

Abstract
Prion diseases such as Creutzfeldt–Jakob disease (CJD) are incurable and rapidly fatal neurodegenerative diseases. Because prion protein (PrP) is necessary for prion replication but dispensable for the host, we developed the PrP–FRET-enabled high throughput assay (PrP–FEHTA) to screen for compounds that decrease PrP expression. We screened a collection of drugs approved for human use and identified astemizole and tacrolimus, which reduced cell-surface PrP and inhibited prion replication in neuroblastoma cells. Tacrolimus reduced total cellular PrP levels by a nontranscriptional mechanism. Astemizole stimulated autophagy, a hitherto unreported mode of action for this pharmacophore. Astemizole, but not tacrolimus, prolonged the survival time of prion-infected mice. Astemizole is used in humans to treat seasonal allergic rhinitis in a chronic setting. Given the absence of any treatment option for CJD patients and the favorable drug characteristics of astemizole, including its ability to cross the blood–brain barrier, it may be considered as therapy for CJD patients and for prophylactic use in familial prion diseases. Importantly, our results validate PrP-FEHTA as a method to identify antiprion compounds and, more generally, FEHTA as a unique drug discovery platform.
Keywords: protein misfolding, high-throughput screening, prion therapeutics
Prion diseases are lethal infectious neurodegenerative diseases affecting humans and animals. In humans, Creutzfeldt–Jakob disease (CJD) is sporadic, affecting mainly people over 60 y; iatrogenic; or genetic with high penetrance. Prion diseases are characterized by the accumulation, in brain and lymphoid tissue of PrPSc, a misfolded, aggregated form of the host prion protein (PrP) (1). PrPSc is thought to be the main or only constituent of the prion, the infectious agent. Prion replication is based on the conformational conversion of the host prion protein into its pathogenic misfolded counterpart. In the sporadic form of CJD, the initial misfolding of PrP may be a rare, stochastic event. In familial CJD, this event is triggered by certain mutations in the gene encoding PrP. When the disease is transmitted, PrPSc acts as a template and “seeds” the misfolding of the host PrP. Prion diseases are not curable, and the survival time is typically 4–12 mo after the onset of symptoms. Bovine spongiform encephalopathy (BSE), or mad cow disease, is transmissible to humans (2, 3). Although 224 individuals are known to have succumbed to “human BSE” (variant CJD; ref. 4), the number of infected but as yet asymptomatic people is unknown because there is no validated preclinical diagnostic test for prion diseases and the incubation time may extend to decades; this uncertainty is of particular concern because the disease is transmissible by blood transfusion (5).
Finding a prevention or cure for these lethal, ubiquitous diseases is urgent. Drugs from various molecular families (such as polyanionic, tetrapyrrolic or tricyclic compounds, polyene antibiotics, tetracyclins, β-sheet breaker peptides, Congo red, and others) have been found to impede prion replication, but none of them are of practical use because of efficacy, pharmacology, or toxicity issues (6–8). Screening of compound libraries for antiprion therapeutics has been implemented by using PrPSc-based assays (9–14). Because PrP is essential for prion propagation, we decided to develop a procedure allowing high-throughput screening for drugs capable of reducing PrP expression. Technically, this procedure implies an assay that does not require any manipulation other than addition of reagents to a well, in contrast to the conventional immunoassays that include washing steps to remove excess antibodies.
Mice devoid of PrP are resistant to prion diseases, suggesting that drug-mediated depletion of PrP may be of therapeutic value (15–17). Depletion of PrP by conditional knockout prevents onset of clinical disease in established neuroinvasive prion infection in a mouse model and reverses early pathology (18). Lentivirus-mediated administration of RNAi or PrP-specific single-chain variable fragment antibodies expressed by an adeno-associated viral vector prolonged the survival time of infected animals (19, 20). Moreover, transgenic mice (16, 17, 21, 22) and chimeric mice derived from lentivector-transduced embryonic stem cells (23) have shown that progression of disease and neuropathological alterations depend on PrP in a concentration-dependent manner. Thus, even incomplete PrP reduction can considerably mitigate clinical disease in prion-infected mice. However, PrP appears to be nonessential for the host, perhaps because of functional redundancy with other protein(s). The function of PrP is still unknown as evidenced by the plethora of seemingly unrelated proposals (24, 25). Mice, cattle, and goats harboring a knocked-out PrP gene develop normally and do not present behavioral abnormalities (26–29), although a role for PrP in myelination and neuroprotection in ischemic brain injury has been described (25, 30). Conditional PrP knockout during adulthood did not lead to deleterious consequences, showing that PrP dispensability does not require compensatory mechanisms taking place during development (22, 31). Collectively, available data indicate that reducing cellular PrP levels is an ideal therapeutic target.
PrP is a GPI-anchored protein of 254 amino acids attached to the cell surface in microdomains known as lipid rafts (32). During biogenesis, PrP is directed cotranslationally into the lumen of the ER, undergoes posttranslational modifications such as the addition of the GPI anchor and two N-glycosylations, and is transported and exposed at the cell surface. PrP is internalized and degraded in the endolysosomal pathway with a half-life of approximately 3–6 h (33). Removal of PrP from the cell surface by phosphatidylinositol-specific phospholipase C (PIPLC) treatment is sufficient to cure prion-infected cells (34).
Therefore, we aimed to screen for small molecules reducing cell surface PrP. Current cellular protein detection techniques such as direct or indirect immunofluorescence, or ELISA, require washing off unbound antibody. To develop a homogeneous assay obviating the need for washes and compatible with high-throughput screening (HTS) platforms, we adapted FRET to the quantification of a cellular protein. Two antibodies recognizing different epitopes of the protein of interest were added simultaneously. Double antibody occupancy generated a FRET signal proportional to the protein level, obviating the need to remove unbound antibodies. We called our HTS-ready assay the PrP–FRET-enabled high throughput assay (PrP-FEHTA). Moreover, this approach does not require prion-infected cells, thereby enabling screening of hundreds of thousands of compounds on ultra-HTS screening platforms. Using PrP-FEHTA, we performed a pilot screen by using a small collection of drugs approved for clinical use. This study led to the discovery of antiprion properties of the drugs tacrolimus and astemizole. Both inhibit prion replication in cell cultures. Moreover, astemizole, currently used to treat allergy, prolonged the survival time of prion-infected mice, thereby constituting a candidate for the treatment of human prion diseases.
Results
Screening and Hit Validation.
We adapted FRET to HTS quantification of a cell surface protein. To this purpose, instead of using two labeled antibodies directed against two different proteins, which is a traditional FRET setting allowing detection of protein–protein interactions, we used two antibodies recognizing distinct domains of PrP. The assay was called PrP-FEHTA. The best signal was obtained with anti-PrP antibodies SAF32 (amino acids 53–93) and D18 (amino acids 133–157) labeled with the donor and acceptor fluorophores, respectively (Fig. S1). Because free PrP molecules shed into the culture medium by neuroblastoma cells would be detected by PrP-FEHTA leading to background signal, we used LD9 cells that exhibit minimal, if any, PrP shedding. Assay development and optimization in the 96- and 384-well formats was performed by monitoring the Z′ factor, an indicator of assay robustness (defined in SI Materials and Methods) (35). The assay meets accepted performance criteria for HTS (Z′ = 0.7, coefficient of variation < 10%; Fig. S2). Brefeldine A, a compound that blocks progression of proteins from the ER to the Golgi apparatus, was chosen as a pharmacological control (Fig. S2).
We screened the US Drug Collection that comprises 1,280 drugs approved for use in humans. All compounds are included in the USP Dictionary (36). The screen yielded nine hits (threshold set at 50% PrP reduction and ≤10% toxicity in the counterscreen) (Fig. 1). Interestingly, one of them was tannic acid, independently found in a screen performed by others with scrapie-infected cells (9). We selected two hits, tacrolimus and astemizole, based on their activity in orthogonal assays (cell-surface immunofluorescence and high-content quantification in neuroblastoma cells) (Fig. 2).
Fig. 1.

Candidate hits from PrP-FEHTA screen and counterscreen. (A) PrP expression levels as a percentage of the DMSO control measured by PrP-FEHTA. DMSO control is shown for each plate (plates shown as separate graphs). Brefeldine A (BFA) control reduced PrP to background in all plates. Z′ was 0.7 for all four plates. (B) Cell viability measured by CellTiter-Glo (counterscreen). Cutoffs (50% for PrP expression, ≤10% toxicity) are indicated by dotted lines. Surprisingly and interestingly, some drugs from this FDA-approved collection exhibited toxicity in vitro at 20 µM.
Fig. 2.
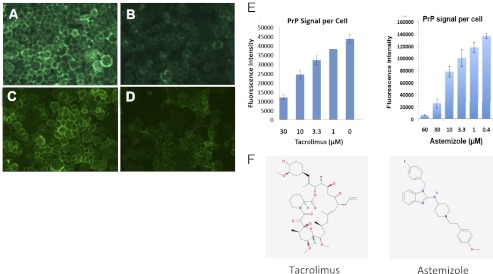
Hit validation by immunofluorescence on neuroblastoma cells and high-content analysis. N2a cells were treated with DMSO 0.5% (controls) or with the drug for 24 h. (A–D) PrP staining with D18 mAb on living cells, postfixation, and epifluorescence analysis. (A and C) Controls. (B) Tacrolimus, 20 µM. (D) Astemizole, 20 µM. (E) Quantitation of the dose–response to treatment by tacrolimus or astemizole using the high-content fluorescent imaging system InCell 1000. (F) Structures of tacrolimus and astemizole.
Tacrolimus and Astemizole Inhibit Prion Replication in Cell Culture.
Our working hypothesis was that drugs reducing cell-surface PrP levels by more than 50% would significantly inhibit prion replication. We tested this hypothesis by infecting drug-treated PK1 neuroblastoma cells by either Rocky Mountain Laboratories (RML) or 22L prions and testing them for PrPSc by Western blot 9 d after infection. Treatment was stopped 12 d after infection, and cells were tested again for PrPSc 6 d later. Tacrolimus at 20 µM (i.e., the screening concentration), but not 6.7 µM, strongly inhibited the replication of both RML and 22L prions. Astemizole blocked replication of both strains at 2 µM (Fig. 3). No rebound was observed by 6 d after ending the treatment (Fig. 3).
Fig. 3.

Tacrolimus and astemizole inhibit replication of RML and 22L prions in neuroblastoma cells without rebound after treatment cessation. PK1 neuroblastoma cells were pretreated for 3 d with the indicated doses of drugs and infected with RML (A) or 22L (B) prions. Treatment was continued for 12 d after infection (p.i.). Cells were analyzed for proteinase K-resistant PrP by Western blot 9 and 18 d p.i (i.e., for the latter 6 d after treatment cessation). Ast, astemizole; Ctrl, vehicle-treated cells; PPS, 16 µg/mL pentosan polysulfate; Tac, tacrolimus. Drug concentrations are indicated in micromolars.
Proposed Mechanism of Action of Tacrolimus and Astemizole.
We then set out to determine the mechanism of action of the drugs. These investigations were performed at the prion inhibitory doses (PID) rather than the screening dose. Indeed, the efficacy of astemizole at 2 µM, a dose that reduced cell surface PrP by ≤20% (Fig. 2), suggested that astemizole inhibits prion propagation by a mechanism other than cell surface PrP reduction. Analysis of cell surface versus intracellular PrP by cell surface biotinylation and Western blot showed that tacrolimus treatment reduced both membrane and intracellular PrP by >70% (Fig. 4A), whereas PrP mRNA levels were unaffected (Fig. 4B). These results suggested that tacrolimus inhibits PrP translation, although accelerated PrP degradation cannot be ruled out. Cell surface and intracellular levels of PrP as well as PrP mRNA were not significantly affected by astemizole treatment (Fig. 4 A and B). These data show that astemizole, at the PID, inhibited prion replication by a mechanism independent of PrP expression.
Fig. 4.

Effect of tacrolimus and astemizole on PrP transcription and translation. (A) Tacrolimus reduces both intracellular and cell membrane levels of PrPc, whereas astemizole has no effect on PrP levels at prion inhibitory doses (PID). PK1 cells were treated for 3 d with tacrolimus at 20 µM or astemizole at 2 µM, doses that prevented prion replication (Fig. 3). Cells were then cell surface biotinylated, and cell-surface and intracellular PrP were analyzed. Gel loading was standardized by cell number. The three blots represent three different exposure times of the same blot. (B) Tacrolimus and astemizole do not significantly alter PrP mRNA levels at PID. RT-PCR analysis of PrP mRNA levels in cells treated for 3 d with tacrolimus or astemizole at 20 and 2 µM, respectively. mRNA levels were standardized by using GAPDH mRNA.
Tacrolimus, also referred to as FK506, shares with rapamycin its intracellular target FKBP12 involved in calcineurin (CaN) and mammalian target of rapamycin (mTOR) signaling (37), which is key in autophagy induction. We therefore assessed whether tacrolimus induces autophagy in PK1 neuroblastoma cells. We used the conversion of the cytosolic protein LC3-I to the conjugated, autophagosome-bound form LC3-II as an indicator of autophagy induction. Astemizole was tested along with tacrolimus. Tacrolimus did not significantly modify the LC3-II/I ratio. Surprisingly, the ratio doubled after astemizole treatment, indicating autophagy induction (Fig. 5).
Fig. 5.

Astemizole activates autophagy. LC3II/I ratios are elevated in astemizole, but not tacrolimus-treated, cells. Cells were treated for 3 d with 20 and 2 µM of tacrolimus or astemizole, respectively. Forty or 20 µg of total proteins were loaded onto the gel.
Collectively, these data suggest that the antiprion effect of tacrolimus is linked to a nontranscriptional regulation of PrP steady-state levels, whereas astemizole acts by enhancing autophagic function and, thereby, prion clearance (38, 39).
Therapeutic Effect of Astemizole in Prion-Infected Mice.
Mice were intracerebrally infected with RML prions and treated from 20 to 50 d after infection (dpi) by i.p. injection of tacrolimus at 1.5 mg/kg or astemizole at 3 mg/kg. Intracerebral prion infection combined with a short i.p. drug application represents a stringent model allowing detection of only the most effective antiprion drugs. The Kaplan–Meier curve showed a clear increase in survival time of the astemizole-treated mice (Fig. 6; P = 0.06 in the log-rank test, hazard ratio of 4, 95% CI of ratio 1–21). No increase in survival times was observed in the tacrolimus-treated group.
Fig. 6.

Astemizole prolongs the survival time of RML prion-infected mice. Kaplan–Meier curve showing the percentages of survival of RML-inoculated mice treated from 20 to 50 d after inoculation with AST or not treated (Control). Median survival times were 149 ± 2 SEM and 155 ± 3 SEM for the control and treated group, respectively.
Discussion
We have developed a drug screening approach for prion diseases with ultra-HTS capability (i.e., throughput of more than 100,000 compounds per day, the standard for automated screening platforms). To achieve this goal, we devised a strategy that does not require the handling of infectious prions. Because even partial reduction of cellular PrP levels considerably impedes prion propagation, yet is safe for the host, we developed a FRET-based HTS assay, PrP-FEHTA, for the identification of drugs that diminish the level of cell-surface PrP.
Using a 384-well PrP-FEHTA format, we screened a small collection of 1,280 Food and Drug Administration (FDA)-approved drugs to establish proof of principle and, if possible, find antiprion activity for drugs established in the clinic. After counterscreening for toxicity and hit validation in a classical immunofluorescence assay, we selected two compounds, tacrolimus and astemizole, for testing of antiprion activity in vitro. Both compounds inhibited replication in cell culture of two different prion strains, RML and 22L, chosen for their different sensitivity to other pharmacological agents (40). Because antiprion activity in cultured cells does not necessarily translate into a beneficial effect in vivo, we then tested the drug’s antiprion activity in RML-infected mice. We used a 30-d treatment regimen, which is short relative to the total infection time of ∼150 d, to single out drugs with high antiprion activity in vivo. Tacrolimus showed no effect, perhaps because of its high PID of 20 µM, difficult to achieve in the brain, and/or to the short treatment duration. However, astemizole, with a PID of 2 µM, prolonged the survival of RML-infected mice. It is likely that this therapeutic effect could be improved by a longer treatment regimen and using a peripheral rather than the intracerebral route of infection.
Tacrolimus, also known as FK506, is an immunosuppressant widely used in organ transplant (41). It is also administered to ameliorate Myasthenia gravis (42). Additionally, it has been reported to slow down neurodegeneration in a murine model of prion disease (43). Tacrolimus, like rapamycin, is a canonical ligand of FK506 binding proteins (FKBPs), of which FKBP12 is the prototype. Although the rapamycin/FKBP12 complex inhibits mTOR, a controller of autophagy and cell growth, the FK506/FKBP12 complex interacts with the phosphatase CaN (37). We found that tacrolimus does not inhibit PrP transcription, yet reduces both intracellular and cell-surface PrP levels. Tacrolimus therefore regulates steady-state levels of PrP by a nontranscriptional mechanism. It remains to be determined whether tacrolimus impedes PrP translation or accelerates PrP degradation. However, it might be difficult to use tacrolimus as an antiprion drug in vivo, because of its high PID, narrow therapeutic index, and its reported neurotoxicity (44).
Conversely, we consider that astemizole may rapidly be made available to CJD patients given, on the one hand, the proof of principle of its antiprion activity in vivo and its excellent drug characteristics and, on the other hand, the rapidly fatal outcome of CJD. Astemizole is a second-generation selective histamine H1-receptor antagonist used in humans to treat benign seasonal allergic rhinitis in a chronic setting at doses as high as 18.6 mg⋅m−2 (45), corresponding to 6 mg/kg in mice, according to the assumptions and constants of Freireich et al. (46). It also possesses antifungal (47) and antimalarial activity (48) and can be used for in vivo brain imaging in Alzheimer’s disease because of its affinity for Tau fibrils and good brain penetration and persistence (49). Following the launch of next generation antihistamine H1-receptor drugs, astemizole was withdrawn voluntarily by the manufacturer in 1999 and 2003, in the United States and Europe, respectively, but generic astemizole is sold in more than 30 countries (48, 50). Astemizole underwent all pre- and postmarketing studies and has therefore an extremely well known safety and drug interaction profile. Moreover, astemizole has been an over-the-counter drug approved for pediatric and adult use for more than a decade and used for self-medication of hay fever over extended time periods (51). It has been removed from the over-the-counter market because of extremely rare occurrences of cardiac arrhythmias when overdosed (52, 53). Overall, given the suitability of astemizole for chronic administration and the extensive knowledge regarding its safe use, it seems particularly suited for prolonged preventive use, albeit under strict medical surveillance, in individuals carrying highly penetrant mutations of the PrP gene associated with familial forms of CJD, fatal familial insomnia, or Gerstmann–Sträussler–Scheinker syndrome.
Finally, besides astemizole’s prion inhibitory effect, we describe a hitherto unreported effect of the drug on stimulation of autophagy. Autophagy is involved in several protein misfolding neurodegenerative diseases (PMNDs) (54), and recent studies showed that enhanced autophagy counteracts cellular prion infection (38, 39). It is therefore likely that the antiprion activity of astemizole is linked to its activity on autophagy, and future studies aimed at determining the exact effect of astemizole on the autophagic pathway may reveal new therapeutic targets for prion diseases and other PMNDs.
In short, our study proposes a candidate for the treatment of prion diseases and establishes FEHTA as a unique drug discovery platform for prion and other diseases where modulation of the level of a cellular protein is beneficial. Moreover, because there is a plethora of biochemical steps whose inhibition could lead to depletion of cell surface PrP, PrP-FEHTA has the potential to identify various classes of active compounds that could be valuable probes to study PrP biosynthetic and intracellular trafficking pathways.
Last but not least, abrogation of cell surface PrP expression is potentially an innovative approach to treat Alzheimer’s disease (AD). AD is hallmarked by the brain deposition of plaques constituted mainly of hyperphosphorylated tau protein and amyloid β (Aβ). Inasmuch as PrP is a cell surface receptor for Aβ peptide oligomers and mediates their synaptotoxic effects (55), neuronal death (56), and memory impairment in transgenic Alzheimer’s mice (57), molecules inhibiting PrP expression at the neuronal surface may exhibit neuroprotective properties in AD.
Materials and Methods
SI Materials and Methods contains a detailed description of PrP-FEHTA, TR-FRET data analysis, assay performance analysis, cell viability measurements, immunocytochemistry, and high-content analysis. It also describes the methods used to analyze the compounds mode of action such as biotinylation of cell surface proteins and Western blot analysis to quantify cell surface and intracellular PrP, and RT-PCR. Details are given about animal treatments, cell treatments, cell culture, and statistical analysis.
Supplementary Material
Acknowledgments
We thank Nicole Salès for help with immunocytochemistry, Franck Madoux for advice on FRET, Patricia McDonald for support with high-content immunofluorescence analysis, and Shannon Sunday for help with animal experiments. We thank the Alafi Foundation for its generous financial support. This work was supported by the Scripps Research Institute, the Alafi Foundation, and National Institutes of Health Grant MH084512 (to P.H., P.C., and T.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303510110/-/DCSupplemental.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Will RG, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347(9006):921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 3.Lasmézas CI, et al. BSE transmission to macaques. Nature. 1996;381(6585):743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 4. World Health Organization (2012) Variant Creutzfeldt-Jakob disease. Available at http://www.who.int/mediacentre/factsheets/fs180/en/. Accessed March 14, 2013.
- 5.Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: Results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 2006;91(3):221–230. doi: 10.1111/j.1423-0410.2006.00833.x. [DOI] [PubMed] [Google Scholar]
- 6.Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129(Pt 9):2241–2265. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 7.Weissmann C, Aguzzi A. Approaches to therapy of prion diseases. Annu Rev Med. 2005;56:321–344. doi: 10.1146/annurev.med.56.062404.172936. [DOI] [PubMed] [Google Scholar]
- 8.Brown P. An historical perspective on efforts to treat transmissible spongiform encephalopathy. CNS Neurol Disord Drug Targets. 2009;8(5):316–322. doi: 10.2174/187152709789541989. [DOI] [PubMed] [Google Scholar]
- 9.Kocisko DA, et al. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77(19):10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertsch U, et al. Systematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets. J Virol. 2005;79(12):7785–7791. doi: 10.1128/JVI.79.12.7785-7791.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heal W, et al. Library synthesis and screening: 2,4-diphenylthiazoles and 2,4-diphenyloxazoles as potential novel prion disease therapeutics. J Med Chem. 2007;50(6):1347–1353. doi: 10.1021/jm0612719. [DOI] [PubMed] [Google Scholar]
- 12.Kimata A, et al. New series of antiprion compounds: Pyrazolone derivatives have the potent activity of inhibiting protease-resistant prion protein accumulation. J Med Chem. 2007;50(21):5053–5056. doi: 10.1021/jm070688r. [DOI] [PubMed] [Google Scholar]
- 13.Ghaemmaghami S, May BC, Renslo AR, Prusiner SB. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2010;84(7):3408–3412. doi: 10.1128/JVI.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geissen M, et al. From high-throughput cell culture screening to mouse model: identification of new inhibitor classes against prion disease. ChemMedChem. 2011;6(10):1928–1937. doi: 10.1002/cmdc.201100119. [DOI] [PubMed] [Google Scholar]
- 15.Büeler H, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 16.Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration. 1994;3(4):331–340. [PubMed] [Google Scholar]
- 17.Sakaguchi S, et al. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J Virol. 1995;69(12):7586–7592. doi: 10.1128/jvi.69.12.7586-7592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallucci G, et al. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 19.White MD, et al. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci USA. 2008;105(29):10238–10243. doi: 10.1073/pnas.0802759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wuertzer CA, Sullivan MA, Qiu X, Federoff HJ. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol Ther. 2008;16(3):481–486. doi: 10.1038/sj.mt.6300387. [DOI] [PubMed] [Google Scholar]
- 21.Büeler H, et al. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med. 1994;1(1):19–30. [PMC free article] [PubMed] [Google Scholar]
- 22.Tremblay P, et al. Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc Natl Acad Sci USA. 1998;95(21):12580–12585. doi: 10.1073/pnas.95.21.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfeifer A, et al. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest. 2006;116(12):3204–3210. doi: 10.1172/JCI29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasmézas CI. Putative functions of PrP(C) Br Med Bull. 2003;66:61–70. doi: 10.1093/bmb/66.1.61. [DOI] [PubMed] [Google Scholar]
- 25.Béland M, Roucou X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem. 2012;120(6):853–868. doi: 10.1111/j.1471-4159.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- 26.Büeler H, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356(6370):577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 27.Manson JC, et al. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8(2-3):121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 28.Richt JA, et al. Production of cattle lacking prion protein. Nat Biotechnol. 2007;25(1):132–138. doi: 10.1038/nbt1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu G, et al. Generation of goats lacking prion protein. Mol Reprod Dev. 2009;76(1):3. doi: 10.1002/mrd.20960. [DOI] [PubMed] [Google Scholar]
- 30.Bremer J, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13(3):310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 31.Mallucci GR, et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53(3):325–335. doi: 10.1016/j.neuron.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Vey M, et al. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA. 1996;93(25):14945–14949. doi: 10.1073/pnas.93.25.14945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caughey B, Race RE, Ernst D, Buchmeier MJ, Chesebro B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol. 1989;63(1):175–181. doi: 10.1128/jvi.63.1.175-181.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA. 2001;98(16):9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 36. US Pharmacopeia l Convention (2005) USP Dictionary.
- 37.Romano S, et al. FK506 binding proteins as targets in anticancer therapy. Anticancer Agents Med Chem. 2010;10(9):651–656. doi: 10.2174/187152010794479816. [DOI] [PubMed] [Google Scholar]
- 38.Aguib Y, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5(3):361–369. doi: 10.4161/auto.5.3.7662. [DOI] [PubMed] [Google Scholar]
- 39.Heiseke A, Aguib Y, Riemer C, Baier M, Schätzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109(1):25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- 40.Oelschlegel AM, Fallahi M, Ortiz-Umpierre S, Weissmann C. The extended cell panel assay characterizes the relationship of prion strains RML, 79A, and 139A and reveals conversion of 139A to 79A-like prions in cell culture. J Virol. 2012;86(9):5297–5303. doi: 10.1128/JVI.00181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krämer BK, et al. Tacrolimus-based, steroid-free regimens in renal transplantation: 3-year follow-up of the ATLAS trial. Transplantation. 2012;94(5):492–498. doi: 10.1097/TP.0b013e31825c1d6c. [DOI] [PubMed] [Google Scholar]
- 42. Imai, T, et al. (2012) Early effect of tacrolimus in improving excitation-contraction coupling in myasthenia gravis. Clin Neurophysiol 123(9):1886–1890. [DOI] [PubMed]
- 43.Mukherjee A, et al. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 2010;6(10):e1001138. doi: 10.1371/journal.ppat.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venkataramanan R, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29(6):404–430. doi: 10.2165/00003088-199529060-00003. [DOI] [PubMed] [Google Scholar]
- 45.Richards DM, Brogden RN, Heel RC, Speight TM, Avery GS. Astemizole. A review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1984;28(1):38–61. doi: 10.2165/00003495-198428010-00003. [DOI] [PubMed] [Google Scholar]
- 46.Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50(4):219–244. [PubMed] [Google Scholar]
- 47. Vu K, Gelli A (2010) Astemizole and an analogue promote fungicidal activity of fluconazole against Cryptococcus neoformans var. grubii and Cryptococcus gattii. Med Mycol 48(2):255–262. [DOI] [PubMed]
- 48.Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2(8):415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 49.Rojo LE, Alzate-Morales J, Saavedra IN, Davies P, Maccioni RB. Selective interaction of lansoprazole and astemizole with tau polymers: Potential new clinical use in diagnosis of Alzheimer’s disease. J Alzheimers Dis. 2010;19(2):573–589. doi: 10.3233/JAD-2010-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. World Health Organization (1999) Astemizole: Voluntary Withdrawal: Janssen, USA. WHO Pharmaceuticals Newsletter Nos. 7 and 8. Available at http://apps.who.int/medicinedocs/fr/d/Js2269e/5.html. Accessed March 14, 2013.
- 51.Janssens MM. Astemizole. A nonsedating antihistamine with fast and sustained activity. Clin Rev Allergy. 1993;11(1):35–63. doi: 10.1007/BF02802293. [DOI] [PubMed] [Google Scholar]
- 52. World Health Organization (1999) Astemizole: OTC Formulation Withdrawn: UK. WHO Pharmaceuticals Newsletter Nos. 1 and 2. Available at http://apps.who.int/medicinedocs/en/d/Js2268e/6.3.html. Accessed March 14, 2013.
- 53.Lindquist M, Edwards IR. Risks of non-sedating antihistamines. Lancet. 1997;349(9061):1322. doi: 10.1016/S0140-6736(97)26018-6. [DOI] [PubMed] [Google Scholar]
- 54.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007;6(4):352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 55.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457(7233):1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kudo W, et al. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet. 2012;21(5):1138–1144. doi: 10.1093/hmg/ddr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gimbel DA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30(18):6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.