

Background: The ability of coenzyme Q10 (CoQ10) to reduce ethanol (EtOH)-induced corneal fibroblast apoptosis has been investigated.
Results: Caspase-2 is activated as an initiator caspase in EtOH-induced apoptosis. By blocking caspase-2 activity, CoQ10 protects the cells from apoptosis.
Conclusion: CoQ10 rescues apoptotic response through caspase-2 inhibition toward EtOH treatment.
Significance: CoQ10 may decrease EtOH-induced apoptosis.
Keywords: Apoptosis, Caspase, Cornea, Cytochrome c, Mitochondrial Apoptosis, Caspase 2, Coenzyme Q10, Corneal Fibroblasts, Ethanol
Abstract
Recent studies indicate that caspase-2 is involved in the early stages of apoptosis, particularly before the occurrence of mitochondrial damage. Here we report the important role of the coenzyme Q10 (CoQ10) on the activity of caspase-2 upstream of mitochondria in ethanol (EtOH)-treated corneal fibroblasts. After EtOH exposure, cells produce excessive reactive oxygen species formation, p53 expression, and most importantly, caspase-2 activation. After the activation of the caspase-2, the cells exhibited hallmarks of apoptotic pathway, such as mitochondrial damage and translocation of Bax and cytochrome c, which were then followed by caspase-3 activation. By pretreating the cells with a cell-permeable, biotinylated pan-caspase inhibitor, we identified caspase-2 as an initiator caspase in EtOH-treated corneal fibroblasts. Loss of caspase-2 inhibited EtOH-induced apoptosis. We further found that caspase-2 acts upstream of mitochondria to mediate EtOH-induced apoptosis. The loss of caspase-2 significantly inhibited EtOH-induced mitochondrial dysfunction, Bax translocation, and cytochrome c release from mitochondria. The pretreatment of CoQ10 prevented EtOH-induced caspase-2 activation and mitochondria-mediated apoptosis. Our data demonstrated that by blocking caspase-2 activity, CoQ10 can protect the cells from mitochondrial membrane change, apoptotic protein translocation, and apoptosis. Taken together, EtOH-induced mitochondria-mediated apoptosis is initiated by caspase-2 activation, which is regulated by CoQ10.
Introduction
It has been known that ethanol (EtOH) may induce apoptosis, programming cell death in a variety of tissues, including the corneal epithelial cells (1, 2), corneal fibroblasts (3), liver (4), and brain (5). In ophthalmology, it is one of the methods used in the removal of the epithelium during the procedures of refractive surgery, such as photorefractive surgery (PRK) and laser subepithelial keratomileusis (LASEK) (6, 7). The molecular mechanism of EtOH-induced apoptosis in corneal fibroblasts remains to be determined. To date, pharmacological efforts to control early corneal fibroblast apoptosis have not been successful. However, ongoing investigations are still being performed to identify agents that can regulate this phenomenon and the wound healing process during treatment.
Apoptosis is mainly executed by cysteine proteases known as caspases. The apoptotic cascade of caspases is initiated by the activation of apical (initiator) caspases that include caspase-2, caspase-8, caspase-9, and caspase-10 (8–10). In response to noxious stimuli and related cellular stress situations, initiator caspases directly or indirectly activate the executioner caspases (such as caspase-3 and caspase-7), which in turn orchestrate apoptotic cell death (11). Among all initiator caspases, caspase-2, the second mammalian caspase identified, is the most evolutionarily conserved (12). Therefore, it has been suggested that caspase-2 might play a critical role in apoptosis in mammals and that it may act upstream or downstream of mitochondria to promote cytochrome c release in the apoptotic pathway in response to various stimuli (13–19). The activation of caspase-2 occurs in the complex that contains the p53-induced death domain-containing protein and the adaptor protein RAIDD (ribosome-inactivating protein (RIP)-associated ICH-1/CED-homologous protein with death domain) (20).
Ubiquinone Q10 (coenzyme Q10 (CoQ10)3) is a well known electron transporter in complexes I (NADH-ubiquinone oxidoreductase), II (succinate-ubiquinone oxidoreductase), and III (ubiquinone-cytochrome c oxidoreductase) of the mitochondrial respiratory chain (21, 22). CoQ10 serves as a critical regulator of mitochondrial apoptosis, functioning as a ubiquitous free radical scavenger or control of the mitochondrial transition pore opening (21, 23–28). CoQ10 reduces the number of apoptotic keratocytes produced in response to excimer laser irradiation to a much greater extent than do other free radical scavengers, such as ascorbic acid and vitamin E (27). A recent study indicated that CoQ10 can inhibit mitochondrial depolarization, caspase activation, and cell apoptosis after ethanol exposure in the corneal fibroblasts (29). There is strong evidence that suggests ethanol (EtOH) treatment facilitates the mitochondrial dysfunction (30–32). Interestingly, CoQ10 supplements decreased p53-dependent cell death in response to oxidative DNA damage in elderly patients (33).
We previously demonstrated that CoQ10 pretreatment can inhibit caspase-2 and caspase-3 activation during EtOH-induced apoptosis (29). To determine the therapeutic approaches for EtOH-inducing cell apoptosis during refractive surgery, it is important to gain a better understanding of the cell death mechanisms induced by EtOH treatment. In this study we further determined the role of caspase-2 in EtOH-induced corneal fibroblast apoptosis by using a technique that traps the initiator caspases in situ, and we identified caspase-2 as an initiator caspase and as an upstream modulator of mitochondria to mediate EtOH-induced apoptosis. Furthermore, CoQ10 pretreatment plays a crucial role in protecting the cells against EtOH-induced caspase-2 activation, subsequent mitochondrial damage, caspase-3 activation, and apoptosis.
EXPERIMENTAL PROCEDURES
Primary Culture of Corneal Fibroblasts
Primary corneal fibroblasts were obtained from fresh bovine corneas by collagenase digestion modified from the methods described by Funderburgh et al. (34). Briefly, the central portions of fresh bovine corneas were incubated at 37 °C in 2.4 units of dispase II (Roche Applied Science)/ Dulbecco's modified Eagle's medium (DMEM; Invitrogen) solution containing antibiotics (penicillin, 50 μg/ml; streptomycin, 50 μg/ml; amphotericin B, 2.5 μg/ml) at 37 °C for 3 h to remove the corneal epithelium and endothelium. After dispase II digestion, serial scraping with a plastic spatula (Cell Scraper, TPP, Switzerland) was performed to remove the epithelial cells in phosphate-buffered saline (PBS). Corneal endothelial cells and Descemet's membrane were peeled away in a sheet from the periphery to the center of the inner surface of the cornea with fine forceps.
The tissue was rinsed twice with DMEM medium containing antibiotics, then minced into several small parts (2–3 mm) and incubated in a volume of 1 ml per corneal stoma of 2 mg/ml (w/v) collagenase A (Roche Applied Science) in DMEM with antibiotics at 37 °C for 12 h until the complete disruption of the tissue was achieved. Nylon mesh (40 mm; Cell Strainer, Falcon) was used to filter the cell suspension. The filtered cell suspension was incubated in 75-ml flasks at 37 °C with 10% fetal bovine serum (FBS; Invitrogen) in 95% air, 5% CO2. The samples were serially trypsinized and passaged three times for the experiments.
Treatment
Treatment with 10 μm CoQ10 dissolved in 0.04% Lutrol F217 was commenced 2 h before the application of EtOH (29). Lutrol F217 was used as the vehicle to ensure the cellular uptake of CoQ10 (35). Corneal fibroblasts cultured to ∼90% confluence were pretreated with or without CoQ10 and then exposed to EtOH (0.004–20%) for 20 s. EtOH was diluted in distilled water to yield the indicated concentrations of EtOH solution. In addition, 20 μm caspase-2 inhibitor (z-VDVAD-fmk; BioVision) were used 2 h before EtOH exposure when indicated. Cells in the control group were treated with medium only.
Analysis of Cell Viability
To measure cell viability, we used the CellTiter-Fluor Cell Viability assay (Promega Corp., Madison, WI). Cell viability was analyzed after the cells were exposed to EtOH (0.004–20%) for 20 s. Briefly, cells (7000 cells/well) were plated in 96-well flat-bottomed plates. After incubation with EtOH, 40 μl of CellTiter-Fluor reagent was added to each well and incubated 1.5–2 h at 37 °C. Fluorescence, which is proportional to cell viability, was measured with a FL600 fluorimeter.
Identification of Apoptosis Induced by Ethanol
To examine the apoptosis in EtOH-exposed (0.004–20%, 20 s) cells with or without 2 h of CoQ10 pretreatment, the cells were washed twice with PBS after exposure and then incubated for 4, 8, or 12 h (1, 2). The cells were simultaneously subjected to annexin V and propidium iodide (PI) assays. An annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (Serotec, Oxford, UK) was used to bind phosphatidylserine, which is translocated to the outer leaflet of the plasma membrane during the early stages of cell apoptosis (36). Therefore, the apoptotic cells were only stained with annexin V-FITC, whereas the necrotic cells were double- stained for both annexin V-FITC and PI. The cells were suspended in binding buffer at a final cell concentration of 1 × 105 cells/ml and incubated with both annexin V-FITC and PI for 15 min in the dark. The exposed phosphatidylserine was measured using fluorescence-activated cell sorter analysis.
Determination of Reactive Oxygen Species (ROS)
Intracellular ROS were measured based on the intracellular peroxide-dependent oxidation of 2′,7′-dichlorodihydrofluorescein diacetate (Molecular Probes) to form the fluorescent compound 2′,7′-dichlorofluorescein (DCF), as previously described (37). The ROS levels were assessed early after ethanol exposure and before the occurrence of apoptosis as examined by flow cytometry. The cells were seeded onto 48-well plates at a density of 2 × 104 cells/well and cultured for 48 h. After the cells were washed twice with PBS, fresh medium with or without 10 μm CoQ10 was added. The cells were incubated for 2 h and then exposed to EtOH (20%, 20 s). DCF diacetate (20 μm) was added to the cells, which were incubated for 30 min at 37 °C. After the indicated incubation periods (1–120 min), the cells were harvested and resuspended in 50 mm HEPES buffer (5 mm HEPES, pH 7.4, 5 mm KCl, 140 mm NaCl, 2 mm CaCl2, 1 mm MgCl2, and 10 mm glucose). The fluorescence intensity was determined at 485 nm, and emission was determined at 530 nm through flow cytometric analysis.
Measurement of Changes in Mitochondrial Membrane Potential (ΔΨm)
To determine ΔΨm, the cells were pretreated with or without CoQ10 and then exposed to EtOH (20%, 20 s). After the incubation periods (0.5–8 h), corneal fibroblasts were loaded with 2 μm 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl-carbocyanine iodide (JC-1; Molecular Probes) for 30 min to follow ΔΨm. In normal healthy cells, JC-1 undergoes aggregation within the mitochondrial matrix when the ΔΨm is >−140 mV. In apoptotic cells exhibiting a dissipated ΔΨm, this dye dose not stain mitochondria and remains located in the cytosol in a monomeric form. After the removal of the JC-1, the cells were washed with PBS, harvested through trypsinization, and resuspended in PBS. For flow cytometry, JC-1 has polychromatic fluorescence emission: a green fluorescence in a monomeric state and a red one in an aggregate state. The amount of JC-1 retained by 10,000 cells per sample was measured at 530 nm (green fluorescence) and 590 nm (red fluorescence) using a flow cytometer and a Cell Quest Alias software for analysis. The changes in green fluorescence were evaluated to determine ΔΨm (38).
In Situ Labeling of Active Caspases
To capture the active initiator caspase, corneal fibroblasts were pretreated with cell-permeable b-VAD-fmk (50 μm) for 2 h in the conditional media (39, 40). Cells were treated with EtOH, and samples were collected at 2 h. The corneal fibroblasts were pelleted and lysed by freeze-thawing (5–6 times) in 500 μl of CHAPS lysis buffer (150 mm KCl, 50 mm HEPES, 0.1% CHAPS, and 0.1% Nonidet P-40 with pH 7.4 also containing the protease inhibitors (2 μg/ml) and phenylmethylsulfonyl fluoride). The supernatant was collected after centrifugation at 15,000 × g for 10 min, and the streptavidin-agarose (30 μl) was then added to the supernatants to capture the b-VAD-fmk-bound active caspases. After overnight rotation at 4 °C, the agarose beads were extensively washed in lysis buffer. The biotinylated proteins were eluted from the beads by the addition of SDS sample buffer and incubated at 95 °C for 10 min. Whole cell lysates (WCL) (25 μg) that did not go through streptavidin precipitation or the eluted biotinylated proteins (25 μl) were resolved for using SDS-PAGE. The endogenous biotinylated protein acetyl-CoA carboxylase functioned as a control for both pulldown efficiency and for loading.
Caspase Activity Assay
To measure the levels of caspase activity, cells that were pretreated with or without CoQ10 were exposed to EtOH (20%, 20 s). The caspase levels were measured with caspase-3/CPP32, caspase-2, caspase-8, and caspase-9 Colorimetric Assay kits (BioVision, Palo Alto, CA) at 0.5, 1, 1.5, 2, 4, and 8 h after EtOH exposure. The cells were resuspended in 100 μl of chilled lysis buffer and incubated on ice for 10 min. After incubation, the cells were centrifuged for 1 min in a microcentrifuge (10,000 × g). The supernatant (cytosol extract) was transferred to a fresh tube and left on ice. After assessment of the protein concentration (bicinchoninic acid (BCA) method), each cytosolic extract was diluted to a concentration of 50–200 μg of protein/50 μl cell lysis buffer (1–4 mg/ml). The samples were measured and aliquoted to provide a 2× reaction buffer into a glass tube (assuming 50 μl of 2× reaction buffer per sample). A volume of 2× reaction buffer containing 10 mm DTT (50 μl) and 5 μl of the appropriate caspase substrate (4 mm) was added to each sample (final concentration 200 μm). The substrates used for the different caspases include DEVD-p-nitroanilide (pNA) for caspase-3, VDVAD-pNA for caspase-2, IETD-pNA for caspase-8, and LEHD-pNA for caspase-9. The samples were incubated for 2 h in the dark. The absorbance was measured on a plate reader at 400 nm (or 405 nm).
Western Blot Analysis
For the Western blot analysis, the corneal fibroblasts were treated with or without CoQ10 and then exposed to EtOH (20%) for 20 s. After the indicated incubation period (p53: 30, 60, 90, and 120 min; Bax: 90 min; cytochrome c: 120 min; caspase-2 and caspase-3: 2, 4, and 8 h), the cytosolic fractions were prepared as reported previously (41). The total protein concentration of each sample was measured using the BCA method. The total cell protein (60 μg) was resolved by SDS-PAGE on 12% acrylamide gels and blotted onto polyvinylidene difluoride membranes. The blotted membrane was incubated with PBS, Tween 20 (0.05%) solution containing 2% skim milk to block nonspecific antigens. Then it was incubated with goat anti-human p53 (R&D Systems, Minneapolis, MN), mouse anti-Bax (BD Pharmingen), mouse anti-cytochrome c (BD Pharmingen), rabbit anti-human caspase-3 (Cell Signaling, Danvers, MA), rabbit anti-human/mouse caspase-2 antibody (R&D Systems, Minneapolis, MN), or mouse anti-GAPDH antibody (Abcam) at 4 °C overnight. After the primary antibody reaction, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG antibody (Cell Signaling), horseradish peroxidase-conjugated anti-mouse IgG antibody (Amersham Biosciences), and horseradish peroxidase-conjugated anti-goat IgG antibody (Sigma), detected using Chemiluminescence Reagent Plus (PerkinElmer Life Sciences), and exposed to film (BioMax MR, Kodak). The intensity of each band was scanned and quantified using a densitometer linked to computer software (ImageQuant; Amersham Biosciences). For examining the existence of caspase-2 dimer formation, SDS sample buffer was supplemented either with β-mercaptoethanol for a reducing condition or without β-mercaptoethanol for a nonreducing condition (42).
Preparation of Cytosolic and Mitochondrial Fractions
The corneal fibroblasts were washed with ice-cold PBS (1 ml). The cells were centrifuged at 600 × g for 5 min at 4 °C. The supernatant was removed, and the cells were resuspended in 500 μl of mitochondria isolation buffer (0.25 m sucrose, 0.5 mm EGTA, 3 mm HEPES-NaOH, protease inhibitors mixture, pH 7.2), after which it was incubated on ice for 60 min. Afterward, the cells were homogenized in an ice-cold Dounce tissue grinder. This task was performed with the grinder on ice (300 up and down). The homogenate was transferred to a 1.5-ml microcentrifuge tube and centrifuged at 700 × g for 10 min at 4 °C. The supernatant was transferred to a fresh, 1.5-ml tube and centrifuged at 10,000 × g for 25 min at 4 °C. The supernatant was collected (this is cytosolic fraction), and the pellet was resuspended in 10–30 μl of mitochondria lysis buffer (50 mm HEPES-NaOH, 1% maltoside, 10% glycerol, 1 mm EGTA, 1 mm EDTA, protease inhibitors mixture, pH 7.4).
RNA Knockdown by Lentivirus-based Short-hairpin RNA (shRNA) Delivery
shRNAs were obtained from the National RNAi Core Facility located at the Institute of Molecular Biology/Genomic Research Center, Academia Sinica, Taiwan. The TRC numbers of shRNA clones for caspase-2 used were TRCN0000003505 (clone 3505), TRCN0000003506 (clone 3506), TRCN0000003507 (clone 3507), TRCN0000003508 (clone 3508), and TRCN0000003509 (clone 3509). The TRC numbers of shRNA clones for caspase-3 used were TRCN0000003549 (clone 3549), TRCN0000003550 (clone 3550), TRCN0000003551 (clone 3551), TRCN0000003552 (clone 3552), and TRCN0000010798 (clone 10798). Lentiviruses containing different shRNA plasmids were generated according to the protocol of the National RNAi Core Facility. A multiplicity of infection equal to 1 was used for all lentiviruses to infect cells. After infection for 72 h, cells were subjected to Western blot analysis.
Statistical Analysis
Values are expressed as the means ± S.D. The statistical analysis was performed with one-way analysis of variance followed by Scheffe test where appropriate. The statistical significance was determined at the 0.05 level.
RESULTS
EtOH Induces Apoptosis in Corneal Fibroblasts
We investigated the dose- and time-dependent cytotoxic effect of EtOH in corneal fibroblast cultures. The cells were treated with different concentrations of EtOH (0.004 −20%) for 20 s, and then the EtOH was removed (washed by PBS twice). The cells were further incubated for 4, 8, and 12 h. The viability was determined using the CellTiter-Fluor Cell Viability Assay. EtOH exposure alone decreased cell viability at indicated concentrations (Fig. 1A). Cell viability was significantly reduced at 4 h in the 4 and 20% EtOH group and at 8 and 12 h in the 0.004, 0.04, 0.4, 2, 4, and 20% EtOH groups. One important finding is that 20% EtOH exposure, which was frequently applied in the cornea during refractive surgery, resulted in decreased cell survival (77.81 ± 4.67%) at 4 h. The fraction of cell survival continued to decrease at 8 and 12 h (18.27 ± 1.66% and 5.77 ± 2.68%, respectively). To identify the mode of cell death, we analyzed cell apoptosis using annexin V-FITC and PI double staining methods after incubation for 4, 8, or 12 h (Fig. 1B). Treatment of EtOH at concentrations of 0.004–20% showed a significant increase in apoptotic cells over the untreated cells after incubating for 4, 8, and 12 h (Fig. 1B), particularly at the concentration of 20%. Therefore, 20% EtOH was used for further experiments.
FIGURE 1.
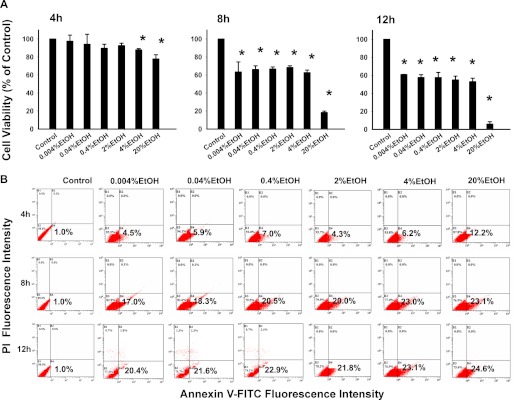
EtOH induces apoptotic cell death in corneal fibroblasts. Primary corneal fibroblasts were treated with EtOH (0.004–20%, 20 s). A, shown is effect of EtOH on cell viability. Cell viability was measured by the CellTiter-Fluor Cell viability assay. B, shown is the effect of EtOH on cell apoptosis. The cells were treated with different concentrations of EtOH (0.004–20%) for 20 s, and then the EtOH was removed (washed by PBS twice). The cells were further incubated for 4, 8, and 12 h and stained with annexin V-PI for flow-cytometric analysis.
EtOH Induces ROS Production and p53 Expression in Corneal Fibroblasts
The results of the DCF assay indicated that EtOH exposure (20%, 20 s) caused an increase in the intracellular levels of ROS. The relative DCF fluorescence intensities that were measured after EtOH exposure at 1, 5, 10, 20, 30, 60, 90, and 120 min were as follows: 1 min, 1.4 ± 0.4%; 5 min, 1.3 ± 0.1%; 10 min, 0.9 ± 0.2%; 20 min, 1.6 ± 0.3%; 30 min, 8.1 ± 0.8%; 60 min, 2.8 ± 0.7%; 90 min, 2.1 ± 0.5%; 120 min, 1.4 ± 0.3%. EtOH exposure caused an increase of at least 8-fold in intracellular ROS levels at 30 min (Fig. 2A). Previous studies have shown that oxidative stress can induce the activation of p53 in stem cell-derived dopaminergic neurons upstream of mitochondrial permeabilization, cytochrome c release, and caspase-3 activation (43, 44). Hence, the time-dependent p53 activation after EtOH exposure was determined using Western blot analysis in the corneal fibroblasts. Our data revealed the activation of p53 in a time-dependent manner (Fig. 2B). The maximal expression of p53 was detected 90 min after EtOH exposure, and the time point was later than the maximal increase of ROS (i.e. 30 min after EtOH exposure). The result suggested that EtOH-induced p53 activation occurred downstream of the ROS formation.
FIGURE 2.

EtOH induces ROS production, p53 expression, and mitochondria-mediated apoptosis in corneal fibroblasts. A, ROS levels in cells treated with EtOH (20%, 20 s) were measured by analyzing the DCF intensity after 1, 5, 10, 20, 30, 60, 90, and 120 min. Data represent the results of three independent experiments performed in triplicate (means ± S.D.; *, p < 0.05 compared with the control group). B, Western blots analysis using p53 antibody demonstrated time-dependent p53 activation after EtOH exposure. Protein expression of GAPDH was used as an internal control. C, the mitochondrial MPT was determined in cells treated with EtOH (20%, 20 s) after intervals of 0.5, 1, 1.5, 2, 4, and 8 h. The bar diagram shows the relative JC-1 green fluorescence after normalization to control at all intervals. Data represent the results of three independent experiments performed in triplicate (means ± S.D.; *, p < 0.05 compared with the control group). D, upon MPT, cytochrome c release can be identified by translocation to cytosol at 2 h. Data from Western blotting are summarized (means ± S.D.) from three separate experiments quantified by densitometry after normalization to GAPDH (a cytosolic internal control) or Cox IV (cytochrome c oxidase IV, a mitochondrial internal control). *, p < 0.05 compared with the control group.
EtOH Induces Mitochondria-mediated Apoptosis in Corneal Fibroblast
To further investigate the involvement of mitochondrial damage in EtOH-induced cell apoptosis, the ΔΨm was determined. Using JC-1 staining, we found that EtOH induced mitochondrial membrane potential transition (MPT) in a time-dependent manner (Fig. 2C). A flow-cytometric analysis of MPT demonstrated that the relative green signals of the EtOH-exposed cells at 0.5, 1, 1.5, 2, 4, and 8 h were 19.7 ± 3.0, 16.6 ± 2.6, 17.5 ± 1.20, 29.65 ± 3.46, 25.75 ± 2.05, and 17.65 ± 2.62%, respectively. EtOH exposure caused a significant change in the mitochondrial membrane potential (0.5 h, p = 0.03; 1 h, p = 0.02; 1.5 h, p = 0.03; 2 h, p = 0.01; 4 h, p = 0.02; 8 h, p = 0.03). In addition, an increase in cytosolic cytochrome c expression was also observed (Fig. 2D).
Caspase-2 Is the Initiator Caspase Activated during EtOH-induced Apoptosis
Caspase-2, caspase-8, and caspase-9 are known initiator caspases in various types of cellular stress (8–10). However, the initiator caspase of EtOH-induced apoptosis remained to be determined. To identify the initiator caspase, we performed the in situ trapping approach (40). We bound active caspase by using a biotinylated form of the caspase inhibitor (b-VAD-fmk) that can bind to activated caspase irreversibly. With the use of streptavidin, activated caspase can be isolated from the cell lysates. The b-VAD-fmk pretreatment of cells before EtOH exposure leads to trapping of the active initiator caspase and also inhibits the activation of downstream caspases. Therefore, the caspase that was bound to the b-VAD-fmk can be identified as the initiator caspase. We treated the corneal fibroblasts with b-VAD-fmk (50 μm) for 2 h before exposure to EtOH. After EtOH exposure, cells were harvested at 0, 1, 2, and 4 h. The initiator caspase was precipitated using streptavidin, and the bound caspase was further identified by Western blotting. The membrane was probed with known initiator caspase antibodies, i.e. anti-caspase-2, anti-caspase-8, and anti-caspase-9 antibodies; the results for the pulldown assay are shown in Fig. 3A. Those for the WCL that did not undergo streptavidin capture are shown in Fig. 3B. The results demonstrate that caspase-2 was pulled down by streptavidin precipitation within hours of EtOH treatment, suggesting its function as the initiator caspase. At 2 h of incubation, we observed the increase in the precursor form of caspase-2, indicating its activation (Fig. 3A). Interestingly, in the WCL the precursor from of caspase-2 was cleaved at 2 h of incubation (Fig. 3B). On the other hand, we did not detect the increase in the precursor form of caspase-8 and caspase-9 in the immunoblots (performed on streptavidin pulldown assay samples) even after a longer time of exposure. There was no cleavage or reduction in the precursor form of caspase-8 or caspase-9 in the WCL samples (Fig. 3B), suggesting that these caspases are not the initiators during EtOH-induced apoptosis in corneal fibroblasts.
FIGURE 3.
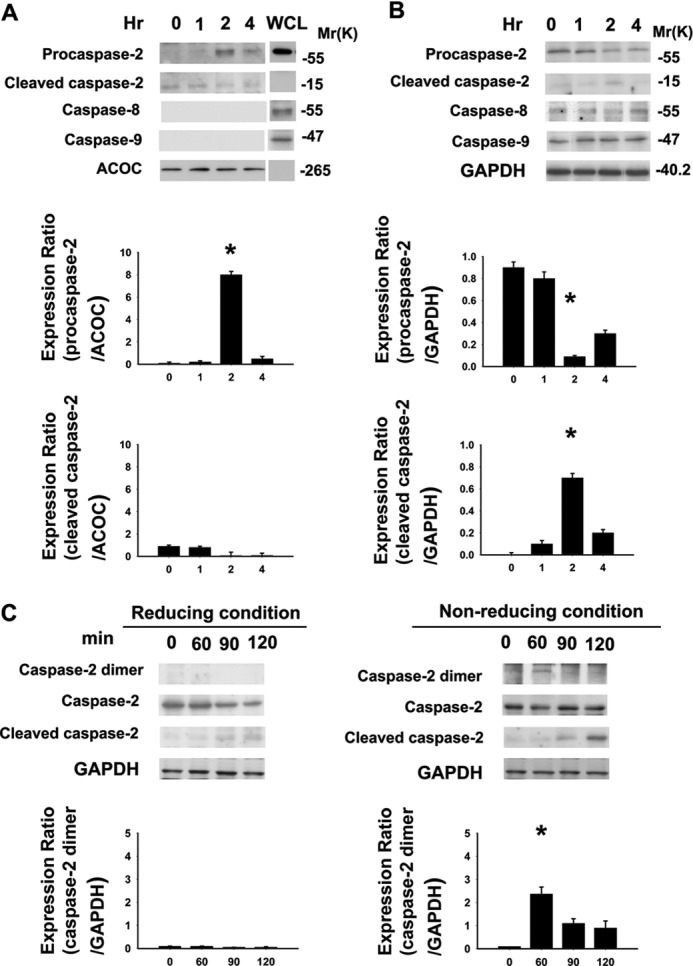
Caspase-2 is the initiator caspase in EtOH-exposed primary corneal fibroblasts. A and B, to identify the initiator caspase, corneal fibroblasts were pretreated with b-VAD-fmk (50 μm) for 2 h, then treated with EtOH (20%, 20s). After EtOH exposure, EtOH was removed (washed by PBS twice). The cells were further incubated for the indicated incubation times (0, 1, 2, and 4 h). Lysates were either subjected to streptavidin pulldown assay (A) or WCL were prepared (B), and Western blot analysis was performed for caspase-2, caspase-8, and caspase-9 using specific antibodies, respectively. WCL of untreated corneal fibroblasts that were not underwent streptavidin pull-down assay acted as a positive control for immunoblotting in the pull-down assay. The same blots were subsequently probed, and acetyl-CoA carboxylase (ACOC) or GAPDH (for WCL) was used as a control for protein loading. Experiments were performed three times, and representative blots with similar results are shown. C, protein samples were analyzed using SDS-PAGE in a reducing and a nonreducing condition. Data from Western blotting are summarized (means ± S.D.) from three separate experiments quantified by densitometry after normalization to GAPDH (internal control). *, p < 0.05 compared with the data at 0 h.
Initiator caspases are present in the cells as inactive monomers, and their activation is promoted by dimerization (45). Dimerization results when the initiator caspases are recruited to large molecular weight protein complexes that act as signaling platforms (9). Evidence shows that dimerization is the initiating step of caspase-2 activation (8). To examine the existence of caspase-2 dimer formation, we perform the reducing and nonreducing SDS-PAGE analysis of the samples. The results confirm that protein expression of caspase-2 dimer (∼120 kDa) was detected in the nonreducing condition after EtOH treatment (Fig. 3C). Thus, dimerization of casapse-2 does occur in the EtOH induced caspase-2 activation process.
To further explore the time-dependent activation of different caspases, we examined the catalytic activation and expression of the caspases (caspase-2, caspase-3, caspase-8, and caspase-9) in the cells that were exposed to EtOH (20%, 20 s) at 0.5- 8 h (see Fig. 8, A–D). In the EtOH-exposed cells, the enzymatic activation of caspase-2 and caspase-3 began at 1.5 and 2 h after treatment, respectively (Fig. 8, A and B). There was an increase of ∼2-fold in caspase-2 and caspase-3 activity at 2 h after EtOH exposure (caspase-2, p = 0.005; caspase-3, p = 0.01). However, there was no significant difference in the activity of caspase-8 or caspase-9 during different incubation time after EtOH exposure (Fig. 8, C and D). Thus, our results identify caspase-2 as an initiator caspase in EtOH-induced apoptosis in primary corneal fibroblasts.
FIGURE 8.
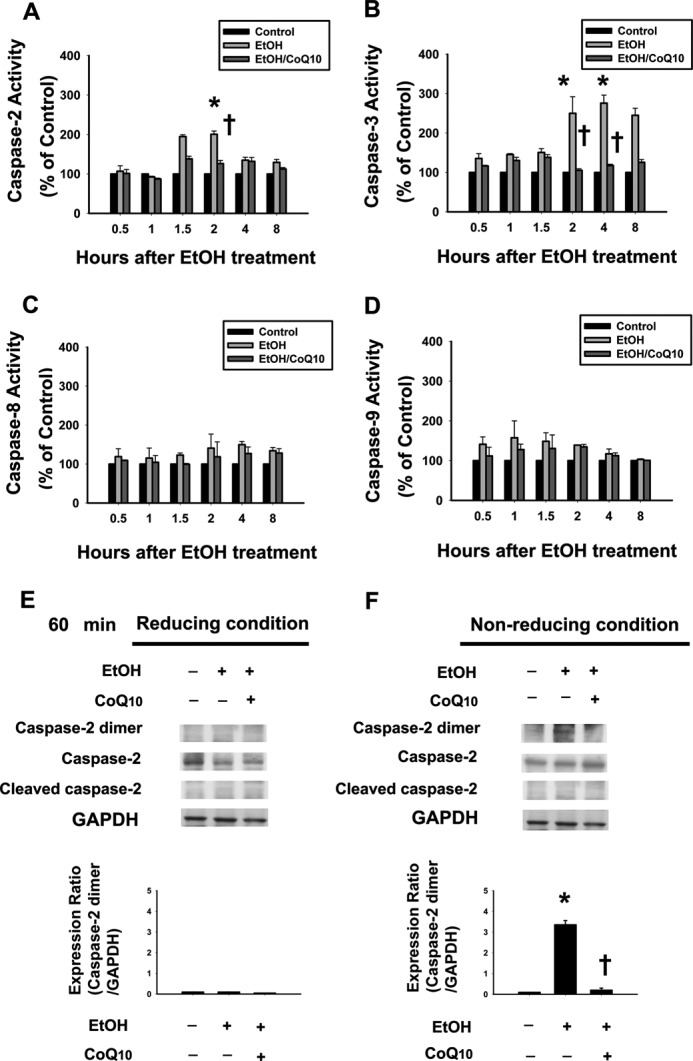
Pretreatment of corneal fibroblasts for 2 h with 10 μm CoQ10 prevents the activation of caspase-2 and caspase-3 induced by EtOH exposure. The activity of caspase-2 (A), caspase-3 (B), caspase-8 (C), and caspase-9 (D) in cells pretreated with or without CoQ10 followed by EtOH treatment (20%, 20 s) was examined by colorimetric assay after intervals of 0.5, 1, 1.5, 2, 4, and 8 h. Protein samples were analyzed using SDS-PAGE in reducing (E) and nonreducing (F) conditions. Data from Western blotting are summarized (means ± S.D.) from three separate experiments quantified by densitometry after normalization to GAPDH (internal control). *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group.
Decreased EtOH-induced Apoptotic Cell Death with Caspase-2 Inhibitor
To examine whether caspase-2 was required for the mitochondrial intrinsic pathway of apoptosis, we inactivated caspase-2 in the corneal fibroblasts through pretreatment with the inhibitor z-VDVAD-fmk (caspase-2 inhibitor). First, we tested the substrate cleavage activity of caspase-2 after z-VDVAD-fmk pretreatment during EtOH-induced apoptosis (Fig. 4A). The z-VDVAD-fmk significantly abrogated the substrate cleavage activity of caspase-2 at all incubation periods (2, 4, 8, and 12 h) after EtOH exposure (Fig. 4A). On the other hand, the z-VDVAD-fmk did not inhibit substrate cleavage activity of caspase-3 at 2 and 4 h (Fig. 4B). However, the z-VDVAD-fmk may exert some inhibition on the substrate cleavage activity of caspase-3 at 8 and 12 h. Therefore, we examined the inhibition of cell death (Fig. 4C), cell apoptosis (Fig. 4D), and the change in ΔΨm (Fig. 4E) after z-VDVAD-fmk pretreatment in response to EtOH stimulation at the indicated incubation period (less than 4 h).
FIGURE 4.

EtOH-induced cell death is inhibited with caspase-2 inhibitor. The cultured corneal fibroblasts were pretreated with or without caspase-2 inhibitor followed by EtOH (20%, 20 s) exposure. The inhibition of EtOH-induced caspase-2 substrate-cleavage activity with caspase-2 inhibitor (A) and the direct influence of caspase-2 inhibitor on caspase-3 cleavage activity (B) were determined. Cell viability (C) and apoptosis (D) was determined by CellTiter-Fluor Cell Viability assay and stained with annexin V-PI for flow-cytometric analysis, respectively. Data were derived from three independent experiments (means ± S.D.; *, p < 0.05 compared with the control group; #, p < 0.05 compared with the EtOH-treated group). E, loss of mitochondrial membrane potential was demonstrated by the change in JC-1 derived fluorescence from red (high potential as JC-1 aggregates) to green (low potential as JC-1 monomer). With or without caspase-2 inhibitor (20 μm), MPT was determined at 0.5, 1, 1.5, and 2 h. Data represent the results of three independent experiments performed in triplicate (means ± S.D.; *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group).
The EtOH-induced apoptosis is shown in nearly 20% of the cultured corneal fibroblasts after 4 h. However, the z-VDVAD-fmk pretreated cells showed a significant decrease of cell apoptosis at 4 h after EtOH exposure (p = 0.01; Fig. 4D). To determine whether the cells were dying via an alternative (nonapoptotic) mechanism after z-VDVAD-fmk pretreatment, we measured cell survival using the CellTiter-Fluor Cell Viability assay. Interestingly, after EtOH exposure, the z-VDVAD-fmk-pretreated cells showed a survival advantage at 4 h (p = 0.01; Fig. 4C). EtOH treatment resulted in a time-dependent change in ΔΨm. By z-VDVAD-fmk pretreatment, the results showed a significant decrease in ΔΨm (0.5 h, 5.7 ± 1.2%, p = 0.02; 1 h, 4.2 ± 0.6%, p = 0.008; 1.5 h, 9.8 ± 1.1%, p = 0.03; 2 h, 10.6 ± 1.6%, p = 0.01; Fig. 4E). Collectively, the data may suggest that inhibition of caspase-2 resulted in increased cell viability and cell survival. However, the nonspecific inhibition of caspase-3 may occur by using the z-VDVAD-fmk (46). We further use a molecular approach by knocking down the caspase-2 to examine the role of caspase-2 in EtOH-induced apoptosis.
Knockdown of Caspase-2 Expression Decreases EtOH-induced Apoptotic Cell Death
To address potential concerns as to the specificity of the caspase inhibitors (46), the RNA interference (RNAi) technique was used to further identify the role of caspase-2 (47). RNAi is a natural, evolutionarily conserve regulatory mechanism that is mediated by the introduction of dsRNA into the cytoplasm of a host cell (48). It has provided a unique tool for sequence-specific silencing to develop practical strategies for studying gene function, biological processes, and pathway analysis (47). shRNA is a sequence of RNA that makes a tight hairpin turn that silences gene expression via RNAi (49). Corneal fibroblasts with specific gene knockdown were generated by transfection of the cells with lentivirus vectors expressing gene specific shRNA (50). Five caspase-2 knockdown clones and one control clone were selected based on enhanced green fluorescent protein fluorescence detection. The protein expression of caspase-2 was further monitored by Western blot analysis (Fig. 5A). The control clone cells did not exhibit any difference in procaspase-2 expression compared with the original cells. The expression levels of procasapse-2 protein in five caspase-2 knockdown clones decreased by 15.23–81.22% (% of control). When treated with EtOH (20%, 20s), the control clones demonstrated a similar rate of cell survival compared with the original cells (54.5 ± 2.12 versus 53.0 ± 1.23%, Fig. 5B). In contrast, caspase-2 knockdown clones showed a higher survival rate (Fig. 5B). Of the 5 caspase-2 knockdown clones, clone 3507 had the significantly higher rate of cell survival and significantly lower rate of apoptosis (p < 0.05, Fig. 5B) and is correlated with the lowest procaspase-2 expression (Fig. 5A). These results confirm that EtOH-induced apoptosis is mediated specifically by caspase-2. We further examined the cell-protective effect of caspase-3 knockdown clones upon EtOH exposure. First we tested the procaspase-3 expression among the knockdown clones and found one of the caspase-3 knockdown clones, clone 3551, had the lowest procaspase-3 expression (Fig. 5A). Clone 3551 also exhibited the higher rate of cell viability and lower rate of apoptotic cell death (p < 0.05, Fig. 5C). However, the cell-protective effect of clone 3551 was not as significant as the caspase-2 knockdown clone (clone 3507).
FIGURE 5.

EtOH-induced cell death and mitochondrial membrane potential change (ΔΨm) are inhibited by caspase-2 RNA knockdown. Corneal fibroblasts with or without caspase-2 shRNA were treated with EtOH (20%, 20 s). A, caspase-2 or caspase-3 expression in different clones was detected by Western blot analysis. The bar graphs represent the amount of procaspase-2 that were quantified by densitometry and normalized to the GAPDH data. The cell viability assay and annexin V-PI analysis were used to detect cell survival and apoptotic rate in different caspase-2 knockdown clones (B) and caspase-3 knockdown clones (C), respectively. The asterisk (*) represents significant difference of cell survival or apoptosis from different clones compared with that from the parent corneal fibroblasts cells. Data are shown as the means ± S.D. from three independent experiments. D, the ΔΨm was investigated at 0.5, 1, 1.5, and 2 h in the original cells, control clone, caspase-2, and caspase-3 knockdown clones (clone 3507 and clone 3551, respectively).
In addition, we compared the effect of caspase-2 or caspase-3 knockdown on the ΔΨm after EtOH treatment. Based on the expression levels of procaspase-2 and procaspase-3, clone 3507 (caspase-2 knockdown) and clone 3551 (caspase-3 knockdown) were chosen to follow the ΔΨm due to the lowest expression in the protein levels of procaspase-2 or -3, respectively. Significant ΔΨm reduction was found in the cells of caspase-2 knockdown (clone 3507) (0.5 h, 16.8 ± 0.5% versus 35.6 ± 0.9%, p = 0.03; 1 h, 18.0 ± 0.6% versus 50.1 ± 0.7%, p = 0.01; 1.5 h, 20.6 ± 0.9% versus 59.0 ± 0.6%, p = 0.02; 2 h, 27.9 ± 0.4% versus 75.0 ± 0.8%, p = 0.008; Fig. 5D). On the other hand, there was no significant change in the ΔΨm in the cells of control clone or capsase-3 knockdown clones (clone 3551) (p > 0.05; Fig. 5D). These findings demonstrated that the loss of caspase-2 can protect the cells against mitochondrial dysfunction in the pathway of EtOH-induced apoptosis.
Caspase-2 Acts Upstream of EtOH-induced Mitochondrial Apoptotic Events
We further investigated how caspase-2 is involved in EtOH-induced apoptosis. Caspase-2 has been shown to induce mitochondrial outer membrane permeabilization (MOMP) that leads to the release of pro-apoptotic molecules from mitochondria (13, 15, 16, 51). It is well documented that cytochrome c is released from the mitochondria after MOMP induction and, therefore, can be a reliable index of MOMP (52). To examine the effect of caspase-2 activation on the release of cytochrome c from mitochondria, we performed Western blotting in the EtOH-exposed cell with or without the z-VDVAD-fmk pretreatment. We found that EtOH-induced cytochrome c release was reduced with the z-VDVAD-fmk pretreatment (Fig. 6B).
FIGURE 6.
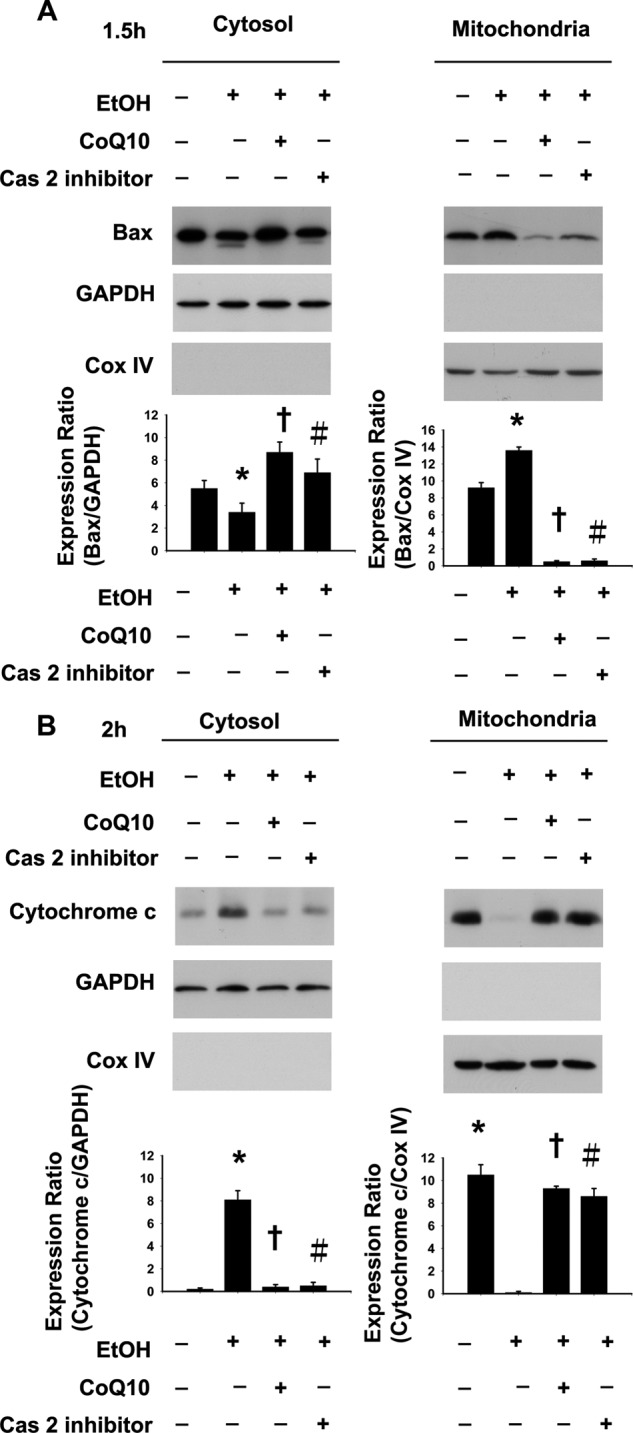
EtOH induces caspase-2 mediated mitochondrial protein translocation. EtOH-induced Bax (A) and cytochrome c (B) with or without caspas-2 inhibitor was determined by Western analysis. Pretreatment of corneal fibroblasts for 2 h with 10 μm CoQ10 prevented the caspase-2 mediated translocation of mitochondrial proteins induced by EtOH exposure (20%, 20 s). Western blot analysis was performed to determine the effect of CoQ10 on caspase-2 mediated translocation of Bax (A) and cytochrome c (B) upon EtOH exposure. Protein expression of GAPDH and Cox IV (cytochrome c oxidase IV) was used as a cytosolic and mitochondrial internal control, respectively. Each bar graph shows summarized data (means ± S.D.) from three separate experiments by densitometry after normalization to GAPDH or Cox IV. *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group; #, p < 0.05 compared with the EtOH-treated group.
One particular way by which caspase-2 can induce MOMP is by activating Bax (13, 53). We examined whether EtOH leads to mitochondrial Bax translocation and, if so, whether this translocation is affected by the inhibition of caspase-2. In the corneal fibroblasts, EtOH treatment induced Bax activation, as determined by Western blotting (Fig. 6A). However, the mitochondrial translocation of Bax was reduced in the EtOH-exposed cells that were pretreated with z-VDVAD-fmk, suggesting an involvement of caspase-2 in the EtOH-induced Bax activation (Fig. 6A).
CoQ10 Blocks Caspase-2-mediated EtOH-induced Apoptosis
CoQ10 acts as an anti-apoptotic factor that blocks death signals, including those from EtOH (29, 33, 35, 54). To investigate the relation between the CoQ10 and caspase-2 activity during EtOH-induced apoptosis, we pretreated the cells with CoQ10. The pretreatment with CoQ10 significantly increased the cell viability at 8 and 12 h in 0.004, 0.04, 0.4, 2, 4, and 20% CoQ10/EtOH groups (Fig. 7A). Similarly, the pretreatment with CoQ10 significantly decreased the percentage of the apoptotic cells that were induced by EtOH at 8- and 12-h time points (Fig. 7B). As shown in Fig. 7C, the percentages of annexin V-FITC-positive cells after EtOH exposure (20%, 20 s) were as follows: 4 h, 12.2 ± 3.4%; 8 h, 23.1 ± 7.3%; 12 h, 24.6 ± 3.67% (one-way analysis of variance, p < 0.05). The percentages of EtOH-induced apoptosis were attenuated by pretreatment with CoQ10 (4 h, 9.3 ± 4.1%; 8 h, 17.4 ± 2.1%; 12 h, 11.5 ± 2.5%). CoQ10 pretreatment significantly reduced cell apoptosis at 8 and 12 h after EtOH exposure (4 h, p = 0.1; 8 h, p = 0.01; 12 h, p < 0.0001, Fig. 7C).
FIGURE 7.

Pretreatment of corneal fibroblasts for 2 h with 10 μm CoQ10 inhibits the caspase-2-mediated cell apoptosis, elevated ROS levels, and p53 expression induced by EtOH exposure. A, the cells were pretreated with 10 μm CoQ10 for 2 h. Thereafter, EtOH (0.004, 0.04, 0.4, 2, 4, and 20%) at indicated concentrations were added for 20s. After the incubation period (4, 8, or 12 h), the EtOH was removed (washed by PBS twice). The cells were further incubated for 4, 8, and 12 h, and the cell viability was determined by CellTiter-Fluor Cell Viability assay. B, the apoptotic cells were stained with annexin V-PI for flow-cytometric analysis at 4, 8, and 12 h. The bar diagram shows the comparison of the relative fluorescence of annexin V-FITC fluorescence intensity at different times. C, shown are flow cytometric histograms of apoptotic cells pretreated with or without CoQ10, followed by EtOH treatment (20%, 20 s). D, ROS levels in cells pretreated with or without CoQ10 followed by EtOH treatment (20%, 20 s) were measured by analyzing the DCF intensity after 30 min. Data represent the results of three independent experiments performed in triplicate (means ± S.D.; *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group). E, Western blot analysis of p53 was done to investigate the effect of CoQ10 on the time-dependent p53 expression after EtOH exposure (20%, 20 s). Each bar graph shows summarized data (means ± S.D.) from three separate experiments by densitometry after normalization to GAPDH (internal control). *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group.
In addition, CoQ10 pretreatment reduced the ROS levels in the EtOH-exposed cells at all time points (1 min, 1 ± 0.2%; 5 min, 1.2 ± 0.4%; 10 min, 0.5 ± 0.1%; 20 min, 1.2 ± 0.5%; 30 min, 4.2 ± 0.7%; 60 min, 2.5 ± 0.2%; 90 min, 1.8 ± 0.3%; 120 min, 1 ± 0.4%; Fig. 7D). The intracellular ROS levels were significantly lower in cells that were pretreated with CoQ10 at 30 min after exposure to EtOH (30 min, p < 0.0001; 60 min, p = 0.05; 90 min, p = 0.06; 120 min, p = 0.05). Furthermore, CoQ10 pretreatment decreased p53 expression at 90 min after EtOH exposure (Fig. 7E).
CoQ10 Blocks Caspase-2-mediated EtOH-induced Mitochondrial Damage
To determine the effect of CoQ10 on caspase-2-mediated EtOH-induced cell apoptosis, we investigated the change in caspase-2 activity with or without CoQ10 pretreatment after EtOH exposure. A caspase substrate activity assay revealed that CoQ10 pretreatment significantly reduced the activation of caspase-2 (2 h) and caspase-3 (2 and 4 h) after EtOH exposure (caspase-2, p = 0.01; caspase-3, 2, 4 h; p = 0.04) (Fig. 8, A and B). On the other hand, CoQ10 pretreatment has no significant effects on caspase-8 and caspase-9 activation (Fig. 8, C and D). Because caspase-2 is activated by dimerization, we investigated the effect of CoQ10 on the caspase-2 dimer formation by using reducing and nonreducing SDS-PAGE analysis. The results showed that CoQ10 inhibited caspase-2 dimer formation after EtOH exposure at 60 min (Fig. 8, E and F).
Using JC-1 staining, the ΔΨm reduction with CoQ10 pretreatment at 0.5, 1, 1.5, and 2 h was 3.3 ± 0.5, 6.9 ± 0.1, 8.9 ± 0.5, and 6.1 ± 0.21%, respectively (Fig. 9). Pretreatment with CoQ10 significantly reduced caspase-2-mediated MPT in the corneal fibroblasts at 0.5 h (p < 0.001), 1 h (p < 0.001), 1.5 h (p = 0.04), and 2 h (p = 0.01). As shown in Fig. 6, we used Western blot analysis to study the effect of CoQ10 on EtOH-induced caspase-2-mediated mitochondrial Bax and cytosolic cytochrome c translocation. Our results showed that caspase-2-mediated Bax translocation and cytochrome c release from mitochondria after EtOH exposure were not detected in cells that were pretreated with CoQ10 (Fig. 6). These results indicate that the application of CoQ10 could interfere with the caspase-2-mediated and the mitochondria-dependent apoptosis induced by EtOH (Fig. 10).
FIGURE 9.

Pretreatment of corneal fibroblasts for 2 h with 10 μm CoQ10 reduces the caspase-2-mediated mitochondrial membrane potential change induced by EtOH exposure. The mitochondrial MPT was determined in cells pretreated with or without CoQ10 followed by EtOH treatment (20%, 20 s) after intervals of 0.5 (A), 1 (B), 1.5 (C), and 2 h (D). Loss of mitochondrial membrane potential was demonstrated by the change in JC-1-derived fluorescence from red (high potential as JC-1 aggregates) to green (low potential as JC-1 monomer). With or without caspase-2 inhibitor (20 μm), MPT was determined at 0.5, 1, 1.5, and 2 h. E, the bar diagram shows relative JC-1 green fluorescence after normalization to control at all intervals. Data represent the results of three independent experiments performed in triplicate (means ± S.D.; *, p < 0.05 compared with the control group; †, p < 0.05 compared with the EtOH-treated group; #, p < 0.05 compared with the EtOH-treated group).
FIGURE 10.
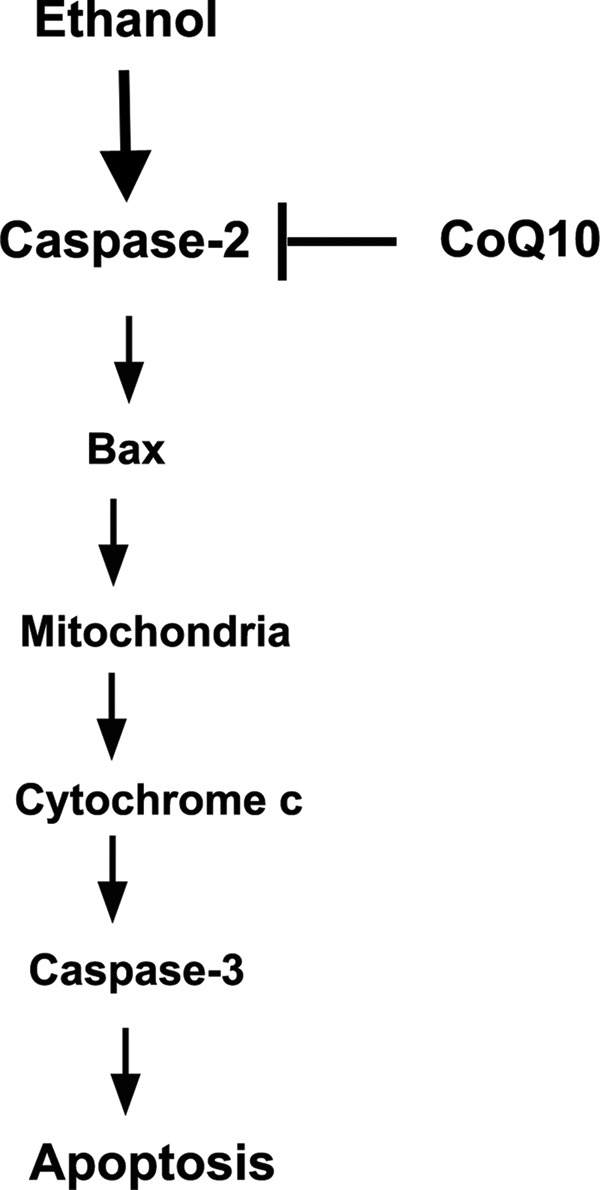
Schematic illustration of the proposed pathways of ethanol-induced cell death in corneal fibroblasts. Caspase-2 is activated as an initiator caspase to induce apoptosis upstream of mitochondria. Coenzyme Q10 rescues apoptotic response through caspase-2 inhibition toward ethanol treatment. In this case the downstream cell death pathways are suppressed, including Bax translocation, mitochondrial dysfunction, cytochrome c release, and caspase-3 activation.
DISCUSSION
In this study we have investigated the mechanism of EtOH-induced cell death in the primary culture of corneal fibroblasts. EtOH-induced oxidative stress causes p53 accumulation and, consequently, caspase-2 activation, which in turn initiates cell apoptosis via the mitochondria-mediated caspase-dependent pathway. We further identified the apical role of caspase-2 in the EtOH-induced cell death and revealed that CoQ10 may exert the anti-apoptotic effect through the inhibition of the ROS, p53 expression, and most importantly, caspase-2 activation to protect the corneal cells from EtOH-induced cell death. CoQ10 pretreatment inhibited caspase-2 activation, mitochondrial damage, expression of Bax and cytochrome c, caspase-3 activation, and cell apoptosis. This study, therefore, addresses the novel anti-apoptotic mechanism of CoQ10. CoQ10 pretreatment blocks caspase-2 activation, although the underlying mechanism remains unclear. Our results also demonstrated the following points; 1) caspase-2 served as an initiator caspase during the mitochondria-dependent pathway of apoptosis upon EtOH exposure, 2) caspase-2 acts upstream of mitochondria during EtOH-induced apoptosis, and 3) the loss of caspase-2 protects the corneal fibroblasts from apoptosis as well as cell death.
Caspase-2 is one of the most conserved caspases. It has been recognized as both an initiator and an effector caspase depending upon the cell type and type of stressor (12, 55–59). In the case of corneal fibroblasts, the initiator role of caspase-2 remained ambiguous. Previous studies have defined the role of caspase-2 as an initiator caspase in neuronal apoptosis during serum deprivation (60), β-amyloid-mediated toxicity (18), and oxidative stress-induced apoptosis of neuronal stem cells (44) where it plays an important role in apoptosis induction. On the other hand, other studies have suggested that caspase-2 may be activated in response to different stimuli in various cell types but may not be essential for the induction of apoptosis (58, 61). In the present study the initiator role of caspase-2 during EtOH-induced apoptosis was identified by using the in situ trapping of initiator caspase approach. Furthermore, employing the caspase-2 knockdown clones, our findings indicate that caspase-2 plays a crucial role mediating EtOH-induced mitochondria-mediated apoptosis.
In the apoptotic signaling cascades, two different initiation machineries play major roles in a variety of cell types: extrinsic death receptor-mediated signaling and intrinsic caspase family cysteine-protease-mediated signaling (54, 62). Intrinsic pathway of apoptosis, such as mitochondria-dependent cell apoptosis, can be caused by mutations or the exposure to toxic agents. It can lead to cellular abnormalities, such as decreased adenosine triphosphate (ATP) synthesis, which may result in cell apoptosis or death (63). Previous studies have been performed to understand the mechanism of EtOH-induced apoptosis in various models. The results showed that EtOH mainly engaged the mitochondria-dependent pathway of apoptosis (30–32). Ishii and co-workers (32) reported that acute EtOH treatment promotes apoptosis in primary hepatocyte cultures in vitro accompanied by ROS formation and mitochondrial depolarization, which is characteristic of MPT activation. Chronic EtOH consumption would induce caspase-2-mediated apoptosis in aged animals (64). Mitochondrial dysfunction in apoptotic signaling after exposure to EtOH has been specifically addressed in the context of apoptosis in the corneal fibroblasts as well as in the corneal epithelium (2, 29). In agreement with previous observations, our results also demonstrate that corneal fibroblasts engage the intrinsic pathway of apoptosis. In addition, the present study demonstrates that caspase-2 activation is an upstream event that engages the mitochondrial-dependent apoptotic pathway by inducing the release of cytochrome c from the mitochondria. One way by which caspase-2 has been shown to modulate the mitochondrial release of apoptogenic proteins is by modulating the proapoptotic members of the Bcl-2 protein family, namely, Bid and Bax (13, 53). In response to DNA damage or other cellular stress signaling, p53 activates cell death through the BH3-only protein p53 up-regulated modulator of apoptosis (PUMA, also known as BBC3) and Bax or Bak activation, leading to MOMP and cytochrome c release and in turn initiates the caspase activation cascade (59). In an alternative pathway caspase-2 acts upstream of MOMP and is possibly activated by different complexes, such as the PIDDosome or DISC (65). The caspase-2 activation cleaves and activates Bid, and this causes Bax activation, MOMP, and cytochrome c release (53). In the present study we did find an inhibition of the EtOH-induced Bax activation in cells that were pretreated with caspase-2 inhibitor, further suggesting that caspase-2 induces MOMP in the corneal fibroblasts after EtOH treatment.
In the present study we found that caspase-2 plays a critical role in the ethanol-induced apoptosis of corneal fibroblasts. We also demonstrated the inhibitory effects of CoQ10 on caspase-2 but not on caspase-8 or caspase-9. Based on the literature, the structure of caspase-2 carries the maximum number of cysteines among the caspase family, which includes a cysteine at the processing site and also a central disulfide bridge that leads to dimer stabilization in caspase-2 (18). Cysteine is an oxidative target because of the reactivity of the thiol group that is susceptible to modification by free radicals that may modulate the activity of these proteins, thus making caspase-2 a target for oxidation-based regulation (39). The activation of caspase-2 has been suggested to occur in a high molecular complex, the so-called PIDDosome complex, which contains RAIDD (ribosome-inactivating protein (RIP)-associated ICH/CED3 homologous protein with death domain), and PIDD (p53-inducible protein with death domain), in which expression is highly regulated by p53 (66). Previous studies have demonstrated that the activation of p53 is an upstream of, and required for, the activation of caspase-2 and subsequently, the activation of the mitochondria-mediated apoptotic pathway under oxidative stress (44, 57, 59, 67). In our study we demonstrate that activation of caspase-2 is the major determinant for cell death in EtOH-induced mitochondria dependent pathway. Caspase-2 is activated as an initiator caspase to induce apoptosis upstream to mitochondria, which then leads to the intrinsic pathway, including Bax translocation, mitochondrial dysfunction, cytochrome c release, and caspase-3 activation. On the other hand, the activation of caspase-9 is not significant in the EtOH-induced apoptosis (Fig. 8D). These findings may suggest that caspase-9 pathway is primarily an ancillary pathway in EtOH-induced apoptosis. Cell apoptosis can also proceed through the direct caspase activation cascade (caspase-2 → caspase-3) (68). This is followed by the cleavage of the executioner caspase-3, which is the major player of downstream event of apoptosis (68). In addition, activated effector caspase (caspase-3) can also process the caspase-2 precursor, providing an amplification loop, but this process is context-dependent (59). We observed an increase of EtOH-induced caspase-2 activity at 2 h, and then caspase-2 activity decreased time-dependently (Fig. 4A). However, a sustained increase of caspase-3 activity was found at 2, 4, and 8 h. These observations suggest that the caspase-2 is not activated by caspase-3 during EtOH-induced caspase-2 activation. Taken together, our findings further support the apical role of caspase-2 as an initiator caspase in the EtOH-induced apoptotic pathway.
CoQ10 or ubiquinone is an endogenously synthesized lipid that shuttles electrons from complexes I (NDSH:ubiquinone reductase) and II (succinate:ubiquinone reductase) and from the oxidation of fatty acids and branched-chain amino acids (via flavin-linked dehydrogenases) to complex III (ubiquinol cytochrome c oxidase) of the mitochondrial respiratory chain (electron transport chain) (69). CoQ10 also has antioxidant properties, which allow it to protect membrane lipids and proteins as well as mitochondrial deoxyribonucleic acid (mtDNA) against oxidative damage (70). Previous studies have demonstrated that the Ca2+-dependent opening of the mitochondrial permeability transition pore in isolated mitochondria can be prevented by two synthetic quinine analogues (28, 71). Our study demonstrates for the first time that CoQ10 exerts its anti-apoptotic effects by preventing caspase-2 activation, abrogating mitochondrial dysfunction in ethanol-treated cells. In addition, we found that CoQ10 inhibited EtOH-induced ROS formation and p53 expression, both of which could further prevent the activation of caspase-2. Whether CoQ10 and caspase-2 can directly interact with each other or whether adapter proteins are necessary for CoQ10 and caspase-2 activation requires further investigation. However, these findings shed light on the role of CoQ10 in the inhibition of caspase-2 before mitochondrial damage upon EtOH exposure.
The present study identified caspase-2 as an initiator caspase in EtOH-induced apoptosis in the cultures of corneal fibroblasts. When the cultured cells were pretreated with CoQ10, the phenomenon of EtOH-induced cell apoptosis was abrogated. In cells that have the potential to produce corneal scar and reduce visual acuity, such as the corneal fibroblasts, the prevention of any kind of damage or loss is of utmost importance. Thus, understanding the mechanism of cell death and the cell-protective measures is crucial in defining the pharmaceutical approaches to prevent corneal cell damage upon EtOH exposure during refractive surgery.
Acknowledgments
We thank Feng-Yen Lin of the Department of Anesthesiology, School of Medicine, Taipei Medical University for helpful discussions. We also thank Tzu-Hua Huang for expert technical assistance.
This work was supported in part by Taiwan National Science Council Grants NSC95-2314-B002-149-MY3, NSC96-2627-B002-011, NSC97-2627-B002-007, NSC98-2627-B002-004, NSC98-3112-B002-040, NSC99-3112-B002-029, NSC99-2314-B002-039-MY3, NSC99-2314-B002-040-MY3, NSC100-2314-B002-061-MY3, Department of Health, Executive Yuan, R.O.C. (Taiwan), DOH100-TD-PB-111-TM005, and National Taiwan University Hospital NTUH.98-S1105, NTUH.99-MSN 01, NTUH.100-001637, and NTUH.VN101-06.
- CoQ10
- coenzyme Q10
- ROS
- reactive oxygen species
- z-VDVAD-fmk
- benzyloxycarbonyl-VDVAD-fluoromethyl ketone
- b-VAD-fmk
- biotinyl-VAD- fluoromethyl ketone
- DCF
- 2′,7′-dichlorofluorescein
- WCL
- whole cell lysates
- MPT
- membrane potential transition
- MOMP
- mitochondrial outer membrane permeabilization
- PI
- propidium iodide.
REFERENCES
- 1. Chen C. C., Chang J. H., Lee J. B., Javier J., Azar D. T. (2002) Human corneal epithelial cell viability and morphology after dilute alcohol exposure. Invest Ophthalmol. Vis. Sci. 43, 2593–2602 [PubMed] [Google Scholar]
- 2. Sosne G., Siddiqi A., Kurpakus-Wheater M. (2004) Thymosin-β4 inhibits corneal epithelial cell apoptosis after ethanol exposure in vitro. Invest. Ophthalmol. Vis. Sci. 45, 1095–1100 [DOI] [PubMed] [Google Scholar]
- 3. Kim T. I., Tchah H., Cho E. H., Kook M. S. (2004) Evaluation for safety of cultured corneal fibroblasts with cotreatment of alcohol and mitomycin C. Invest. Ophthalmol. Vis. Sci. 45, 86–92 [DOI] [PubMed] [Google Scholar]
- 4. Nanji A. A. (1998) Apoptosis and alcoholic liver disease. Semin. Liver Dis. 18, 187–190 [DOI] [PubMed] [Google Scholar]
- 5. Zhang F. X., Rubin R., Rooney T. A. (1998) Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J. Neurochem. 71, 196–204 [DOI] [PubMed] [Google Scholar]
- 6. Stein H. A., Stein R. M., Price C., Salim G. A. (1997) Alcohol removal of the epithelium for excimer laser ablation. Outcomes analysis. J. Cataract. Refract. Surg. 23, 1160–1163 [DOI] [PubMed] [Google Scholar]
- 7. Abad J. C., An B., Power W. J., Foster C. S., Azar D. T., Talamo J. H. (1997) A prospective evaluation of alcohol-assisted versus mechanical epithelial removal before photorefractive keratectomy. Ophthalmology 104, 1566–1574; discussion 1574–1565 [DOI] [PubMed] [Google Scholar]
- 8. Baliga B. C., Read S. H., Kumar S. (2004) The biochemical mechanism of caspase-2 activation. Cell Death Differ. 11, 1234–1241 [DOI] [PubMed] [Google Scholar]
- 9. Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J. E., Edris W. A., Sutherlin D. P., Green D. R., Salvesen G. S. (2003) A unified model for apical caspase activation. Mol. Cell 11, 529–541 [DOI] [PubMed] [Google Scholar]
- 10. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Budihardjo I., Oliver H., Lutter M., Luo X., Wang X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290 [DOI] [PubMed] [Google Scholar]
- 12. Lamkanfi M., Declercq W., Kalai M., Saelens X., Vandenabeele P. (2002) Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 9, 358–361 [DOI] [PubMed] [Google Scholar]
- 13. Guo Y., Srinivasula S. M., Druilhe A., Fernandes-Alnemri T., Alnemri E. S. (2002) Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J. Biol. Chem. 277, 13430–13437 [DOI] [PubMed] [Google Scholar]
- 14. Paroni G., Henderson C., Schneider C., Brancolini C. (2002) Caspase-2 can trigger cytochrome c release and apoptosis from the nucleus. J. Biol. Chem. 277, 15147–15161 [DOI] [PubMed] [Google Scholar]
- 15. Robertson J. D., Enoksson M., Suomela M., Zhivotovsky B., Orrenius S. (2002) Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem. 277, 29803–29809 [DOI] [PubMed] [Google Scholar]
- 16. Lassus P., Opitz-Araya X., Lazebnik Y. (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297, 1352–1354 [DOI] [PubMed] [Google Scholar]
- 17. Kumar S., Vaux D. L. (2002) Apoptosis. A cinderella caspase takes center stage. Science 297, 1290–1291 [DOI] [PubMed] [Google Scholar]
- 18. Schweizer A., Briand C., Grutter M. G. (2003) Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway. J. Biol. Chem. 278, 42441–42447 [DOI] [PubMed] [Google Scholar]
- 19. Troy C. M., Ribe E. M. (2008) Caspase-2. Vestigial remnant or master regulator? Sci. Signal 1, pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tinel A., Tschopp J. (2004) The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846 [DOI] [PubMed] [Google Scholar]
- 21. Lenaz G., Fato R., Castelluccio C., Genova M. L., Bovina C., Estornell E., Valls V., Pallotti F., Parenti Castelli G. (1993) The function of coenzyme Q in mitochondria. Clin. Investig. 71, S66–S70 [DOI] [PubMed] [Google Scholar]
- 22. Valls V., Castelluccio C., Fato R., Genova M. L., Bovina C., Saez G., Marchetti M., Parenti Castelli G., Lenaz G. (1994) Protective effect of exogenous coenzyme Q against damage by adriamycin in perfused rat liver. Biochem. Mol. Biol. Int. 33, 633–642 [PubMed] [Google Scholar]
- 23. Alleva R., Tomasetti M., Andera L., Gellert N., Borghi B., Weber C., Murphy M. P., Neuzil J. (2001) Coenzyme Q blocks biochemical but not receptor-mediated apoptosis by increasing mitochondrial antioxidant protection. FEBS Lett. 503, 46–50 [DOI] [PubMed] [Google Scholar]
- 24. Kelso G. F., Porteous C. M., Coulter C. V., Hughes G., Porteous W. K., Ledgerwood E. C., Smith R. A., Murphy M. P. (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells. Antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596 [DOI] [PubMed] [Google Scholar]
- 25. López-Lluch G., Barroso M. P., Martín S. F., Fernández-Ayala D. J., Gómez-Díaz C., Villalba J. M., Navas P. (1999) Role of plasma membrane coenzyme Q on the regulation of apoptosis. Biofactors 9, 171–177 [DOI] [PubMed] [Google Scholar]
- 26. Tomasetti M., Alleva R., Borghi B., Collins A. R. (2001) In vivo supplementation with coenzyme Q10 enhances the recovery of human lymphocytes from oxidative DNA damage. FASEB J. 15, 1425-1427 [DOI] [PubMed] [Google Scholar]
- 27. Brancato R., Schiavone N., Siano S., Lapucci A., Papucci L., Donnini M., Formigli L., Orlandini S. Z., Carella G., Carones F., Capaccioli S. (2000) Prevention of corneal keratocyte apoptosis after argon fluoride excimer laser irradiation with the free radical scavenger ubiquinone Q10. Eur. J. Ophthalmol. 10, 32–38 [DOI] [PubMed] [Google Scholar]
- 28. Fontaine E., Eriksson O., Ichas F., Bernardi P. (1998) Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex i. J. Biol. Chem. 273, 12662–12668 [DOI] [PubMed] [Google Scholar]
- 29. Chen C. C., Liou S. W., Chen C. C., Chen W. C., Hu F. R., Wang I. J., Lin S. J. (2011) Coenzyme Q10 reduces ethanol-induced apoptosis in corneal fibroblasts. PLoS ONE 6, e19111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoek J. B., Cahill A., Pastorino J. G. (2002) Alcohol and mitochondria. A dysfunctional relationship. Gastroenterology 122, 2049–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kroemer G., Reed J. C. (2000) Mitochondrial control of cell death. Nat. Med. 6, 513–519 [DOI] [PubMed] [Google Scholar]
- 32. Higuchi H., Adachi M., Miura S., Gores G. J., Ishii H. (2001) The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology 34, 320–328 [DOI] [PubMed] [Google Scholar]
- 33. Gutierrez-Mariscal F. M., Perez-Martinez P., Delgado-Lista J., Yubero-Serrano E. M., Camargo A., Delgado-Casado N., Cruz-Teno C., Santos-Gonzalez M., Rodriguez-Cantalejo F., Castano J. P., Villalba-Montoro J. M., Fuentes F., Perez-Jimenez F., Lopez-Miranda J. (2012) Mediterranean diet supplemented with coenzyme Q10 induces postprandial changes in p53 in response to oxidative DNA damage in elderly subjects. Age (Dordr) 34, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Funderburgh J. L., Funderburgh M. L., Mann M. M., Corpuz L., Roth M. R. (2001) Proteoglycan expression during transforming growth factor β-induced keratocyte-myofibroblast transdifferentiation. J. Biol. Chem. 276, 44173–44178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papucci L., Schiavone N., Witort E., Donnini M., Lapucci A., Tempestini A., Formigli L., Zecchi-Orlandini S., Orlandini G., Carella G., Brancato R., Capaccioli S. (2003) Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 278, 28220–28228 [DOI] [PubMed] [Google Scholar]
- 36. Pepper C., Thomas A., Tucker H., Hoy T., Bentley P. (1998) Flow cytometric assessment of three different methods for the measurement of in vitro apoptosis. Leuk. Res. 22, 439–444 [DOI] [PubMed] [Google Scholar]
- 37. Kanno S., Tomizawa A., Ohtake T., Koiwai K., Ujibe M., Ishikawa M. (2006) Naringenin-induced apoptosis via activation of NF-κB and necrosis involving the loss of ATP in human promyeloleukemia HL-60 cells. Toxicol. Lett. 166, 131–139 [DOI] [PubMed] [Google Scholar]
- 38. Shim H. Y., Park J. H., Paik H. D., Nah S. Y., Kim D. S., Han Y. S. (2007) Acacetin-induced apoptosis of human breast cancer MCF-7 cells involves caspase cascade, mitochondria-mediated death signaling and SAPK/JNK1/2-c-Jun activation. Mol. Cells 24, 95–104 [PubMed] [Google Scholar]
- 39. Tiwari M., Lopez-Cruzan M., Morgan W. W., Herman B. (2011) Loss of caspase-2-dependent apoptosis induces autophagy after mitochondrial oxidative stress in primary cultures of young adult cortical neurons. J. Biol. Chem. 286, 8493–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tu S., McStay G. P., Boucher L. M., Mak T., Beere H. M., Green D. R. (2006) In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat. Cell Biol. 8, 72–77 [DOI] [PubMed] [Google Scholar]
- 41. Rutjes S. A., van der Heijden A., Utz P. J., van Venrooij W. J., Pruijn G. J. (1999) Rapid nucleolytic degradation of the small cytoplasmic Y RNAs during apoptosis. J. Biol. Chem. 274, 24799–24807 [DOI] [PubMed] [Google Scholar]
- 42. Srimathi T., Robbins S. L., Dubas R. L., Hasegawa M., Inohara N., Park Y. C. (2008) Monomer/dimer transition of the caspase-recruitment domain of human Nod1. Biochemistry 47, 1319–1325 [DOI] [PubMed] [Google Scholar]
- 43. Nair V. D. (2006) Activation of p53 signaling initiates apoptotic death in a cellular model of Parkinson's disease. Apoptosis 11, 955–966 [DOI] [PubMed] [Google Scholar]
- 44. Tamm C., Zhivotovsky B., Ceccatelli S. (2008) Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis 13, 354–363 [DOI] [PubMed] [Google Scholar]
- 45. Bouchier-Hayes L., Green D. R. (2012) Caspase-2. The orphan caspase. Cell Death Differ 19, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McStay G. P., Salvesen G. S., Green D. R. (2008) Overlapping cleavage motif selectivity of caspases. Implications for analysis of apoptotic pathways. Cell Death Differ 15, 322–331 [DOI] [PubMed] [Google Scholar]
- 47. Chang K., Elledge S. J., Hannon G. J. (2006) Lessons from nature. MicroRNA-based shRNA libraries. Nat. Methods 3, 707–714 [DOI] [PubMed] [Google Scholar]
- 48. Chen F., He Y. (2009) Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS ONE 4, e6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen Y., Cheng G., Mahato R. I. (2008) RNAi for treating hepatitis B viral infection. Pharm. Res. 25, 72–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fewell G. D., Schmitt K. (2006) Vector-based RNAi approaches for stable, inducible. and genome-wide screens. Drug Discov. Today 11, 975–982 [DOI] [PubMed] [Google Scholar]
- 51. Zhivotovsky B., Orrenius S. (2005) Caspase-2 function in response to DNA damage. Biochem. Biophys. Res. Commun. 331, 859–867 [DOI] [PubMed] [Google Scholar]
- 52. Kluck R. M., Bossy-Wetzel E., Green D. R., Newmeyer D. D. (1997) The release of cytochrome c from mitochondria. A primary site for Bcl-2 regulation of apoptosis. Science 275, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 53. Bonzon C., Bouchier-Hayes L., Pagliari L. J., Green D. R., Newmeyer D. D. (2006) Caspase-2-induced apoptosis requires bid cleavage. A physiological role for bid in heat shock-induced death. Mol. Biol. Cell 17, 2150–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thornberry N. A., Lazebnik Y. (1998) Caspases. Enemies within. Science 281, 1312–1316 [DOI] [PubMed] [Google Scholar]
- 55. Kumar S., Kinoshita M., Noda M., Copeland N. G., Jenkins N. A. (1994) Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 β-converting enzyme. Genes Dev. 8, 1613–1626 [DOI] [PubMed] [Google Scholar]
- 56. Wang X., Yang C., Chai J., Shi Y., Xue D. (2002) Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298, 1587–1592 [DOI] [PubMed] [Google Scholar]
- 57. Bouchier-Hayes L. (2010) The role of caspase-2 in stress-induced apoptosis. J Cell Mol. Med. 14, 1212–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Troy C. M., Rabacchi S. A., Hohl J. B., Angelastro J. M., Greene L. A., Shelanski M. L. (2001) Death in the balance. Alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J. Neurosci. 21, 5007–5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kumar S. (2009) Caspase 2 in apoptosis, the DNA damage response and tumour suppression. Enigma no more? Nat. Rev. Cancer 9, 897–903 [DOI] [PubMed] [Google Scholar]
- 60. Chauvier D., Lecoeur H., Langonné A., Borgne-Sanchez A., Mariani J., Martinou J. C., Rebouillat D., Jacotot E. (2005) Upstream control of apoptosis by caspase-2 in serum-deprived primary neurons. Apoptosis 10, 1243–1259 [DOI] [PubMed] [Google Scholar]
- 61. Bergeron L., Perez G. I., Macdonald G., Shi L., Sun Y., Jurisicova A., Varmuza S., Latham K. E., Flaws J. A., Salter J. C., Hara H., Moskowitz M. A., Li E., Greenberg A., Tilly J. L., Yuan J. (1998) Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 12, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cryns V., Yuan J. (1998) Proteases to die for. Genes Dev. 12, 1551–1570 [DOI] [PubMed] [Google Scholar]
- 63. Keating D. J. (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis, and their relevance to neurodegenerative diseases. J. Neurochem. 104, 298–305 [DOI] [PubMed] [Google Scholar]
- 64. Madesh M., Zong W. X., Hawkins B. J., Ramasamy S., Venkatachalam T., Mukhopadhyay P., Doonan P. J., Irrinki K. M., Rajesh M., Pacher P., Thompson C. B. (2009) Execution of superoxide-induced cell death by the proapoptotic Bcl-2-related proteins Bid and Bak. Mol. Cell. Biol. 29, 3099–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Olsson M., Vakifahmetoglu H., Abruzzo P. M., Högstrand K., Grandien A., Zhivotovsky B. (2009) DISC-mediated activation of caspase-2 in DNA damage-induced apoptosis. Oncogene 28, 1949–1959 [DOI] [PubMed] [Google Scholar]
- 66. Krumschnabel G., Sohm B., Bock F., Manzl C., Villunger A. (2009) The enigma of caspase-2. The laymen's view. Cell Death Differ. 16, 195–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kitevska T., Spencer D. M., Hawkins C. J. (2009) Caspase-2. Controversial killer or checkpoint controller? Apoptosis 14, 829–848 [DOI] [PubMed] [Google Scholar]
- 68. Chan K. M., Rajab N. F., Siegel D., Din L. B., Ross D., Inayat-Hussain S. H. (2010) Goniothalamin induces coronary artery smooth muscle cells apoptosis. The p53-dependent caspase-2 activation pathway. Toxicol. Sci. 116, 533–548 [DOI] [PubMed] [Google Scholar]
- 69. Mancuso M., Orsucci D., Volpi L., Calsolaro V., Siciliano G. (2010) Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr. Drug Targets 11, 111–121 [DOI] [PubMed] [Google Scholar]
- 70. Dallner G., Sindelar P. J. (2000) Regulation of ubiquinone metabolism. Free Radic. Biol. Med. 29, 285–294 [DOI] [PubMed] [Google Scholar]
- 71. Petronilli V., Penzo D., Scorrano L., Bernardi P., Di Lisa F. (2001) The mitochondrial permeability transition, release of cytochrome c, and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276, 12030–12034 [DOI] [PubMed] [Google Scholar]