

Abstract
Background. Artemisinin resistance, a long parasite clearance half-life in response to artemisinin, has been described in patients with Plasmodium falciparum malaria in southeast Asia. Few baseline half-lives have been reported from Africa, where artemisinins were recently introduced.
Methods. We treated P. falciparum malaria in 215 Malian children aged 0.5–15 years with artesunate (0, 24, 48 hours) and amodiaquine (72, 96, 120 hours). We estimated half-life by measuring parasite density every 6 hours until undetectable and evaluated the effects of age, sex, ethnicity, and red blood cell (RBC) polymorphisms on half-life. We quantified the proportion of parasitized RBCs recognized by autologous immunoglobulin G (IgG).
Results. The geometric mean half-life was 1.9 hours (95% confidence interval, 1.8–2.0) and did not correlate with parasite ex vivo susceptibility to artemisinins. In a linear model accounting for host factors, half-life decreased by 4.1 minutes for every 1-year increase in age. The proportion of parasitized RBCs recognized by IgG correlated inversely with half-life (r = −0.475; P = .0006).
Conclusions. Parasite clearance in response to artesunate is faster in Mali than in southeast Asia. IgG responses to parasitized RBCs shorten half-life and may influence this parameter in areas where age is not an adequate surrogate of immunity and correlates of parasite-clearing immunity have not been identified.
Clinical Trials Registration. NCT00669084.
Keywords: Plasmodium falciparum, malaria, artemisinin, parasite clearance, antimalarial immunity, Mali
Artemisinins rapidly kill all asexual blood stages of Plasmodium falciparum [1], including young ring forms before they sequester in the microvessels of vital organs, and are thus more effective than quinolines in reducing malaria morbidity and mortality [2, 3]. For this reason, artemisinin-based combination therapies (ACTs) are first-line treatments for P. falciparum malaria worldwide [4]. ACTs are the use of artemisinin or one of its derivatives—artesunate, artemether, dihydroartemisinin (DHA)—in combination with a partner drug. DHA, the active metabolite of all artemisinins, has a short half-life (approximately 1–3 hours) in plasma; therefore, partner drugs with longer half-lives (ie, approximately 7–21 days) are needed to eliminate residual parasites [5, 6]. Parasite resistance to nearly every commonly used partner drug is entrenched or emerging in western Cambodia [7].
Reports of artemisinin resistance in western Cambodia [8, 9] and western Thailand [10]—where antimalarial-resistant parasites have previously emerged and spread to Africa [11–13]—are thus extremely worrisome. This phenotype manifests as a slow parasite clearance rate in response to an artemisinin taken orally to treat uncomplicated malaria [14]. Unlike quinoline resistance, artemisinin resistance does not associate with known molecular markers of drug resistance or reduced parasite drug susceptibility in vitro [8, 9, 15]. Slow parasite clearance may be partly defined as a parasite-heritable trait [9, 10, 16] and was recently associated with a major region of the parasite genome [17]. Microsatellite-defined parasite genetics, however, has not been able to account for all the variation in parasite clearance rates in Cambodia and Thailand [9, 10, 16].
Few studies have explored how host factors, such as naturally acquired immunity and red blood cell (RBC) polymorphisms, influence parasite clearance rates in response to artemisinins. A retrospective analysis of 18 699 patients with P. falciparum malaria treated with artemisinins found that parasite clearance was slowest in low-transmission settings and relatively fast in high-transmission settings, suggesting that acquired immunity accelerates parasite clearance [18]. A recent prospective study suggested that hemoglobin (Hb) E is associated with slow parasite clearance rate in Pursat, western Cambodia [9], but the effect of acquired immunity on this parameter is not well defined. This is because, in southeast Asia, age is an inadequate surrogate of immunity to malaria and no in vitro correlate of parasite-clearing immunity has been identified. Few parasite clearance rates have been reported from Africa [19], where ACTs were recently introduced and artemisinin monotherapies have been used for 15 years in some areas [20].
The proposed mechanism of action of artemisinins involves endoperoxide-derived, free radicals that alkylate and oxidize the proteins and lipids of intraerythrocytic parasites [21]. Artemisinin-treated parasites undergo pyknosis rapidly in vivo and are cleared from the bloodstream by “pitting” in the spleen, which returns previously infected, intact RBCs back into circulation (Figure 1A) [22–25]. Although largely determined by the rates of pyknosis and pitting, the parasite clearance rate is nonetheless a complex phenotype produced by a combination of parasite and host factors. Parasite expression of cytoadherence ligands, for example, can determine when late ring-stage parasites sequester in microvessels and disappear from peripheral blood (Figure 1C) [26]. If parasites sequester en masse [26] while parasite clearance is being monitored, parasite densities may decrease markedly by an artemisinin-independent mechanism. Host antibody responses that prevent sequestration and opsonize parasitized RBCs may also accelerate parasite clearance (Figure 1B).
Figure 1.

Clearance of ring-stage Plasmodium falciparum parasites from peripheral blood during a parasite clearance rate study. Dihydroartemisinin, the active metabolite of all artemisinins, causes ring-stage parasites to undergo pyknosis (A). These circulating pyknotic forms are eventually “pitted” from red blood cells (RBCs) as they pass through endothelial slits in the spleen, which returns the previously infected, intact RBCs to the peripheral blood. This process occurs in all patients treated with artesunate and is likely the predominant mechanism of parasite clearance in most cases. Depending on the maturity of ring-stage parasites, however, 2 additional processes may also work to reduce parasite density in an artemisinin-independent manner. Ring-stage parasites that are sufficiently mature (indicated by a thicker ring of light blue cytoplasm in the figure) may place PfEMP1-laden knobs on the surface of their host RBCs at the same time artesunate is consumed and begins to kill parasites (B and C). If these PfEMP1-expressing parasitized RBCs are traveling through a microvessel at this time, they may successfully sequester there and disappear from the peripheral blood (C). On the other hand, if these parasitized RBCs are traveling through other blood vessels where sequestration does not occur, they become targets for antibodies against PfEMP1 and other surface antigens (B). By preventing parasitized RBCs from sequestering and opsonizing them for phagocytosis in the spleen, these antibodies may contribute to the clearance of ring-stage parasites. Abbreviation: DHA, dihydroartemisinin.
To obtain baseline surveillance data for the emergence or spread of artemisinin resistance in Africa and to investigate the role of acquired immunity in parasite clearance, we measured parasite clearance rates in response to artesunate in a high transmission area of Mali, where ACTs were introduced only 2 years previously and where children rapidly acquire antimalarial immunity with age.
METHODS
Study Site and Patients
In May 2008, we initiated a 4-year study of malaria incidence in Kenieroba, a village 75 km southwest of Bamako where artemisinins were not previously used. When children presented to clinic with malaria symptoms, we used findings from history taking and physical examination, along with screening parasite density and Hb and glucose levels (Hemocue) to diagnose them with uncomplicated or severe P. falciparum malaria. Uncomplicated malaria was defined as fever (axillary temperature ≥37.5°C or history of fever in the previous 24 hours) and any P. falciparum density. These children did not have severe malaria, defined by any P. falciparum density and any 1 of the following: coma (Blantyre coma score ≤2), convulsions (witnessed), severe prostration, severe anemia (Hb level ≤5 g/dL), respiratory distress, hypoglycemia (glucose ≤40 mg/dL), jaundice, shock, cessation of eating and drinking, repetitive vomiting, or other etiologies of fever discernible on clinical examination.
In the 2010 transmission season, we diagnosed 1050 uncomplicated malaria episodes in 636 children within a cohort of 1325 children. In 2011, we diagnosed 926 uncomplicated malaria episodes in 565 children within a cohort of 1287 children. Within this cohort study, we conducted a parasite clearance rate substudy in 2010 and 2011. Inclusion criteria for this substudy were uncomplicated P. falciparum malaria, age 0.5–15 years, screening parasite density of 10 000/µL–100 000/µL, negative pregnancy test (if girl aged 14–15 years), and no history of antimalarial use for present symptoms. Because of space limitations and intensity of monitoring parasitemia, we enrolled up to 4 patients on any given day.
After children were evaluated and informed consent was obtained, typically 1–3 hours after the screening parasite density was counted, an initial parasite density was counted before the first artesunate dose was given (t = 0 hours). We treated children with artesunate 4 mg/kg (0, 24, 48 hours) and amodiaquine 10 mg/kg (72, 96, 120 hours) as directly observed oral therapy. Parasite densities were quantified from thick blood films by counting the number of ring forms until 300 leukocytes were also counted, then multiplying this number by 25 (which assumes an average number of 7500 leukocytes/µL). For each child, we determined Hb phenotype and α-thalassemia and G6PD deficiency genotypes, as described [27, 28].
The Ethics Committee of the Faculty of Medicine, Pharmacy and Odontostomatology at the University of Bamako and the Institutional Review Board at the National Institute of Allergy and Infectious Diseases approved protocol activities. The parents or guardians of children gave written informed consent. The protocol is registered at Clinicaltrials.gov (NCT00669084).
Parasite Clearance Rates
We counted the initial parasite density before giving artesunate (0 hours) and then every 6 hours until parasite density was undetectable or until 48 hours elapsed. Parasite clearance curves were derived from these parasite counts [29], and parasite clearance half-life was calculated using the Parasite Clearance Estimator (http://www.wwarn.org/research/parasite-clearance-estimator). This tool calculates the parasite clearance rate constant, based on the linear part of the loge parasite density–time profile [30].
Ex Vivo Parasite Drug Response Assay
From each child, we collected 5 mL of venous blood into a sodium heparin Vacutainer before giving the first artesunate dose. Using a conventional drug assay [31], we tested the ex vivo susceptibility of parasite isolates to chloroquine (CQ), amodiaquine (AQ), monodesethylamodiaquine (MDAQ), quinine (QN), artesunate, and DHA. All drugs were obtained from Sigma, except for MDAQ (kindly provided by Chris Lourens, WWARN). Stock drug solutions were prepared in water (CQ), dimethyl sulfoxide (AQ, MDAQ, QN, and artesunate), or ethanol (DHA). A ×4 working solution of each drug was prepared in complete medium (Roswell Park Memorial Institute 1640, 25 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 7.5% sodium bicarbonate, 50 μg/mL gentamicin, 50 mg/mL hypoxanthine, and 5 g/L Albumax). From these solutions, 1:3 serial dilutions were made to cover a range of concentrations for each drug: CQ (0.2–4000 nM), AQ (0.2–2000 nM), MDAQ (0.3–2000 nM), quinine (0.2–5000 nM), artesunate (0.02–300 nM), and DHA (0.02–300 nM). Serial dilutions at ×4 were distributed in triplicate (50 µL/well) in opaque 96-well plates. Four batches of plates containing drug dilutions were prepared, stored at −20°C, and used within 1 month. Each batch was tested by obtaining the median inhibitory concentration (IC50) values for the P. falciparum clones 3D7 (CQ-sensitive) and Dd2 (CQ-resistant) [32, 33].
To carry out the drug assay, blood samples were centrifuged at 2500 rpm × 10 minutes to separate plasma and peripheral blood mononuclear cells. RBCs were washed 3 times in complete medium within 4–6 hours of the blood draw. We determined parasitemias by counting 1000 RBCs in thin blood smears and adjusted samples to 1% parasitemia and 2.4% hematocrit by adding uninfected donor RBCs and complete medium. For few samples with <1% parasitemia, we performed the assay at 0.5% parasitemia. These cell suspensions were added to previously prepared, drug-coated 96-well plates (150 µL/well) to achieve ×1 drug concentrations in each well. Plates were incubated at 37°C in an atmosphere of 5% carbon dioxide for 48 hours, and parasite development monitored at 24 and 48 hours in extra drug-lacking wells plated expressly for this purpose. Plates were stored at −20°C for up to 3 weeks before staining with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Parasite DNA was quantified in each well using freshly-prepared DAPI. Original DAPI reagent was reconstituted in culture-grade water to 5 mg/mL and stored at 4°C in the dark. Working solutions were prepared by diluting DAPI 1:38 000 in phosphate-buffered saline. Plates were thawed for 1 hour at ambient temperature, centrifuged at 3500 rpm for 30 minutes, and gently inverted to remove supernatants. DAPI solution (100 µL/well) was then added and incubated for 30 minutes in the dark. Plates were centrifuged again at 3500 rpm for 30 minutes, and pellet DNA was resuspended in 200 µL phosphate-buffered saline. DAPI signal was measured by reading plates at Ex/Em 358 nm/461 nm using a Fluoroskan Ascent reader.
Surface Immunoglobulin G (IgG) Reactivity Assay
Plasma samples were heated at 56°C for 30 minutes and stored at −80°C until use. Parasite isolates were cultured at 2% hematocrit in complete media until they matured to late trophozoites, as determined by microscopy of Giemsa-stained thin blood films [34]. Parasitized RBCs were washed once in 2% fetal bovine serum in phosphate-buffered saline, adjusted to 1% hematocrit in 2% fetal bovine serum/phosphate-buffered saline, and plated (200 µL/well) in 96-well plates. Parasitized RBCs were incubated with plasma dilutions (1:5, 1:10, 1:20, 1:40) for 30 minutes at ambient temperature with constant agitation. After 3 washes, samples were incubated with Alexa Fluor 488-conjugated goat anti-human IgG (2 mg/mL), Alexa Fluor 594-conjugated goat anti-human immunoglobulin M (IgM; 2 mg/mL), and SYTO® 61 red fluorescent nucleic acid stain (5 mM in dimethyl sulfoxide) for 30 minutes with constant agitation. After 3 washes, parasitized RBCs were analyzed by an Accuri C6 Flow Cytometer.
Statistical Analysis
Correlations were performed using the Spearman method. Inferences for change in Hb level over time were done by a paired t test and the appropriate confidence intervals on log-transformed Hb values. To compare parasite clearance half-lives between Mali and Cambodia, we used a t test and related confidence intervals on the log-transformed values and back-transformed the difference in the means of the logs into a ratio. Parasite drug responses were analyzed using the nonlinear regression model [Y = 100/(1 + 10^((LogIC50−X)×HillSlope))] to calculate IC50 values, where DAPI signals for each drug were normalized to 100% of the maximum value. IC50 values from regression models with R2 ≥ 0.9 were analyzed. For plasma dilution studies, we fit a linear model for each plasma sample using number of doublings of the dilution to predict the log-transformed proportion of IgG-positive parasitized RBCs. We performed a one-sample t test on the slopes and back-transformed them into average fold-change.
In a linear regression model, we defined the following variables for RBC polymorphisms: HbC type (HbAA or HbAS or HbSS = 0, HbAC = 1), HbS type (HbAA or HbAC = 0, HbAS or HbSS = 1), α-thalassemia−3.7 kb genotype (wild type = 0, heterozygote = 1, homozygote = 2), and G6PD∗A- genotype (wild type = 0, heterozygote = 1, hemizygote = 2). We also defined 2 binary variables: male vs female and Fulani vs non-Fulani ethnicity [35]. Analyses were performed using GraphPad Prism software version 5 or R (http://www.R-project.org). A P value < .05 was considered statistically significant.
RESULTS
In 2010 and 2011, we studied parasite clearance in 12.5% (n = 131 of 1050) and 14.0% (n = 130 of 926) of uncomplicated malaria episodes occurring in 636 and 565 children, respectively. The 261 children were aged 0.5–15 years, were permanent residents of Kenieroba, and had parasite densities of 10 000–100 000/µL at screening. For 38 children enrolled twice (ie, in 2010 and 2011), we randomly selected 1 of 2 malaria episodes for analysis. We then excluded 8 children from analysis because of missing genotype data. The remaining 215 children were 53% male, were 81.4% Malinke, and had a median age of 5 years (Table 1). The geometric mean Hb level of the children significantly dropped by 12.0% (95% confidence interval [CI], 10.3–13.7; P < .0001) from 10.8 g/dL at presentation to 9.7 g/dL at 72 hours. The proportions of children who had Hb variants (HbS or HbC), α-thalassemia, or G6PD deficiency were 21.4%, 28.8%, and 14.9%, respectively (Table 1). A majority (53%) of children had ≥1 RBC polymorphism (Supplementary Figure 1). At presentation, children had a median screening parasite density of 25 200/µL (interquartile range [IQR], 16 700/µL–37 600/µL) (Table 1).
Table 1.
Patient Characteristics and Parasitological Data
| Patient demographic and clinical information | |
| Patients, No. | 215 |
| Male sex, No. (%) | 114 (53) |
| Age, years, median (IQR) | 5 (3.5–8) |
| Ethnicity, No. Malinke (%)a | 175 (81.4) |
| Hb at 0 hours, g/dL, median (IQR) | 10.8 (9.8–12.1) |
| Hb at 72 hours, g/dL, median (IQR)b | 9.7 (8.4–10.9) |
| Erythrocyte polymorphisms, No. (%) | |
| Hb type | |
| AA | 169 (78.6) |
| AS or SSc | 25 (11.6) |
| AC | 21 (9.8) |
| α-Thalassemia | |
| αα/αα | 153 (71.2) |
| −α/αα | 60 (27.9) |
| −α/−α | 2 (0.9) |
| G6PD deficiency (A−) | |
| Wild type | 183 (85.1) |
| Heterozygote | 23 (10.7) |
| Hemizygote | 9 (4.2) |
| Homozygote | 0 |
| Parasitemia, median (IQR; range) | |
| Screening Pf density, per µL | 25 200 (16 700–37 600; 10 000–91 675) |
| Initial Pf density, per µL | 28 200 (18 875–45 150; 3375–175 325) |
| Parasite clearance parameters | |
| Parasite clearance time, h, median (IQR; range)d | 24 (24–30; 12–48) |
| Half-life of parasite clearance, h, geometric mean (95% CI; range) | 1.9 (1.8–2.0; 1.0–5.3) |
Abbreviations: CI, confidence interval; Hb, hemoglobin; Pf, P. falciparum.
a Of the 40 non-Malinke children, 26 were Fulani, 13 were Bambara, and 1 was Sarakole.
b Hemoglobin level was significantly lower at 72 hours compared with time of presentation (P < .0001, paired t test).
c Only 1 child of the 25 had hemoglobin type SS.
d Three children remained parasitemic at 48 hours (parasite densities = 100, 100, and 75/µL), so the parasite clearance time could not be established. All 3 children had an undetectable parasite density at 72 hours.
Because a time interval elapsed between screening and determining patient eligibility for the study, we also counted the initial (t = 0 hours) parasite density just before administering the first artesunate dose. During a 48-hour period, we counted parasite density every 6 hours until it was undetectable by microscopy. The median parasite density was 28 200/µL (IQR, 18 875–45 150/µL) (Table 1). The median parasite clearance time, the time it takes for parasitemia to become undetectable, was 24 hours (range, 12–48) (Table 1). Because parasite clearance times correlated significantly with initial parasite densities (r = 0.197; P = .004), we measured parasite clearance rates. To estimate these, we plotted loge-transformed parasite densities vs time for each child and used a parasite clearance estimator [30] to identify the linear portion of the parasite clearance curve [29]. From the slope of this line, we calculated the parasite clearance half-life. Half-lives ranged 1.0–5.3 hours (Figure 2A), and the geometric mean half-life was 1.9 hours (95% CI, 1.8–2.0; Table 1 and Figure 2A). By comparison, this value was 3.0 times longer (95% CI, 2.8–3.3, P < .0001) than the geometric mean half-life (5.9 hours) in western Cambodia [9]. As observed in Cambodian patients [9], half-life did not correlate with initial parasite densities (r = −0.092; P = .179) in Malian children.
Figure 2.
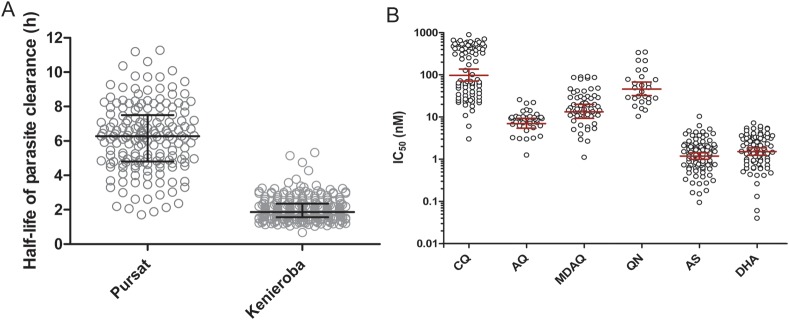
In vivo and ex vivo responses of parasites to artemisinin derivatives. A, Distribution of parasite clearance half-lives in 215 children with uncomplicated Plasmodium falciparum malaria in Kenieroba, Mali, compared with that of 168 (mostly adult) patients in Pursat, western Cambodia. The geometric mean half-life in Pursat (5.9 hours) is 3.0 times longer (95% confidence interval [CI], 2.8–3.3; P < .0001) than in Kenieroba (1.9 hours). The geometric mean parasite clearance half-life 95% CIs are indicated by horizontal lines and whiskers. B, Ex vivo responses of P. falciparum isolates to antimalarial drugs. The geometric mean (GM) IC50 values for chloroquine (CQ; n = 87), amodiaquine (AQ; n = 41), monodesethylamodiaquine (MDAQ; n = 57), quinine (QN; n = 27), artesunate (AS; n = 96), and dihydroartemisinin (DHA; n = 87) were 119 nM, 7.79 nM, 16.7 nM, 49.9 nM, 1.17 nM, and 1.50 nM, respectively. The GM IC50 and 95% CIs are indicated by horizontal lines and whiskers. This assay showed that approximately 50% of parasite isolates have reduced susceptibility to CQ (IC50 > 100 nM) but remain susceptible to the other antimalarial drugs tested. These data are consistent with the only other in vitro responses of parasite isolates reported from Mali [36]. The IC50 values for CQ correlated with those for AQ (r = 0.608; P = .0008) and MDAQ (r = 0.707; P < .0001). The only other IC50 correlation among the 6 drugs was that observed for QN and AS (r = 0.585; P = .002). Abbreviations: AQ, amodiaquine; AS, artesunate; CQ, chloroquine; DHA, dihydroartemisinin; IC50, median inhibitory concentration; MDAQ, monodesethylamodiaquine; QN, quinine.
To explore whether half-life associates with parasite susceptibility to artemisinins, we measured the ex vivo responses of parasite isolates to artesunate and DHA. We simultaneously measured parasite responses to CQ, AQ, MDAQ, and QN to validate assay performance. We found that 44 of 87 (50.6%) parasite isolates showed reduced ex vivo susceptibility to CQ (IC50 > 100 nM) (Figure 2B). Few parasite isolates showed reduced susceptibility to AQ (IC50 > 20 nM; 3 of 41, 7.32%), MDAQ (IC50 > 60 nM; 6 of 57, 10.5%), and QN (IC50 > 200 nM; 4 of 27, 14.8%) (Figure 2B). These data are consistent with drug-response profiles expected for parasites in Mali [36], where only CQ has been used historically. The geometric mean IC50 values for artesunate and DHA were very low: 1.17 nM (95% CI, 0.98–1.40 nM; n = 96) and 1.50 nM (95% CI, 1.22–1.83 nM; n = 87), respectively (Figure 2B). Half-life did not correlate with IC50 for artesunate (r = −0.151; 95% CI, −.346 to .057; n = 96; P = .14) or DHA (r = 0.169; 95% CI, −.049 to .372; n = 87; P = .12), indicating that this parameter is independent of parasite susceptibility to artemisinins, as measured by a conventional ex vivo drug response assay.
To investigate whether half-life depends on host factors, we used a linear regression model that accounted for the effects of age, sex, ethnicity, RBC polymorphisms, and parasite density. In this model, only age associated with half-life (P < .0001; Figure 3A). Age inversely correlated with half-life (r = −0.312; P < .0001), and a linear model that included only age predicted a 4.1-minute shortening of half-life for every 1-year increase in age (95% CI, 2.6–5.7; P < .0001; Figure 3B). These data suggest that age-associated acquired immunity accelerates parasite clearance. To investigate whether artemisinin-independent clearance of ring-parasitized RBCs occurs while parasite clearance is monitored, we correlated screening and initial parasite densities (typically counted 1–3 hours apart). Although screening and initial parasite densities strongly correlated (r = 0.747; P < .0001), the parasite densities of many children dropped before the first artesunate dose was consumed (Figure 4A). This finding may be due to the often-synchronous sequestration of late ring-parasitized RBCs in microvessels [26] and opsonic removal of parasitized RBCs (Figure 1B and 1C), both of which may decrease half-life.
Figure 3.

Influence of host factors on parasite clearance half-life. A, Results from a linear model in which the response is half-life in hours and the independent variables are age, sex (female reference), ethnicity (non-Fulani reference), hemoglobin (Hb) AC, HbAS, G6PD deficiency, α-thalassemia, and loge-transformed initial parasite density. Each effect is given as the predicted change in half-life while holding all other variables constant. Hemoglobinopathy effects compare heterozygous with wild type (with homozygous and hemizygous having double effects), whereas age and ln (Pf day 0) are compared with adding 1 unit to that variable. Without correcting for multiple comparisons, we see that only the age effect is significant. The age parameter states that for every 1-year increase in age, the half-life changes by −0.0686 hours (95% confidence interval [CI], −.0946 to −.0425). The parameters with 95% CIs (uncorrected for multiple comparisons) are plotted. B, Half-life is inversely correlated with age (r = −0.312; P < .0001), a surrogate of naturally acquired immunity in our study population. The linear model estimates that there is a 4.1-minute (95% CI, 2.6–5.7; P < .0001) shortening of half-life for every 1-year increase in age. Abbreviations: Hb, hemoglobin; HE, heterozygote; Pf day 0, initial P. falciparum density.
Figure 4.

Autologous immunoglobulin G (IgG) responses against the surface of Plasmodium falciparum–infected red blood cells (RBCs) obtained directly from Malian children with malaria and cultured to the trophozoite-stage. A, Correlation between screening and initial parasite densities. Although screening and initial parasite densities correlated (r = 0.736; P < .0001), the parasite densities of many children dropped before the first artesunate dose was taken (data points below the line of equality). This finding may be due to the often-synchronous sequestration of late ring-stage parasitized RBCs in microvessels [26] and opsonic removal of parasitized RBCs, both of which may decrease half-life. B, Representative flow cytometry scatterplot showing that 20.6% of parasitized RBCs in this child were recognized by autologous IgG. C, The median proportions of IgG-positive parasitized RBCs were 21.0% (interquartile range [IQR], 11.7–37.2), 10.8% (IQR, 6.92–23.7), and 5.94% (IQR, 4.04–17.6) for plasma tested at 1:10 (n = 48), 1:20 (n = 48), and 1:40 (n = 22) dilutions, respectively. For each dilution doubling, the proportion of IgG-positive parasitized RBCs changed on average by a factor of 0.61 (95% CI, .54–.69; P < .0001). D, The plot shows an inverse correlation between IgG responses and half-life (r = −0.475; P = .0006), suggesting they are involved in clearing ring-stage parasites from peripheral blood. Abbreviations: IgG, immunoglobulin; FL1-H, fluorescence channel 1-height; FL2-H, fluorescence channel 2-height.
We thus hypothesized that the proportion of parasitized RBCs recognized by autologous antibody correlates with the parasite clearance rate. From 48 children, we obtained parasite isolates and cultured them ex vivo until they matured to trophozoites expressing PfEMP1, the parasite's main cytoadherence ligand, and other variant surface antigens. We then measured each child's antibody response by quantifying the proportion of parasitized RBCs that bound autologous IgM or IgG. Whereas an IgM response was detected in only 1 child, autologous IgG identified a subpopulation of parasitized RBCs in all 48 children (Figure 4B). The proportion of IgG-positive parasitized RBCs increased in a plasma dose-dependent manner (1:5 plasma dilutions resulted in agglutination of parasitized RBCs; Figure 4C). The median proportions of IgG-positive parasitized RBCs were 21.0% (IQR, 11.7–37.2), 10.8% (IQR, 6.92–23.7), and 5.94% (IQR, 4.04–17.6) for plasma tested at 1:10 (n = 48), 1:20 (n = 48), and 1:40 (n = 22) dilutions, respectively. For each doubling of the dilution, the proportion of IgG-positive parasitized RBCs changed on average by a factor of 0.61 (95% CI, .54–.69; P < .0001). At 1:10 dilutions of plasma, IgG responses ranged 4%–76% and correlated inversely with half-life (r = −0.475; P = .0006; Figure 4D), suggesting that antibodies promote clearance of parasitized RBCs from peripheral blood. IgG responses also correlated positively with age (r = 0.383; P = .007) and inversely with log-transformed initial parasite densities (r = −0.400; P = .005).
DISCUSSION
Here we report rapid parasite clearance rates in response to artesunate in a high-transmission area of Mali. We found that half-lives are markedly shorter in Mali than in southeast Asia [8–10], as recently reported by Maiga et al [19], and similarly do not correlate with ex vivo susceptibility of parasites to artesunate or DHA [9]. In a linear model accounting for host factors, half-life decreases with age, suggesting that naturally acquired immunity accelerates parasite clearance in response to artesunate [19]. The proportion of a child's parasitized RBCs recognized by autologous IgG inversely correlates with both half-life and initial parasite density, suggesting that these IgG responses clear parasitized RBCs and account for the age-associated suppression of parasite densities commonly observed in African children. Although the IgG responses we measured were directed at trophozoite-infected RBCs expressing PfEMP1 and other variant surface antigens, we believe that some of these IgG responses may be the same as those against the late ring-parasitized RBCs that cytoadhere in vivo. Determining whether IgG responses directed against ring-stage parasites accelerate parasite clearance will require further studies. To this end, we are now serially monitoring each child's IgG response to autologous parasites ex vivo—from the early rings that freely circulate in the bloodstream to the late rings that sequester or are opsonized by immune IgG.
Results from the surface IgG reactivity assay are likely to be more informative about parasite-clearing immunity than those from related assays [37–39], which test an individual's IgG cross-reactivity to panels of heterologous parasites (ie, short-term-adapted parasite isolates from other patients or long-term-adapted parasite lines). By quantifying autologous IgG responses to parasitized RBCs in the first cycle of parasite development ex vivo, before antigenic switching, our assay directly models how a child's existing IgG repertoire reacts to the parasite population being cleared by artemisinin-independent mechanisms. Measuring these IgG responses in southeast Asia may improve our understanding of the artemisinin resistance phenotype by accounting for the effects of acquired immunity on half-life and may test whether waning immunity in human populations in western Cambodia and western Thailand contributes to the lengthening of half-life over time [10]. Our assay may also be useful in parasite clearance studies currently underway in sub–Saharan Africa to monitor for the emergence of artemisinin resistance, particularly in areas where artemisinin monotherapies are commonly used and immunity may be waning.
Studying antimalarial immunity is difficult in endemic areas of Africa, partly because no in vitro correlate of parasite-clearing immunity has been identified. Typically, investigators evaluate specific immune responses by measuring them before a malaria season and then associating the titer and breadth of these responses with subsequent malaria incidence [28, 37, 40, 41]. Such studies are informative but are expensive, time consuming, and confounded by parasite diversity, levels of immunity, and RBC polymorphisms (eg, HbS) [28]. Age-associated clearance of drug-resistant parasites has been proposed as a model system for studying immunity in African children [42]. However, such studies can no longer be conducted because ACTs are now first-line treatments for malaria. Also, the recrudescence of drug-resistant parasites may be influenced by the proportion of parasites that is drug resistant, initial parasite density, variable pharmacokinetic profiles of antimalarials, and the sensitivity of detecting low parasitemias by microscopy. In contrast, estimating half-life in response to artemisinins is ethical and facile and not strongly influenced by initial parasite density or presence of parasite subpopulations resistant to partner drugs. Measuring autologous IgG responses to parasitized RBCs and investigating their ability to block cytoadherence, opsonize, and activate complement may improve our understanding of IgG-mediated reductions in parasitemia [43]. The parasite targets of autologous IgG responses that associate most significantly with half-life may be promising vaccine candidate antigens.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. The authors wish to acknowledge Ismaila Coulibaly, Seydou Doumbia, Aminata Famanta, Robert Gwadz, Kazutoyo Miura, Sam Moretz, Erika S. Phelps, Dick Sakai, Ibrahim Sanogo, Papa Diogoye Sene, Karambe Souleymane, Cheick Traore, Greg Tullo, Sarah Volkman, Thomas Wellems, and Dyann Wirth for their efforts in support of this work.
Financial support. This work was supported by the Intramural Research Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Jiang JB, Li GQ, Guo XB, Kong YC, Arnold K. Antimalarial activity of mefloquine and qinghaosu. Lancet. 1982;2:285–8. doi: 10.1016/s0140-6736(82)90268-9. [DOI] [PubMed] [Google Scholar]
- 2.Dondorp A, Nosten F, Stepniewska K, Day N, White N. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–25. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp AM, Fanello CI, Hendriksen IC, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–57. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Guidelines for the treatment of malaria. 2nd ed. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 5.Nosten F, White NJ. Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg. 2007;77:181–92. [PubMed] [Google Scholar]
- 6.Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7:864–74. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. Geneva, Switzerland: World Health Organization; 2010. Global report on antimalarial efficacy and drug resistance: 2000–2010. [Google Scholar]
- 8.Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaratunga C, Sreng S, Suon S, et al. Artemisinin-resistant Plasmodium falciparum in Pursat province, western Cambodia: a parasite clearance rate study. Lancet Infect Dis. 2012;12:851–8. doi: 10.1016/S1473-3099(12)70181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phyo AP, Nkhoma S, Stepniewska K, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toshihiro M, Venkatesan M, Ohashi J, et al. Limited geographical origin and global spread of sulfadoxine-resistant dhps alleles in Plasmodium falciparum populations. J Infect Dis. 2011;204:1980–8. doi: 10.1093/infdis/jir664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 13.Ariey F, Fandeur T, Durand R, et al. Invasion of Africa by a single pfcrt allele of South East Asian type. Malar J. 2006;5:34. doi: 10.1186/1475-2875-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairhurst RM, Nayyar GM, Breman JG, et al. Artemisinin-resistant malaria: research challenges, opportunities, and public health implications. Am J Trop Med Hyg. 2012;87:231–41. doi: 10.4269/ajtmh.2012.12-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim P, Alker AP, Khim N, et al. Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malar J. 2009;8:11. doi: 10.1186/1475-2875-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson TJ, Nair S, Nkhoma S, et al. High heritability of malaria parasite clearance rate indicates a genetic basis for artemisinin resistance in western Cambodia. J Infect Dis. 2010;201:1326–30. doi: 10.1086/651562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheeseman IH, Miller BA, Nair S, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stepniewska K, Ashley E, Lee SJ, et al. In vivo parasitological measures of artemisinin susceptibility. J Infect Dis. 2010;201:570–9. doi: 10.1086/650301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiga AW, Fofana B, Sagara I, et al. No evidence of delayed parasite clearance after oral artesunate treatment of uncomplicated falciparum malaria in Mali. Am J Trop Med Hyg. 2012;87:23–8. doi: 10.4269/ajtmh.2012.12-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton PN, Green MD, Mildenhall DC, et al. Poor quality vital anti-malarials in Africa—an urgent neglected public health priority. Malar J. 2011;10:352. doi: 10.1186/1475-2875-10-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Neill PM, Barton VE, Ward SA. The molecular mechanism of action of artemisinin—the debate continues. Molecules. 2010;15:1705–21. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chotivanich K, Udomsangpetch R, Dondorp A, et al. The mechanisms of parasite clearance after antimalarial treatment of Plasmodium falciparum malaria. J Infect Dis. 2000;182:629–33. doi: 10.1086/315718. [DOI] [PubMed] [Google Scholar]
- 23.Newton PN, Chotivanich K, Chierakul W, et al. A comparison of the in vivo kinetics of Plasmodium falciparum ring-infected erythrocyte surface antigen-positive and -negative erythrocytes. Blood. 2001;98:450–7. doi: 10.1182/blood.v98.2.450. [DOI] [PubMed] [Google Scholar]
- 24.Chotivanich K, Udomsangpetch R, McGready R, et al. Central role of the spleen in malaria parasite clearance. J Infect Dis. 2002;185:1538–41. doi: 10.1086/340213. [DOI] [PubMed] [Google Scholar]
- 25.Buffet PA, Milon G, Brousse V, et al. Ex vivo perfusion of human spleens maintains clearing and processing functions. Blood. 2006;107:3745–52. doi: 10.1182/blood-2005-10-4094. [DOI] [PubMed] [Google Scholar]
- 26.Beaudry JT, Fairhurst RM. Microvascular sequestration of Plasmodium falciparum. Blood. 2011;117:6410. doi: 10.1182/blood-2010-09-305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007;4:e66. doi: 10.1371/journal.pmed.0040066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crompton PD, Traore B, Kayentao K, et al. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. J Infect Dis. 2008;198:1265–75. doi: 10.1086/592224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White NJ. The parasite clearance curve. Malar J. 2011;10:278. doi: 10.1186/1475-2875-10-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flegg JA, Guerin PJ, White NJ, Stepniewska K. Standardizing the measurement of parasite clearance in falciparum malaria: the parasite clearance estimator. Malar J. 2011;10:339. doi: 10.1186/1475-2875-10-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ndiaye D, Patel V, Demas A, et al. A non-radioactive DAPI-based high-throughput in vitro assay to assess Plasmodium falciparum responsiveness to antimalarials—increased sensitivity of P. falciparum to chloroquine in Senegal. Am J Trop Med Hyg. 2010;82:228–30. doi: 10.4269/ajtmh.2010.09-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellems TE, Panton LJ, Gluzman IY, et al. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345:253–5. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]
- 33.Walliker D, Quakyi IA, Wellems TE, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–6. doi: 10.1126/science.3299700. [DOI] [PubMed] [Google Scholar]
- 34.Silamut K, Phu NH, Whitty C, et al. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999;155:395–410. doi: 10.1016/S0002-9440(10)65136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modiano D, Petrarca V, Sirima BS, et al. Different response to Plasmodium falciparum malaria in west African sympatric ethnic groups. Proc Natl Acad Sci U S A. 1996;93:13206–11. doi: 10.1073/pnas.93.23.13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaddouri H, Djimde A, Dama S, et al. Baseline in vitro efficacy of ACT component drugs on Plasmodium falciparum clinical isolates from Mali. Int J Parasitol. 2008;38:791–8. doi: 10.1016/j.ijpara.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Staalsoe T, Giha HA, Dodoo D, Theander TG, Hviid L. Detection of antibodies to variant antigens on Plasmodium falciparum–infected erythrocytes by flow cytometry. Cytometry. 1999;35:329–36. doi: 10.1002/(sici)1097-0320(19990401)35:4<329::aid-cyto5>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- 38.Warimwe GM, Keane TM, Fegan G, et al. Plasmodium falciparum var gene expression is modified by host immunity. Proc Natl Acad Sci U S A. 2009;106:21801–6. doi: 10.1073/pnas.0907590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diatta AM, Marrama L, Tall A, et al. Relationship of binding of immunoglobulin G to Plasmodium falciparum–infected erythrocytes with parasite endemicity and antibody responses to conserved antigen in immune individuals. Clin Diagn Lab Immunol. 2004;11:6–11. doi: 10.1128/CDLI.11.1.6-11.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bull PC, Lowe BS, Kortok M, Molyneux CS, Newbold CI, Marsh K. Parasite antigens on the infected red cell surface are targets for naturally acquired immunity to malaria. Nat Med. 1998;4:358–60. doi: 10.1038/nm0398-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackintosh CL, Mwangi T, Kinyanjui SM, et al. Failure to respond to the surface of Plasmodium falciparum–infected erythrocytes predicts susceptibility to clinical malaria amongst African children. Int J Parasitol. 2008;38:1445–54. doi: 10.1016/j.ijpara.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Djimde AA, Doumbo OK, Traore O, et al. Clearance of drug-resistant parasites as a model for protective immunity in Plasmodium falciparum malaria. Am J Trop Med Hyg. 2003;69:558–63. [PubMed] [Google Scholar]
- 43.Cohen S, Mc GI, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. 1961;192:733–7. doi: 10.1038/192733a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.