

Background: Somatic cell reprogramming is an inefficient process because of the existence of roadblocks.
Results: Class IIa histone deacetylases and MEF2 proteins increase during mouse fibroblast reprogramming and differentially regulate the expression of Tgfβ cytokines.
Conclusion: This interplay regulates the mesenchymal-to-epithelial transition phase of reprogramming.
Significance: Our findings help understand the mechanisms of reprogramming and may have implications in other contexts.
Keywords: Epithelial Cell, Epithelial-to-Mesenchymal Transition, Histone Deacetylase, Induced Pluripotent Stem Cells, Stem Cells, Transforming Growth Factor β (TGFβ)
Abstract
Class IIa histone deacetylases (HDACs) and myocyte enhancer factor 2 (MEF2) proteins compose a signaling module that orchestrates lineage specification during embryogenesis. We show here that this module also regulates the generation of mouse induced pluripotent stem cells by defined transcription factors. Class IIa HDACs and MEF2 proteins rise steadily during fibroblast reprogramming to induced pluripotent stem cells. MEF2 proteins tend to block the process by inducing the expression of Tgfβ cytokines, which impairs the necessary phase of mesenchymal-to-epithelial transition (MET). Conversely, class IIa HDACs endeavor to suppress the activity of MEF2 proteins, thus enhancing the MET and colony formation efficiency. Our work highlights an unexpected role for a developmental axis in somatic cell reprogramming and provides new insight into how the MET is regulated in this context.
Introduction
The reprogramming of somatic cells to iPSCs3 requires the removal of a series of endogenous (somatic cell genetic program) and exogenous (extracellular milieu) barriers that prevent the transition from intermediate states to pluripotency (1, 2). An improved understanding of these barriers is important because it may facilitate a higher fidelity of the conversion through experimental manipulation. In this regard, we and others have shown that a mesenchymal-to-epithelial transition (MET) is an early requisite step of fibroblast reprogramming (3, 4). This discovery helps explain why inhibiting Tgfβ signaling enhances reprogramming (5, 6) because Tgfβ cytokines are highly secreted by fibroblasts and induce/sustain the epithelial-to-mesenchymal transition phenotype (7). Yet, the initiation of the MET by the reprogramming factors is still poorly understood, except for the repression of Tgfβ-related genes by c-Myc (plus Sox2/Oct4) (3, 8) and specific microRNAs (4, 9–11) and the activation of an epithelial program by Klf4 (3).
In our search for signaling pathways that regulate reprogramming, we focused on class IIa HDACs because of their fundamental role in tissue specification during embryogenesis (12–15). This subfamily of HDACs consists of isoforms 4, 5, 7, and 9, and compared with other HDACs they have little or no catalytic activity (16, 17). Instead, they function by binding to and controlling the activity of transcription factors, including the MEF2 family of proteins (15, 18, 19). MEF2 was originally identified as a transcription factor capable of binding to the muscle creatine kinase promoter (20) but is now known to comprise four isoforms (A, B, C, and D) in vertebrates and is considered a master regulator of metazoan development (15, 21). Class IIa HDACs suppress the activity of MEF2 proteins by preventing the binding of coactivators (e.g. p300) and inducing the recruitment of corepressor complexes through their catalytic domains (15, 22). Class IIa HDACs also shuttle between nucleus and cytoplasm in response to extracellular signals, thus fine-tuning MEF2 activity (12–15). Despite the fact that this signaling pathway has been studied extensively studied in development, little is known about other contexts. Here we show that the interplay between class IIa HDACs and MEF2 proteins determines the efficiency of somatic cell reprogramming by controlling the expression of Tgfβ cytokines.
EXPERIMENTAL PROCEDURES
Cell Culture and Reprogramming Experiments
OG2 embryonic fibroblasts were used in all reprogramming experiments unless mentioned otherwise. They were obtained by crossing OG2 male mice with 129/sv female mice (23). Embryonic fibroblasts, tail tip, and mammary fibroblasts were isolated as described (23, 24). These cells and HEK293T cells were maintained in DMEM (Hyclone) supplemented with 10% FBS (Hyclone), l-glutamine, non-essential amino acids, and penicillin/streptomycin. 20,000 cells were transduced twice in 12-well dishes using viral supernatants generated with PlatE cells (24, 25). The medium was changed to mouse ESC medium (DMEM supplemented with 15% FBS (Invitrogen), l-glutamine, non-essential amino acids, sodium pyruvate, penicillin/streptomycin, β mercaptoethanol, and 100 units/ml leukemia inhibitory factor (Millipore)) on day 2 post-infection and renewed daily. Cells were not split on feeders except for colony expansion and characterization. Feeder layers consisted of mouse embryonic fibroblasts treated with mitomycin C. Doxycycline (Sigma) was added at 1 μg/ml for the indicated times. GFP+ colonies were visualized and counted using a Zeiss SteREO Lumar V12 microscope. iPSCs generated in this study or produced in a previous report (23), and also mouse ESCs (generated by us from OG2 mice), were routinely cultured on feeders in KSR medium (contains the same recipe as mouse ESC medium but FBS is substituted by knockout serum replacement (Invitrogen)). Karyotype analysis, DNA methylation analysis, and chimeric mouse production with newly generated iPSCs were done as described (3, 23, 26). Tgfβ receptor 1 (TgfβR1) inhibitor and Tgfβ3 cytokine were purchased from Tocris and R&D Systems, respectively, and vitamin C was purchased from Sigma.
Plasmids
pMXs vectors expressing the Yamanaka factors were purchased from Addgene. All other vectors were made by us using either cDNA obtained from mouse fibroblasts or purchased from Fulengene. The doxycycline-inducible lentiviral system was also described before (26). All newly generated vectors have a FLAG tag in the carboxy terminal end of the protein for ease of detection. DNA mutagenesis/deletion was produced using suitable oligos and a PCR-based method. shRNA inserts were cloned into the pRetroSuper vector. The sequences were as follows (5′-3′): MEF2A, GCAGTTATCTCAGGGTTCAAA and GATTG AAATACTGGTGCAAA; MEF2C, GCCTCAG TGATACAGTATAAA and CCATCAGTGAAT CAAAGGATA; MEF2D, CACATCAGCATCA AGTCAGAA and GCGAATCACTGATGAAC GGAA; HDAC4, GCAGAGGATCCACCAGTT AAG and GGTACAATCTCTCTGCCAAAT; HDAC5, GACGCCTCCCTCCTACAAATT and CATCGCTGAGAACGGCTTTAC; and HDAC7, A GACAAGAGCAAGCGAAGT and CCATGTT TCTGCCAAATGTTT. A sequence that targets the firefly luciferase gene transcript was used as a control (3). Retroviral supernatants containing these constructs were produced as for the pMXs plasmids. The infection efficiency was near 100% (on the basis of the use of a control GFP retroviral vector), but we added puromycin at day 3 post-transduction (it was maintained for 3 days) for selecting only cells that contained the shRNA vectors. All new plasmids were verified by sequencing before use. The MEF2-responsive reporter was purchased from Panomics. Luciferase activity was measured using the Dual-Glo luciferase assay system (Promega). A Renilla luciferase plasmid was used for normalization.
PCR Analysis, Immunofluorescence, Western Blotting, and Immunoprecipitation
qPCR analysis was performed using SYBR Green (Takara) and an ABI 7300 machine. Items were run in triplicate, and values were normalized on the basis of β-actin values. Primers used in this study were as follows (5′-3′): HDAC4, AAACCTGCTGAGAAGAG ATCTGA (forward) and CTGAGCTTCAAGACA GACAAACA (reverse); HDAC5, GGACGCCTC CCTCCTACAAATTG (forward) and AGTTGGG TTCCGAGGCCGTTTTAC (reverse); HDAC7, GTGGCGAGGGCTTCAATGTCAACG (forward) and TCGGGCAATGGGCATCACCACTA (reverse); MEF2A, CAGGTGGTGGCAGTCTTG G (forward) and TGCTTATCCTTTGGGCATTC AA (reverse); MEF2C, ATCCCGATGCAGACG ATTCAG (forward) and AACAGCACACAATCT TTGCCT (reverse); MEF2D, CGAGATCGCGC TCATCATCTT (forward) and AGCCGTTGAAA CCCTTCTTCC (reverse); Tgfβ1, CTCCCGTG GCTTCTAGTGC (forward) and GCCTTAGTTT GGACAGGATCTG (reverse); Tgfβ2, TCGACA TGGATCAGTTTATGCG (forward) and CCCTG GTACTGTTGTAGATGGA (reverse); and Tgfβ3, CA GGCCAGGGTAGTCAGAG (forward) and ATTT CCAGCCTAGATCCTGCC (reverse). Primers for iPSC characterization (qPCR), semi-quantitative RT-PCR, and bisulfite sequencing were described before (23). Immunofluorescence microscopy was performed using a Leica TCS SP2 spectral confocal microscope. Western blotting was performed using ECL and ECL Plus (Amersham Biosciences). Antibodies were purchased from the following suppliers: HDAC4, -5, and -7 and MEF2A-D from Abcam; FLAG and β-actin from Sigma; SSEA-1 from R&D Systems; and E-cadherin and β-catenin from BD Biosciences. The Nanog antibody was made by us. DAPI was purchased from Sigma. Immunoprecipitation was done using monoclonal anti-FLAG M2 affinity gel and FLAG peptide (Sigma).
Statistical Analysis
Student's t test was used throughout the manuscript.
RESULTS
Class IIa HDACs Regulate Somatic Cell Reprogramming
Given the fundamental role of class IIa HDACs in development (12–14), we speculated that they might regulate reprogramming as well. Supporting this idea, we observed by Western blotting that mouse ESCs/iPSCs display higher levels of HDAC4, -5, and -7 than fibroblasts (Fig. 1A). We did not study HDAC9 because qPCR analysis showed low expression (data not shown). This prompted us to test whether overexpressing HDAC4, -5, and -7 influences fibroblast reprogramming (see the validation of the expression vectors in Fig. 1B). We used embryonic fibroblasts bearing a transgenic Oct4 promoter that produces GFP as an indication of completed reprogramming (3, 27). We also employed both three (Oct4, Klf4, and Sox2 or OKS) and four (the same plus c-Myc or OKSM) Yamanaka factors (1) with the aim of identifying potential differences between the two methods. Reprogramming with OKS is less efficient than with OKSM, which is caused at least in part by the existence of a stronger MET phase in the latter (3). Yet, OKS is preferred because of the higher risk of tumor formation using OKSM, which is related to the oncogenic effects of c-Myc (28). The three class IIa HDACs enhanced GFP+ colony formation with OKS (∼10-fold) but not with OKSM (Fig. 1C). HDAC1 and -6, which belong to the class I and IIb HDAC subfamilies, respectively (15), were used as controls and had no effect (Fig. 1C). Importantly, iPSC colonies generated with OKS and class IIa HDACs (only HDAC7 was tested) were bona fide according to standard characterization procedures (including qPCR analysis, bisulfite sequencing, and normal karyotype) and produced chimeric mice with germ line transmission (Fig. 2, A–G). Similarly, we confirmed that HDAC7 improves reprogramming with OKS using two additional types of mouse fibroblasts (adult tail tip and mammary) (Fig. 1D). Hence, exogenous class IIa HDACs enhance fibroblast reprogramming using OKS but not OKSM.
FIGURE 1.

Class IIa HDACs regulate somatic cell reprogramming. A, representative Western blot analysis for endogenous HDAC4, -5, and -7 using lysates from the indicated cell types. β-actin was used as the loading control. B, representative Western blot analysis with anti-FLAG shows adequate overexpression of the indicated class IIa HDACs, HDAC1, and -6. C, number of GFP+ colonies produced in fibroblasts reprogrammed with OKS/OKSM and HDAC4, -5, and -7. An empty vector was used as a control (Ctrl, also in D) along with HDAC1 and -6. Colonies were counted at day 18 (also hereafter for other colony formation experiments unless indicated otherwise). Mean values ± S.D. of a representative experiment with items in triplicate are shown (also hereafter for other colony formation experiments). Double asterisks indicate p value < 0.01. D, number of GFP+ colonies in tail tip fibroblasts (TTFs) and mammary (MaFs) transduced with OKS and HDAC7.
FIGURE 2.

Characterization of iPSCs generated by HDAC7 and OKS. A, phase contrast and immunofluorescence photographs of a representative iPSC clone produced with OKS and HDAC7. Scale bars = 50 μm. B, semiquantitative PCR showing integration of the exogenous transgenes in the genome of selected iPSC clones. Untransduced fibroblasts and pMXs plasmids containing the corresponding cDNA were used as controls (ctrl). C, qPCR for endogenous (Endo) ESC-like markers in the indicated iPSC clones and mouse ESCs compared with fibroblasts. D, qPCR for the exogenous (Exo) transgenes in the indicated iPSC clones. ESCs and reprogramming fibroblasts extracted at day 6 were the controls. E, normal karyotype of a selected iPSC clone generated with OKS and HDAC7. F, DNA methylation status of the Oct4 proximal promoter in the indicated cell types. ○ represent demethylated 5′-cytosine-phosphodiester-guanine sites. G, photograph of a representative chimeric mouse (blue arrowhead) generated with the same iPSC clone. Transmission to the germ line can be observed in the baby mouse with agouti skin color (red arrowhead).
Next, we envisaged that the expression of HDAC4, -5, and -7 should augment during reprogramming to mimic the profile of ESCs/iPSCs (see Fig. 1A). As anticipated, all three isoforms increased steadily, but this was more noticeable in OKSM than OKS (Fig. 3A). Knocking down HDAC4, -5, and -7 using shRNA vectors (see the knockdown efficiency in Fig. 3B) reduced the reprogramming efficiency with OKS and OKSM (C). Therefore, endogenous class IIa HDACs are required for OKS and OKSM reprogramming. The fact that their individual knockdown decreases reprogramming efficiency reflects that their effect is synergistic. These experiments also suggest that the higher basal expression of class IIa HDACs in OKSM reprogramming makes the system less sensitive to overexpression (see Fig. 1B).
FIGURE 3.

Knockdown of endogenous class IIa HDACs impairs reprogramming. A, representative Western blot analysis for HDAC4, -5, and -7 using lysates of a time course reprogramming experiment with OKS/OKSM in fibroblasts. An empty vector was used as a control (Ctrl). D, day. B, shRNA vectors (depicted as sh followed by the target gene) for HDAC4, -5, and -7 were infected in fibroblasts, and the knockdown efficiency was measured by qPCR. C, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and the indicated shRNA vectors. shRNA against firefly luciferase was used as a control (also hereafter in similar experiments). Single asterisk indicates p value < 0.05.
Class IIa HDACs Regulate the MET of Reprogramming
Although the above explanation of why exogenous class IIa HDACs fail to enhance OKSM reprogramming may be partly correct, we aimed to investigate additional possibilities. We considered a putative effect of exogenous class IIa HDACs on cell proliferation because this is an important aspect in determining reprogramming efficiency (29) and is significantly lower in OKS than OKSM (30). However, class IIa HDACs only changed this parameter modestly with OKS (Fig. 4A). Interestingly, we also noticed that they significantly increase epithelial-like morphology during the first days of OKS reprogramming (Fig. 4B, upper panel), suggestive of an accelerated MET. This phenomenon also existed in OKSM but was less obvious, in agreement with the MET happening more quickly and being more substantial in this setting (3). To confirm our idea, we stained fibroblasts reprogrammed with OKS/OKSM and HDAC4 or -7 for two relevant cell-cell adhesion proteins (E-cadherin and β-catenin). Class IIa HDACs considerably increased the distribution of both molecules at intercellular junctions in cells reprogrammed with OKS compared with the control and less notably with OKSM (Fig. 4B, center and lower panels). This differential effect was verified by Western blotting for E-cadherin (Fig. 4C). Moreover, shRNA for HDAC4 and -7 decreased the accumulation of E-cadherin in OKS and OKSM reprogramming (Fig. 4D), demonstrating that endogenous class IIa HDACs are required for the MET phase of both methods. To further validate our hypothesis, we reprogrammed mammary epithelial cells with OKS and HDAC7, as in this context the MET phase is not necessary and the effect of HDAC7 should be redundant (3). As predicted, we observed no synergistic effect in GFP+ colony formation (Fig. 4E). We also verified that the HDAC7 transgene had integrated effectively in the resulting colonies (Fig. 4F).
FIGURE 4.

Class IIa HDACs enhance reprogramming by accelerating the MET phase. A, measurement of proliferation at different times in fibroblasts reprogrammed as indicated. An empty vector was added to OKS as a control for HDAC7. A representative experiment with triplicate items counted in duplicate (after detaching cells with trypsin) is shown. D, day. B, representative phase contrast and immunofluorescence photographs of fibroblasts reprogrammed with OKS/OKSM and HDAC4 or -7 or empty vector. Scale bars = 50 μm. E-cad, E-cadherin; Ctrl, control. C and D, representative Western blot analyses for E-cadherin using lysates of fibroblasts reprogrammed with OKS/OKSM and the indicated expression vectors or shRNA constructs. Lysates were taken at day 6 in all cases. E, number of GFP+ colonies in mammary epithelial cells (MECs) reprogrammed with OKS and HDAC7 or empty vector. Colonies were counted at day 21. F, semiquantitative PCR showing integration of the exogenous HDAC7 transgene in the genome of selected iPSC clones produced as in E. Untransduced MECs and pMXs plasmids containing the corresponding cDNA were used as controls. G, Western blot analysis of fibroblasts infected with iHDAC7-Dox and treated with doxycycline (Dox). H, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and iHDAC7-Dox. Doxycycline was administered at the indicated time points. Double asterisks indicate p value < 0.01.
We then wished to determine the time window of sensitivity to class IIa HDACs, as one would expect that it is restricted to the early phase. To test this, we prepared a vector producing HDAC7 in a doxycycline-inducible manner (iHDAC7-Dox) (Fig. 4G). Treatment with doxycycline between days 3 and 9 produced a sharp increase (∼6-fold) of GFP+ colonies in cells transduced with OKS and iHDAC7-Dox, but there was a plateau when doxycycline was administered longer (Fig. 4H, left panel). We also detected a reproducible increase (∼2-fold) when doxycycline was added from days 3–7 to cells reprogrammed with OKSM and iHDAC7-Dox (Fig. 4H, right panel). Yet this increase was reduced to base-line levels when doxycycline was maintained during the whole process (Fig. 4H, right panel). Hence, overexpression of exogenous class IIa HDACs only during the MET phase also potentiates OKSM reprogramming, albeit less remarkably than OKS. However, their sustained overexpression abrogates this response in OKSM by an unknown mechanism. It is possible that the different reprogramming kinetics between OKS and OKSM explain this phenomenon, as the molecular mechanisms induced in both settings are similar but not identical (31).
Class IIa HDACs Regulate the Expression of Tgfβ Cytokines during Reprogramming
We investigated how class IIa HDACs regulate the MET of reprogramming with a focus on Tgfβ signaling. We chose this pathway because shutting down Tgfβ signals is a major mechanism used by c-Myc to enhance the MET in OKSM reprogramming (3). Notably, our qPCR analysis showed a decrease of Tgfβ cytokines (particularly Tgfβ2) in fibroblasts transduced with HDAC4 or -7 (Fig. 5A). We also detected a reduction of all three Tgfβ cytokines in fibroblasts reprogrammed with OKS and HDAC7 (Fig. 5B). Moreover, addition of recombinant Tgfβ3 (Tgfβ1, 2, and 3 have been reported to block reprogramming (3, 6)) neutralized the improved reprogramming efficiency caused by HDAC7 in OKS (Fig. 5C). These results explain why class IIa HDACs potentiate fibroblast reprogramming but do not demonstrate how this is mediated.
FIGURE 5.

Class IIa HDACs modulate the expression of Tgfβ cytokines. A, qPCR for Tgfβ cytokines using RNA lysates from fibroblasts transduced as indicated. An empty vector was used as a control (Ctrl, also in C). Double asterisks indicate p value < 0.01 (also in B and C). B, qPCR for Tgfβ cytokines using RNA lysates from fibroblasts reprogrammed with OKS and either empty vector or HDAC7. D, day. Single asterisk indicates p value < 0.05. C, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and HDAC7. Tgfβ3 was added as shown.
MEF2 Proteins Are Downstream Targets of Class IIa HDACs in Reprogramming
Next, we searched for putative effectors of class IIa HDACs that could explain the reduction of Tgfβ cytokines in reprogramming. We centered on MEF2 proteins because they are well studied partners of class IIa HDACs (Fig. 6A) and have a prodifferentiation effect (15, 21). We observed a progressive increase of MEF2 isoforms during reprogramming with OKS or OKSM (Fig. 6B). To define whether MEF2 proteins play a role in this process, we first created mutated forms of HDAC4, -5, and -7 that bear amino acid substitutions in the MEF2 binding site (32) and a version of HDAC7 with a deleted MEF2 binding site (24) (see schematic in Fig. 6A). These mutated forms lost the ability to bind to MEF2, as verified by coexpression with MEF2D and immunoprecipitation (Fig. 6C), and could not repress the activation of a MEF2-responsive luciferase reporter by MEF2D (D). As anticipated, the MEF2 binding mutants of all three class IIa HDACs failed to enhance OKS reprogramming (Fig. 6E). These findings advocate but do not demonstrate that class IIa HDACs regulate reprogramming by suppressing MEF2 proteins.
FIGURE 6.

MEF2 proteins are downstream targets of class IIa HDACs during reprogramming. A, schematic depicting the structure of HDAC4, -5, and -7, the location of the MEF2 binding site and the catalytic domain, and the corresponding modifications were performed to abolish their function. B, representative Western blot analysis for MEF2 proteins using lysates of a time course reprogramming experiment with OKS/OKSM in fibroblasts. An empty vector was used as a control (Ctrl, also in D, E, and G). D, day. C, immunoprecipitation of MEF2D after coexpression with the indicated FLAG-tagged class IIa HDAC variants in HEK293T cells. IP, immunoprecipitated samples; Ctrl, control. D, cotransfection of a MEF2 luciferase reporter gene and the indicated expression vectors in HEK293T cells. Activity was measured 48 h post-transfection. Items were measured in triplicate, and the mean values ± S.D. of a representative experiment are shown. E, number of GFP+ colonies in fibroblasts reprogrammed with OKS and wild-type or mutated forms of HDAC4, -5, and -7. F, shRNA vectors for MEF2 proteins were infected in fibroblasts, and the knockdown efficiency was measured by qPCR. Double asterisks indicate p value < 0.01 (also in G). G, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and the indicated shRNA vectors. Single asterisk indicates p value < 0.05.
To determine a direct role for MEF2 proteins in reprogramming, we reduced MEF2 expression levels by using shRNA vectors (Fig. 6F). This enhanced the number of GFP+ colonies in OKS and OKSM (Fig. 6G), in agreement with the observation (see Fig. 4H) that restricted (inducible) overexpression of class IIa HDACs in the MET phase potentiates both types of reprogramming. Yet, knockdown of MEF2 proteins boosted OKS reprogramming less significantly than overexpressing class IIa HDACs (see Fig. 1C, right panel). This apparent discrepancy is perhaps due to the circumstance that, besides preventing the binding of coactivators to MEF2 proteins, class IIa HDACs recruit chromatin regulators to induce repressive modifications near MEF2 binding sites (22).
Elevated MEF2 Proteins Block Reprogramming by Inducing Tgfβ Cytokines
We prepared expression vectors for each of the four isoforms of MEF2 (Fig. 7A) and overexpressed them individually in OKS/OKSM reprogramming. MEF2A, B, C, and, more potently, MEF2D, reduced GFP+ colony formation with either factor combination (Fig. 7B). MEF2 proteins could also block reprogramming in the presence of vitamin C (Fig. 7C), a natural compound that enhances colony formation efficiency significantly (23). Importantly, simultaneous overexpression of class IIa HDACs counteracted the reprogramming blockade triggered by exogenous MEF2 proteins (in the presence or absence of vitamin C) without altering their expression levels (Fig. 7, D–F). Therefore, excessive MEF2 activity prevents fibroblast reprogramming to iPSCs.
FIGURE 7.
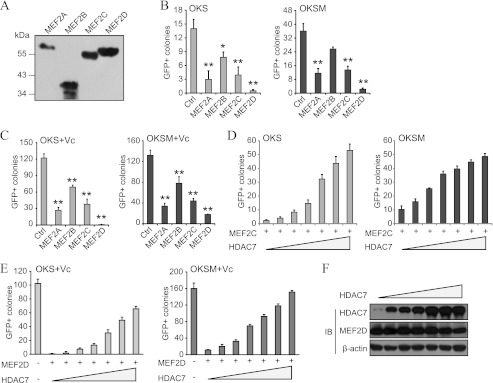
MEF2 proteins derail reprogramming by impairing the MET phase. A, representative Western blot analysis with anti-FLAG showing adequate overexpression of the indicated MEF2 proteins. B, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and MEF2 proteins. An empty vector was used as a control (Ctrl, also in C). Single asterisk indicates p value < 0.05, and double asterisks indicate p value < 0.01 (also in C). C, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and MEF2 isoforms. Cells were treated with vitamin C (Vc) to enhance the number of colonies and thus magnify differences (also in E). D, coexpression of HDAC7 rescues the reprogramming blockade in fibroblasts transduced with OKS/OKSM and MEF2C. A dose-response experiment is shown (also in E). The total amount of viruses (as per volume) was kept constant by using an empty vector (also in E). E, coexpression of HDAC7 rescues the reprogramming blockade in fibroblasts transduced with OKS/OKSM and MEF2D. F, Western blot analysis for HDAC7 and MEF2D using lysates from a dose-response experiment as indicated in E. Lysates were prepared at day 6 after infection. IB, immunoblot.
Considering the role of class IIa HDACs in the MET of reprogramming, we envisaged that MEF2 should play an opposite role in this process. As expected, overexpression of MEF2A or MEF2D reduced the MET in OKS and OKSM reprogramming (Fig. 8, A and B). Moreover, qPCR analysis showed an increase of Tgfβ cytokines (particularly Tgfβ2) in fibroblasts transduced with MEF2A or MEF2D (Fig. 8C) or reprogrammed with OKS and MEF2D (D). Importantly, the addition of a TgfβR1 inhibitor restored MET-like morphology changes and iPSC generation in fibroblasts reprogrammed with OKS/OKSM and MEF2D (Fig. 8, E and F). We also observed that MEF2D overexpression prevents the increase in E-cadherin triggered by Klf4 alone (3), which could be relieved by using a TgfβR1 inhibitor (Fig. 8G). Therefore, MEF2 proteins block the MET phase of reprogramming by inducing Tgfβ cytokines, and this, subsequently, impairs colony formation efficiency.
FIGURE 8.
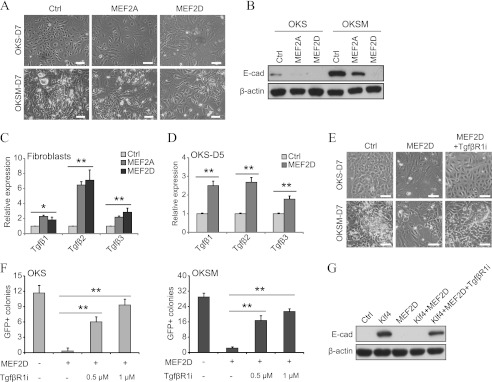
MEF2 proteins impair the MET of reprogramming by inducing Tgfβ cytokines. A, representative phase contrast photographs at day 7 post-infection of fibroblasts transduced with OKS/OKSM and MEF2A or MEF2D. An empty vector was used as a control (Ctrl) (also in B–G). Scale bars = 50 μm (also in E). D, day. B, representative Western blot analyses for E-cadherin (E-cad) using lysates of fibroblasts reprogrammed as indicated. Lysates were taken at day 7 in all cases. C, qPCR for Tgfβ cytokines using RNA lysates from fibroblasts transduced with MEF2A or MEF2D. Single asterisk indicates p value < 0.05 and double asterisks indicates p value < 0.01 (also in D and F). D, qPCR for Tgfβ cytokines of fibroblasts reprogrammed with OKS and MEF2A or MEF2D. E, representative phase contrast photographs of fibroblasts reprogrammed with OKS/OKSM and MEF2D treated or untreated with TgfβR1 inhibitor (TgfβR1i). F, number of GFP+ colonies in fibroblasts reprogrammed with OKS/OKSM and MEF2D. TgfβR1 inhibitor was added as indicated. G, Western blot analysis for E-cadherin of lysates from fibroblasts transduced with Klf4 and MEF2D treated or untreated with TgfβR1 inhibitor.
DISCUSSION
From a conceptual perspective, reprogramming can be viewed as a reversal of tissue specification during embryogenesis. However, if this were true, it would imply that developmental programs must become switched on during reprogramming. This in turn would tend to derail the epigenetic transformation and perhaps help explain the low efficiency of the process. In support of such an idea, we have shown that MEF2 proteins, a family of transcription factors with a key role in organogenesis (15, 21), augment during reprogramming and impair iPSC generation (Fig. 9). This negative effect is counteracted in a rheostat-like manner (using the analogy of an electrical circuit) by a steady increase in class IIa HDACs, which are well known regulators of MEF2 proteins (15, 21) (Fig. 9).
FIGURE 9.

Schematic depicting how class IIa HDACs/MEF2 proteins regulate the expression of Tgfβ cytokines during reprogramming.
MEF2 proteins block reprogramming by restraining the epithelial-like program driven by Klf4 (3, 33), which is mediated at least in part by increasing the expression of Tgfβ cytokines (Fig. 9). This suggests that MEF2 proteins bind to DNA consensus sites regulating the transcription of Tgfβ cytokines, but an indirect mode of action is also plausible. A connection between MEF2 proteins and the MET/epithelial-to-mesenchymal transition had not yet been explored, except for the induction of MEF2 by Twist during mesoderm specification in Drosophila and the observation that MEF2 is needed for proper craniofacial formation after neural crest delamination (21). It has been reported previously that MEF2 proteins and Tgfβ-induced Smads interact with each other in myoblasts (34, 35). Smad2 binds to and activates MEF2A (34), favoring myoblast terminal differentiation, whereas Smad3 binds to MEF2C and prevents it (35). We have shown here that MEF2 proteins induce Tgfβ cytokines in mouse fibroblasts, further adding complexity to the reported cross-talk. Given the numerous functions of MEF2 proteins in development (15, 21), it seems probable that they impair reprogramming through additional prodifferentiation pathways. This phenomenon might help us to understand the paradoxical observation that on one side the Yamanaka factors drive cells to a pluripotent cell fate and that on the other, they can be used to instruct specific lineages (e.g. neural and cardiomyocytes) (36, 37). Favoring such an idea, MEF2C is among the transcription factors used to induce the direct transdifferentiation of fibroblasts into cardiomyocytes (38). It is also interesting to think that a putative activation of lineage-specific signals by MEF2 proteins makes the resulting iPSCs more prone to retain epigenetic memory of those lineages (39). Likewise, it is possible that during reprogramming class IIa HDACs target other transcription factors besides MEF2 proteins (18, 19), further increasing the complexity of the presented network and its putative consequences.
Notably, our work also ascribes a positive role for a subfamily of HDACs in reprogramming. This contrasts with the general assumption that HDACs are negative in this process, which is on the basis of the positive effect of valproic acid in iPSC generation (40). However, the discrepancy can be explained by interference with a different type of HDACs (e.g. class I), as class IIa HDACs are not responsive to valproic acid (41). In this regard, we have observed that treatment with valproic acid synergizes with class IIa HDACs in OKS reprogramming (data not shown).
In summary, we have demonstrated that a signaling module that controls embryogenesis plays a fundamental role in somatic cell reprogramming by regulating the MET phase (Fig. 9). It is tempting to speculate that developmental transitions driven by class IIa HDACs/MEF2 proteins are also orchestrated through changes in Tgfβ expression and that this regulatory node participates in normal tissue homeostasis, fibrosis, and tumorigenesis.
Acknowledgments
We thank Jiekai Chen, Ronghui Li, and Ting Zhou for providing advice and/or critical comments. We also thank Hong Song, Shilong Chu, Xiangjie Zhao, and Keyu Lai for technical assistance.
This work was supported by Strategic Priority Research Program of the Chinese Academy of Sciences Grant XDA01020106, by Ministry of Science and Technology of China 973 Program Grants 2011CB965200, and by National Natural Science Foundation of China Grant 31071309.
- iPSC
- induced pluripotent stem cell
- MET
- mesenchymal-to-epithelial transition
- HDAC
- histone deacetylase
- ESC
- embryonic stem cell
- TgfβR1
- Tgfβ receptor 1
- qPCR
- quantitative PCR
- OKS
- Oct4, Klf4 and Sox2
- OKSM
- Oct4, Klf4, Sox2 and c-Myc
- iHDAC7-Dox
- doxycycline inducible HDAC7.
REFERENCES
- 1. Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 2. Plath K., Lowry W. E. (2011) Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 12, 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li R., Liang J., Ni S., Zhou T., Qing X., Li H., He W., Chen J., Li F., Zhuang Q., Qin B., Xu J., Li W., Yang J., Gan Y., Qin D., Feng S., Song H., Yang D., Zhang B., Zeng L., Lai L., Esteban M. A., Pei D. (2010) A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 [DOI] [PubMed] [Google Scholar]
- 4. Samavarchi-Tehrani P., Golipour A., David L., Sung H. K., Beyer T. A., Datti A., Woltjen K., Nagy A., Wrana J. L. (2010) Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 [DOI] [PubMed] [Google Scholar]
- 5. Ichida J. K., Blanchard J., Lam K., Son E. Y., Chung J. E., Egli D., Loh K. M., Carter A. C., Di Giorgio F. P., Koszka K., Huangfu D., Akutsu H., Liu D. R., Rubin L. L., Eggan K. (2009) A small-molecule inhibitor of TGF-β signaling replaces Sox2 in reprogramming by inducing nanog. Cell Stem Cell 5, 491–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maherali N., Hochedlinger K. (2009) TGFβ signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr. Biol. 19, 1718–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mikkelsen T. S., Hanna J., Zhang X., Ku M., Wernig M., Schorderet P., Bernstein B. E., Jaenisch R., Lander E. S., Meissner A. (2008) Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liao B., Bao X., Liu L., Feng S., Zovoilis A., Liu W., Xue Y., Cai J., Guo X., Qin B., Zhang R., Wu J., Lai L., Teng M., Niu L., Zhang B., Esteban M. A., Pei D. (2011) MicroRNA Cluster 302–367 Enhances Somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J. Biol. Chem. 286, 17359–17364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Subramanyam D., Lamouille S., Judson R. L., Liu J. Y., Bucay N., Derynck R., Blelloch R. (2011) Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 29, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Z., Yang C. S., Nakashima K., Rana T. M. (2011) Small RNA-mediated regulation of iPS cell generation. EMBO J. 30, 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vega R. B., Matsuda K., Oh J., Barbosa A. C., Yang X., Meadows E., McAnally J., Pomajzl C., Shelton J. M., Richardson J. A., Karsenty G., Olson E. N. (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119, 555–566 [DOI] [PubMed] [Google Scholar]
- 13. Chang S., Young B. D., Li S., Qi X., Richardson J. A., Olson E. N. (2006) Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126, 321–334 [DOI] [PubMed] [Google Scholar]
- 14. Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology. Implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lahm A., Paolini C., Pallaoro M., Nardi M. C., Jones P., Neddermann P., Sambucini S., Bottomley M. J., Lo Surdo P., Carfí A., Koch U., De Francesco R., Steinkühler C., Gallinari P. (2007) Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang X. J., Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases. From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., Shaw R. J. (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang B., Moya N., Niessen S., Hoover H., Mihaylova M. M., Shaw R. J., Yates J. R., 3rd, Fischer W. H., Thomas J. B., Montminy M. (2011) A hormone-dependent module regulating energy balance. Cell 145, 596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gossett L. A., Kelvin D. J., Sternberg E. A., Olson E. N. (1989) A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol. 9, 5022–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Potthoff M. J., Olson E. N. (2007) MEF2. A central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 22. Fischle W., Dequiedt F., Hendzel M. J., Guenther M. G., Lazar M. A., Voelter W., Verdin E. (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9, 45–57 [DOI] [PubMed] [Google Scholar]
- 23. Esteban M. A., Wang T., Qin B., Yang J., Qin D., Cai J., Li W., Weng Z., Chen J., Ni S., Chen K., Li Y., Liu X., Xu J., Zhang S., Li F., He W., Labuda K., Song Y., Peterbauer A., Wolbank S., Redl H., Zhong M., Cai D., Zeng L., Pei D. (2010) Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 6, 71–79 [DOI] [PubMed] [Google Scholar]
- 24. Qin D., Gan Y., Shao K., Wang H., Li W., Wang T., He W., Xu J., Zhang Y., Kou Z., Zeng L., Sheng G., Esteban M. A., Gao S., Pei D. (2008) Mouse meningiocytes express Sox2 and yield high efficiency of chimeras after nuclear reprogramming with exogenous factors. J. Biol. Chem. 283, 33730–33735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morita S., Kojima T., Kitamura T. (2000) Plat-E. An efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 26. Chen J., Liu J., Yang J., Chen Y., Chen J., Ni S., Song H., Zeng L., Ding K., Pei D. (2011) BMPs functionally replace Klf4 and support efficient reprogramming of mouse fibroblasts by Oct4 alone. Cell Res. 21, 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J. B., Zaehres H., Wu G., Gentile L., Ko K., Sebastiano V., Araúzo-Bravo M. J., Ruau D., Han D. W., Zenke M., Schöler H. R. (2008) Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 454, 646–650 [DOI] [PubMed] [Google Scholar]
- 28. Nakagawa M., Koyanagi M., Tanabe K., Takahashi K., Ichisaka T., Aoi T., Okita K., Mochiduki Y., Takizawa N., Yamanaka S. (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106 [DOI] [PubMed] [Google Scholar]
- 29. Hanna J., Saha K., Pando B., van Zon J., Lengner C. J., Creyghton M. P., van Oudenaarden A., Jaenisch R. (2009) Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462, 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wernig M., Meissner A., Cassady J. P., Jaenisch R. (2008) c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2, 10–12 [DOI] [PubMed] [Google Scholar]
- 31. Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., Liang H., Chen Y., Chen J., Wu Y., Guo L., Zhu J., Zhao X., Peng T., Zhang Y., Chen S., Li X., Li D., Wang T., Pei D. (2013) H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45, 34–42 [DOI] [PubMed] [Google Scholar]
- 32. Han A., He J., Wu Y., Liu J. O., Chen L. (2005) Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J. Mol. Biol. 345, 91–102 [DOI] [PubMed] [Google Scholar]
- 33. Yori J. L., Johnson E., Zhou G., Jain M. K., Keri R. A. (2010) Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J. Biol. Chem. 285, 16854–16863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinn Z. A., Yang C. C., Wrana J. L., McDermott J. C. (2001) Smad proteins function as co-modulators for MEF2 transcriptional regulatory proteins. Nucleic Acids Res. 29, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu D., Kang J. S., Derynck R. (2004) TGF-β-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 23, 1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Efe J. A., Hilcove S., Kim J., Zhou H., Ouyang K., Wang G., Chen J., Ding S. (2011) Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 13, 215–222 [DOI] [PubMed] [Google Scholar]
- 37. Kim J., Efe J. A., Zhu S., Talantova M., Yuan X., Wang S., Lipton S. A., Zhang K., Ding S. (2011) Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. U.S.A. 108, 7838–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ieda M., Fu J. D., Delgado-Olguin P., Vedantham V., Hayashi Y., Bruneau B. G., Srivastava D. (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim K., Doi A., Wen B., Ng K., Zhao R., Cahan P., Kim J., Aryee M. J., Ji H., Ehrlich L. I., Yabuuchi A., Takeuchi A., Cunniff K. C., Hongguang H., McKinney-Freeman S., Naveiras O., Yoon T. J., Irizarry R. A., Jung N., Seita J., Hanna J., Murakami P., Jaenisch R., Weissleder R., Orkin S. H., Weissman I. L., Feinberg A. P., Daley G. Q. (2010) Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huangfu D., Maehr R., Guo W., Eijkelenboom A., Snitow M., Chen A. E., Melton D. A. (2008) Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26, 795–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Göttlicher M., Minucci S., Zhu P., Krämer O. H., Schimpf A., Giavara S., Sleeman J. P., Lo Coco F., Nervi C., Pelicci P. G., Heinzel T. (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]