

Abstract
Chk1 is the effector kinase of the G2 DNA damage checkpoint. Chk1 homologs possess a highly conserved N-terminal kinase domain and a less conserved C-terminal regulatory domain. In response to DNA damage, Chk1 is recruited to mediator proteins assembled at lesions on replication protein A (RPA)-coated single-stranded DNA (ssDNA). Chk1 is then activated by phosphorylation on S345 in the C-terminal regulatory domain by the PI3 kinase-related kinases ATM and ATR to enforce a G2 cell cycle arrest to allow time for DNA repair. Models have emerged in which this C-terminal phosphorylation relieves auto-inhibitory regulation of the kinase domain by the regulatory domain. However, experiments in fission yeast have shown that deletion of this putative auto-inhibitory domain actually inactivates Chk1 function. We show here that Chk1 homologs possess a kinase-associated 1 (KA1) domain that possesses residues previously implicated in Chk1 auto-inhibition. In addition, all Chk1 homologs have a small and highly conserved C-terminal extension (CTE domain). In fission yeast, both of these motifs are essential for Chk1 activation through interaction with the mediator protein Crb2, the homolog of human 53BP1. Thus, through different intra- and intermolecular interactions, these motifs explain why the regulatory domain exerts both positive and negative control over Chk1 activation. Such motifs may provide alternative targets to the ATP-binding pocket on which to dock Chk1 inhibitors as anticancer therapeutics.
Keywords: checkpoint, Chk1, DNA damage, KA1 domain, protein kinase
Introduction
The G2 DNA damage checkpoint functions to prevent mitotic entry in the presence of DNA damage, thereby allowing time for DNA repair to reassemble intact chromosomes.1 Failure of checkpoint signaling leads to the segregation of chromosomes containing unrepaired lesions, resulting in gross chromosomal rearrangements and/or cell death. A large number of proteins collaborate in this signaling process, and culminate in the activation of the highly conserved effector protein kinase, Chk1. This activation requires Chk1 to be recruited to complexes of checkpoint proteins assembled at the sites of primary lesions that have subsequently been converted into DNA and then coated by RPA. There, Chk1 transiently binds BRCT-domain mediator proteins, which enables Chk1 to be phosphorylated on S345 in its C-terminal regulatory domain by the resident PI3K-related protein kinases (PIKKs) ATM and ATR, activating its kinase activity.2-4 Active Chk1 then elicits a G2 cell cycle delay by upregulation of the kinase (Wee1) and downregulation of the phosphatase (Cdc25) that control inhibitory tyrosine-15 phosphorylation of the mitotic cyclin-dependent kinase Cdc2.1,5,6
Although S345 phosphorylation is required for Chk1 activation,3 and its dephosphorylation is both necessary and sufficient to inactivate Chk1 once repair is completed,7 mechanistic details of how this modification regulates Chk1 activity are not known. All Chk1 homologs possess an N-terminal kinase domain and a C-terminal regulatory domain of ~200 amino acids.8 Removal of the regulatory domain substantially stimulates the activity of the kinase domain when assayed in vitro, suggesting that the regulatory domain is auto-inhibitory.9,10 Within this domain are two small regions that are conserved from the yeasts to humans, and consistent with an auto-inhibitory model, specific amino acid substitutions in these domains can activate Chk1 without apparent DNA damage or S345 phosphorylation.11-13 However, in the fission yeast Schizosaccharomyces pombe, several other mutations in these domains functionally inactivate Chk1,13 and in the budding yeast Saccharomyces cerevisiae, mutations in these domains ablate an interaction with the mediator protein Rad9.14 Further, a series of C-terminal truncations in S. pombe Chk1, ranging from the final 11 residues through to the entire regulatory domain, are all non-functional proteins when assayed in vivo by their ability to mount a checkpoint response in cells exposed to DNA damaging agents.13 These data suggest that the C-terminal regulatory domain may be both inhibitory and yet also required for Chk1 activation and/or function in the cell.
Loss of G1/S checkpoint signaling through the p53 tumor-suppressor pathway is commonplace in cancer cells.15 On the contrary, the G2 checkpoint is rarely (if ever) lost, and many studies have shown that tumor cells actually require Chk1 and the G2 checkpoint for viability, particularly if challenged by genotoxins.16,17 For this reason, a number of small-molecule inhibitors of Chk1 have been identified and are in various stages of clinical and preclinical development.18,19 The majority of these inhibitors are ATP-competitive molecules, and thus run the risk of off-target effects. Despite this, interest in inhibiting Chk1 in combination with genotoxic therapy remains high, and design of inhibitory strategies would benefit greatly from a more detailed understanding of mechanisms of Chk1 activation.
The deletion of chk1 is functionally equivalent to deleting its specific mediator,20 which in S. pombe is known as Crb2 and Rad9 in S. cerevisiae. The Crb2/Rad9 homolog in humans is 53BP1,21 though additional mediator proteins exist in vertebrates that also contribute to Chk1 activation, such as MDC122-24 and Claspin.25-27 Conversely, other components of the checkpoint cascade have additional roles in DNA repair, lesion bypass, replication fork stability and telomere maintenance.17,28-30 Thus, inhibiting checkpoint signaling upstream of the mediators and Chk1 may be very toxic to cells due to these other functions in genome integrity, which for DNA repair and telomere maintenance would not require active cell cycling.
A viable alternative strategy to inhibit Chk1 signaling would be to block its DNA damage-induced activation, either through its interaction with Crb2/53BP1, or by blocking the activating effects promoted by S345 phosphorylation. With this in mind, we sought to define small regions of the regulatory domain that are required for Chk1 activation, which could potentially serve as docking sites for such allosteric inhibitors. We describe here an analysis of a kinase-associated 1 (KA1) domain in the C-terminus of Chk1, plus an additional highly conserved motif present in an unique C-terminal extension (CTE), both of which are required for Chk1 activation. The presence of these motifs provides a rationale as to the dual nature of the regulatory domain, and presents an alternative target for small-molecule inhibition.
Results
In silico analysis of the regulatory domain of Chk1
A large number of mutations in the regulatory domain of S. pombe Chk1 have been identified that ablate function,8 though whether these alleles cause a specific and informative change in Chk1 regulation or a general change in the fold and/or stability of the protein is not known. However, rare alleles that are mis-sense mutations in the only highly conserved regions outside the kinase domain are gain-of-function in both the yeasts and in Xenopus,11-13 suggesting these regions are indeed critical to Chk1 regulation. Limited homology between the most N-terminal motif (RMTRFFT in human Chk1) and a 37 amino acid protein phosphatase interacting (PPI) domain of a number of protein kinases in Arabidopsis thaliana has been previously noted.31 However, subsequent structural and phylogenetic analyses have indicated that this PPI domain is larger (80–90 amino acids), and is at the extreme C-terminus of a number of protein kinases found in a wide variety of species.32,33 This domain has been renamed the kinase-associated 1 (KA1) domain and is a compact structure with a hydrophobic concave surface constrained by a βαββββα fold. Such a structure is consistent with this domain functioning as a protein-protein interaction module. Importantly, KA1 domains have been shown to function as autoinhibitory domains in the mouse MELK (maternal embryonic leucine zipper kinase)34 and yeast Kin1 kinases,35 both relatives of the Par-1 kinase of Caenorhabditis elegans. Based on these findings, and the proposal that the C-terminus of Chk1 is autoinhibitory, we searched for similar regulatory motifs in Chk1.
First, we investigated predictions of protein order and disorder across S. pombe Chk1. While the kinase domain is predictably highly structured, it is immediately followed by a disordered region of ~100 amino acids. However, the C-terminal 100 residues, which include the conserved regions of homology, are predicted to adopt an ordered structure (Fig. 1A). We then compared the predicted protein fold of this domain to that determined for the solution structure of the KA1 domain of the mouse MARK3 kinase,32 another member of the Par-1 family. Both the S. pombe and human Chk1 sequences are predicted to form the same βαββββα fold of the KA1 with analogous spacing to that of MARK3 (Fig. 1B and C). The two highly conserved regions within the regulatory domain in which both activating and inactivating mutations have been identified comprising β1 and α2 regions of the KA1 domain. The disordered region between the kinase and KA1 domains could presumably provide the flexibility to enable these domains to interact intramolecularly, which is in keeping with the autoinhibitory model of Chk1 regulation.10 However, the existence of many inactivating mutations and deletions in the KA1 domain shows that this cannot be the only function for this region of the regulatory domain.

Figure 1. The C-terminal region of Chk1 contains a predicted kinase-associated 1 (KA1) domain with a unique C-terminal extension (CTE). (A) DISOPRED generated disorder plot of S. pombe Chk1. The horizontal line at 5% represents the order/disorder threshold. (B) PSIPRED secondary structure prediction of the KA1 domain (βαββββα) for S. pombe Chk1, (C) human Chk1, (D) Mouse MARK3 kinase. Note that Chk1 has an extension containing β6. α-helices (H) are green cylinders, β-sheets (E) are yellow arrows and black lines are coiled regions (C). Cyan bars represent the confidence of the prediction (0–100%). The Chk1 residues boxed by the red line are the most conserved residues that when mutated can either activate or inactivate Chk1 homologs.
Importantly, unlike other KA1-containing kinases, all Chk1 homologs contain a small C-terminal extension (CTE domain) with a predicted β-sheet containing an isoleucine-valine (IV) motif that is conserved from S. pombe to humans (Fig. 1B and C). The IV motif is present as a related VV motif in some species, such as mice, chickens and Xenopus. A short but not conserved region follows this motif, again in all Chk1 homologs. The conservation of these KA1 and CTE domains across all Chk1 homologs makes them likely candidates as important regions of Chk1 regulation, which we have dissected for the S. pombe enzyme.
KA1 and CTE domains are essential for Chk1 function
To interrogate the importance of the KA1 and CTE domains in Chk1 function, a number of mutant alleles were constructed at the endogenous chk1 locus. Notably, although a previously constructed truncation series did delete the predicted KA1 domain,13 this series simultaneously deleted the CTE domain in each allele, hence, we have analyzed these separately in this study.
First, an internal deletion of the entire KA1 domain leaving the CTE intact (ΔKA1) inactivates Chk1 function, as does a smaller deletion of the C-terminal half of the KA1 domain (Δ½KA1). This is evidenced by sensitivity to UV-C irradiation (Fig. 2A) and the alkylating agent methyl methanesulfonate (MMS) (Fig. 2B). Like all other chk1 alleles identified to date, this sensitivity extends to all DNA damaging agents so far tested. This sensitivity is accompanied by an inability to delay mitotic entry after irradiation, resulting in the “cut” phenotype where the division septum bisects the damaged chromosomes that are unable to segregate (arrowed in Fig. 2C), again seen with a diverse range of DNA damaging agents. Thus, the KA1 domain is required for Chk1 function in vivo. This result does not support the hypothesis that the KA1 domain is solely autoinhibitory, but does not rule out an additional autoinhibitory function as proposed for the Par-1 homologs.

Figure 2. Mutations within the C-terminal regulatory domain ablate Chk1 function. (A) Clonogenic survival to UV-C irradiation for the indicted strains. (B) Ten-fold serial dilutions of the indicated strains were spotted onto YES agar containing no drug and 0.01% MMS and grown at 30°C for 4 d. The MMS and UV-C sensitive strains are similarly sensitive to all DNA damaging agents tested. (C) Non-functional chk1 alleles display the “cut” phenotype (arrowed) after treatment with DNA damaging agents, which is characteristic of checkpoint failure; chk1Δ, Δ½KA1 and IV488–489AA mutants are shown as examples. Under the same conditions, wild-type cells elongate due to the Chk1-dependent cell cycle arrest. Agents used are UV-C (150 J/m2), 2 h after irradiation and a 4 h incubation in the following drugs: 0.01% MMS, 0.5 mU Bleomycin (Bleo), 10 µM Camptothecin (CPT) and 1mM mechlorethamine hydrochloride (HN2).
Second, we made a number of mutations C-terminal to the KA1 domain (Fig. 2). As seen before, deletion of the entire region (Δ485–496) inactivates Chk1. Moreover, deletion of the CTE domain (Δ485–491), or the conserved IV motif (Δ488–489) within the CTE domain also inactivated Chk1. Similarly, mutation of the IV to alanines (IV488–489AA) also creates an inactive chk1 allele. However, deletion of the non-conserved amino acids after the CTE domain at the extreme C-terminus (Δ492–496) had no measurable effect on Chk1 function. Therefore, the CTE domain of Chk1, which appears to be unique among protein kinases with KA1 domains, is absolutely required for function.
KA1 and CTE domains are required for Chk1 activation through interaction with Crb2
We next assayed expression of these Chk1 mutants by western blotting in untreated cells, and in cells irradiated with 150 J/m2 UV-C. DNA damage induces ATR- (Rad3 in S. pombe) catalyzed phosphorylation on S345, which activates Chk1’s kinase activity and results in a mobility shift on SDS-PAGE.3 As expected, functional Chk1 alleles (wild-type and Δ492–496) were expressed and phosphorylated in UV-C-irradiated cells (Fig. 3A). Conversely, none of the non-functional alleles were phosphorylated, and the expression of both the ΔKA1 and Δ488–489 proteins was not detectable (Fig. 3A and B) despite normal levels of mRNA for these alleles (Fig. 3C). This is not the case for amino acid substitutions in these domains, and suggests a profound effect on protein folding leading to degradation.
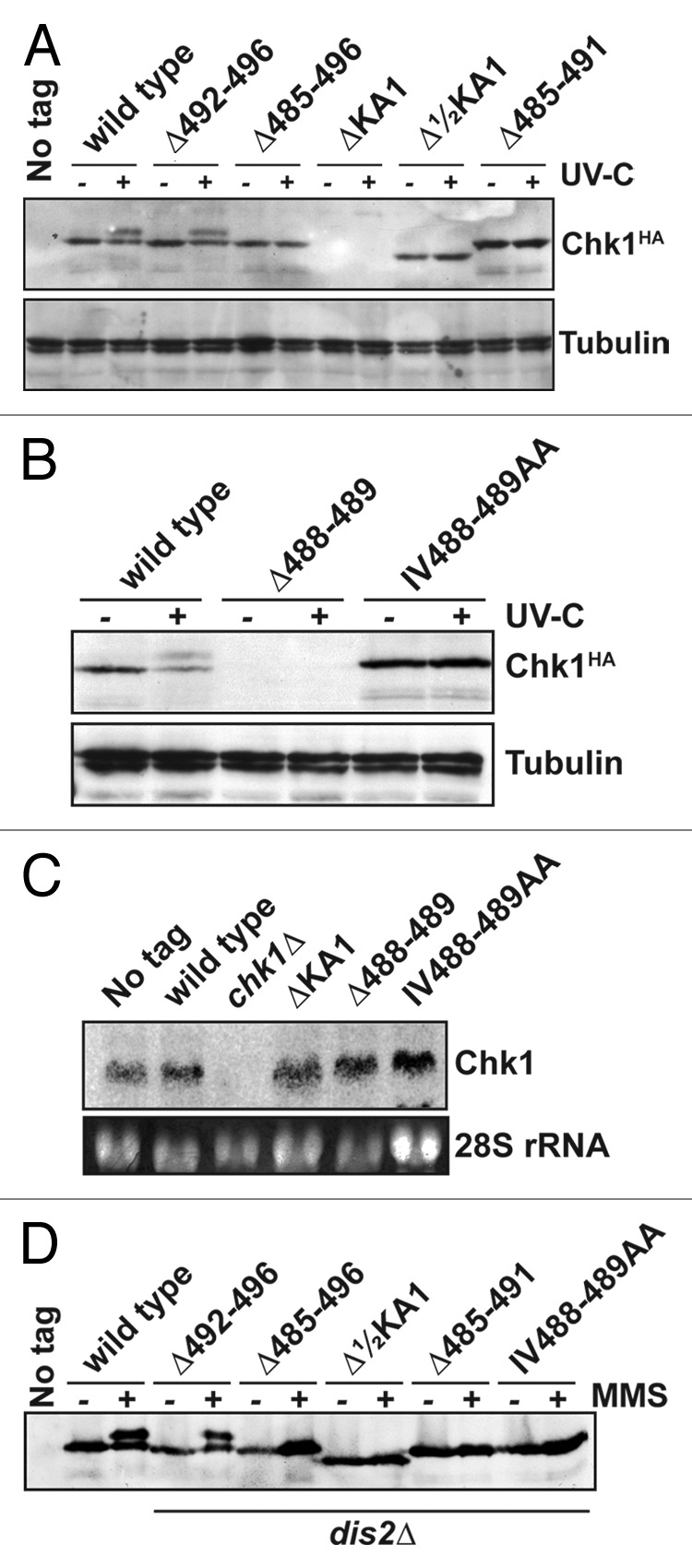
Figure 3. Non-functional Chk1 mutants fail to undergo activating phosphorylation. (A and B) Western blotting (anti-HA for Chk1, anti-tubulin as a loading control) for the indicated strains either mock irradiated, or irradiated with 150 J/m2 UV-C. Activating phosphorylation is seen as a retarded mobility on SDS-PAGE. Note that the ΔKA1 and Δ488–489 mutants show no detectable protein expression, but (C) show normal mRNA levels by northern blotting (28S rRNA is shown as a loading control). (D) The indicated Chk1 mutants were expressed in cells lacking the Chk1 S345 phosphatase Dis2, and either mock-treated or incubated with 0.01% MMS for 7 h. The non-functional alleles still fail to show activating phosphorylation.
The lack of activating S345 phosphorylation on non-functional Chk1 mutants may be due to an inability to be phosphorylated or, given the interactions between KA1 domains and protein phosphatases,31,36 an enhanced dephosphorylation. Dis2, a type 1 protein phosphatase is both necessary and sufficient for Chk1 dephosphorylation in S. pombe.7,37,38 We therefore assayed phosphorylation of the mutant Chk1 proteins in dis2∆ cells, and used chronic exposure to the alkylating agent MMS to further increase the chance to trap Chk1 in its phosphorylated and active state. Again, only the functional Δ492–496 mutant was phosphorylated (Fig. 3D), and so we found no evidence of an enhanced dephosphorylation of the non-functional mutants.
Therefore, these data suggest that the mutant Chk1 proteins fail to be phosphorylated by Rad3. The phospho-acceptor site, S345, is intact in all the expressed mutant proteins, thus it is unlikely that ATR would fail to recognize them, so long as the proteins were in close enough proximity for catalysis to occur. In the S. pombe cell, this is achieved in the presence of DNA damage via a transient interaction between Chk1 and Crb2 (53BP1 in humans) at sites of DNA damage,4 at which Rad3 is also anchored by its partner protein Rad26 (ATR and ATRIP in humans).39 The transient nature of this interaction means that the physical association of Chk1 and Crb2 cannot be detected unless one or both of the proteins are overexpressed.4 However, the interaction has been confirmed in multiple species, and by two-hybrid interaction assays.12,40 and is defective in S. cerevisiae mutants in the conserved regions of the KA1 domain identified herein.14 Therefore the overexpression allows detection of a bona fide interaction that activates Chk1, but rapidly releases it to phosphorylate Wee1 and Cdc25 homologs to elicit a G2 cell cycle arrest.
To assay a Chk1-Crb2 interaction, strains expressing wild-type Chk1, the Δ½KA1 mutant and the IV488–489AA substitution mutant in CTE domain were transformed with plasmids expressing GST or GST-Crb2 from the strong, thiamine-repressible nmt1 promoter.41 Following induction of GST and GST-Crb2, cells were treated with MMS to activate the checkpoint cascade and lysates prepared for glutathione-mediated capture on magnetic beads. GST-Crb2, but not GST, pulled down wild-type Chk1 at ~1–2% efficiency (Fig. 4). However, neither of the Chk1 mutants were detected in the GST-Crb2 pull downs. This suggests that these Chk1 proteins are non-functional due to an inability to interact with Crb2; hence, they are unable to undergo activating phosphorylation within the checkpoint complexes at sites of DNA damage. Hence, both the KA1 and CTE domains promote this interaction, though it is not necessarily the case that both do so directly.
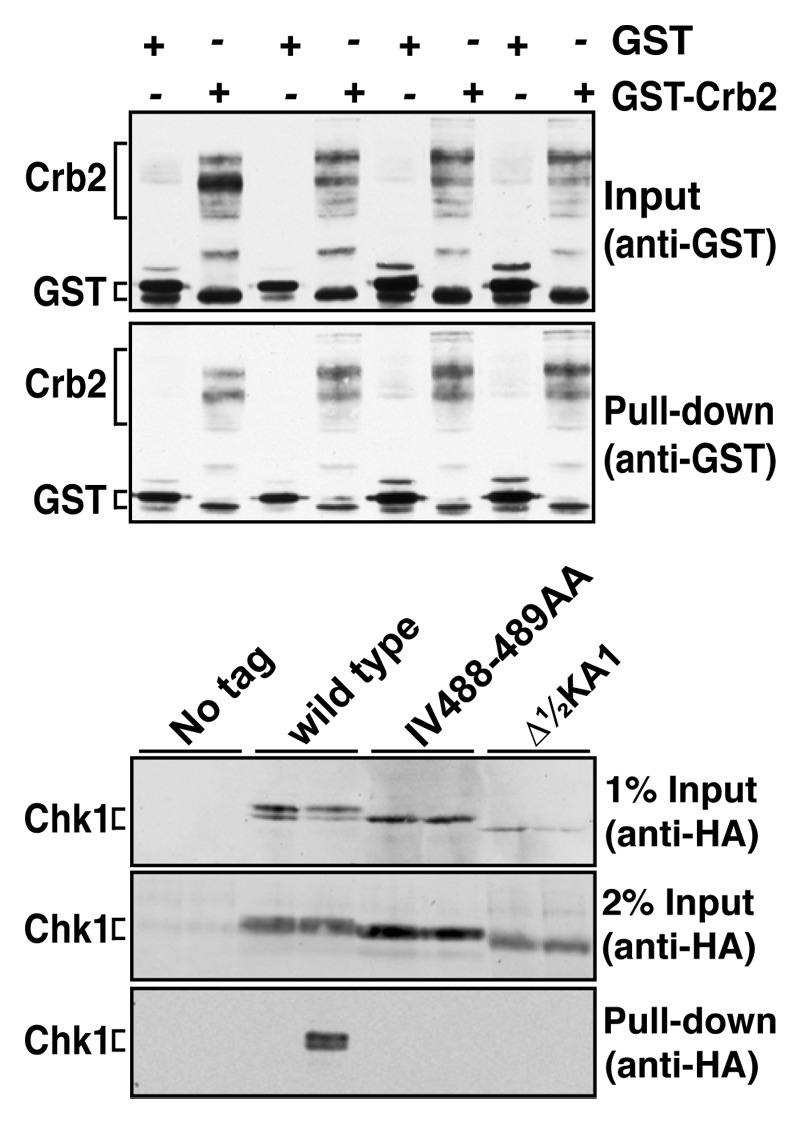
Figure 4. Non-functional Chk1 mutants fail to interact with Crb2. Strains expressing the indicated Chk1 mutants were transformed with plasmids expressing GST or GST-Crb2 from the nmt1 promoter. Expression from nmt1 was induced by thiamine withdrawal for 12 h at 30°C, whereupon cells were then incubated with 0.01% MMS for a further 7 h. The top panels show the input (50 µg total extract) and recovered (on glutathione magnetic beads) GST and GST-Crb2, as well as the endogenous S. pombe GST that runs slightly faster than the exogenous GST. Lower panels show 1% or 2% of the input for Chk1 (50 µg or 100 µg of total extract) and the Chk1 (wild-type only) recovered on glutathione magnetic beads.
Discussion
Based on the structural predictions, sequence conservation and genetic dissection in S. pombe, we conclude that the KA1 and CTE domains are important determinants in Chk1 regulation. The activating phosphorylation sites in Chk1 are within the disordered spacer between the kinase and KA1 domains, and their phosphorylation may alter protein flexibility to regulate the ability for the KA1 domain to interact with and inhibit the kinase domain. Presumably the activating mutations in the KA1 domain11-13 would disrupt this interaction, thereby mimicking the effects of S345 phosphorylation.
Chk1 is unique among the KA1 domain kinases in that the CTE domain follows the KA1 domain, whereas KA1 domains are normally located at the extreme C-terminus of protein kinases. The alanine substitutions show the IV motif is clearly required for Chk1 activation via Crb2 interaction. Although the resulting instability of the ∆IV mutant suggests the CTE domain is critical for the overall protein fold, it is notable that deletion of the entire domain (Δ485–491) does not affect stability. Structural data for Chk1 is currently limited to the kinase domain,9 and understanding the arrangement of the C-terminus relative to the kinase domain would be illuminating in understanding precise mechanisms of Chk1 regulation. However, the unstructured spacer will make this very challenging and is presumably why structural data outside the kinase domain is currently lacking. Nevertheless, the data presented here may enable this to be pursued in a modular fashion, perhaps together with gene replacement studies in human cells to confirm the generality of our conclusions.
Our findings are consistent with the CTE domain physically contacting Crb2, perhaps providing specificity to this interaction with spacing and/or orientation provided by the KA1 domain. Such an association may give access to S345 to be phosphorylated, and lock Chk1 into an active configuration. However, given the transient nature of the Crb2-Chk1 interaction, directly demonstrating the CTE-Crb2 interaction with purified domains will also be very challenging. Nevertheless, the CTE domain only confers positive regulation to Chk1, where the KA1 domain appears to confer both positive (Crb2-interacting) and negative (autoinhibition) regulation. Therefore, the CTE domain may provide specificity for small-molecule inhibitors of human Chk1 activation by blocking the activating interaction with 53BP1. Alternatively, molecules that interact with 53BP1 and block CTE interaction would have the same effect. Such molecules could act as alternatives to those currently under development as anticancer therapeutics that target the ATP-binding pocket of Chk1’s kinase domain.
Materials and Methods
Structural predictions
Predictions of protein disorder with S. pombe Chk1 were performed using the Database of Protein Disorder (Disprot).42 Analysis of the C-termini of S. pombe Chk1, human Chk1 and Mouse Mark3 was performed using the PSIPRED server.43
S. pombe methods
The propagation and genetic manipulation of S. pombe was performed using standard methods described previously.44,45 All strains are derivatives of 972h− and 975h+. Alleles of chk1 were constructed on a genomic clone of the chk1 locus including a C-terminal HA3-epitope tag by the method of Kunkel46 (oligonucleotides are listed in Table 1). All resulting mutant clones were sequenced, integrated at the chk1 locus by marker replacement in a chk1::ura4 strain and confirmed by Southern blotting and DNA sequencing of the chk1 locus. Total RNA for northern blotting was extracted using Trizol Reagent (Invitrogen), separated on MOPS/formaldehyde gels and transferred to Zeta-Probe (Bio Rad).
Table 1. Oligonucleotides used to construct chk1 alleles.
| ∆KA1: |
GTAAGAACAATCGGCTTCCCCTTTCAGGAGGACAAATTTC |
| ∆½KA1: |
GTAAGAACAATCGGCTTCCCTTTCCGTTTATCATGTAAAT |
| ∆492–496: |
GGGTAAAAGATGCGGCCGCCTGTAAGAACAATCGGCTTCC |
| ∆485–496: |
GGGTAAAAGATGCGGCCGCCTATTGAACTGACAACGTTCT |
| ∆485–491: |
CCGCCATTTTGTGAAACATCTATTGAACTGACAACGTTCT |
| ∆488–489: |
TTTTGTGAAACATCTGTAAGCGGCTTCCCTATTGAACTGA |
| IV488–489AA: | TGTGAAACATCTGTAAGTGCTGCCGGCTTCCCTATTGAAC |
Protein analysis
For straight western blotting, extracts were made from exponentially growing cells by bead beating in Urea Lysis Buffer [8 M urea, 100 mM sodium phosphate, 10 mM Tris.HCl, pH7.2]. Chk1 was detected using mouse monoclonal 12CA5 (Roche) at 2 µg/ml. Alpha-tubulin was detected with mouse monoclonal B-5-1-2 (Sigma) at 0.2 µg/ml. Immune complexes were detected with HRP-conjugated donkey anti-mouse IgG (1/2,000) and ECL reagent (GE Healthcare). For GST and GST-Crb2 pull-down experiments, extracts were made from exponentially growing cells that had been grown in media lacking thiamine for 19 h, including 7 h in 0.01% MMS, in native lysis buffer [50 mM Tris.HCl pH 8, 250 mM NaCl, 10% glycerol, 1 mM EDTA, 0.1% IGEPAL CA-630, 0.1mM NaVO4, 10 mM β-glycerophosphate, 30 mM Na2H2P2O7, 50 mM NaF, 1 mM DTT, 5× protease inhibitor cocktail (Sigma P8340)]. Twenty µl of Pierce Glutathione Magnetic Beads (Thermo Scientific) was added to 5 mg of clarified extract, and GST and GST-Crb2 was collected for 4 h at 4°C. The beads were collected by magnetism, and washed 3× in lysis buffer. HA-tagged Chk1 was detected as above, and GST and GST-Crb2 (plus the endogenous S. pombe GST) were detected using mouse monoclonal B-14 (Santa Cruz) at 2 µg/ml. Electrophoretic and western transfer methods were as described.47
Acknowledgments
We are grateful to Fernando Bazan (Genentech) for communication of unpublished results, and to the members of our laboratory for fruitful discussions. This work was supported by NIH grant GM087326.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23881
References
- 1.O’Connell MJ, Walworth NC, Carr AM. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/S0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- 2.O’Connell MJ, Cimprich KA. G2 damage checkpoints: what is the turn-on? J Cell Sci. 2005;118:1–6. doi: 10.1242/jcs.01626. [DOI] [PubMed] [Google Scholar]
- 3.Capasso H, Palermo C, Wan S, Rao H, John UP, O’Connell MJ, et al. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci. 2002;115:4555–64. doi: 10.1242/jcs.00133. [DOI] [PubMed] [Google Scholar]
- 4.Mochida S, Esashi F, Aono N, Tamai K, O’Connell MJ, Yanagida M. Regulation of checkpoint kinases through dynamic interaction with Crb2. EMBO J. 2004;23:418–28. doi: 10.1038/sj.emboj.7600018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raleigh JM, O’Connell MJ. The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J Cell Sci. 2000;113:1727–36. doi: 10.1242/jcs.113.10.1727. [DOI] [PubMed] [Google Scholar]
- 6.Sørensen CS, Syljuåsen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–86. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.den Elzen NR, O’Connell MJ. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 2004;23:908–18. doi: 10.1038/sj.emboj.7600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tapia-Alveal C, Calonge TM, O’Connell MJ. Regulation of chk1. Cell Div. 2009;4:8. doi: 10.1186/1747-1028-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–92. doi: 10.1016/S0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 10.Katsuragi Y, Sagata N. Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol Biol Cell. 2004;15:1680–9. doi: 10.1091/mbc.E03-12-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang SX, Dunphy WG. Activation of Xenopus Chk1 by mutagenesis of threonine-377. FEBS Lett. 2000;487:277–81. doi: 10.1016/S0014-5793(00)02370-X. [DOI] [PubMed] [Google Scholar]
- 12.Pereira E, Chen Y, Sanchez Y. Conserved ATRMec1 phosphorylation-independent activation of Chk1 by single amino acid substitution in the GD domain. Cell Cycle. 2009;8:1788–93. doi: 10.4161/cc.8.11.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosoy A, O’Connell MJ. Regulation of Chk1 by its C-terminal domain. Mol Biol Cell. 2008;19:4546–53. doi: 10.1091/mbc.E08-04-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Caldwell JM, Pereira E, Baker RW, Sanchez Y. ATRMec1 phosphorylation-independent activation of Chk1 in vivo. J Biol Chem. 2009;284:182–90. doi: 10.1074/jbc.M806530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 16.Koniaras K, Cuddihy AR, Christopoulos H, Hogg A, O’Connell MJ. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene. 2001;20:7453–63. doi: 10.1038/sj.onc.1204942. [DOI] [PubMed] [Google Scholar]
- 17.Kuntz K, O’Connell MJ. The G(2) DNA damage checkpoint: could this ancient regulator be the Achilles heel of cancer? Cancer Biol Ther. 2009;8:1433–9. doi: 10.4161/cbt.8.15.9081. [DOI] [PubMed] [Google Scholar]
- 18.Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv. 2011;11:133–40. doi: 10.1124/mi.11.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–16. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–8. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 22.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–6. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–6. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 24.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–61. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 25.Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem. 2003;278:30057–62. doi: 10.1074/jbc.M301136200. [DOI] [PubMed] [Google Scholar]
- 26.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–49. doi: 10.1016/S1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Kumagai A, Dunphy WG. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell. 2003;11:329–40. doi: 10.1016/S1097-2765(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 28.Kai M, Wang TS. Checkpoint responses to replication stalling: inducing tolerance and preventing mutagenesis. Mutat Res. 2003;532:59–73. doi: 10.1016/j.mrfmmm.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Dahlen M, Olsson T, Kanter-Smoler G, Ramne A, Sunnerhagen P. Regulation of telomere length by checkpoint genes in Schizosaccharomyces pombe. Mol Biol Cell. 1998;9:611–21. doi: 10.1091/mbc.9.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindsay HD, Griffiths DJF, Edwards RJ, Christensen PU, Murray JM, Osman F, et al. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–95. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohta M, Guo Y, Halfter U, Zhu JK. A novel domain in the protein kinase SOS2 mediates interaction with the protein phosphatase 2C ABI2. Proc Natl Acad Sci USA. 2003;100:11771–6. doi: 10.1073/pnas.2034853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tochio N, Koshiba S, Kobayashi N, Inoue M, Yabuki T, Aoki M, et al. Solution structure of the kinase-associated domain 1 of mouse microtubule-associated protein/microtubule affinity-regulating kinase 3. Protein Sci. 2006;15:2534–43. doi: 10.1110/ps.062391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moravcevic K, Mendrola JM, Schmitz KR, Wang YH, Slochower D, Janmey PA, et al. Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell. 2010;143:966–77. doi: 10.1016/j.cell.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beullens M, Vancauwenbergh S, Morrice N, Derua R, Ceulemans H, Waelkens E, et al. Substrate specificity and activity regulation of protein kinase MELK. J Biol Chem. 2005;280:40003–11. doi: 10.1074/jbc.M507274200. [DOI] [PubMed] [Google Scholar]
- 35.Elbert M, Rossi G, Brennwald P. The yeast par-1 homologs kin1 and kin2 show genetic and physical interactions with components of the exocytic machinery. Mol Biol Cell. 2005;16:532–49. doi: 10.1091/mbc.E04-07-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akaboshi M, Hashimoto H, Ishida H, Saijo S, Koizumi N, Sato M, et al. The crystal structure of plant-specific calcium-binding protein AtCBL2 in complex with the regulatory domain of AtCIPK14. J Mol Biol. 2008;377:246–57. doi: 10.1016/j.jmb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 37.den Elzen N, Kosoy A, Christopoulos H, O’Connell MJ. Resisting arrest: recovery from checkpoint arrest through dephosphorylation of Chk1 by PP1. Cell Cycle. 2004;3:529–33. [PubMed] [Google Scholar]
- 38.Calonge TM, O’Connell MJ. Turning off the G2 DNA damage checkpoint. DNA Repair (Amst) 2008;7:136–40. doi: 10.1016/j.dnarep.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 40.Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maundrell K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 1993;123:127–30. doi: 10.1016/0378-1119(93)90551-D. [DOI] [PubMed] [Google Scholar]
- 42.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, et al. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35(Database issue):D786–93. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38(Web Server issue):W563–8. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calonge TM, Eshaghi M, Liu J, Ronai Z, O’Connell MJ. Transformation/transcription domain-associated protein (TRRAP)-mediated regulation of Wee1. Genetics. 2010;185:81–93. doi: 10.1534/genetics.110.114769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calonge TM, O’Connell MJ. Antagonism of Chk1 signaling in the G2 DNA damage checkpoint by dominant alleles of Cdr1. Genetics. 2006;174:113–23. doi: 10.1534/genetics.106.060970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–82. doi: 10.1016/0076-6879(87)54085-X. [DOI] [PubMed] [Google Scholar]
- 47.Tapia-Alveal C, O’Connell MJ. Methods for studying checkpoint kinases - Chk1. Methods Mol Biol. 2011;782:171–9. doi: 10.1007/978-1-61779-273-1_12. [DOI] [PubMed] [Google Scholar]