass spectrometry from cells exposed to a variety of conditions has confirmed the phosphorylation of 13 of the 16 consensus Cdk sites in human pRB. Dephoure et al.51 and Olsen et al.53 mapped the cell cycle stage specific phosphoproteome in HeLa cells arrested in G1 by a double thymidine block or in M phase by nocodazole. In both studies, extensive pRB phosphorylation was observed in M phase, but only S249 and T252 were observed to be phosphorylated in G1. 51 In M phase pRB was determined to be phosphorylated on 10 of the known Cdk phosphorylation sites (S249, T252, T356, T373, S608, S612, S807, S811, T821, and T826).51,53 Huttlin et al.50 compiled an atlas of murine protein phosphorylation in specific tissues—namely brain, brown fat, heart, kidney, liver, lung, pancreas, spleen, and testis—and identified 12 of the known Cdk sites commonly shared between human and rodent (corresponding to S249, T252, T350, T373, S608, S612, S780, S795, S807, S811, T821, and T826) as being phosphorylated. They also identified murine T364 corresponding to P370 in human pRB as being phosphorylated.50 Proteomic analysis by others, of both human and murine cell lines, is consistent with the studies discussed above.54-57 Thus, phosphoproteomic approaches are revealing many of the same phosphorylation events as mutational analysis and phosphopeptide mapping, the exceptions being T5, S230, and S567.
A number of studies have attempted to identify critical phosphorylation sites that regulate pRB-E2F interactions. Brown et al.44 attempted to address this by expressing combinatorial pRB mutants in pRB null C33A cancer cells. It was concluded that the effect of phosphorylation on pRB was cumulative, with no single site or combination of sites predominating. Others have observed that the central pocket region (or small pocket) of pRB spanning amino acids 379-792 is sufficient to confer transcriptional repression on E2F and that this is not affected by co-expression of this region with cyclin D1 and cyclin E.58,59 In contrast, transcriptional repression was relieved when the pocket domain including the C-terminal region (large pocket) was expressed with Cdk 4 and Cdk 2, suggesting an important regulatory role for the C-terminal Cdk phosphosites.59 In agreement with these findings, Knudsen and Wang60 observed that phosphorylation of the C-terminal sites inhibited binding of the large pocket to E2F, and Rubin et al.61 observed that phosphorylation of S788 and S795 and of T821 and T826 induce allosteric changes to pRB that destabilize the interaction with E2F.61 Moreover, these conformational changes unmask other phosphorylation sites which, upon phosphorylation, induce further alterations in secondary structure, ultimately resulting in the loss of the binding interface required by E2F.19,45,49,61 Phosphorylation at T821 and T826 also disrupts binding of the histone deacetylases HDAC1 and HDAC2, which facilitate transcriptional repression by helping to maintain chromatin in a closed conformation.49,62,63 In contrast to the C-terminus alone, pRB mutants in which all the C-terminal and linker phosphorylation sites were mutated, leaving just the amino terminal Cdk sites (T5, S230, S249, T252, T356, and S567), were still capable of G1 arrest, and binding to E2Fs was unaffected,60 indicating the importance of the carboxyl-terminal Cdk phosphorylation sites in regulating the pRB-E2F interaction.
In another approach, the roles of specific Cdk phosphorylation sites were determined by measuring the relative binding affinities of purified phosphorylated pRB Cdk mutants for an E2F derived peptide. In this case, a pRB mutant phosphorylated at S608/S612 in the linker region and T356/T373 in the amino-terminal region displayed a binding affinity for E2F similar to that of a fully phosphorylated wild-type.19,20 Phosphorylation of these sites results in intramolecular rearrangements within the pocket domain that occlude E2F and prevent binding.19,20 This suggests that discrete aspects of pRB-E2F interactions are competed unambiguously by single phosphorylation modifications. In summary, phosphorylation of the C-terminal Cdk sites by cyclin D/Cdk4/6 initiates structural changes to pRB resulting in the establishment of intramolecular interactions leading to the exposure of secondary Cdk phosphorylation sites. As the cells enter late G1, the exposed Cdk sites at T373 and S612 are phosphorylated by cyclin E/Cdk2, leading to conformational changes within pRB and resulting in a loss of the E2F binding cleft within the pocket domain. Taken together, the picture that is emerging indicates that different combinations of phosphorylation events catalyzed by Cdks inactivate pRB by changing the binding affinity for E2Fs and possibly other interacting proteins as well.
Phosphorylation by Cdk9
Cdk9 is another cyclin dependent kinase related to Cdk1, but it is regulated by 2 different cyclins, cyclin T and cyclin K, neither of which exhibits cell cycle regulation.64 Consequently, Cdk9 activity does not fluctuate with the cell cycle.64 The function of Cdk9 differs depending on which cyclin it is associated with. With cyclin T, Cdk9 regulates transcription by forming a complex with and stabilizing Pol II as the positive transcription elongation factor b complex.64 When in a complex with cyclin K, Cdk9 functions in response to replicative stress where it accumulates on chromatin to mitigate the effects of accumulating single stranded DNA.65 Cdk9 phosphorylates pRB exclusively on serine residues66,67 subsequently mapped to S795, S807, and S811.68 The functional significance of Cdk 9 phosphorylation of pRB is also not known since these phosphorylation sites were identified by in vitro phosphorylation assays of pRB by Cdk9. Because Cdk9 does not undergo cell cycle regulation and levels of active enzyme and cyclin T are high in terminally differentiated cells,64,69 it is unlikely to be involved in proliferative control of actively cycling cells.
Regulation of pRB function by Cdk3 phosphorylation during G0 to G1 phase
Under conditions such as nutrient deprivation, cells exit the cell cycle and move into a state of quiescence termed G0.21 The role of pRB in the G0-G1 transition was suggested in experiments in which knockdown of pRB in quiescent fibroblasts initiated a rapid reentry into the cell cycle.70 In G0, pRB is considered to be nonphosphorylated,41,42 but hypophosphorylation of pRB has been proposed to be necessary in order to initiate G1.41 However, while levels cyclin D are low at the onset of G1, but expression of cyclin C peaks at the transition between G0 and G1.71 In an extensive study to determine the function of pRB in the transition from G0 to G1, Ren and Rollins72 observed phosphorylation by cyclin C/Cdk3 on S807/S811. Moreover, phosphorylation of pRB at these sites was shown to be necessary for an efficient G0-G1 transition, as SAOS-2 cells expressing a double S807/S811 mutant transitioned to G1 at a much lower frequency than cells overexpressing wild-type pRB or cyclin C.72 Although these sites are also targets for Cdk4/6 and Cdk2, pRB phosphorylated in vitro by Cdk3 and microinjected into cells cannot inactivate E2F function,52 suggesting that when present, cyclin C/Cdk3 phosphorylation of S807/S811 mediates pRB functions specific for G0 exit.72
pRB phosphorylation by Cdk5 in neurons
Reentry of neurons into the cell cycle results in apoptosis and cell death and neurodegenerative diseases such as Alzheimer’s and Parkinson’s have been linked to aberrant cell cycle regulation.73-75 The mechanism as to how neurons lose their postmitotic status and enter the cell cycle is somewhat controversial; however, the atypical Cdk5 appears to play a central role. This kinase is unique in that although structurally similar to other Cdks, it is not regulated by any known cyclin but rather is regulated by p35 and p39 and is localized primarily in the cytoplasm.76-79 The regulatory protein p35 is normally myristoylated and membrane associated; however, under neurotoxic conditions such as high ROS, p35 is cleaved into a soluble 25 kDa fragment that binds and activates Cdk5,79 and a number of reports have linked p25/Cdk5 with cell cycle dysregulation and neurotoxicity.77,80-82 In neuronal-like SY5Y cells compelled to reenter the cell cycle by overexpressing p25, pRB was phosphorylated at S795 and S807/811.83 Phosphorylation was shown to be Cdk5 dependent, as opposed to Cdk4 and Cdk6, in vivo by differential inhibition and by in vitro kinase reactions.83 In another study, a pool of nuclear Cdk5 was shown to phosphorylate all the C-terminal pRB phosphosites (S780, S788, S795, S807, S811, T821, and T826) in mouse cerebellar and cortical neurons, and this correlated with increased E2F1 transcriptional activity.84 Thus, it appears that dysregulation of pRB function by overactive p35 or p25, or by mislocalization of Cdk5 to the nucleus, results in activation of E2F and reentry into the cell cycle.
Phosphorylation by Other Serine/Threonine Kinases
p38 MAP kinase
In addition to undergoing Cdk mediated phosphorylation, pRB is also phosphorylated by p38 MAPK. The p38 MAPK pathway may be activated by mitogenic stimulation85 but is mostly associated with pathways activated in response to cell stress such as oxidative stress and DNA damage.86-88 In studies of nonmitogenic stimuli on pRB function, Wang and colleagues observed pRB to be directly phosphorylated by p38.25,26 In addition, phosphorylation of pRB in vivo following TNFα stimulation of Jurkat T cells was independent of Cdk activity and corresponded with elevated levels of p38 activity and transcription of E2F1 target genes.25,26 Subsequently, others have observed p38 phosphorylation and inactivation of pRB in response to nonmitogenic cues in other cell lines.27,28,89,90
Although it appears that p38 phosphorylates pRB directly, only limited information exists concerning the location of these sites. Faust et al,85 using a phosphospecific antibody, observed pRB to be phosphorylated on S807/S811 in a p38 dependent fashion under mitogenic conditions. Again, using a phosphospecific antibody, Yeste-Valasco et al.28 and Cho et al.90 observed S780 to be a target for direct phosphorylation by p38 in cerebellar granular neurons and the hepatocyte cell line AML12, respectively. Similarly, Delston et al.89 observed p38 mediated phosphorylation of S567 of pRB using an antibody specific for phosphorylation of this site. Phosphorylation of S567 of pRB resulted in an association with the E3 ubiquitin ligase Mdm2 resulting in pRB degradation, loss of E2F1 repression, and active transcription of proapoptotic genes.89 This latter result is surprising in the sense that a previous report suggested that this site only becomes exposed upon Cdk mediated phosphorylation of C-terminal phosphorylation sites.49 However, p38 phosphorylation of S567 occurred independently of C-terminal phosphorylation suggesting that alternate mechanisms must exist to facilitate p38 phosphorylation of S567. Thus, pRB phosphorylation by p38 is part of a proapoptotic signal.
All the p38 MAPK phosphorylation sites identified are also targets for Cdk phosphorylation. This contrasts with the observations of Nath et al,26 who indicated that p38 MAPK phosphorylation of pRB occurred at sites distinct from those of Cdk. Like other members of the MAPK family, p38 MAPK is a proline directed kinase91,92 and phosphorylates sequences lying within the general consensus of P-X-S/T-P and S/T-P; however it may also phosphorylate sequences that diverge from this generally accepted consensus.93-95 In addition, upstream docking sites may be more important in determining the site of phosphorylation than the individual consensus sequence.96 Therefore, the possibility exists that p38 phosphorylation of pRB is not restricted to specific MAPK consensus sequences. This variability of sites that are phosphorylated raises the question of how they are selected and contribute to this distinct cellular outcome. More work is clearly needed in this area.
Phosphorylation by Chk1/2
The checkpoint kinases Chk1 and Chk2 play an integral role as regulators of cell survival under conditions of genotoxic stress. These kinases are activated by upstream “sensor” kinases ATM and ATR in response to double stranded DNA breaks, or replicative stress, which may lead to the accumulation of single stranded DNA.97,98 The Chk kinases function in the S and G2 phases and initiate growth arrest in response to DNA damage.97,98 Inoue et al.99 observed an increase in S612 phosphorylation in pRB in cells that had undergone growth arrest due to genotoxic stress. Although S612 is phosphorylated by Cdk4/6, reduced phosphorylation at other Cdk4/6 or Cdk2 phosphorylation sites suggested that S612 was being phosphorylated by another kinase activated in response to DNA damage.99 Further experiments indicated that both Chk1 and Chk2 phosphorylate pRB at S612 and that Chk2 phosphorylation was ATM dependent.99 E2F1 is involved in transcription of proapoptotic genes in response to DNA damage, and E2F1-pRB complexes have been observed in cells that have undergone growth arrest due to DNA damage.22,24 Phosphorylation of S612 by Chk2 increases the affinity for pRB toward E2F1, and alanine substitution at S612 both decreased the pRB/E2F1 interaction and compromised the E2F1 dependent transcription of proapoptotic genes in DNA damaged cells.99 It is interesting that phosphorylation of S612 by Chk1/2 promotes pRB/E2F1 interactions under these conditions, whereas at the G1-S checkpoint, Cdk phosphorylation of S612, in combination with phosphorylation of S608 and T356/T373, reduced the affinity of the E2F1 transactivation domain for pRB by about 250-fold.19 These differing results indicate that although different kinases phosphorylate the same sites on pRB, it is the cellular context and combination of phosphorylated sites, along with potentially other posttranslational modifications, that dictate pRB function for a given physiological state.22
Phosphorylation by Aurora kinase
In addition to having a role in G1-S checkpoint control, pRB functions in the maintenance of chromosome stability. Indeed, chromosomal instability is a characteristic feature of most solid tumors,100 a primary consequence of which is aneuploidy that arises as a result of the failure of chromosomes to segregate properly.100 Loss of pRB has been observed to cause polyploidy or aneuploidy in a number of cell types,101,102 and one function of pRB is the recruitment of chromatin modifying proteins to the centromere.101,103,104 The serine/threonine Aurora b kinase is a member of the Chromosomal Passenger Complex and functions in regulating the attachment of the mitotic spindle to the centromere.105 Transcription of Aurora b is cell cycle dependent, beginning in G2 106 and continuing through mitosis. Aurora b shows dynamic changes in localization as the cell progresses through mitosis associating with chromosomes, inner centromeres, microtubules at the central spindle, the cleavage furrow, and finally the midbody.106 As a key regulator of mitotic fidelity, Aurora b misregulation has been implicated in a variety of cancers.106,107 Aurora b phosphorylates a large array of proteins including pRB.105,108 In a series of experiments using a combination of siRNA and selective Aurora b inhibitors, Nair et al.108 observed that pRB hypophosphorylation could be uncoupled from Cdk inhibition under conditions of pseudo-G1 cell cycle arrest.108 Inhibition of Aurora b led to polyploidy, which was mitigated by knockdown of pRB and inhibition or pRB dephosphorylation.108 The suggestion that pRB was a potential downstream substrate of Aurora b kinase was confirmed by both in vitro and in vivo kinase assays identifying S780 as the target residue. Expression of a phosphomimetic S780D pRB mutant in SaOS2 cells provided a measure of protection against polyploidy induced by Aurora b inhibition, suggesting that the function of pRB in preventing endoreduplication is dependent on Aurora b.108 Interestingly, the primary effect of phosphorylation of pRB on S780 by Aurora b was to stabilize the E2F1-pRB interaction.108 In forming this stable interaction, it was argued that phosphorylation of pRB by Aurora b serves as a checkpoint after a failed mitosis to prevent cell cycle reentry and polyploidy.108
Interaction with Raf1 and Ask1 kinases
Mitogenic signals propagated by activated cell surface receptors converge on Ras and require Cdk mediated inactivation of pRB for cell cycle progression.109-113 The serine/threonine kinase c-Raf1 functions to activate the MAPK/Erk pathway in response to growth factors as a downstream effector of Ras.114 Upon activation by Ras, Raf1 interacts directly with pRB early in G1 preceding cyclin D/Cdk 4/6 binding.115,116 This interaction is necessary to alleviate the repressor activity exerted by pRB on E2F, partly by disrupting the LXCXE mediated interaction between Brg1 and pRB,116 and is dependent upon Raf1 kinase activity.115-118 Despite this interaction, phosphorylation of pRB by Raf1 has only been observed in vitro and the sites of phosphorylation have not been assigned.115
The apoptosis signal-regulating kinase 1, Ask1, is also a member of the MAPKKK family of serine/threonine kinases.119 Unlike Raf1, Ask1 does not activate the MAPK/Erk pathway but rather is involved in the activation of stress response pathways mediated by JNK and p38 and as such plays a role in apoptosis and other cellular stress responses.119 In addition, Ask1 has been linked to a number of malignancies such as colon, skin, and gastric tumors.120 In human gastric cancer cells, Ask1 regulates cyclin D1 expression and knockdown of Ask1 arrests cells in G1.120 Ask1 interacts with pRB through the LXCXE binding motif in response to apoptotic signals and phosphorylates pRB in vitro in an LXCXE dependent fashion, although the sites of phosphorylation have not been identified.121 In addition, binding of Ask1 alleviates the pRB mediated repression of E2F1 by destabilizing the pRB/E2F1 interaction,121,122 and knockdown of Ask1 or disruption of the pRB/Ask1 interaction mitigates the apoptotic effects of TNFα in human aortic epithelial cells.122 The fact that Ask1 mutants that are incapable of binding pRB could not induce apoptosis suggests that this interaction contributes to a balance between the proapoptotic function of E2F1 and the antiapoptotic functions of pRB.121,122 It is interesting to note that both Raf1 and Ask1 compete for binding to pRB in vitro, but binding of these kinases in vivo is governed by different pathways indicating a level of specificity.121
Tyrosine Phosphorylation
Although much is known about serine/threonine phosphorylation of pRB, little is known about tyrosine phosphorylation. Tyrosine phosphorylation in response to external stimuli serves a fundamental regulatory role in almost all facets of cellular physiology. Many tyrosine kinases are rendered oncogenic by mutations that lead to dysregulation,123 and because of this many protein tyrosine phosphatases are tumor suppressors.124 Unlike serine/threonine phosphorylation, which is to a large part structural, tyrosine phosphorylation is mostly regulatory and a large number of proteins bind phosphotyrosine motifs.123 There are 28 tyrosine residues in pRB, of which 8 are phosphorylated under a variety of conditions (Fig. 3).57 Most of these sites are uncharacterized, and little is known as to their function. This part of the review focuses on what is known about tyrosine phosphorylation of pRB and discusses possible functions for it.
Figure 3.
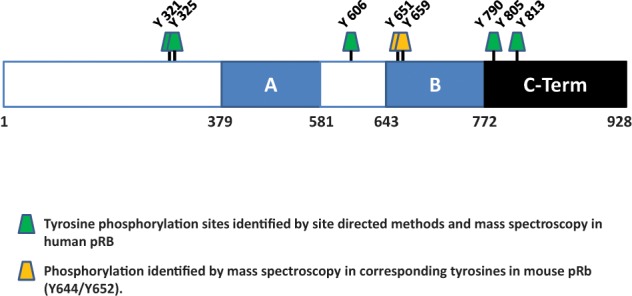
Identified tyrosine phosphorylation sites in pRB. Sites of tyrosine phosphorylation in pRB identified by site specific methods or mass spectrometry are indicated. Tyrosine phosphorylation at Y651/Y659 has yet to be observed in human pRB, but phosphorylation of the corresponding tyrosines (Y644/Y652) in murine pRb has been identified by mass spectrometry (see text). The site at Y805 in pRB is a target of the c-Abl tyrosine kinase.
Phosphorylation by c-Abl
The non–receptor tyrosine kinase c-Abl is ubiquitously expressed and is found in both the cytosol and the nucleus. Observations that pRB and c-Abl interact in a phosphorylation dependent manner125,126 and that overexpression of c-Abl induces cell cycle arrest suggested that c-Abl functions as a negative growth regulator through pRB.126-128 The interaction between pRB and c-Abl is mediated through the C-terminus of pRB interacting with the kinase domain of c-Abl126 and is sensitive to phosphorylation of S807/S811 but not T821/T826, suggesting that it is not mediated through an LXCXE motif.60,129 Although Abl can drive either transformation or growth arrest, depending on the cellular context,128 it also functions in apoptosis and is positively regulated by ATM during genotoxic stress in proliferating cells but not in growth arrested cells.130-132 In G0-G1 cells, Abl is negatively regulated by pRB where binding inhibits Abl activity by masking the kinase domain.125,126
Nagano et al.133 observed constitutive tyrosine phosphorylation on Y805 of pRB in chronic myelogenous tumor (CML) cells expressing a constitutively active Bcr/Abl fusion protein. From a functional aspect, phosphorylation of Y805 by Abl in these cells plays a role in cell survival. These CML cells are dependent on Abl to activate signaling pathways necessary for survival, and inhibition of Abl activity induces apoptosis.133 However, nuclear localization of Bcr/Abl induces apoptosis134 and, as pRB is a nuclear protein, it can be inferred that in these CML cells Bcr/Abl must at least in part be nuclear, suggesting that phosphorylation of Y805 of pRB must mediate an antiapoptotic signal. Indeed, expression of a phosphomimetic Y805D pRB mutant reduced the apoptotic index relative to wild-type pRB, suggesting that when phosphorylated on Y805, pRB is capable of exerting an antiapoptotic effect. This argument is further supported by observations that apoptosis is induced in CML cells upon knockdown of endogenous pRB.133
It is puzzling that binding of Abl to pRB blocks Abl kinase activity, and yet in Abl dependent CML cells, pRB is constitutively phosphorylated by Abl. Indeed, Nagano et al.133 observed virtually no phosphotyrosine immunoreactivity in pRB/Abl co-immunoprecipitates from CML cells upon overexpression of pRB, indicating a lack of Abl kinase activity. However, the Bcr/Abl fusion protein, while retaining Abl tyrosine kinase activity, displays inherently different properties than Abl alone.130 Therefore, the loss of Abl activity upon binding to pRB observed in these cells must occur only when pRB is in excess. Indeed, the cells used in this study are dependent upon Abl activity for survival, suggesting that binding to pRB does not impede tyrosine kinase activity.133 How phosphorylation of Y805 provides protection against apoptosis is open to speculation. However, although Y805 phosphorylation of pRB has been observed by others,57,135 it is not universal133 and has been observed primarily in cancer cell lines. Bearing this in mind, it was suggested that phosphorylation of Y805 by Abl might represent a form of dysregulation of pRB and may facilitate tumorigenesis by protecting transformed cells from apoptosis.133
Acetylation and Methylation of pRB
Acetylation
Another means of posttranslational regulation of pRB function involves acetylation and methylation of lysine and arginine residues. Recruitment of pRB into the p300/CPB transcriptional co-activator complex by binding the viral oncoprotein E1A leads to the acetylation of pRB on K873/K874 in the C-terminus by the histone acetyltransferase activity inherent to this complex (Fig. 4).30 Acetylation at these sites increases the affinity of pRB for Mdm2 and exerts a negative influence on pRB cyclin/Cdk dependent phosphorylation, thus maintaining pRB in an active state.136 Such a state of activation is desirable in cells that are terminally differentiated. Indeed, it was noted that acetylation of pRB increased in differentiating U927 cells.136
Figure 4.

Nonphosphorylation posttranslational modifications of pRB. Sites of acetylation, methylation, ubiquitinylation, and SUMOylation in pRB identified by a combination of site specific methods and mass spectrometry are indicated. *Ubiquitinylation of the corresponding lysine in mouse pRb (K57) has been identified by mass spectrometry (see text). Ubiquitinylation of this site in human pRB has yet to be observed.
Similar to Chan et al,30 Nguyen et al.31 observed acetylation of pRB in differentiating myocytes. In this instance, the initiation of myogenesis in CC42 cells by serum withdrawal of confluent cultures induced P/CAF, the p300/CBP associated factor, to associate with and acetylate pRB on K873/874. In addition, acetylation of pRB was necessary for proper transactivation of MyoD and thus for cell cycle withdrawal and differentiation in CC42 muscle precursor cells.31 Increased P/CAF dependent acetylation of pRB on K873/874 has also been observed in differentiating keratinocytes.32 Similar to the results described previously with CC42 cells, keratinocyte differentiation was impeded by an acetylation defective mutant of pRB in which K873/K874 were replaced with arginine,32 despite the fact that this mutant still retained the ability to arrest growth in SaOS cells.32
Acetylation of pRB at K873/K874 is induced by DNA damage, and although it is expected to protect pRB from phosphorylation as outlined above, it also acts to reduce the interaction between pRB and E2F1.137 Interestingly, a distinct pRB/E2F1 population exists following DNA damage that excludes acetylation.22,24,99 In addition, pRB phosphorylated at S612 by Chk2 appears to also be part of a separate subpopulation of pRB/E2F1 complexes that arises following DNA damage. Taken together, current evidence suggests that a series of pRB containing multiprotein complexes are formed in response to DNA damage and that each is distinguished by unique modifications.22,24,99 In addition to these site specific studies, a proteomic study of global protein acetylation identified acetylated lysine residues at K427, K548, K640, K652, and K896 on pRB (Fig. 4).138 Of these sites, 2 (K640 and K652) lie within or close to the boundary of the B domain of the large pocket, 2 (K427 and K548) lie within the A domain, and K896 is in the C-terminus. Another site at K925 in the C-terminus has also been identified by mass spectrometry.57 In both of these studies, cells were treated with HDAC inhibitors before harvesting, making it difficult to ascertain the state of the cells and therefore the context in which acetylation of these new sites occurs.
Methylation
In contrast to acetylation, which occurs only on lysine residues, methylation occurs on both lysine and arginine residues.139 Methylation has been implicated in transcriptional regulation primarily through the study of histones and chromatin, and in many ways methylation of histones defines the degree of transcriptional activity taking place in given regions of the genome.140 Many nonhistone proteins are also methylated, and like other posttranslational modifications, these influence interactions and function.141-143 Methyltransferases have previously been shown to bind to pRB both directly and indirectly and to regulate E2F transcriptional activity, but it was not clear whether binding of these proteins resulted in pRB methylation.144-148 Recently, the mono-methyltransferase Set7/9 was shown to actively methylate pRB at K873149 and at K81033 (Fig. 4). When pRB is methylated at K873, a docking site for the heterochromatin binding protein HP1 is created.149 HP1 binds methylated histones and functions to repress transcriptional activity by regulating the structure of chromatin.150 Methylation of either K873 or K810 on pRB by SET 7/9 enhances cell cycle arrest,33,149 and K810 methylation exerts a negative effect on global Cdk dependent phosphorylation.33 The effect of K873 methylation on Cdk phosphorylation has yet to be determined. The correlation between increased pRB methylation and cell cycle arrest is consistent with observations that increased K873 methylation in differentiating myoblast cell lines is concurrent with increased pRB and HP1 occupancy at E2F regulated promoters.149 Moreover, decreases in E2F1 transcriptional activity in cells undergoing genotoxic stress are also associated with increased Set 7/9 dependent pRB methylation at K810.33 Recently, pRB has been shown to be methylated on K860 by SMYD2 (Fig. 4),34 a mono-methyltransferase that also methylates p53 resulting in repression of p53 responsive genes.151 Methylation of K860 by SMYD2 allows for the binding to pRB of the malignant brain tumor protein L3MBTL1, a transcriptional repressor that binds to mono and dimethylated histones and silences gene expression by influencing chromatin structure.34 As with Set7/9 methylation at K873 and K810, methylation at K860 increases as cells withdraw from active proliferation.34
In contrast to the studies described above which indicate that methylation of pRB is involved in gene silencing and senescence, in human bladder cancer cells, SMYD2 dependent methylation of pRB at K810 has a positive effect on proliferation by promoting pRB inactivation through phosphorylation of S807/S811.152 In addition, knockdown of SMYD2 in these cells inhibits proliferation.152 Overexpression of SMYD2 has been observed in bladder, colon, prostate, and breast cancer cells and correlates with a poor prognosis with respect to patient survival.152,153 Such a correlation has been observed with another closely related methyltransferase, SMYD3.154,155 It was not determined whether in these cells methylation of pRB results in the recruitment of chromatin remodeling proteins, although given the positive response on proliferation, one might presume not. These data indicate that similar posttranslational modifications can exert opposing effects depending upon the context and that the observed effect is dependent on which pathways are active within a given paradigm.
Ubiquitinylation
Ubiquitin conjugation of lysine residues and targeting of proteins for degradation constitute an acute means of regulation, constantly remodeling the cellular proteome to meet changing physiological needs. It has been shown that pRB may be targeted for proteasomal degradation by interaction with viral proteins such as human papilloma virus E7, which then facilitate pRB proteolysis by recruiting ubiquitin ligases.156-159 For example, the Epstein-Barr nuclear antigen 3C (EBNA3C) binds to pRB and mediates its degradation by recruiting the Skp1/Cul1/F-box (SCFSkp2) ubiquitin ligase complex.159 It is believed that targeting pRB by viral oncogenes plays an integral part in the tumorigenic properties of these viruses.160,161 Ubiquitin dependent proteasomal degradation of pRB is also mediated through interactions with the Mdm2 oncoprotein.35,36 Structurally, Mdm2 contains a C-terminal RING finger that has been shown to function as a ubiquitin E3 ligase for p53.162 Mdm2 overexpression has been implicated in many cancers, and its oncogenic functions revolve largely around its ability to target the ubiquitin dependent degradation of p53.163,164 However, part of the oncogenic functions of Mdm2 may also involve inappropriate regulation of the pRB pathway. An inverse relationship between pRB and Mdm2 has been observed in non–small cell lung cancer,36,165 and the reduced level of pRB has been linked to Mdm2 mediated degradation.36 Genotoxic stress has also been shown to induce Mdm2 facilitated pRB degradation. In this instance, phosphorylation of S567 by stress activated p38 promotes Mdm2 binding and degradation of pRB.89 Under these conditions, degradation of pRB leads to E2F1 mediated transcription of proapoptotic genes, resulting in cell death.89 However, Mdm2 can promote ubiquitin dependent and independent protein degradation,166 and this was not determined for pRB degradation in the context of S567 phosphorylation.
Analysis by mass spectrometry reveals that human pRB contains 4 ubiquitinylation sites, at K143, K265, K574, and K810 (Fig. 4).57,167,168 K57 in mouse hypophosphorylated murine retinoblastoma protein (pRb) (K63 in human) has also been identified as a ubiquitinated lysine, and it is likely that K63 in pRB is also a target for ubiquitinylation.57 In an examination of the ubiquitome in HEK293T and HCT116 cells treated with proteasomal inhibitors, Kim et al.167 observed K143 to be the only modified lysine residue in pRB. In a similar study with HEK293T and MV4-11 cells treated with a different proteasomal inhibitor, MG-132, Wagner et al.168 found K810 to be modified by ubiquitin. Whether distinct lysine residues are key for pRB degradation in different physiological contexts, or whether these discrepancies are more related to the extent of coverage possible in most mass spectrometric experiments, has yet to be established.
SUMOylation
Similar to ubiquitinylation, SUMOylation involves the covalent attachment of a small protein moiety to specific lysines on a target protein. Attachment of SUMO (Small Ubiquitin-like Modifier) to a target protein can have profound effects on protein function, from influencing protein-protein interactions to directing proteins for ubiquitin-dependent proteasomal degradation.169 There are 4 distinct human SUMO paralogues designated SUMO-1 to SUMO-4 with varying levels of similarity.170 Hypophosphorylated pRB is SUMOylated by SUMO-137 and by SUMO-2/3.39 The site of SUMOylation on pRB was mapped to K720, which lies in a cluster of lysine residues surrounding the LXCXE binding cleft in the “B” domain of the small pocket (Fig. 4).37 Binding of LXCXE containing proteins blocked SUMOylation at K720 but SUMOylation did not block the binding of these proteins, indicating that the structure of the LXCXE binding cleft was not compromised by SUMOylation.37 In addition, SUMOylation enhanced the ability of pRB to repress E2F transcription,37 and overexpression of the SUMO E3 ligase PIASy or processed forms of SUMO2/3 induced senescence in a pRB dependent manner concomitant with an increase in the occupancy at E2F target promoters by pRB.38,39 However, although induction of a senescent phenotype by overexpression of SUMO2/3 involved SUMOylation of pRB,39 it was not clear whether PIASy did likewise, although evidence indicated that PIASy interacted with the small pocket domain of pRB and was found with pRB to occupy E2F target promoters in senescent cells.38 In this sense, although the outcome of SUMO2/3 and PIASy overexpression is the same, the mechanism may be slightly different.
Phosphorylation of pRB in the Absence of Known Function
Serine/threonine phosphorylation
As discussed above, phosphorylation by multiple kinases plays a pivotal role in the function of pRB, and many of these phosphorylation sites have been identified. Recent data obtained by proteomic analysis of global posttranslational modifications in cells exposed to various conditions have validated much of the information regarding pRB phosphorylation but have also shown that phosphorylation of pRB is more extensive than previously thought (Fig. 5). Although much can be learned from these data, most of this information is collated with minimal functional context. In addition, because of the scale of many of these studies, the kind of rigorous analysis more typical of site-specific approaches is impossible. Therefore, it is important that phosphosites identified as part of phosphoproteomic studies be independently confirmed. With that said, phosphoproteomic approaches have identified additional phosphorylation sites that we have divided into 2 categories based on observational frequency and are summarized in Fig. 5. In mapping the mitotic phosphoproteome, Dephoure et al.51 and Olsen et al.53 reported phosphorylation of 10 of the known Cdk target sites and 3 additional sites not previously described—S37, T823, and S855—none of which are proline directed and therefore are unlikely to be targets of Cdks.51,53 Phosphorylation of these residues has been identified in other proteomic studies.50,54-57,171-174 In compiling an atlas of murine tissue specific protein phosphorylation, Huttlin et al.50 identified previously undocumented phosphosites corresponding to S601, T778, S794, and S855 in pRB. These phosphorylation sites have also been identified by others.51,57,175,176 Of the other undocumented phosphorylation sites of note, S624 and T841 have also been identified in multiple studies (Fig. 5).54,56,57
Figure 5.

Non-Cdk targeted serine/threonine phosphorylation sites on pRB identified in proteomic studies. Information regarding the sites of phosphorylation was obtained from the literature and from the PhosphoSite database (www.phosphosite.org).
Although the above phosphosites have been observed multiple times, other sites have been observed only a minimal number of times. Phosphorylation of T583 and S588 was observed in studies on nuclear phosphorylation dynamics during DNA damage in differentiating stem cells173,174 and S625, S838, T842, and S882 were observed in Jurkat T lymphocytes.56 Phosphorylation of S838 and T842 was also observed by Hoffert et al.177 in glomerular kidney cells. The PhosphoSite database (www.phosphosite.org) maintained by Cell Signaling Technology (CST)57 offers a comprehensive list of all the phosphorylation sites obtained from published reports and proteomic studies, including in-house experiments conducted by CST. Many of the studies conducted by CST are antibody based PTMScan validation studies using a variety of established cell lines, many of them cancer cell lines. A search through this database reveals additional unpublished sites including T140, S163, S347, S350, T605, S618, S829, S895, and T905 (Fig. 5). Many of these sites do not conform to any kinase consensus recognition sequence, making them novel phosphorylation motifs.178 In addition, many of these sites were identified in Jurkat T leukemia cells under conditions of phosphatase inhibition, raising the possibility that phosphorylation of these sites may be transitory and under these conditions phosphorylation may be forced. However, the fact that phosphorylation of these sites is observed is interesting and merits further investigation.
Tyrosine phosphorylation
As noted previously, the only characterized pRB phosphotyrosine site is the c-Abl phosphosite at Y805. However, phosphoproteomic analysis reveals 8 tyrosine sites, including Y805, as being targets of phosphorylation on pRB (Fig. 3). Tyrosine kinases function in the regulation of cell growth, differentiation, and survival. Dysregulation of these kinases is commonly associated with malignant transformation and cancer through inappropriate activation of downstream pathways. Moreover, phosphotyrosine residues serve as docking sites for SH2 and PTB domains and in this way function as scaffolds in multiprotein signaling complexes. Because of this, studies have been undertaken with a view to understand the ramifications of dysregulated tyrosine kinase activity in malignancy. Phosphorylation of Y790, Y606, and Y813 has been observed in several studies.50,57,176,179 Other sites, Y644 (Y651 in human pRB) and Y652 (Y659 in human), have been observed in untreated C2C12 mouse myoblasts.57
Two separate studies identified Y321 and Y325 to be phosphorylated in non–small cell lung cancer cells and in highly metastatic hepatocellular carcinoma cells.180,181 However, although these studies profiled their phosphorylation targets to match specific oncogenic tyrosine kinases such as FER and the EGFR, the sequences at Y321 and Y325 do not match those of any known tyrosine kinase.182 Phosphorylation of tyrosine residues may create binding sites for SH2 and PTB domain containing proteins, many of which are themselves kinases. With this in mind, sequence analysis of Y321 when phosphorylated conforms strongly to a SH2 binding site for the Src family tyrosine kinase Fyn.182 These data suggest that an investigation of tyrosine phosphorylation of pRB might uncover novel aspects of regulation that may be important in a number of processes, especially proliferation and cancer.
Concluding Remarks
In this review we have summarized the posttranslational modifications that regulate the function of pRB. By far the most common of these is phosphorylation. For instance, we have seen how phosphorylation by Cdks regulates the interaction of pRB with the E2F family of transcription factors.
But proteomic analysis has revealed pRB to contain many more phosphosites than those targeted by the Cdks. In the context of the G1-S checkpoint control, phosphorylation of pRB by Cdks inactivates pRB in the sense that it no longer binds to E2F, allowing for progression into S phase. However, observations that partially phosphorylated forms of pRB may still associate with E2F, especially E2F1,22,24,41,42 have led to the idea that different functions of pRB are regulated by variations in its phosphorylation state. Evidence supporting this has been supplied by proteomic analysis of peptides isolated within a given tissue or cell line that indicates considerable variation exists with respect to phosphorylation. For example, Stokes et al.176 identified 12 individual molecular species within a single peptide sequence based entirely on differential phosphorylation. Given that pRB contains 51 identified phosphorylation sites, the number of combinations that could arise is enormous, giving rise to considerable regulatory potential. In addition to phosphorylation, pRB is also subject to methylation, acetylation, SUMOylation, and ubiquitinylation, which create many more binding sites. These modifications, in concert with phosphorylation, add to the complexity of pRB regulation.
It has been suggested that posttranslational modifications in proteins can be explained in terms of a “code,” where proteins are modified by “writers”; recognized by “readers,” effector proteins that bind the modified site; and then subject to “erasers” that remove the modification and restore the system back to the ground state.18 Although the concept was originally formulated to address histone modifications in the context of chromatin structure and DNA dynamics,183 the idea has been extended to include other biological processes including phosphorylation and posttranslational modifications in proteins other than histones and may represent a universal mechanism for biochemical regulation.184,185 We have seen how acetylation and methylation at specific sites in pRB create docking sites for other proteins such as L3MBTL1.34 Thus far it is not clear whether SUMOylation creates additional docking sites and what proteins might be involved. With respect to phosphorylation the writers are the kinases, the readers are proteins that recognize the modified sequence. and the erasers are the phosphatases. In this context it is interesting to note that some of the Cdk phosphorylation sites are also targeted by other kinases (Table 1) and in so doing confer specific functions to pRB. For example, S567 phosphorylation by p38 creates a binding site for Mdm2, which then targets pRB for degradation.89 In addition, Chk2 phosphorylation of S612 under conditions of genotoxic stress strengthens the interaction between pRB and E2F1 and prevents the transcription of proapoptotic genes.99 Thus, although the site is the same, different “writers” may influence profoundly different “readers” and therefore direct protein function within a given set of physiological parameters.
Table 1.
Summary of Documented Phosphorylation Sites on Prb
| Site | Kinase | Reference |
|---|---|---|
| T5 | Cdk2/Cdk4 | 46 |
| S230 | N/D | |
| S249 | Cdk2/Cdk4/Cdk6 | 46, 48, 52 |
| T252 | Cdk2/Cdk4/Cdk6 | 46, 48, 52 |
| T356 | Cdk2/Cdk4 | 46, 52 |
| T373 | Cdk2/Cdk4 | 46, 48, 52 |
| S567 | Cdk2/p38 | 49, 89 |
| S608 | Cdk2/Cdk4 | 46 |
| S612 | Cdk2/Chk1/2 | 46, 99 |
| S780 | Cdk4/Cdk5/p38/Aurora b | 28, 52, 84, 90, 108 |
| S788 | Cdk2/Cdk4/Cdk5 | 46, 48, 52, 84 |
| S795 | Cdk2/Cdk4/Cdk5/Cdk9 | 46, 52, 68, 83, 84 |
| S807 | Cdk2/Cdk3/Cdk5/Cdk9/p38 | 46, 48, 68, 72, 83, 84, 85 |
| S811 | Cdk2/Cdk3/Cdk5/Cdk9/p38 | 46, 48, 72, 83, 84, 85 |
| T821 | Cdk2/Cdk5 | 46, 52, 84 |
| T826 | Cdk4/Cdk5 | 46, 84 |
Although pRB is highly phosphorylated, little is known about potential adapter proteins that may bind to the phosphosites and how they might influence pRB function. Numerous proteins with key regulatory functions contain domains shown to bind phosphoserine and phosphothreonine residues,185,186 and members of the 14-3-3 family of phosphoserine/phosphothreonine binding proteins have a role in checkpoint control.187 Similarly phosphotyrosine residues may be targets for SH2 and PTB domain containing proteins.185,188 Amanchy et al.182 have established a database of collected information regarding phosphorylation motifs that allows for the prediction of kinases responsible for a given modification (www.hprd.org). However, many phosphorylation sites do not conform to known kinase consensus sequences. Using a bioinformatics approach, Amanchy et al.178 examined phosphorylation data accumulated from the Human Protein Reference Database and identified 1167 novel phosphorylation motifs, of which 299 were statistically significant. Thus, although consensus sequence similarities are useful for their predictive value, the findings of Amanchy et al. expose limitations to these predictions based solely on consensus sequence similarity and suggest a great deal of latitude regarding recognition sequences for individual kinases. With respect to pRB, the Cdk phosphosites are proline directed, and we have seen how they are targets for other proline directed kinases such as members of the MAP kinase family. However, most of the additional phosphorylation sites are found within sequences that either conform loosely to consensus sequences corresponding to any one of a number of kinases or have no known consensus sequence, making it difficult to predict new upstream regulators of pRB. Initially, phosphorylation of pRB served to explain E2F transcriptional repression within the context of G1-S phase checkpoint control. Now, phosphorylation by other kinases has entered the picture, revealing more aspects for phosphorylation based regulation. With the advent of proteomics, a whole new group of phosphosites with no known function and many with unique sequences has been introduced. Functional studies on these sites promise to introduce new insights into the regulatory roles played by pRB in normal cell physiology and cancer.
Acknowledgments
The authors wish to thank colleagues in the Dick laboratory for encouragement during the preparation of this manuscript. The authors apologize to those who have made contributions in the field but whose work was omitted due to space constraints.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research on pRB in the authors’ laboratory is supported by grants from the Canadian Institutes of Health Research (MOP-89765 and MOP-64253) and the Canadian Cancer Society Research Institute (700720). F.A.D. is the Wolfe Senior Research Fellow in Tumour Suppressor Genes.
References
- 1. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274(5293):1664-72 [DOI] [PubMed] [Google Scholar]
- 2. Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672-7 [DOI] [PubMed] [Google Scholar]
- 3. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220-7 [DOI] [PubMed] [Google Scholar]
- 4. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245-62 [DOI] [PubMed] [Google Scholar]
- 5. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910-7 [DOI] [PubMed] [Google Scholar]
- 6. DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997;94(14):7245-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med. 2006;6:739-48 [DOI] [PubMed] [Google Scholar]
- 8. Ho A, Dowdy SF. Regulation of G1 cell-cycle progression by oncogenes and tumor suppressor genes. Curr Opin Genet Dev. 2002;12(1): 47-52 [DOI] [PubMed] [Google Scholar]
- 9. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103-12 [DOI] [PubMed] [Google Scholar]
- 10. Chauveinc L, Mosseri V, Quintana E, et al. Osteosarcoma following retinoblastoma: age at onset and latency period. Ophthalmic Genet. 2001;22(2):77-88 [DOI] [PubMed] [Google Scholar]
- 11. Sachdeva UM, O’Brien JM. Understanding pRb: toward the necessary development of targeted treatments for retinoblastoma. J Clin Invest. 2012;122(2):425-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wickenheiser-Brokamp KA. Retinoblastoma pathway in lung cancer. Curr Mol Med. 2006;6:783-93 [DOI] [PubMed] [Google Scholar]
- 13. Kaye FJ, Harbour JW. For whom the bell tolls: susceptibility to common adult cancers in retinoblastoma survivors. J Natl Cancer Inst. 2004;96(5):342-3 [DOI] [PubMed] [Google Scholar]
- 14. Gordon G, Du W. Conserved RB functions in development and tumor suppression. Protein Cell. 2011;2(11):864-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paternot S, Bockstaele L, Bisteau X, Kooken H, Coulonval K, Roger P. Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK-activating kinase. Cell Cycle. 2010;9(4):689-99 [DOI] [PubMed] [Google Scholar]
- 17. Xin F, Radivojac P. Posttranslational modifications induce significant yet not extreme changes to protein structure. Bioinformatics. 2012; 28(22):2905-2913.. [DOI] [PubMed] [Google Scholar]
- 18. Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: is there a code of conduct[quest]. Oncogene. 2012;31(40):4343-52 [DOI] [PubMed] [Google Scholar]
- 19. Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein Inhibit E2F transactivation domain binding. J Biol Chem. 2010;285(21):16286-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012;26(11):1156-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coller HA. What’s taking so long? S-phase entry from quiescence versus proliferation. Nat Rev Mol Cell Biol. 2007;8(8):667-70 [DOI] [PubMed] [Google Scholar]
- 22. Ianari A, Natale T, Calo E, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15(3):184-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cecchini MJ, Dick FA. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem J. 2011;434(2):297-308 [DOI] [PubMed] [Google Scholar]
- 24. Carnevale J, Palander O, Seifried LA, Dick FA. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol Cell Biol. 2012;32(5):900-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang S, Nath N, Minden A, Chellappan S. Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J. 1999;18(6):1559-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nath N, Wang S, Betts V, Knudsen E, Chellappan S. Apoptotic and mitogenic stimuli inactivate Rb by differential utilization of p38 and cyclin-dependent kinases. Oncogene. 2003;22(38):5986-94 [DOI] [PubMed] [Google Scholar]
- 27. Hou ST, Xie X, Baggley A, Park DS, Chen G, Walker T. Activation of the Rb/E2F1 pathway by the nonproliferative p38 MAPK during Fas (APO1/CD95)-mediated neuronal apoptosis. J Biol Chem. 2002;277(50):48764-70 [DOI] [PubMed] [Google Scholar]
- 28. Yeste-Velasco M, Folch J, Pallàs M, Camins A. The p38MAPK signaling pathway regulates neuronal apoptosis through the phosphorylation of the retinoblastoma protein. Neurochem Int. 2009;54(2):99-105 [DOI] [PubMed] [Google Scholar]
- 29. Dick F. Structure-function analysis of the retinoblastoma tumor suppressor protein—is the whole a sum of its parts? Cell Division. 2007;2(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, Thangue NBL. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol. 2001;3(7):667-74 [DOI] [PubMed] [Google Scholar]
- 31. Nguyen DX, Baglia LA, Huang S-M, Baker CM, McCance DJ. Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J. 2004;23(7):1609-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pickard A, Wong P-P, McCance DJ. Acetylation of Rb by PCAF is required for nuclear localization and keratinocyte differentiation. J Cell Sci. 2010;123(21):3718-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carr SM, Munro S, Kessler B, Oppermann U, La Thangue NB. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 2011;30(2):317-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saddic LA, West LE, Aslanian A, et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 2010;285(48):37733-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchida C, Miwa S, Kitagawa K, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005;24(1):160-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miwa S, Uchida C, Kitagawa K, et al. Mdm2-mediated pRB downregulation is involved in carcinogenesis in a p53-independent manner. Biochem Biophys Res Comm. 2006;340(1):54-61 [DOI] [PubMed] [Google Scholar]
- 37. Ledl A, Schmidt D, Muller S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene. 2005;24(23):3810-8 [DOI] [PubMed] [Google Scholar]
- 38. Bischof O, Schwamborn K, Martin N, et al. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol Cell. 2006;22(6):783-94 [DOI] [PubMed] [Google Scholar]
- 39. Li T, Santockyte R, Shen R-F, et al. Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. J Biol Chem. 2006;281(47):36221-7 [DOI] [PubMed] [Google Scholar]
- 40. Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell. 1989;58(6):1097-105 [DOI] [PubMed] [Google Scholar]
- 41. Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei MC, Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci U S A. 1997;94(20):10699-704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G1 cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21(14):4773-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ubersax JA, Ferrell Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8(7):530-41 [DOI] [PubMed] [Google Scholar]
- 44. Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol. 1999;19(5):3246-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lents NH, Gorges LL, Baldassare JJ. Reverse mutational analysis reveals threonine-373 as a potentially sufficient phosphorylation site for inactivation of the retinoblastoma tumor suppressor protein (pRB). Cell Cycle. 2006;5(15):1699-707 [DOI] [PubMed] [Google Scholar]
- 46. Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272(19):12738-46 [DOI] [PubMed] [Google Scholar]
- 47. Kitagawa M, Higashi H, HK J, et al. The consensus sequence for phosphorylation by CyclinD1-Cdk4 is different from that for phosphorylation by Cyclin A/E-Cdk2. EMBO J. 1996;15(24):7060-9 [PMC free article] [PubMed] [Google Scholar]
- 48. Lees JA, Buchkovich KJ, Marshak DR, Anderson CW, Harlow E. The retinoblastoma protein is phopshorylated on multiple sites by human cdc2. EMBO J. 1991;10(13):4179-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harbour JW, Luo RX, Santi AD, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98(6):859-69 [DOI] [PubMed] [Google Scholar]
- 50. Huttlin EL, Jedrychowski MP, Elias JE, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dephoure N, Zhou C, Villén J, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105(31):10762-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997;8(2):287-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. [DOI] [PubMed] [Google Scholar]
- 54. Choudhary C, Olsen JV, Brandts C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36(2):326-39 [DOI] [PubMed] [Google Scholar]
- 55. Old WM, Shabb JB, Houel S, et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34(1):115-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mayya V, Lundgren DH, Hwang S-I, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2(84):ra46-. [DOI] [PubMed] [Google Scholar]
- 57. Hornbeck PV, Kornhauser JM, Tkachev S, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined posttranslational modifications in man and mouse. Nucleic Acids Res. 2012;40(D1):D261-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chow KN, Dean DC. Domains A and B in the Rb pocket interact to form a transcriptional repressor motif. Mol Cell Biol. 1996;16(9):4862-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chow KN, Starostik P, Dean DC. The Rb family contains a conserved cyclin-dependent-kinase-regulated transcriptional repressor motif. Mol Cell Biol. 1996;16(12):7173-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17(10):5771-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rubin SM, Gall A-L, Zheng N, Pavletich NP. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123(6):1093-106 [DOI] [PubMed] [Google Scholar]
- 62. Takaki T, Fukasawa K, Suzuki-Takahashi I, Hirai H. Cdk-mediated phosphorylation of pRB regulates HDAC binding in vitro. Biochem Biophys Res Comm. 2004;316(1):252-5 [DOI] [PubMed] [Google Scholar]
- 63. Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391(6667):597-601 [DOI] [PubMed] [Google Scholar]
- 64. Romano G, Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle. 2008;7(23):3664-8 [DOI] [PubMed] [Google Scholar]
- 65. Yu DS, Zhao R, Hsu EL, et al. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010;11(11):876-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Luca A, Esposito V, Baldi A, et al. CDC2-related kinase PITALRE phosphorylates pRb exclusively on serine and is widely expressed in human tissues. J Cell Physiol. 1997;172(2):265-73 [DOI] [PubMed] [Google Scholar]
- 67. Graña X, De Luca A, Sang N, et al. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc Natl Acad Sci U S A. 1994;91(9):3834-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Simone C, Bagella L, Bellan C, Giordano A. Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene. 2002;21(26):4158-65 [DOI] [PubMed] [Google Scholar]
- 69. Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12(5):755-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424(6945):223-8 [DOI] [PubMed] [Google Scholar]
- 71. Liu Z-J, Ueda T, Miyazaki T, et al. A critical role for cyclin C in promotion of the hematopoietic cell cycle by cooperation with c-Myc. Mol Cell Biol. 1998;18(6):3445-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ren S, Rollins BJ. Cyclin C/Cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117(2):239-51 [DOI] [PubMed] [Google Scholar]
- 73. Bonda DJ, Bajić VP, Spremo-Potparevic B, et al. Cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol. 2010;36(2):157-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bonda DJ, Lee H-p, Kudo W, Zhu X, Smith MA, Lee H-g. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev Mol Med. 2010;12: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee H-g, Casadesus G, Zhu X, et al. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem Int. 2009;54(2):84-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang J, Cicero SA, Wang L, Romito-DiGiacomo RR, Yang Y, Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci U S A. 2008;105(25):8772-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang J, Li H, Yabut O, Fitzpatrick H, D’Arcangelo G, Herrup K. Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1–DP1 complex. J Neurosci. 2010;30(15):5219-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang J, Herrup K. Nucleocytoplasmic Cdk5 is involved in neuronal cell cycle and death in postmitotic neurons. Cell Cycle. 2011;10(8):1208-14 [DOI] [PubMed] [Google Scholar]
- 79. Dhavan R, Tsai L-H. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2(10):749-59 [DOI] [PubMed] [Google Scholar]
- 80. Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F·DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999;274(27):19011-6 [DOI] [PubMed] [Google Scholar]
- 81. Ranganathan S, Scudiere S, Bowser R. Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer’s disease and amyotrophic lateral sclerosis. J Alzheimers Dis. 2001;3(4):377-85 [DOI] [PubMed] [Google Scholar]
- 82. Zhang J, Krishnamurthy PK, Johnson GVW. Cdk5 phosphorylates p53 and regulates its activity. J Neurochem. 2002;81(2):307-13 [DOI] [PubMed] [Google Scholar]
- 83. Hamdane M, Bretteville A, Sambo A-V, et al. p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci. 2005;118(6):1291-8 [DOI] [PubMed] [Google Scholar]
- 84. Fatasugi A, Utreras E, Rudrabhatla P, Jaffe H, Pant H, Kulkami AB. Cyclin-dependent kinase 5 regulates E2F transcription factor through phosphorylation of Rb protein in neurons. Cell Cycle. 2012;11(8):1603-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Faust D, Schmitt C, Oesch F, et al. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun Signal. 2012;10(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15(8):369-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Thornton TM, Rincon M. Non-classical p38 MAP kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5(1):44-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pillai MS, Sapna S, Shivakumar K. p38 MAPK regulates G1-S transition in hypoxic cardiac fibroblasts. Int J Biochem Cell Biol. 2011;43(6):919-27 [DOI] [PubMed] [Google Scholar]
- 89. Delston RB, Matatall KA, Sun Y, Onken MD, Harbour JW. p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene. 2011;30(5):588-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cho HJ, Park S-M, Hwang EM, et al. Gadd45b mediates Fas-induced apoptosis by enhancing the interaction between p38 and retinoblastoma tumor suppressor. J Biol Chem. 2010;285(33):25500-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100-12 [DOI] [PubMed] [Google Scholar]
- 92. Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005;118(16):3569-72 [DOI] [PubMed] [Google Scholar]
- 93. Bulavin DV, Higashimoto Y, Popoff IJ, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001; 411(6833):102-7 [DOI] [PubMed] [Google Scholar]
- 94. Cox DM, Du M, Marback M, et al. Phosphorylation motifs regulating the stability and function of myocyte enhancer factor 2A. J Biol Chem. 2003;278(17):15297-303 [DOI] [PubMed] [Google Scholar]
- 95. Cao X, Rui L, Pennington PR, et al. Serine 209 resides within a putative p38(MAPK) consensus motif and regulates monoamine oxidase-A activity. J Neurochem. 2009;111(1):101-10 [DOI] [PubMed] [Google Scholar]
- 96. Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J Biol Chem. 2008;283(28):19511-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Stracker TH, Usui T, Petrini JHJ. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair. 2009;8(9):1047-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sørensen CS, Syljuåsen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40(2):477-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007;26(8):2083-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. F BS, A CD. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122(4):1138-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sage J, Straight AF. RB’s original CIN? Genes Dev. 2010;24(13):1329-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Iovino F, Lentini L, Amato A, Di Leonardo A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol Cancer. 2006;5(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24(13):1364-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Coschi CH, Martens AL, Ritchie K, et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24(13):1351-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. van der Waal MS, Hengeveld RCC, van der, Horst A, Lens SMA. Cell division control by the chromosomal passenger complex. Exp Cell Res. 2012;318(12):1407-20 [DOI] [PubMed] [Google Scholar]
- 106. Vader G, Lens SMA. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786(1):60-72 [DOI] [PubMed] [Google Scholar]
- 107. Farag SS. The potential role of Aurora kinase inhibitors in haematological malignancies. Br J Haematol. 2011;155(5):561-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nair JS, Ho AL, Tse AN, et al. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell. 2009;20(8):2218-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mittnacht S, Paterson H, Olson MF, Marshall CJ. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr Biol. 1997;7(3):219-21 [DOI] [PubMed] [Google Scholar]
- 110. Peeper DS, Upton TM, Ladha MH, et al. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature. 1997;386(6621):177-81 [DOI] [PubMed] [Google Scholar]
- 111. Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol. 1996;16(12):6917-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Williams JP, Stewart T, Li B, Mulloy R, Dimova D, Classon M. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006;26(4):1170-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Takahashi C, Bronson RT, Socolovsky M, et al. Rb and N-ras function together to control differentiation in the mouse. Mol Cell Biol. 2003;23(15):5256-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases. Genes Cancer. 2011;2(3):232-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang S, Ghosh RN, Chellappan SP. Raf-1 Physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol Cell Biol. 1998;18(12):7487-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dasgupta P, Sun J, Wang S, et al. Disruption of the Rb-Raf-1 interaction inhibits tumor growth and angiogenesis. Mol Cell Biol. 2004;24(21):9527-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kinkade R, Dasgupta P, Carie A, et al. A small molecule disruptor of Rb/Raf-1 interaction inhibits cell proliferation, angiogenesis, and growth of human tumor xenografts in nude mice. Cancer Res. 2008;68(10):3810-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Davis R, Pillai S, Lawrence N, Sebti S, Chellappan SP. TNF-α-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle. 2012;11(1):109-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Comm Signal. 2009;7(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hayakawa Y, Hirata Y, Nakagawa H, et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc Natl Acad Sci U S A. 2011;108(2):780-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dasgupta P, Betts V, Rastogi S, et al. Direct binding of apoptosis signal-regulating kinase 1 to retinoblastoma protein. J Biol Chem. 2004;279(37):38762-9 [DOI] [PubMed] [Google Scholar]
- 122. Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNF-[alpha] response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19(2):274-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21(2):140-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Julien SG, Dubé N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11(1):35-49 [DOI] [PubMed] [Google Scholar]
- 125. Welch PJ, Wang JYJ. A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell. 1993;75(4):779-90 [DOI] [PubMed] [Google Scholar]
- 126. Welch PJ, Wang JY. Abrogation of retinoblastoma protein function by c-Abl through tyrosine kinase-dependent and -independent mechanisms. Mol Cell Biol. 1995;15(10):5542-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-abl negatively regulates cell growth. Cell. 1994;77(1):121-31 [DOI] [PubMed] [Google Scholar]
- 128. Chen Y, Knudsen ES, Wang JYJ. Cells arrested in G1 by the v-Abl tyrosine kinase do not express cyclin A despite the hyperphosphorylation of RB. J Biol Chem. 1996;271(33):19637-40 [DOI] [PubMed] [Google Scholar]
- 129. Knudsen ES, Wang JYJ. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271(14):8313-20 [DOI] [PubMed] [Google Scholar]
- 130. Wang JYJ. Regulation of cell death by Abl tyrosine kinase. Oncogene. 2000;19(49):5643-50 [DOI] [PubMed] [Google Scholar]
- 131. Baskaran R, Wood LD, Whitaker LL, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387(6632):516-9 [DOI] [PubMed] [Google Scholar]
- 132. Shafman T, Khanna KK, Kedar P, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387(6632):520-3 [DOI] [PubMed] [Google Scholar]
- 133. Nagano K, Itagaki C, Izumi T, et al. Rb plays a role in survival of Abl-dependent human tumor cells as a downstream effector of Abl tyrosine kinase. Oncogene. 2005;25(4):493-502 [DOI] [PubMed] [Google Scholar]
- 134. Vigneri P, Wang JYJ. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7(2):228-34 [DOI] [PubMed] [Google Scholar]
- 135. Wang Y-T, Tsai C-F, Hong T-C, et al. An informatics-assisted label-free quantitation strategy that depicts phosphoproteomic profiles in lung cancer cell invasion. J Proteome Res. 2010;9(11):5582-97 [DOI] [PubMed] [Google Scholar]
- 136. Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114(13):2363- 73 [DOI] [PubMed] [Google Scholar]
- 137. Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006;7(2):192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834-40 [DOI] [PubMed] [Google Scholar]
- 139. Ng S, Yue W, Oppermann U, Klose R. Dynamic protein methylation in chromatin biology. Cell Mol Life Sci. 2009;66(3):407-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693-705 [DOI] [PubMed] [Google Scholar]
- 141. Shi X, Kachirskaia I, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27(4):636-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kurash JK, Lei H, Shen Q, et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29(3):392-400 [DOI] [PubMed] [Google Scholar]
- 143. Dormann D, Madl T, Valori CF, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31(22):4258-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25(3):338-42 [DOI] [PubMed] [Google Scholar]
- 145. Steele-Perkins G, Fang W, Yang X-H, et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein–methyltransferase superfamily. Genes Dev. 2001;15(17):2250-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol. 2001;21(19):6484-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Nielsen SJ, Schneider R, Bauer U-M, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412(6846):561-5 [DOI] [PubMed] [Google Scholar]
- 148. Yoshimoto T, Boehm M, Olive M, et al. The arginine methyltransferase PRMT2 binds RB and regulates E2F function. Exp Cell Res. 2006;312(11):2040-53 [DOI] [PubMed] [Google Scholar]
- 149. Munro S, Khaire N, Inche A, Carr S, La Thangue NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29(16):2357-67 [DOI] [PubMed] [Google Scholar]
- 150. Hediger F, Gasser SM. Heterochromatin protein 1: don’t judge the book by its cover! Curr Opin Genet Dev. 2006;16(2):143-50 [DOI] [PubMed] [Google Scholar]
- 151. Ferguson Andrew D, Larsen Nicholas A, Howard T, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19(9):1262-73 [DOI] [PubMed] [Google Scholar]
- 152. Cho H-S, Hayami S, Toyakawa G, et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation Neoplasia. 2012;14(6):476-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Komatsu S, Imoto I, Tsuda H, et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis. 2009;30(7):1139-46 [DOI] [PubMed] [Google Scholar]
- 154. Hamamoto R, Furukawa Y, Morita M, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004;6(8):731-40 [DOI] [PubMed] [Google Scholar]
- 155. Hamamoto R, Silva FP, Tsuge M, et al. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Science. 2006;97(2):113-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Boyer SN, Wazer DE, Band V. E7 Protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56(20):4620-4 [PubMed] [Google Scholar]
- 157. Kalejta RF, Shenk T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A. 2003;100(6):3263-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Berezutskaya E, Yu B, Morozov A, Raychaudhuri P, Bagchi S. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differ. 1997;8(12):1277-86 [PubMed] [Google Scholar]
- 159. Knight JS, Sharma N, Robertson ES. Epstein–Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc Natl Acad Sci U S A. 2005;102(51):18562-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25(38):5277-85 [DOI] [PubMed] [Google Scholar]
- 161. Bellacchio E, Paggi MG. Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J Cell Physiol. 2013:228(2):285-91 [DOI] [PubMed] [Google Scholar]
- 162. Ying H, Xiao Z-XJ. Targeting retinoblastoma protein for degradation by proteasomes. Cell Cycle. 2006;5(5):506-8 [DOI] [PubMed] [Google Scholar]
- 163. Marine JC, Lozano G. Mdm2-mediated ubiquitinylation: p53 and beyond. Cell Death Differ. 2009;17(1):93-102 [DOI] [PubMed] [Google Scholar]
- 164. Manfredi JJ. The Mdm2–p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24(15):1580-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Tang Y-A, Lin R-K, Tsai Y-T, et al. MDM2 Overexpression deregulates the transcriptional control of RB/E2F leading to DNA methyltransferase 3A overexpression in lung cancer. Clin Cancer Res. 2012;18(16):4325-33 [DOI] [PubMed] [Google Scholar]
- 166. Sdek P, Ying H, Chang DLF, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20(5):699-708 [DOI] [PubMed] [Google Scholar]
- 167. Kim W, Bennett Eric J, Huttlin Edward L, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wagner SA, Beli P, Weinert BT, et al. A proteome-wide, quantitative survey of in vivo ubiquitinylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Praefcke GJK, Hofmann K, Dohmen RJ. SUMO playing tag with ubiquitin. Trends Biochem Sci. 2012;37(1):23-31 [DOI] [PubMed] [Google Scholar]
- 170. Da Silva-Ferrada E, Lopitz-Otsoa F, Lang V, Rodrguez MS, Matthiesen R. Strategies to identify recognition signals and targets of SUMOylation. Biochem Res Int. 2012;2012:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hsu PP, Kang SA, Rameseder J, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Nagano K, Shinkawa T, Mutoh H, et al. Phosphoproteomic analysis of distinct tumor cell lines in response to nocodazole treatment. Proteomics. 2009;9(10):2861-74 [DOI] [PubMed] [Google Scholar]
- 173. Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010;9(6):1314-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Rigbolt KTG, Prokhorova TA, Akimov V, et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4(164):rs3. [DOI] [PubMed] [Google Scholar]
- 175. Yu Y, Yoon S-O, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035): 1322-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Stokes MP, Farnsworth CL, Moritz A, et al. PTMScan direct: identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol Cell Proteomics. 2012;11(5):187-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hoffert JD, Pisitkun T, Wang G, Shen R-F, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci U S A. 2006;103(18):7159-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Amanchy R, Kandasamy R, Mativanan S, et al. Identification of novel phosphorylation motifs through an integrative computational and experimental analysis of the human phosphoproteome. J Proteomics Bioinform. 2011;4(2):22-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chen R-q, Yang Q-k, Lu B-w, et al. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009;69(6):2663-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Li H, Ren Z, Kang X, et al. Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer. 2009;9(1):366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190-203 [DOI] [PubMed] [Google Scholar]
- 182. Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nat Biotech. 2007;25(3):285-6 [DOI] [PubMed] [Google Scholar]
- 183. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074-80 [DOI] [PubMed] [Google Scholar]
- 184. Sims RJ, Reinberg D. Is there a code embedded in proteins that is based on posttranslational modifications? Nat Rev Mol Cell Biol. 2008;9(10):815-20 [DOI] [PubMed] [Google Scholar]
- 185. Jin J, Pawson T. Modular evolution of phosphorylation-based signalling systems. Philos Trans R Soc Lond B Biol Sci. 2012;367(1602):2540-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Yaffe MB, Elia AEH. Phosphoserine/threonine-binding domains. Curr Opin Cell Biol. 2001;13(2):131-8 [DOI] [PubMed] [Google Scholar]
- 187. Gardino AK, Yaffe MB. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biology. 2011;22(7):688-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yaffe MB. Phosphotyrosine-binding domains in signal transduction. Nat Rev Mol Cell Biol. 2002;3(3):177-86 [DOI] [PubMed] [Google Scholar]