

Abstract
The development of new therapeutics for the treatment of neurodegenerative pathophysiologies currently stands at a crossroads. This presents an opportunity to transition future drug discovery efforts to target disease modification, an area in which much still remains unknown. In this Perspective we examine recent progress in the areas of neurodegenerative drug discovery, focusing on some of the most common targets and mechanisms; N-methyl-d-aspartic acid (NMDA) receptors, voltage gated calcium channels (VGCCs), neuronal nitric oxide synthase (nNOS), oxidative stress from reactive oxygen species, and protein aggregation. These represent the key players identified in neurodegeneration and are part of a complex, intertwined signaling cascade. The synergistic delivery of two or more compounds directed against these targets, along with the design of small molecules with multiple modes of action should be explored in pursuit of more effective clinical treatments for neurodegenerative diseases.
1. Introduction
Neurodegenerative disease is an encompassing term for a set of over 600 diseases in which the nervous system progressively and irreversibly deteriorates. The single greatest risk factor for the development of neurodegenerative disease is advancing age, with associated mitochondrial DNA mutation and oxidative stress damage,1 illustrated by the fact that the majority of these diseases are late-onset. Alzheimer’s Disease (AD), the most prevalent of the neurodegenerative diseases, affects approximately 15 million people worldwide. Estimates expect the number of sufferers to triple in the USA2 and Europe3 by 2050, figures that are repeated for most other forms of neurodegeneration, including Parkinson’s disease (PD) and Huntington’s disease (HD).
The most common forms of neurodegenerative diseases are AD, PD, HD, and amyotrophic lateral sclerosis (ALS), (or in Europe, motor neurone disease, MND).4 Specific aspects of these diseases, including protein aggregation, genetic mutations, and pathophysiology, are discussed in this Perspective. Extensive literature exists detailing each of the pathways highlighted, as well as for the many other neurodegenerative diseases. The interested reader is directed to those excellent reviews and references cited within this manuscript.
A commonality of the neurodegenerative diseases is that they are not diseases attributed to a single gene or even multiple genes; they are far more complicated, involving a myriad of known and unknown signaling cascades, misfolded proteins, protofibril formation, ubiquitin-proteasome dysfunction, oxidative and nitrosative stress, mitrochondrial injury, and many more events.5 Our current understanding of neuroscience has enabled the identification of some key genes involved, but since many cases are seemingly sporadic, we know that mechanisms of neurodegeneration are more than just genetic.6 While they vary in symptoms, neurodegenerative diseases have many pathologic overlaps (Figure 1). In this Perspective, we focus on the major components of this shared pathway: N-methyl-D-aspartic acid (NMDA) receptors, voltage gated calcium channels (VGCCs), neuronal nitric oxide synthase (nNOS), oxidative stress from reactive oxygen species (ROS), and protein aggregation.
Figure 1.

Common targets and mechanisms associated with many neurodegenerative diseases that are highlighted in this Perspective.
NMDA receptors are ligand-gated, voltage-dependent ion channels that respond to the neurotransmitters glutamate and NMDA. NMDA receptor activation leads to Ca2+ influx into post-synaptic cells, which continues signal potentiation by activating signaling cascades. Over-stimulation of NMDA receptors has been implicated in neurodegeneration, in addition to other disease states, and therefore NMDA receptors have been greatly investigated as a drug target (Section 2). Ca2+ influx is also regulated by VGCCs. As discussed in Section 3, one specific channel, Cav1.3, has been identified to play a role in the progression of PD and therefore selective antagonism of Cav1.3 is hypothesized to be potentially neuroprotective in early or presymptomatic stages of PD. Among many other responses, increased intracellular Ca2+ concentration activates nNOS. Overactivation of nNOS produces high intracellular levels of nitrite and superoxide, which react to form ROS including peroxynitrite. This oxidative stress within the cell causes damage to DNA, lipids, and causes protein modifications. Overactivation of nNOS has also been implicated in neurodegeneration and therefore inhibitors of nNOS are sought-after as potential therapeutics (Section 4). Additionally antioxidant therapeutics are designed to directly prevent cellular damage from oxidative species (Section 5). Lastly, protein aggregation is a hallmark of AD and PD, and while it may result from modifications by ROS, other mechanisms, such as mutations, are thought to be involved in protein aggregates observed in AD and PD. Progress towards developing anti-aggregation therapeutics is examined in Section 6.
A separate, but crucially important, pathophysiological pathway that contributes to neurodegeneration is inflammation and immune response.7, 8 Many of the most relevant immunological molecules are produced within the brain, and this leads to the observation that systemic immune responses affect brain function and contribute to neurodegeneration.9 This pathway of inflammation and immune response is intricately connected with several, if not all, of the individual targets discussed in this Perspective. Space does not permit an all-encompassing review of this subject, and the reader is directed to the excellent review articles referenced herein. Expanding research continues to link this key pathophysiological pathway to the individual targets discussed in this Perspective as briefly highlighted below.
Highly regulated by the immune system, the kynurenine pathway is an enzymatic cascade that converts tryptophan to serotonin but also to several other neuroactive compounds. Misregulation of this pathway results in increased or decreased production of these metabolites and contributes to a neurodegenerative effect.10 Kynurenic acid is a competitive antagonist for the NMDA, kainite, and AMPA receptors and forms the structural scaffold for drug discovery efforts mentioned in Section 2. Independent of the antagonistic effect of kynurenic acid is the observation that it is an effective free radical scavenger and can therefore display antioxidant properties similar to compounds discussed in Section 5.11
Further connections from inflammation and immune response to antioxidants and nNOS inhibitors exist via a family of proteins known as toll-like receptors (TLRs). Evidence is increasing for the role of TLRs in sterile inflammation observed in neurodegenerative diseases, such as PD and AD.12 These receptors play an important role in innate immunity because they recognize conserved motifs found in microorganisms. They are expressed in large numbers in various cells within the central nervous system (CNS) and serve to activate microglia. Microglia, the immune cells of the brain, are activated by distress signals from nearby cells. A side effect of this activation is the release of toxic factors, such as nitric oxide (Section 4) and ROS (Section 5), resulting in increased neuronal damage. TLRs are also being explored as targets for the treatment of neurodegenerative diseases.13 Furthermore, polyphenols, discussed in Section 5, acting predominantly as antioxidants, also have activity crossover to the inflammation and inflammatory response; these compounds are known to modulate neuroinflammation by inhibiting the expression of inflammatory genes and the level of intracellular antioxidants.14 Neuroinflammation commonly occurs as a result of oxidative and excitotoxic damage to neurons, and because of mitochondrial dysfunction and is linked to protein aggregation.15 It is therefore envisioned that drugs that combat neuroinflammation might also combat neurodegenerative disease progression.
However, while anti-inflammatory agents may be required to treat neurodegenerative diseases, they may not be sufficient on their own but may be effective as part of a combination therapy.16 Such a combination might include targeting pro-inflammatory factors, such as tumor necrosis factor alpha and Fas ligand. One compound, revlimid, has been reported to modestly reduce these proinflammatory cytokines and show some neuroprotection in an ALS mouse model.17 Use of revlimid in a combinatorial approach with selected antiaggregation agents discussed in Section 6 or potentially any of the agents in this Perspective may increase the neuroprotective effects from ‘modest’ results with a single therapeutic compound to ‘significant’ results with a combination of two or more compounds.
The thesis that AD, PD, HD, and ALS, although distinct disorders, share a common mechanism that progresses neuron death along a ‘neurodegenerative spectrum’ has been advanced by numerous researchers.18, 19
Recent advances have increased our understanding of the processes underlying neurodegeneration.20-23 Drug candidates that target single or dual processes have been advanced to the clinic, but they only provide relief of symptoms, not of the underlying causes (which still remain unknown). Modulation of multiple targets along the same biological pathway could potentially lead to disease modification rather than just control of symptoms. The widely accepted definition of a disease modifying drug is ‘an agent that alters the underlying pathophysiology of the disease in question and demonstrates meaningful reduction in the rate of decline or progression to a defined milestone.’24 In a complex pathway with many drug targets, such as those contributing to neurodegenerative diseases, a drug with a single-target mechanism of action cannot always correct that pathway. The synergistic delivery of a ‘cocktail’ of two or more drugs may deliver enhanced potency in the treatment of neurodegenerative diseases by potentially leading to disease modification.
Polypharmacy, the synergistic combination of two or more drugs acting on different targets, has been successful in treating other diseases, such as hyperlipidemia (high blood cholesterol). The combination of simvastatin (a 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitor) and ezetimibe (an inhibitor of dietary cholesterol uptake) is marketed as Vytorin®.25 This combination therapy works by preventing the body from making its own cholesterol while also inhibiting the absorption of cholesterol from dietary intake. A drug that inhibits only one source of cholesterol is less effective at lowering overall levels, as cholesterol is still produced by the second source. This disease-modifying therapy reduces overall cholesterol levels by targeting both sources. Other disease-modifying synergistic drug combinations have been utilized for the treatment of cancer26, hepatitis C virus,27 and HIV/AIDS.28 supporting the potential strategy of treating other complex diseases in a similar manner.
Alternatively, a single therapeutic agent could be designed to interact at two different targets. An example of a dual-acting drug that is disease-modifying is duloxetine (Cymbalta®), used in the treatment of depression. This compound inhibits both serotonin and norepinephrine reuptake in the CNS.29 Inhibition of the reuptake of both neurotransmitters increases overall levels of the two compounds, known to play an important role in mood. A deficit in either neurotransmitter can cause depression; therefore, increasing levels of both provides a disease-modifying therapy to counteract this deficit.30 An example relevant to the disease pathway outlined in Figure 1 is the polycyclic caged amine compound NGP1-01 (18) (Section 2), which blocks both the NMDA receptor and L-type calcium channel with resulting neuroprotective effects. Blocking only one of these targets allows the continued influx of Ca2+ ions from the other. As excess Ca2+ ion concentration is implicated in excitotoxicity, this would be analogous to trying to stop a bucket leaking from two holes by only stoppering one. Preventing an increase in overall Ca2+ ion concentration, and thereby excitotoxicity, could be considered disease-modifying over simply reducing the concentration.
Additionally, a multi-faceted strategy would further probe the pathophysiological pathway(s) of neurodegeneration and confirm the intricate connectivity that has been proposed between therapeutic targets. Therapeutics developed for one type of neurodegenerative disease, when delivered in combination, may transfer their therapeutic potential to a different disease state, thereby exploiting and expanding the neurodegenerative therapeutic arsenal. Such combination therapy would also have advantages in drug delivery and dosing. Delivery of a significant dose of an inhibitor of Cav1.3-type calcium channels is hindered by concerns of selectivity over other Cav channels and resultant off-target toxicity. Dosing a lower concentration of such an inhibitor in conjunction with a modulator of another target on the pathophysiological pathway would allow for the inhibition of two targets that alone may provide no therapeutic response. This approach would provide the opportunity to use drugs that are active but only have low efficacy because of selectivity problems. As a result, the potential to tune inhibitory responses rather than outright blockage would allow for the use of small molecules previously discarded as non-efficacious.
This Perspective examines the state-of-the-art small molecule therapeutics available for each of the neurodegenerative disease targets depicted in Figure 1. A combination of these molecules or the design of new compounds bearing active moieties that target two or more of the pathophysiological hallmarks of neurodegenerative diseases might be expected to bring about a new era in neuroscience drug discovery while efforts continue toward further elucidation of the underlying causes of neurodegeneration.
2. Alteration of NMDA Receptor Function
NMDA receptors, named after their selective agonist, N–methyl–d–aspartate, are ionotropic receptors that mediate glutamatergic neurotransmission. These receptors, as well as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, kainate cation channel receptors, and metabotropic receptors, respond to glutamate, the major excitatory transmitter in the brain.31, 32 The NMDA receptors have long been studied as potential therapeutic targets because of the numerous CNS functions in which they have been implicated, both in normal physiological function and in disease states.32, 33 However, the numerous roles they play in normal neurological function have led to disappointing clinical outcomes in the development of new drugs as a result of adverse side effects.32, 34 Another complicating factor for the lack of successful therapeutic intervention through modification of NMDA receptor function lies in the permeability of Ca2+ through the channel; excessive ion influx results in excitotoxicity that can lead to neuronal cell death.35, 36
The NMDA receptor has a relatively complex tetrameric subunit organization, and the subunit combination varies in different regions of the brain. This subunit heterogeneity presents additional challenges in drug design since each subunit has distinct functional and pharmacological properties. Seven NMDA receptor subunits have been identified: a GluN1 subunit, four different GluN2 subunits (GluN2 A-D), and two GluN3 subunits (GluN3A,B). The intact receptor consists of two GluN1 subunits and two GluN2 subunits. Glutamate binds to the GluN2 subunit, but for the receptor to be functional, glycine must simultaneously bind to the GluN1 subunit as a co-agonist.37 However, studies involving the CA1 region of rat hippocampus tissue slices have shown that synaptic NMDA receptors, which are important in long-term potentiation and NMDA-induced neurotoxicity, utilize d-serine as co-agonist, whereas extrasynaptic NMDA receptors have a preferential affinity for glycine.38 The intracellular portion of the transmembrane domains connect to a post-synaptic signaling complex known as the postsynaptic density, which contains PSD-95, enzymes such as nNOS or kinases, and other signaling and scaffolding proteins (Figure 2). 39, 40 Excitotoxicity, including the excessive stimulation of NMDA receptors, has long been hypothesized to play a role in the etiology of neurodegenerative disorders, such as PD41 and HD,42, 43 and may contribute, at least in part, to neuronal loss in AD44 and other dementias, ALS,36, 45-47 and possibly multiple sclerosis and prion disease.48
Figure 2.

NMDA receptor showing agonist and antagonist binding site domains
Earlier work on NMDA receptor antagonists has focused on designing compounds targeting four regions of the receptor: the glutamate and the glycine agonist binding domain (ABD) sites, the channel pore, and the N-terminal domain (NTD) region between GluN1 and GluN-B.49 Among the earliest glutamate competitive antagonists to be developed was phosphonic acid (R)-AP5 (1), which showed strong selectivity for NMDA receptors over kainate and AMPA receptors.50, 51 The introduction of rigidity with a piperazine ring provided another phosphonic glutamate site antagonist (2), which relieved parkinsonian symptoms and improved locomotion in animal models of PD when co-administered with l-DOPA, although it was not effective when given alone.52 A drawback of the phosphonic acid class of antagonists is that their polarity makes passage
 through the blood-brain barrier (BBB) and intestinal membranes difficult.53 Another problem with these competitive antagonists is that they generally demonstrate poor subunit selectivity.31 The differences in potency between NMDA and glutamate between the different GluN2 subtypes is less than 4-fold, and an analysis of key residues in the binding pocket shows the presence of a highly conserved binding site.54 Glycine site competitive antagonist 7-chlorokynurenate (3) did prevent NMDA-induced striatal lesions in an animal model as well as in vitro, but the compound targeted kainite receptors as well as NMDA receptors. In addition, the protection afforded by the compound could not be surmounted by co-administration of glycine, but instead by increasing the dose of NMDA, calling into question its actual mechanism of action.55 Since glycine acts at all NMDA subtypes, there may be an issue of tolerability of compounds targeting the glycine binding site, in addition to the higher doses of either glutamate or glycine antagonists that would be needed to surmount the competition with the endogenous ligands for their respective binding sites.39 However, since glycine antagonists act at the GluN1 subunit, they may be associated with fewer side effects, although in the case of 7-chlorokynurenate (3), there has been the added issue of poor BBB penetration.53, 54 A series of 3-amino-1-hydroxypyrrolidin-2-one (HA-966, 4) analogues were synthesized to overcome the limitations of poor solubility and BBB penetration.53
through the blood-brain barrier (BBB) and intestinal membranes difficult.53 Another problem with these competitive antagonists is that they generally demonstrate poor subunit selectivity.31 The differences in potency between NMDA and glutamate between the different GluN2 subtypes is less than 4-fold, and an analysis of key residues in the binding pocket shows the presence of a highly conserved binding site.54 Glycine site competitive antagonist 7-chlorokynurenate (3) did prevent NMDA-induced striatal lesions in an animal model as well as in vitro, but the compound targeted kainite receptors as well as NMDA receptors. In addition, the protection afforded by the compound could not be surmounted by co-administration of glycine, but instead by increasing the dose of NMDA, calling into question its actual mechanism of action.55 Since glycine acts at all NMDA subtypes, there may be an issue of tolerability of compounds targeting the glycine binding site, in addition to the higher doses of either glutamate or glycine antagonists that would be needed to surmount the competition with the endogenous ligands for their respective binding sites.39 However, since glycine antagonists act at the GluN1 subunit, they may be associated with fewer side effects, although in the case of 7-chlorokynurenate (3), there has been the added issue of poor BBB penetration.53, 54 A series of 3-amino-1-hydroxypyrrolidin-2-one (HA-966, 4) analogues were synthesized to overcome the limitations of poor solubility and BBB penetration.53

NMDA channel pore blockers, in particular amantadine (5) and its derivative memantine (6), have been developed for therapeutic use in inhibiting excitotoxicity. One of the earlier NMDA pore blockers to be advanced was dizocilpine (7), a high-affinity, uncompetitive antagonist.56 Co-administration of dizocilpine with l-DOPA completely prevented the progressive reduction in the duration of the l-DOPA response occurring with chronic l-DOPA therapy in parkinsonian rats.57 However, use of dizocilpine has been associated with severe side effects, such as coma,58 and in one study, actually exacerbated the symptoms of MPTP-induced parkinsonism when administered to a monkey.59

Amantadine (5) is an FDA approved drug that has been widely used in the treatment of PD. The predominant inhibitory mechanism of action of amantadine results from increasing the rate of channel closed states when the drug is bound in the channel of NMDA receptors.60 In a retrospective study of PD patients attending a single clinic, improved survival was associated with amantadine use.61 Amantadine, as well as the NMDA channel pore blocker dextromethorphan (8), also improved l-DOPA-associated motor response complications when given as an adjuvant to l-DOPA therapy.62, 63 However, a systematic Cochrane review of randomized controlled trials for amantadine concluded that because of a lack of evidence, it was not possible to determine whether amantadine is a safe and effective treatment for l-DOPA-induced dyskinesias in PD patients. The report also noted that in one study, 8 out of 18 participants had side effects, including confusion and worsening of hallucinations.64 In an unrelated study, NMDA channel blocker remacemide (9), as adjunct therapy with l-DOPA, was not found to significantly improve motor fluctuation symptoms, although the compound was found to be safe and tolerable.65

Memantine (6), another NMDA antagonist, is classified as an uncompetitive, open-channel blocker. The drug was patented by Eli Lilly & Co. in 1968 and was marketed by the German pharmaceutical company Merz to treat PD. The drug has a relatively fast off-rate from channel binding, which contributes to the drug’s low affinity for the channel pore, and, as a result, its clinical efficacy and tolerability. As a result, after several clinical trials, memantine was approved by the European Union in 2002 and the FDA in 2003 for the treatment of moderate to severe Alzheimer’s disease.66 One analysis concluded that the safety profile of memantine makes it particularly suitable for its use in elderly patients, since it is associated with a low overall rate of adverse events and a low potential for drug-drug interactions (because of a low extent of metabolism and protein binding, particularly to cytochrome P450 enzymes).67 A recent review adds that for the treatment of moderate to severe AD, memantine should be offered as either a monotherapy or in conjunction with an acetylcholinesterase inhibitor, but that the use of memantine as a first-line therapy for mild to moderate AD is not supported by current data. Further, insufficient data exist to make a recommendation for its use in PD dementia.68 A paradoxical feature of the action of memantine is that a higher concentration of memantine is needed to alleviate mild dementia than to counteract the damage associated with moderate-to-severe dementia. This apparent incongruity has been explained by the on-rate for channel blockage by memantine being increased by increasing the drug’s concentration, which leads to a greater proportion of channels being blocked.66 To increase the neuroprotective efficacy of memantine, a second-generation derivative has been designed to incorporate a nitric oxide moiety, which should bind to the sulfhydryl group of a cysteine residue in the channel pore that has been found to down-regulate NMDA receptor activity.58, 69
Much work remains for the design of therapeutic agents that can modulate NMDA receptors without adversely affecting normal cellular processes regulated by these channels. One more recent approach toward accomplishing this goal would be to design ligands that are subunit-specific and serve as noncompetitive antagonists.31 The first subunit-selective NMDA receptor antagonist was ifenprodil (10), which inhibits Glu2B-containing receptors with a 200 to 400-fold selectivity over receptors containing Glu2A, Glu2C, and Glu2D subunits, and an IC50 of 0.34 μM for the GluN1/Glu2B receptor.31, 70 The difficulty in designing noncompetitive antagonists for the Glu2B subunit, which may be therapeutically relevant for a number of disorders, is that they also act at other receptors and channels, leading to side effects, causing a sub-efficacious lowering of the dose.71, 72 To improve selectivity, ifenprodil analogue 1173 and traxoprodil (CP-101,606, 12)74 were designed. Analogue 11 was found to be more effective than 10 in preventing toxicity to cortical neurons that mimic ischemic brain damage when exposed to glutamate (IC50 of 0.4 versus 3.5 μM, respectively).73 Compounds 10-12 have antiparkinsonian activity in animal models. In a study of the use of 12 in counteracting dyskinesia and parkinsonism, it was found to reduce the maximum severity of l-DOPA-induced dyskinesia by approximately 30%, although many of the subjects in the study experienced dissociation, abnormal thinking, and amnesia. Compound 12 did not reduce parkinsonism in the study; however, the antidyskinetic effects were maximal at a lower dose, and the adverse effects were found to be dose-responsive.75

Among the recently identified voltage-independent negative allosteric modulators of NMDA receptors is 13, one of the quinazoline-4-one derivatives advanced by Traynelis and co-workers.76 Compound 13 had approximately 50-fold selectivity for recombinant GluN2C/D-containing receptors over GluN2A/B-containing receptors, low micromolar potency, and a novel mechanism that requires binding of glutamate to the GluN2 subunit, but not glycine binding to the GluN1 subunit. The GluN2C/D over GluNA/B subunit selectivity of 13 was determined from ABD residues adjacent to the transmembrane helices.77 Differing subunit selectivity profiles for negative allosteric modulation were also seen in a recent series of naphthalene and phenanthrene derivatives.78 9-Iodophenanthrene-3-carboxylic acid (UBP512, 14) inhibited only GluN1/GluN2C and GluN1/GluN2D receptors, while 6-bromocoumarin-3-carboxylic acid (UBP608, 15) inhibited GluN1/GluN2A receptors (23-fold) over GluN1/GluN2D, with IC50 values in the micromolar range. Consistent with their characterization as allosteric modulators, these compounds were found to be voltage-independent and not competitive with glutamate and glycine agonists. The site of action for 14 and related compounds is thought to be at the dimer interface between the ABDs, and the NTD was not found to be necessary for inhibitory activity.78 Another negative allosteric antagonist, dihydropyrazoloquinoline 16, inhibited GluN2C- and GluN2D-containing NMDA receptors over recombinant GluN2A- and GluN2B-containing receptors, with at least 50-fold difference in IC50 values. The selectivity was attributed to residues in the membrane-proximal lower lobe of the GluN2 ABD.79

Bettini and co-workers reported several sulfonamide derivatives from high-throughput screening that were selective antagonists at GluN1/GluN2A over GluN1/GluN2B.80 Addition of 1 mM glycine, but not 1 mM L-glutamate, overcame the inhibitory effects of the two most promising compounds. It was subsequently determined that one of the compounds, 17, binds to a novel allosteric site at the dimer interface between the GluN1 and GluN2 ABDs, thereby reducing glycine potency (IC50 = 0.1 – 0.32 μM).

To exploit the subunit selectivity of these newer pharmacological tools for therapeutic advantage, it will be necessary to map the relative contribution of each subunit to its function in both normal cellular processes as well as in disease states. Recently, it was found that the NMDA receptor subtype specificity of three crucial channel properties – Mg2+ blockage, relative Ca2+ permeability, and single-channel conductance – were all determined primarily at a single GluN2 subunit residue in the transmembrane region.81 However, because of the challenges involved in obtaining highly selective antagonists, there is still relatively little known about the function and therapeutic potential of the different subunits of the NMDA receptors.49 One confounding issue in this regard is the subunit composition of the NMDA receptor, since two different GluN2 subunits may be present, rather than only the binary combination of GluN1 and one type of GluN2 (or GluN3) subunit, in different regions of the brain.82 Using recombinant heterotrimeric GluN1/GluN2A/GluN2B and GluN1/GluN2A/GluN2C receptors with Zn2+ and ifenprodil antagonism, it was found that each ligand produced only partial inhibition and that maximal inhibition was only achieved with both copies of each GluN2 subunit in the receptor.83 This finding suggested a potential limitation in using ifenprodil and its derivatives, since both GluN1/GluN2B and heterotrimeric GluN1/GluN2A/GluN2B receptors will be inhibited, possibly affecting subunit-specific control of normal neurological processes.
Other considerations may enter into the design of NMDA therapeutic antagonists, such as whether the agents will preferentially target synaptic or extrasynaptic NMDA receptors and the GluN2 subunit composition at those receptor sites. Work on the mechanism of action of memantine has shown that it preferentially targets extrasynaptic over synaptic NMDA receptors in a hippocampal autaptic neuronal preparation. In this study, the synaptic and extrasynaptic NMDA receptors in mature neurons differed in subunit composition, with the GluN2A receptors predominating at the synaptic receptors and the GluN2B subunits at the extrasynaptic receptors.84 When cultured primary striatal or cortical neurons from rat were transfected with the gene coding for mutant huntingtin, a protein with polyglutamine repeat residues implicated in the pathogenesis of HD, and treated with D-APV, memantine, or ifenprodil, there was a significant decrease in the number of neurons expressing aggregated mutant-protein macro inclusions.85 Further studies using a transgenic YAC128 mouse HD model showed that at high concentration of memantine both synaptic and extrasynaptic NMDA-mediated currents were blocked, worsening neurodegeneration; at low concentration of memantine the extrasynaptic receptors were predominately blocked, leading to a neuroprotective effect (reduction of striatal volume loss and motor learning deficits at 12 months post-treatment) from the largely unaffected synaptic activity.85 Milnerwood et al. reported a similar reversal in intracellular signaling and motor learning deficits, when the extrasynaptic NMDA receptors of YAC128 HD mice were pharmacologically blocked with a low dose of memantine, thus providing further evidence for a new therapeutic approach to combating HD.86, 87
In view of the complex interplay of factors affecting NMDA receptor function, an integrative approach involving the modulation of receptor-associated pathways in the disease state may constitute a viable therapeutic strategy for neurodegenerative disorders. Since an excessive influx of Ca2+ ion into neurons is an important factor in the excitotoxic process, blockade of NMDA receptors could potentially be offset by the presence of L-type voltage-gated calcium channels. 88 Thus, design of therapeutic agents that can jointly antagonize both targets may be a desirable goal, and at least one such compound (18) has been developed.89-91 Dimebon (latrepirdine) (19), an antihistamine compound used clinically in Russia for many years, has demonstrated efficacy in Phase II clinical trials for AD and HD,92 although a Phase III trial for use in mild-to-moderate AD gave disappointing results.93 Dimebon was found to inhibit both NMDA receptors (IC50 = 10 μM) and L-type calcium channels (IC50 = 50 μM) in cultured neurons, possibly accounting, at least in part, for its mechanism of action.92, 94, 95 Blockage of NMDA-induced currents was different from that of memantine, suggesting a different site of action for dimebon at the NMDA receptor.95

Influx of calcium via NMDA receptors can induce necrotic or apoptotic cell death, depending on the degree of glutamate stimulation, with the cellular fate influenced by the effect of the resultant intracellular calcium concentration on the mitochondria.96 A therapeutic approach targeting the apoptotic pathway had mixed results when compounds shown to be effective both in vitro and in animal models for several neurodegenerative disorders were tested in human clinical trials.97-99
Another strategy for counteracting neuronal excitotoxicity might involve co-administration of an NMDA antagonist with an inhibitor of glutamate release. One potential candidate, riluzole (20), has been approved for use in treating the symptoms of ALS, although with limited success.48 In addition to its role as an inhibitor for glutamate release, riluzole also protects neurons against NMDA-induced toxicity.100-102 A clinical trial for use of riluzole in early PD showed that it was well tolerated in patients, but with no significant difference relative to the placebo group.103 Similarly, a 3-year randomized controlled study showed no neuroprotective or beneficial symptomatic effect of riluzole in HD.104 An alternative to inhibiting glutamate release might be to use an agonist, such as ceftriaxone (21), for stimulation of glutamate uptake transporters. Ceftriaxone, a third-generation cephalosporin antibiotic, was found to be a potent modulator of glutamate transport through NF-κB-mediated excitatory amino acid transporter-2 (EAAT2) in primary human fetal astrocytes105 and was in a clinical trial for ALS.48

Another important facet of an integrative therapeutic strategy to combat neurodegenerative diseases is the role that metabotropic glutamate receptors (mGluR) may play in the neural excitotoxicity process. An advantage of additionally targeting the mGlu receptors is that they can modulate the activity of voltage-gated calcium channels, while not affecting fast excitatory synaptic transmission.106 Pharmacological blockade of mGlu5 receptors has led to reduced neuronal death in animal models of PD and ALS, and negative allosteric modulators for this receptor, as well as selective mGlu3 receptor agonists, have been in clinical development.107 A recent perspective provides an excellent overview of this subject.108
3 Voltage-Gated Calcium Channels
In the central nervous system, calcium’s conductance properties are principally mediated by two types of receptors: ligand109 and voltage-gated channels.110 While NMDA receptors are involved in the total calcium load in neurons, smaller, but still significant, calcium contributions are mediated primarily through voltage-gated calcium channels.111 Voltage-gated calcium channels (VGCCs) are expressed on the plasma membrane and open in response to depolarizing stimuli (events that lower the resting potential of neurons). In most physiological environments, VGCCs shuttle calcium from the extracellular space into the intracellular space. The accessory subunit and conducting pore (α1-subunit) that constitutes VGCCs is the portion of the channel that conducts calcium, gives rise to the biochemical and biophysical properties in identified channels, and is the major site of pharmacological action. To date, ten types of α1-subunits have been identified, normally classified into five subtypes: L-type (Cav1.1, 1.2, 1.3, 1.4), P/Q-type (Cav2.1), N-type (Cav2.2), R-type (Cav2.3), and T-type (Cav3.1, 3.2, 3.3).112
Cav1.1, Cav1.2, Cav1.3, and Cav1.4 α1-subunits, constituting the family of L-type calcium channels (LTCCs), are central players in neuronal calcium dynamics in both physiological and pathophysiological states. α1-Subunits constituting Cav1.1 and Cav1.4 LTCCs are predominantly expressed outside of the central nervous system, in skeletal muscle and the retina, respectively.109 Functional Cav1.2 and Cav1.3 LTCCs are found in cardiovascular113 and nervous tissue, respectively,114 and play central roles as pharmacological targets in anti-arrhythmia and anti-hypertension therapeutics. 1,4-Dihydropyridines (DHPs), phenylalkylamines (PAAs), and benzodiazepines (BZAs) are among the most common LTCC channel antagonists.115, 116
Cav1.3 LTCCs have been identified as playing a role in the progression of Parkinson’s disease by allowing uncompensated calcium loading in dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc), which subsequently places a heavy and unsustainable metabolic burden on these neurons. It has been shown that by antagonism of these channels non-selectively with isradipine (22), a potent LTCC inhibitor, SNc DA neurons exhibit less metabolic stress and are protected in multiple PD animal models.117 Selective antagonism of Cav1.3 LTCC is therefore hypothesized to be potentially neuroprotective in early or presymptomatic stages of PD.

While promising, isradipine is non-selective; it is also a potent Cav1.2 LTCC inhibitor. Therefore, long-term use of isradipine as a treatment for PD might result in hypotension or peripheral edema. Even if this does not result directly from isradipine use, it is known that during the course of PD, hypotension is common, thereby exacerbating this undesirable side-effect.118 Moreover, the non-selective nature of DHP inhibition of both neuronal types of LTCCs, Cav1.2 and Cav1.3, makes dosing regimens of isradipine, or other non-selective DHPs that might be disease-modifying in PD or other neurodegenerative diseases, an impossibility because the efficacious dose may have a serious effect on the therapeutic index of this strategy.119 None of the Cav1 antagonists in the clinic preferentially selects for Cav1.3 channels.114, 120 Thus, the search for more isoform-selective Cav1.3 LTCC antagonists has garnered recent attention.
Chang et al. described121 a comprehensive structure-activity-relationship (SAR) study of modifications to the structurally simple, active DHP nifedipine122 (23). A collection of 124 chemically diverse 4-substituted 1,4-dihydropyridines was generated; however, little selectivity of Cav1.3 over Cav1.2 was obtained. The most selective compounds generated (24 and 25) possessed Cav1.3 selectivities of only 2.2 and 2.4-fold, respectively, and both demonstrated only micromolar potencies. Observations from the SAR study showed an activity pattern for 4-substituted 1,4-dihydropyridines of substituted phenyl > thienyl > furyl > pyridyl > napthyl > alkyl (cyclic alkyl), with substitution of the phenyl ring at the 2-position the most potent and at the 4-position the least potent. Complete loss of activity was observed when the pyridyl nitrogen was methylated or replaced with oxygen. These data implied that the generation of a high Cav1.3 selective DHP may be unlikely, and attention was focused on the generation of new scaffolds.

Compounds that show Cav1.3 LTCC inhibition, however selective, must maintain good pharmacokinetic properties and structure components with good potential for passing through the BBB. A high-throughput screen was initialized by the Silverman laboratory to identify novel Cav1.3 selective LTCC inhibitors by use of a calcium-sensing fluorescent imaging plate reader (FLIPR) assay. Over 60,000 molecules from a variety of commercial and government libraries were screened for activity with no hits resulting. In parallel with this screen Xia et al. described a series of symmetric pyrimidine-2,4,6-triones (PYT) investigated for applications in anti-aggregation ALS models, possessing good pharmacokinetic properties and BBB penetration.123 About 200 of these compounds and other scaffolds from the Silverman lab that were active in anti-aggregation assays were tested, and the first PYT Cav1.3-selective antagonists were realized, although most of the compounds screened showed Cav1.2 selectivity. The lead compound was modified, providing the first highly selective Cav1.3 antagonist (26),124

4. Neuronal Nitric Oxide Synthase Inhibitors
As shown in the Figure 1 cellular cascade, activation of the NMDA receptor leads to calcium influx, which activates many downstream proteins, including nNOS.125 The signaling molecule, nitric oxide (NO), is a free radical that is produced by nitric oxide synthases (NOSs) from substrate l-arginine, molecular oxygen, and NADPH. There are three isoforms of NOS: neuronal NOS (nNOS), which produces NO as a neurotransmitter; endothelial NOS (eNOS), which produces NO to signal the relaxation of smooth muscle cells in blood vessels; and inducible NOS (iNOS), which produces a burst of NO in response to invading pathogens. The prominent role of NO in the nervous system leads to the possibility of improper regulation of NO and, therefore, various disease pathologies. High levels of NO have been implicated in neurodegenerative diseases including ALS and PD.126 NO itself is a reactive molecule that leads to the formation of other oxidative species, such as superoxide and peroxynitrite. Also, nitrated protein aggregates, which are highly toxic to neurons, are found in patients with neurodegenerative diseases. S-Nitrosylation of proteins is a common feature of Lewy bodies and intraneuronal protein aggregates found in PD.127 Increased nitrosative stress may also compromise the ubiquitin degradation system, which, when impaired, cannot properly degrade proteins, leading to aggregation.125 Parkin, an E3 ubiquitin ligase implicated in PD, has been shown to be S-nitrosylated, which impairs its function and leads to increased protein aggregation.128-130 S-Nitrosylation of Hsp90 compromises its protection abilities as a chaperone, and leads to increased aggregation.131 NO stress can also compromise mitochondrial function. Electrons leak from the electron transport chain (ETC) to react with NO, forming peroxynitrite (ONOO−), which damages lipids, proteins, and DNA.132 S-Nitrosylation of pro-survival and pro-apoptotic proteins, p21, Ras, and Bcl-2 alters their activity.132 Two of the isoforms of NOS, nNOS and eNOS, are constitutively expressed and activated by an increase in intracellular calcium concentration. Calcium binds to calmodulin, a small protein, and this complex binds and activates these NOSs.133
NOS inhibitors as potential therapeutics have been sought since the discovery of NO’s signaling role in the late 1980s. Since crystal structure information was not yet available, early inhibitors began as arginine analogues, shown in Table 1 (27 - 33).134 These inhibitors, however, lacked selectivity. Selectivity has proven difficult to achieve over the past 20 years because of the high homology of the isoforms, especially in the active site. N-Nitro-l-arginine (L-NNA, 27) does show some selectivity, about 250-fold for nNOS over iNOS, but essentially no selectivity for nNOS over eNOS.135 Because of the important role of eNOS in vascular regulation, this poor selectivity over eNOS leads to hypertension in animals.136 L2NNA (27), L-NMA (28), and L-NAME (29) are commonly used in both in vitro and in vivo pharmacology experiments because of their stability, commercial availability, low toxicity, and solubility. Thiocitrulline and methylated thiocitrulline analogues have also been explored, but selectivity over eNOS has been difficult to achieve.134
Table 1.
Structures, potencies, and selectivities of arginine analogues. Data reported from Erdal et al. and references within.134
| Selectivity | |||||||
|---|---|---|---|---|---|---|---|
| Name | R 1 | R 2 |
K
i rat
nNOS (μM) |
nNOS/eNOSa | nNOS/iNOSb | ||

|
27 | L-NNA | NO2 | H | 0.015c | 11 | 293 |
| 28 | L-NMA | CH3 | H | 0.2 | 4.5 | 65 | |
| 29 | L - NAME | CH3 | CH3 | -d | - | - | |
| 30 | L-NAA | NH2 | H | 0.3 c | 8.3 | 10 | |
| 31 | L-ALA |

|
H | 0.2 c | 35 | 11 | |
| 32 | L-NCPA |

|
H | 0.6 | - | 400 | |
| 33 | L-NPA | CH2CH2CH3 | H | 0.55 | 18 | 145 | |
Notes:
Ki bovine eNOS / Ki rat nNOS.
Ki murine iNOS / Ki rat nNOS.
bovine nNOS, not rat nNOS.
L-NAME presumably is hydrolyzed to L-NMA intracellularly or in vivo.
Using L-NNA as a starting point, Silverman and coworkers developed several series of nNOS-selective dipeptides (see Table 2). This approach takes advantage of the potency of the arginine analogue scaffolds, but also provides compounds that can potentially extend out of the heme binding pocket in an attempt to interrogate the isozymes for selective contacts away from the active site. Residue differences between isoforms include: S585 in nNOS is N370 in iNOS, but S356 in eNOS; and D597 nNOS is N368 in eNOS.
Table 2.
Dipeptide ester and amide nNOS inhibitors.
| Selectivity | |||||
|---|---|---|---|---|---|
| R |
K
i rat nNOS
(μM) |
nNOS/eNOSa | nNOS/iNOSb | ||

|
34 137 | 1.9 | 2.6 | 1800 | |

|
35 138 | 0.13 | 1540 | 190 | |

|
36 140 | 0.10 | 1280 | 290 | |

|
|
37 141 | 0.12 | 2600 | 330 |

|
38 142 | 0.05 | 2100 | 70 | |
Notes:
Ki bovine eNOS / Ki rat nNOS.
Ki murine iNOS / Ki rat nNOS.
Early dipeptide esters (34) achieved impressive selectivity over iNOS,137 while more optimization led to 35, which is highly selective over eNOS (1500 fold).138 Modifications of the peptide scaffold included acetylation, benzyloxycarbonylation, amide-methylation or conversion to peptoids, in an attempt to protect against metabolic degradation, but led to drops in selectivity.139 Exploration of conformationally-rigid analogues of 34 led to highly potent, selective dipeptide amides 36,140 37,141 and 38,142 but these compounds have limited BBB permeability because of their tricationic structures.
As crystal structures of NOS became available,143, 144 nNOS inhibitors moved away from the arginine mimetic scaffold. The Silverman group switched their strategy to de novo design using a pharmacophoric approach they developed termed “fragment hopping”.145, 146 This led to a potent, nNOS selective aminopyridine scaffold (±39). Optimization led to potent, selective nNOS inhibitors (±40), and replacement of a nitrogen atom with an oxygen atom increased bioavailability (41).145 Furthermore, (±)-40 was shown to prevent hypoxia-ischemia-induced death in a rabbit model of cerebral palsy.147 Upon maternal administration, (±)-40 and an analogue was able to distribute readily to the fetal brain, inhibit NOS activity and decrease NO concentration in vivo, to be nontoxic, without detrimental cardiovascular effects, and show a remarkable protection of fetal rabbit kits from the HI induced phenotype of cerebral palsy. The rabbit kits from saline-treated dams had a large incidence of fetal/neonatal deaths (16/34; 47%) but no deaths (0/49) were observed from animals treated with (±)-40 and its analogue. Of the kits from saline-treated dams that came to term (18/34), severe neurobehavioral abnormalities occurred in 12/18 (67%) compared to only 7/49 (14%) in those from dams treated with the inhibitors. Furthermore, the inhibitor-treated animals exhibited a remarkably larger number (83%) of normal kits; only 9% (3/34) of the kits from saline-treated dams were born normal.
Crystal structures of single enantiomers 41 and 42 revealed a surprising difference in binding mode.148 Previous crystal structures show aminopyridines bind NOS with the aminopyridine head over the heme, but the (R,R) stereochemistry of 42 induces a flipped binding mode with the fluoro-phenyl tail binding over the heme. Moreover, this flipped binding mode is more potent; the Ki of 42 is 7.2 nM, which is lower than the Ki of 41 (116 nM) for nNOS.149 Bioavailability of these inhibitors is poor as a result of the multiple cationic charges. A successful attempt to increase bioavailability and take advantage of the two binding pockets for the aminopyridine head led to 43,150 which displayed decreased nNOS selectivity. Compound 43 is a lead compound for designing future selective inhibitors of nNOS with increased bioavailability and a synthetically simple structure.

The aminopyridine moiety is a bioisostere of the guanidium group of arginine. 2-Amino-4-methylpyridine (44) was found to be potent both in vitro and in vivo, but is non-selective.151 Simple substituted 2-aminopyridines were studied as NOS inhibitors, but were found to be mostly selective for iNOS.152 6-Phenyl-2-aminopyridines were explored by Pfizer in the early 2000s as nNOS selective inhibitors. Compound 45 had an IC50 of 140 nM for human nNOS, but only modest selectivity over eNOS (6 fold).153 Compound 46 has an IC50 of 70 nM for human nNOS with 50- and 10-fold selectivity over eNOS and iNOS, respectively. The compound possessed a good pharmacokinetic profile and was well tolerated and effective in vivo.154

L-NIO (47) is an arginine analogue known to be a non-selective inactivator of NOS.155 Compound 48,156, is an nNOS selective reversible inhibitor, unlike its homologue 49,157 which is iNOS selective and is an irreversible inhibitor. Compound 48 is selective for nNOS over eNOS (155 fold) and potent (IC50 human nNOS = 40 nM) but is not effective in vivo in rats.156 nNOS selective carbamidines 50 and 51 were reported by Amoroso and coworkers;158 computer modeling was used to rationalize that the selectivity of these bulky inhibitors is the result of the larger heme binding pocket of nNOS compared to that of eNOS and iNOS.

Another successful guanidine-mimetic moiety is the thiophene amidine (thienylcarbamidine) (52). Developed by AstraZeneca, 52 is a selective nNOS inhibitor with an IC50 of 0.035, 5.0 and 3.5 uM for human nNOS, iNOS and eNOS, respectively.159 It has been shown to effectively cross the BBB in animal studies and to be neuroprotective in ischemia models.160 Crystal structures reveal that the amidine nitrogens hydrogen bond to nNOS Glu592, much like arginine, while the thiophene sulfur lies 3.4 Å above the heme iron, but does not coordinate as a sixth ligand.161 NeurAxon has further explored the thienylcarbamidine functionality with 53 and 54; these nNOS selective inhibitors are being developed for the treatment of migraine pain. Compound 53 is a dual-acting inhibitor of nNOS (IC50 of 0.44 μM) and agonist of the μ-opioid receptor (Ki of 5.4 nM), which is involved in pain.162 Compound 54 is a potent, selective nNOS inhibitor without any cardiovascular effects and with a good side effect profile.163

A variety of other scaffolds have been explored as NOS inhibitors, but they are beyond the scope of this perspective. They are described in detail in other excellent, recent review articles.134, 164, 165 The compounds described above have all been active site inhibitors. Inhibitors that disrupt dimerization166 and inhibitors that displace tetrahydrobiopterin167 also have been explored as NOS inhibitors.
5. Antioxidants as therapeutics for neurodegenerative disorders
Misregulation of nNOS production can lead to oxidative stress, a hallmark of neurodegeneration. In addition to unregulated production of nitric oxide (NO) by nNOS, other reactive oxygen species (ROS) exist and have also been implicated in neurodegeneration. A large number of ROS are free radicals or radical-generating derivatives of oxygen such as superoxide (O2•−), hydroxyl radical (•OH), and hydrogen peroxide (H2O2). Cellular damage in the nervous system by free radical species has been implicated in AD,168-173 PD,174-177 HD,178 and ALS.179, 180 Antioxidants are molecules that react with ROS to deactivate them and are of interest as potential therapeutics. Here, we present a summary of the major classes of antioxidants and their known or potential efficacy as treatments of neurodegenerative disease.
Compounds that can directly react with free radical species are referred to as direct antioxidants. These compounds do not generally rely on endogenous cellular mechanisms to wield their primary effect. The major classes of direct antioxidants include phenols, low-molecular weight enzyme mimetics, and polyenes. Because of their ability to quench free radical species, direct antioxidants can halt the procession of potentially damaging radical chain reactions.
Free radical scavenging phenols, which rely on the facile ability of phenols to be oxidized to their corresponding quinones, can be divided into two categories: monophenolic and polyphenolic compounds. Monophenols vitamin E (α-tocopherol, 55) and estrogens (56) have been repeatedly investigated for potential efficacy in treating a number of neurodegenerative diseases. Vitamin E has been shown to offer modest cognitive benefits in some AD patients;181-183 however, other studies have shown no preventative benefit of vitamin E for AD development.182-186 While no significant effects have been observed in PD187-190 or HD,191 vitamin E was found to delay onset as well as slow the progression of ALS.192, 193 The use of estrogen replacement therapy (ERT) is also believed to play a preventative rather than therapeutic role in AD,194-198 and it is likewise the case for PD,199-201 which may be related to the increased disease prevalence observed in men over women.202, 203 Polyphenols, which include well-known compounds resveratrol (57) and curcumin (58), as well as a large family of flavonoids such as quercitin (59) and epicatechin (60), have already been extensively reviewed for their potential therapeutic properties.204-207

Under normal conditions, cellular antioxidant defense mechanisms are present to combat both metabolic and exogenous sources of ROS. These mechanisms include both small molecules and enzymes. The primary small molecule utilized is glutathione (GSH), a tripeptide possessing a sulfhydryl moiety capable of donating electrons to oxidized molecules, followed by regeneration via NADPH.208, 209 As for antioxidant enzymes, superoxide dismutase (SOD) converts O •−2 into H2O2, which can then be rapidly reduced by catalase or glutathione peroxidase (GPx) to H2O and O2.177, 210, 211 The metabolism of O2•− and H2O2 is critical because O2•− can react with NO to form peroxynitrite (ONOO−), and H2O2 can react with Fe2+ to generate •OH, which are highly reactive species that are capable of lipid, protein, and DNA damage.209, 212 A study of potential low-molecular weight mimics of SOD and GPx has identified a small number of potential therapeutics. The small, organoselenium compound ebselen (61) has exhibited notable activity as a GPx mimetic and ONOO− scavenger in vitro213, 214 and has shown significant beneficial effects in a primate model of PD.215 Ebselen’s catalytic ability can be initiated with or without the presence of ROS. In the presence of oxidative species, oxidation to the selenoxide (Se=O) followed by reduction via a thiol electron donor generates a reactive selenenic acid (Se–OH) that readily loses water and converts back to ebselen. Without the presence of ROS, oxidation of a free thiol forms a selenyl sulfide (Se–S-R) leading to the production of a disulfide (R-S–S-R) and a selenol intermediate (Se–H). The selenol can be readily oxidized by ROS to form selenenic acid, which leads to regeneration of ebselen.216 The organoselenium compound 62, similar to ebselen, has shown even higher in vitro GPx-like activity and may also possess therapeutic potential for PD or other neurodegenerative disorders.217, 218 Organoselenium compound 62 is currently in clinical trials for cardiovascular indications.

In addition to the therapeutic potential found in GPx mimetics, Mn-containing complexes have been found to be effective SOD mimetics. Metalloporphyrins 63 and 64 have been found to exhibit high SOD activity, catalase activity, as well as inhibition of lipid peroxidation.219, 220 The antioxidant activity of 63, 64, and similar metalloporphyrins has been extensively studied in a variety of models for oxidative neuronal damage.221 Salen manganese complexes 65 and 66 are also SOD and catalase mimetics. These complexes
 have been shown to confer neuroprotection in both in vitro and in vivo models for PD.222, 223
have been shown to confer neuroprotection in both in vitro and in vivo models for PD.222, 223
Polyene antioxidants are primarily comprised of carotenoids, such as β-carotene (67), lycopene, retinol, and lutein, and are typically of plant origin. These carotenoids are capable of scavenging singlet molecular oxygen (1O2) and peroxyl radicals forming stabilized radicals that can further react with ROS to halt radical chain reaction processes.224 The rate constants for singlet oxygen quenching by carotenoids are in the range of 109 M−1 s−1, which is near diffusion control.225 Peroxyl radical scavenging is also efficient, especially under hypoxic conditions, and is important for the prevention of lipid peroxidation.226 Carotenoids are an important part of the diet of animals because many carotenoids are metabolized to retinol (vitamin A), which cannot be synthesized endogenously. Retinoic acid (RA), an irreversibly oxidized form of retinol, is an important signaling molecule involved in embryonic growth and development. There have been a number of studies that suggest the therapeutic potential of RA for AD prevention,227, 228 but there has been no evidence to suggest a direct antioxidant effect of carotenoids being involved in neurodegenerative prevention. A more recent study, however, has shown that all-trans RA (ATRA, 68) treatment invokes a decrease in brain Aβ deposition in an AD mouse model by inhibition of amyloid precursor protein processing.229

Indirect antioxidants are compounds that do not directly react with ROS, but are involved in cellular management of oxidative species. Many of these compounds are essential cofactors required for cellular oxidative metabolism; however, a number of synthetic compounds have been identified that fall under this classification. These compounds act by easing the secondary metabolic burden of free radicals and therefore diminishing oxidative damage.
Quinones are a class of compounds, often derived from aromatic compounds, containing two carbonyl groups in an unsaturated six membered carbon ring. The general label of vitamin K is applied to a number of related quinone compounds with a polyprenylated naphthoquinone ring structure. The primary role of vitamin K is to act as a necessary enzyme cofactor for a number of processes that include blood coagulation230 and bone metabolism.231 There is growing evidence, however, that vitamin K has important functions in the brain, and that a deficiency may contribute to the pathogenesis of AD.232 Ubiquinone (69), also known as coenzyme Q10, serves as an important cofactor, primarily in the electron transport chain during aerobic cellular respiration. Ubiquinone has been found to exert a protective effect against PD and HD,233, 234 in addition to reducing intracellular Aβ deposition235 and plaque pathology in AD mouse models.236
The mitochondrial-targeted ubiquinone analogues 70 and 71 have shown encouraging results for the treatment of age-related neurodegenerative disease.237 Targeted delivery of these analogues is made possible by the cationic triphenylphosphonium group, which takes advantage of the potential gradient of the inner-mitochondrial membrane. Both have gone to clinical trials.238

Spin trapping compounds are labeled as such because of their ability to form adducts with free radicals via their nitrone functionality. Initially utilized in analytical chemistry, spin traps have exhibited biological activity in a number of ROS implicated disease states.239, 240 Although spin traps are capable of directly quenching ROS, recent evidence suggests that they induce a number of endogenous antioxidants and enzymes involved in the attenuation of oxidative cellular damage.241 α-Phenyl-N-tert-butylnitrone (PBN, 72) has exhibited efficacy for the treatment of neurodegenerative diseases by affecting signal transduction pathways related to neuroinflammatory processes.242, 243 On the other hand, Cerovive (73) was tested in stroke patients in clinical trials and failed to show efficacy, likely because of its poor pharmacokinetic properties.244 The more lipophilic nitrones 74 and 75 have much higher radical scavenging potential over PBN and Cerovive and have both exhibited neuroprotective efficacy in mouse models of PD.245, 246

This final category of antioxidants is composed of a large and mixed group of compounds that are employed by the cell in the transport of reducing equivalents. Prominent examples include ascorbate (76), which has exhibited antioxidant activity in several in vitro studies,247 and the previously mentioned GSH (77). A number of thiol precursors of GSH belong to this category as well. These include the dipeptide CysGly, which is used to generate GSH in neurons,248 as well as cysteine precursors N-acetylcysteine (78) and procysteine (79).249, 250 Despite no known efficacy of these compounds for the treatment or prevention of neurodegenerative disease, brain levels of GSH are depleted by up to 30% in the elderly, suggesting a strong link to age-related disease.
 The nuclear factor E2-related factor 2 (Nrf2) signaled gene expression in response to cellular stress must be considered when discussing potential therapeutic effects of antioxidants. Nrf2 is a transcription factor responsible for the activation of a number of genes whose products are important in the reduction of cellular oxidative stress. These genes, which include glutathione-S-transferase (GST), coenzyme Q10, NAD(P)H:quinone oxidoreductase, superoxide dismutase 1 (SOD1), and many others, contain a common promoter called the antioxidant response element (ARE). Under normal conditions, Nrf2 is bound to repressor protein Keap1 (Kelch ECH associating protein 1) and retained in the cytoplasm. In the Nrf2-Keap1-ARE expression pathway, Keap1 acts as a sensor for oxidative stress through oxidation of or electrophilic addition to its many cysteine residues.251 Alteration of the cysteine residues of Keap1 disturbs Nrf2 binding, which allows Nrf2 to translocate into the nucleus. Once in the nucleus, Nrf2 heterodimerizes with members of the small Maf (sMaf) family of transcription factors, binds to the ARE, and ultimately leads to gene expression (Figure 3). An ever-growing number of molecules able to induce Nrf2 signaling have been identified, including many previously mentioned antioxidants. These molecules, and the varying chemical characteristics that provide their activity, have previously been reviewed in great detail.252, 253
The nuclear factor E2-related factor 2 (Nrf2) signaled gene expression in response to cellular stress must be considered when discussing potential therapeutic effects of antioxidants. Nrf2 is a transcription factor responsible for the activation of a number of genes whose products are important in the reduction of cellular oxidative stress. These genes, which include glutathione-S-transferase (GST), coenzyme Q10, NAD(P)H:quinone oxidoreductase, superoxide dismutase 1 (SOD1), and many others, contain a common promoter called the antioxidant response element (ARE). Under normal conditions, Nrf2 is bound to repressor protein Keap1 (Kelch ECH associating protein 1) and retained in the cytoplasm. In the Nrf2-Keap1-ARE expression pathway, Keap1 acts as a sensor for oxidative stress through oxidation of or electrophilic addition to its many cysteine residues.251 Alteration of the cysteine residues of Keap1 disturbs Nrf2 binding, which allows Nrf2 to translocate into the nucleus. Once in the nucleus, Nrf2 heterodimerizes with members of the small Maf (sMaf) family of transcription factors, binds to the ARE, and ultimately leads to gene expression (Figure 3). An ever-growing number of molecules able to induce Nrf2 signaling have been identified, including many previously mentioned antioxidants. These molecules, and the varying chemical characteristics that provide their activity, have previously been reviewed in great detail.252, 253
Figure 3.

Mechanism of ARE-promoted gene expression by Nrf2. Disruption of the Keap1-Nrf2 complex allows for Nrf2 migration to the nucleus, where it joins with other transcription factors that bind to ARE regions of genes and initiate their transcription.
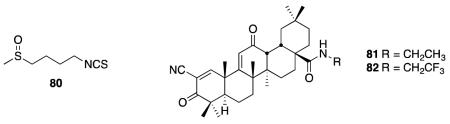
The Nrf2 system has previously been the subject of reviews in regard to its potential as a therapeutic target.254, 255 Activation of Nrf2 has been observed to prevent apoptosis of motor neurons that were co-cultured with the ALS model G93A-SOD1 mutant astrocytes. This prevention of apoptotic signaling is believed to be caused by glutathione-mediated NO detoxification.256 Downregulation of Nrf2 activated genes has also been observed in microarray analysis of mutant SOD transfected motor neuron-like Nsc34 cells.257 It has also been shown that dietary sulforophane (80), a known Nrf2 signaling activator, protects against degeneration of dopaminergic neurons in a Drosophila PD model.258 A study to assess the localization of Nrf2 in hippocampal neurons of AD indicated a loss of nuclear Nrf2 when compared to age-matched control individuals; however, cytoplasmic levels were the same between AD and control cases. This suggests impairment in nuclear trafficking and may limit Nrf2 activation as a potential AD treatment.259 In an N171-82Q transgenic mouse model of HD, upregulation of the Nrf2/ARE pathway by synthetic terpenoids 81 and 82 was found to rescue behavioral deficits, extend survival, and attenuate brain and peripheral pathology.260
6. Antiaggregation Agents
One consequence of high levels of ROS is the damage and aggregation of proteins. Neurodegenerative diseases have been termed ‘protein aggregation diseases’ because protein aggregation is another hallmark of these diseases. Aggregation consists of the formation of identical monomers of proteins self-associated into large oligomeric structures of reduced solubility that directly contribute to the onset and progression of the disease in question. Commonly implicated proteins (Table 3) include tau and amyloid-β in AD, α-synuclein in PD, huntingtin in HD, SOD1 in ALS, and others covering a range of diseases.261 The aggregation process leads to formation of fibrils with defined morphologies or amorphous deposits, or both. Here, we present an overview of therapeutic targets and a selection of molecules displaying anti-aggregation properties. In this section we will concentrate on the most frequently studied protein targets for the development of antiaggregation inhibitors as listed in Table 3. Additional targets are known, and inhibitors are being actively developed; however, space precludes a detailed examination of all potential targets. Comprehensive introductions to the subjects of pathogenicity and potential targets,262 and strategies for the design of anti-aggregation inhibitors263 that have been published previously, are excellent sources of in-depth coverage of the subject matter.
Table 3.
Proteins implicated in misfolding aggregation diseases and respective targets for aggregation inhibitor compound design, as discussed herein.
| Disease | Aggregating Protein | Direct Targets | Indirect Targets |
|---|---|---|---|
| Alzheimer’s | Amyloid-β | BACE-1 α-secretase γ-secretase Metal chelators |
HMG-CoA reductase inhibitors Protein kinases (GSK3, CDK5) Autophagy activators |
| Tau | Physiological tau | ||
| Parkinson’s | α-synuclein | HMG-CoA reductase inhibitors Protein kinases (LRRK2) Autophagy activators Heat Shock Protein |
|
| Huntington’s | Huntingtin | PolyQ aggregates | HMG-CoA reductase inhibitors Autophagy activators Heat Shock Protein |
| Amyotrophic lateral sclerosis |
SOD1 and many others implicated |
Protein Kinases (GSK3) Autophagy activators Heat Shock Protein |
Inhibiting the accrual of misfolded forms of the proteins comprising aggregates is one commonly exploited approach for targeting many neurodegenerative diseases. Kinase inhibitors offer the advantage of allowing inhibition of the transformative process responsible for misfolding over inhibition of the production of the naturally occurring protein, which may be crucial to normal cellular function. Dual inhibition of glycogen synthase kinase 3 (GSK23)264 and cyclin-dependant kinase 5265 is a promising strategy for tau-related diseases such as AD, when hyperphosphorylation of tau protein leads to malfunction and aggregation.266 One such example of a dual-acting inhibitor for both kinases is the natural product hymenialdisine 83, which has been the basis of intense SAR and analogue development to generate potential therapeutic candidates.267 Similarly, GSK23 has been implicated in the pathogenesis of a number of neurodegenerative diseases, such as ALS268 and spinal muscular atrophy (SMA).269 GSK23 inhibitor VII (84) has been shown to significantly delay the onset of symptoms and extend the life span of a G93A-SOD1 mouse model of ALS.270 BIP2135 (85) was found to prolong the median survival of the Δ7 SMA KO mouse model of SMA and elevate survival motor neuron levels in SMA patient-derived fibroblast cells.269 Leucine-rich repeat kinase-2 (LRRK2) may regulate the propensity of α-synuclein to aggregate and, as a result, has garnered much attention as a target for a therapeutic intervention in PD.271

Inhibition of tau aggregation has become an important strategy in targeting pathological tau. The use of small molecules to inhibit self-assembly of tau into oligomeric and/or polymeric species, including the well-defined fibrils comprising neurofibrillary tangles, has met with mixed success. Several compounds demonstrate activity in vitro; however, most also suffer from toxicity and/or limited CNS uptake issues.272 A testament to the difficulty in designing tau inhibitors is the fact that one of the most promising structures is still one of the originally reported compounds having tau aggregation inhibition activity – methylene blue (86).273 While methylene blue itself only possesses weak inhibitory activity, the compound provided a lead scaffold for derivatization. The dimethyl derivative, tolonium chloride, was found to exhibit 30-fold greater activity with a Ki of 69 nM, acting at almost equimolar concentrations with tau. Crucially, the compound did not inhibit the required tau-tubulin binding interaction. Interestingly the inhibitory activity was also observed in hyperphosphorylated tau, bringing into question the need to inhibit kinases to elicit a subsequent inhibition of tau aggregation. Recent advances have identified aminothienopyridazines (ATPZs) as a novel class of tau fibrilization inhibitors with efficacy in in vitro models and favorable drug-like properties. Compound 87 represents the current lead structure, demonstrating good brain penetration, oral bioavailability, and non-specific brain tissue binding. The compound is set to undergo long term in vivo testing.274 Refinement of in vitro models of tauopathy will also have a large impact on drug design targeting tau aggregation. A recently disclosed model using inducible hippocampal brain slices provides a more robust assay environment for further advances.275

Accumulation of oligomeric β-amyloid peptides (Aβ) is characteristic of AD and is the primary agent in the pathogenesis of the disease. Aβ is generated from the amyloid precursor protein (APP) via two proteolytic enzymes, β-secretase and γ-secretase, responsible for the regulation of the first step in amyloidogenic APP metabolism and generation of Aβ, respectively. α-Secretase conducts an alternative proteolytic cleavage that prevents Aβ production and accumulation. Development of β-secretase (BACE-1) inhibitors has been hindered because of poor BBB permeability.276 Recent efforts have moved away from peptide-based compounds to more lipophilic structures such as difluoride 88, which demonstrated a 70% reduction of Aβ in beagle dog CSF up to nine hours after dosing.277 Progression to Phase I trials followed, showing, for the first time, a good correlation with preclinical and clinical efficacy. However, rat toxicology data indicated renal toxicity from an off-target interaction that has yet to be identified. The compound succeeded in demonstrating BACE to be a ‘druggable’ target. Potent compounds inhibiting γ-secretase have been developed; however, inherent toxicity problems still remain as a challenge.278 Avegacestat (89) a potent and selective γ-secretase inhibitor, succeeded in reducing brain levels of Aβ40 in wild-type rats by 59 ± 12% with a 30 mg/kg dose.279 However, the compound has now been dropped from clinical studies by BMS. Modulation of α-secretase is an increasingly attractive target in AD therapy; activation is controlled by the protein phosphorylation signal transduction pathway of protein kinase C (PKC).280 The known PKC activator bryostatin (90) promotes sAPP-α-secretase at sub-nanomolar levels;281 analogue construction to refine pharmacological properties has garnered a great deal of attention in the literature.282

Twenty-five percent of the cholesterol present in humans is synthesized in the brain, where it performs many vital functions, such as myelin formation, structural composition of glial and neuronal membranes, and in neurotransmission. A link between cholesterol levels and the production of APP in Alzheimer’s patients has been suggested by the use of statins, cholesterol-lowering drugs that inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, demonstrating a decreased prevalence of AD.283 In particular, simvastatin showed a marked decrease of both cholesterol and Aβ in guinea pig CSF; however, translation to human patients only showed a small decrease, which may be attributed to the higher concentration of Aβ in plaque formed within the brain and therefore not available to transfer to CSF.284 Interest in the link between cholesterol regulation and neurodegenerative diseases has grown considerably in recent years with implications ranging past AD,285 to PD,286 HD,287 and the prion diseases.288 Attempts to identify new HMG-CoA reductase inhibitors and analogues of statins continue.289
There is increasing evidence that heat shock proteins (HSPs), molecular chaperones that control protein misfolding and aggregation, could counteract the pathological mechanisms that take place during AD, PD, and HD.290 The HSPs can interfere with the misfolded disease-linked proteins, thereby preventing interactions that can lead to formation of toxic oligomers. Moreover, HSPs are expected to interfere with detrimental processes that occur during these diseases, such as oxidative stress, and act in support of the ubiquitin-proteasome degradation process. These factors combine to make HSP activators an important target in combatting neurodegenerative disease.291 Central to the hypothesis of targeting protein misfolding is heat shock transcription factor 1 (HSF1), the main activator of chaperone protein gene expression; activating HSF1 increases the amount of chaperone expression and, in turn, the rate of clearance of misfolded proteins.292 The chaperone protein Hsp90 is responsible for protein folding regulation in many cells and, in addition, is known to bind to HSF1 and impeded activation; therefore, Hsp90 inhibitors have therapeutic potential in many neurodegenerative diseases.293 A representative example of a Hsp90 inhibitor, celastrol (91), which is isolated from a Chinese medicinal herb, is a potent inhibitor of Hsp90 acting by activation of HSF1;294 however, the exact mode of action is poorly understood and is the subject of much investigation to aid in the development of this compound as a potential therapeutic.295 Arimoclomol (92), a coinducer of heat shock proteins, increases median survival in a G93A-SOD1 mouse model of ALS by 22%, illustrating the therapeutic potential of the HSP target in ALS.296 In addition, the closely related protein, Hsp70, is rapidly gaining interest as a target for many diseases, including those contributing to neurodegeneration.297 In addition to HSP inhibitors, several other classes of compounds have been found to elicit an activation effect in autophagy and hence increase the rate of fibril clearance.298 Among the most widely investigated of these compounds are rapamycin (93), which displays neuroprotective effects in many neurodegenerative diseases,299 and the disaccharide trehalose (94), which has been shown to reverse aggregation caused by proteasome inhibition.300 HSPs act as protein folding machinery and work in conjunction with the ubiquitin-proteasome system (UPS),301 failure of which is thought to contribute to the pathogenesis of PD.302

A myriad of additional approaches for therapeutic design and intervention in neurodegenerative disease have been proposed, and a comprehensive discussion is beyond the scope of this perspective; however, several newly emerging areas will be discussed briefly. The use of chemical chaperones, small molecules having the ability to stabilize unfolded monomer conformations and/or to destabilize misfolded oligomers, has been widely explored.303, 304 An alternative approach involves the use of metal chelators, compounds that sequester physiological metal ions, and in so doing, block protein aggregation. Aβ, for example, shows ready binding to metals such as Zn2+ and Cu2+, which induce nucleation and the formation of aggregate plaques. In addition, the interaction between redox active Cu2+ and Aβ can produce neurotoxic reactive oxygen species.305 Recent developments have led to the synthesis of platiniferous chelators, such as 95, containing a Pt(bipyridine)Cl2 group as the Aβ binding portion.306 Compounds of this type show almost complete reversal of Aβ aggregation in turbidimetry experiments conducted using Aβ40. Further compounds, many of them peptidyl in structure, acting as β-sheet blockers, have been investigated.307

Direct inhibition of the oligomerization pathway has been shown to be possible with a number of small molecule inhibitors that disrupt soluble oligomers and interfere with larger structures; however, this approach does not take into account the accumulation of potentially toxic monomers. Such compounds include N-phenylanthranilic acid analogues (96), displaying activity against the Aβ peptide, with an IC50 of 1 μM,308 and benzothiazole 97, with activity to prevent polyQ aggregates, the cause of Huntington’s disease, in vitro with an IC50 of 1.2 μM.309

Despite a rich choice of targets of action, the use of antiaggregation agents in the treatment of neurodegenerative diseases has been limited. Poor BBB penetration, off-target toxicity, distribution in target tissue, and poor kinase selectivity have limited the potential of this broad class of compounds. Developments continue to counteract these limitations, and further optimization has the potential to create an efficacious and clinically useful therapeutic. One such strategy is the development of compounds that interact at multiple targets across the antiaggregation target spectrum.
7. Conclusions
Modulation of one or more of the distinct physiological processes depicted in Figure 1 has been shown to be pathological in neurodegenerative diseases. Small molecule therapeutics have been developed to target each step of the process: voltage-gated calcium channels, NMDA receptors, neuronal nitric oxide synthase, oxidative stress from reactive oxygen species, and protein aggregation. In addition to their potential as therapeutics, small molecules targeting proteins involved in neurodegenerative pathways can provide insight into their roles and connections. Determining how targets interact with each other could reveal previously unknown pathways. However, after more than 100 years of intense research, all current therapies for neurodegeneration are still directed towards symptomatic relief, not disease-modifying effects.
When developing small molecules to combat neurodegenerative diseases, a combination therapy approach, such as those taken to treat cancer, to fight microorganisms, and to remedy hypertension, should be considered. A multi-target therapeutic approach will combat the disease from many pathways, greatly enhancing the probability of disease modification. For example, inhibiting nNOS from producing species that generate ROS and inhibiting protein aggregation provides a dual mode of action by simultaneously reducing the underlying cause of protein misfolding and aggregation (oxidative stress damage) and combating the aggregation caused by ROS that survive to cause protein damage. Second, the potential synergy that may result from a drug combination may allow for the use of therapeutics at lower levels, allowing for the deployment of therapeutics that have small therapeutic windows, off-target effects, poor potency, or high toxicity, thereby expanding the arsenal of drugs available to fight these crippling, fatal diseases.
Successful dual-acting compounds are beginning to be reported; however, such compounds remain scarce in the literature and represent a minute proportion of the overall effort in drug discovery for neurodegeneration.310 Examples designed to target two of the pathways depicted in Figure 1 include compound 18, a dual NMDA and LTCC inhibitor discussed in Section 2. Benzofuran-based compound 98 and analogues demonstrate activity in three pertinent targets for AD; they serve as acetylcholine esterase inhibitors, Aβ aggregation inhibitors, and as inhibitors of Aβ25-35 peptide-induced ROS formation. The potential of these compounds to be disease modifying lies not just with their targets but also with their ability to combat one of the underlying generators of ROS in AD, rather than simply inactivating individual species.311 Imidazole 99 represents the most active compound of a library of analogues screened for nNOS inhibition activity. The compounds were designed as dual-target compounds; in addition to acting as nNOS inhibitors, the compounds are active antioxidants that scavenge free radicals and/or reduce lipid peroxidation.312 While these compounds were designed as a possible therapeutic for cerebral ischemia, they would appear to be ideal compounds for screening in neurodegenerative disease drug discovery.
A final example is the combination of two therapeutic constructs by a linker chain. Huprine-tacrine heterodimers (100) simultaneously block the active and peripheral sites of acetylcholinesterase, resulting in high potency (approximately 1 nM). Additionally, the heterodimers interact at two targets in the pathway of Figure 1, acting as both self-aggregation inhibitors of Aβ (ranging from 39 to 67% inhibition) and BACE-1 inhibitors (5-7 μM). Interestingly, while both of these targets are AD specific, the compounds inhibit the aggregation of a prion peptide that plays a key role in the aggregation of the prion protein.313

A multi-target approach would provide drug combinations and first-in-class molecular scaffolds to allow for the further probing and elucidation of the precise molecular networks and signaling cascades that underlie the nature of neurodegeneration. Given the fact that, at present, there is not yet a single therapy, let alone a combination of therapies, that significantly alters the progression of neurodegeneration, an understanding of the root causes for neurodegeneration, and the knowledge of the important pathways that should be altered to accomplish this, would be welcomed.
This Perspective, based on the commonalities within neurodegeneration (Figure 1), describes druggable targets of action and the compounds developed to interact with them. In so doing we have covered many targets that are specific to one particular disease (e.g., BACE1 inhibitors for AD). It is our intent to bring to the attention of the reader possible target combinations and molecular scaffolds that, when combined, may advance the discovery of the ultimate goal in neuroscience drug discovery – a disease modifying therapeutic.
Acknowledgments
The authors are grateful to the National Institutes of Health (Grants GM049725 and DA030604) and the Department of Defense (Grant W81XWH-10-1-0536) for financial support of our work in neurodegenerative and neurological diseases. We thank Garry Cooper for assistance in writing the section on voltage-gated calcium channels.
Abbreviations used
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- HD
Huntington’s disease
- ALS
amyotrophic lateral sclerosis
- Cav
voltage gated calcium channel
- NMDA
N-methyl-D-aspartic acid
- nNOS
neuronal nitric oxide synthase
- ROS
reactive oxygen species
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CNS
central nervous system
- BBB
blood brain barrier
- ABD
agonist binding domain
- NTD
N-terminal domain
- l-DOPA
l-3,4-dihydroxyphenylalanine
- mGluR
metabotropic glutamate receptor
- VGCC
voltage gated calcium channel
- DHP
1,4-dihydroxypyridine
- PAA
phenylalkylamine
- BZA
benzodiazepines
- DA
dopamine
- SNc
substantia nigra pars compacta
- SAR
structure-activity relationship
- FLIPR
fluorescent imaging plate reader
- PYT
pyrimidine-2,4,6-trione
- NO
nitric oxide
- NADPH
nicotinamide adenine dinucleotide phosphate
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- HSP90
heat shock protein 90
- ETC
electron transport chain
- ONOO−
peroxynitrite
- L-NNA
N-nitro-l-arginine
- GSH
glutathione
- SOD
superoxide dismutase
- GPx
glutathione peroxidase
- RA
retinoic acid
- GSK-3
glycogen synthase kinase-3
- SMA
spinal muscular atrophy
- LRRK2
leucine rich repeat kinase 2
- Aβ
beta amyloid
- APP
amyloid precursor protein
- BACE-1
beta secretase
- CSF
cerebrospinal fluid
- PKC
protein kinase C
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HSPs
heat shock proteins
- HSF1
heat shock factor protein 1
Biography
Paul C. Trippier is assistant professor of medicinal chemistry in the School of Pharmacy at Texas Tech University Health Sciences Center and a member of the Center for Chemical Biology, Department of Chemistry and Biochemistry at Texas Tech University. Paul completed his doctorate with Professor Mark G. Moloney at The University of Oxford working on the total synthesis and biological evaluation of bioactive natural products. He carried out postdoctoral work at the Welsh School of Pharmacy, Cardiff, UK with Professor Chris McGuigan, working on the design and synthesis of antiviral and anticancer compounds. In 2010 he moved to the laboratory of Professor Richard B. Silverman at Northwestern University, working on the synthesis of compounds possessing neuroprotective activity and determination of their intracellular targets of action.
Kristin Jansen Labby recently completed her doctorate in organic chemistry in the laboratory of Richard B. Silverman at Northwestern University. Her dissertation examined substrate analogues of neuronal nitric oxide synthase. She was a 2011-2012 National Science Foundation GK-12 fellow at Northwestern University. Kristin earned her bachelors degree from the University of Wisconsin-Madison in 2007, working under the direction of Hans J. Reich in the Chemistry Department as well as with Andrew F. Bent in the Plant Pathology Department. She is currently a postdoctoral fellow in the Life Sciences Institute at the University of Michigan in the laboratory of Sylvie Garneau-Tsodikova, studying aminoglycoside modifying enzymes and their roles in antibiotic resistance in tuberculosis and other mycobacterium.
Dustin D. Hawker obtained his B.S. degree in Biochemistry from California Polytechnic State University, San Luis Obispo in 2007. In 2006 he was a Summer Undergraduate Research Fellow at California Institute of Technology in the laboratory of Harry B. Gray. He is currently a Ph.D. candidate in the laboratory of Richard B. Silverman at Northwestern University, where his dissertation research is focused on the synthesis of novel substrates and inactivators of GABA aminotransferase.
Jan J. Mataka completed his doctoral work in 2002 under the direction of Professor David Crich at the University of Illinois at Chicago on the stereoselective formation of glycosyl sulfoxides, as well as the development of novel organic bases. Since then, he has been an instructor in chemistry at Northeastern Illinois University, teaching courses in general, organic, and, more recently, toxicological chemistry, in addition to teaching in the Urban Health Program at the University of Illinois at Chicago. Jan is a member of the National Academy of Clinical Biochemistry, and currently is a Visiting Scholar in the laboratory of Richard B. Silverman at Northwestern University, having worked on the development of inhibitors for neuronal nitric oxide synthase as a therapeutic target for Parkinson’s disease.
Richard B. Silverman received his Ph.D. in organic chemistry from Harvard in 1974, did postdoctoral work in enzymology for two years at Brandeis, then joined the chemistry faculty at Northwestern. Since 2004 he has been the John Evans Professor of Chemistry. He is the inventor of Lyrica™, marketed by Pfizer for epilepsy, neuropathic pain, and fibromyalgia. Recent awards include the 2009 Perkin Medal, ACS Medicinal Chemistry Hall of Fame (2009), E.B. Hershberg Award from the ACS (2011), Fellow of the ACS (2011), Sato Memorial International Award of the Japan Pharmaceutical Society (2012), Roland T. Lakey Award from Wayne State (2013), and the BMS-Smissman Award from the ACS (2013). He has published over 300 articles, holds 48 domestic and foreign patents, and has written four books.
References
- 1.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch. Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 3.Wancata J, Musalek M, Alexandrowicz R, Krautgartner M. Number of dementia sufferers in Europe between the years 2000 and 2050. Eur. Psychiatry. 2003;18:306–313. doi: 10.1016/j.eurpsy.2003.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Forman MS, Trojanowski JQ, Lee VMY. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 5.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat. Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 6.Pardo LM, van Duijn CM. In search of genes involved in neurodegenerative disorders. Mutat. Res. 2005;592:89–101. doi: 10.1016/j.mrfmmm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czirr E, Wyss-Coray T. The immunology of neurodegeneration. J. Clin. Invest. 2012;122:1156–1163. doi: 10.1172/JCI58656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 2012;13:465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lugo-Huitron R, Blanco-Ayala T, Ugalde-Muniz P, Carrillo-Mora P, Pedraza-Chaverri J, Silva-Adaya D, Maldonado PD, Torres I, Pinzon E, Ortiz-Islas E, Lopez T, Garcia E, Pineda B, Torres-Ramos M, Santamaria A, La Cruz VP. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011;33:538–547. doi: 10.1016/j.ntt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends Neurosci. 2008;31:176–182. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Drouin-Ouellet J, Cicchetti F. Inflammation and neurodegeneration: the story ‘retolled’. Trends Pharmacol. Sci. 2012;33:542–551. doi: 10.1016/j.tips.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Aquilano K, Baldelli S, Rotilio G, Ciriolo MR. Role of nitric oxide synthases in Parkinson’s disease: a review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008;33:2416–2426. doi: 10.1007/s11064-008-9697-6. [DOI] [PubMed] [Google Scholar]
- 15.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosley RL, Gendelman HE. Control of neuroinflammation as a therapeutic strategy for amyotrophic lateral sclerosis and other neurodegenerative disorders. Exp. Neurol. 2010;222:125. doi: 10.1016/j.expneurol.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neymotin A, Petri S, Calingasan NY, Wille E, Schafer P, Stewart C, Hensley K, Beal MF, Kiaei M. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009;220:191–197. doi: 10.1016/j.expneurol.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenfield S, Vaux DJ. Parkinson’s disease, Alzheimer’s disease and motor neurone disease: Identifying a common mechanism. Neuroscience. 2002;113:485–492. doi: 10.1016/s0306-4522(02)00194-x. [DOI] [PubMed] [Google Scholar]
- 19.Calne DB, Mcgeer E, Eisen A, Spencer P. Alzheimers-Disease, Parkinsons-Disease, and Motoneuron Disease - Abiotropic Interaction between Aging and Environment. Lancet. 1986;2:1067–1070. doi: 10.1016/s0140-6736(86)90469-1. [DOI] [PubMed] [Google Scholar]
- 20.Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 2012;8:518–530. doi: 10.1038/nrneurol.2012.156. [DOI] [PubMed] [Google Scholar]
- 21.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jellinger KA. Recent advances in our understanding of neurodegeneration. J. Neural. Transm. 2009;116:1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- 24.Salloway S, Mintzer J, Weiner MF, Cummings JL. Disease-modifying therapies in Alzheimer’s disease. Alzheimers Dement. 2008;4:65–79. doi: 10.1016/j.jalz.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann GR, Lehar J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov. Today. 2007;12:34–42. doi: 10.1016/j.drudis.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 26.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V, Kalluri R, Normant E, Haigis MC, Manning BD, Wong KK, Macleod KF, Cichowski K. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McHutchison JG, Manns M, Patel K, Poynard T, Lindsay KL, Trepo C, Dienstag J, Lee WM, Mak C, Garaud JJ, Albrecht JK. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology. 2002;123:1061–1069. doi: 10.1053/gast.2002.35950. [DOI] [PubMed] [Google Scholar]
- 28.Ho DD. Time to hit HIV, early and hard. N. Engl. J. Med. 1995;333:450–451. doi: 10.1056/NEJM199508173330710. [DOI] [PubMed] [Google Scholar]
- 29.Kihara T, Ikeda M. Effects of duloxetine, a new serotonin and norepinephrine uptake inhibitor, on extracellular monoamine levels in rat frontal cortex. J. Pharmacol. Exp. Ther. 1995;272:177–183. [PubMed] [Google Scholar]
- 30.Delgado PL, Moreno FA. Role of norepinephrine in depression. J. Clin. Psychiatry. 2000;61(Suppl 1):5–12. [PubMed] [Google Scholar]
- 31.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipton SA, Rosenberg PA. Excitatory Amino-Acids as a Final Common Pathway for Neurologic Disorders. N. Engl. J. Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 33.Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J. Pharmacol. Exp. Ther. 2002;300:717–723. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- 34.Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- 35.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J. Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothstein JD. Excitotoxicity hypothesis. Neurology. 1996;47:19S–26S. doi: 10.1212/wnl.47.4_suppl_2.19s. [DOI] [PubMed] [Google Scholar]
- 37.Hedegaard MK, Hansen KB, Andersen KT, Bräuner-Osborne H, Traynelis SF. Molecular pharmacology of human NMDA receptors. Neurochem. Int. 2012;61:601–609. doi: 10.1016/j.neuint.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 39.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002;5:1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 40.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 42.Coyle J. An animal model for Huntington’s disease. Biol. Psychiatry. 1979;14:251–276. [PubMed] [Google Scholar]
- 43.Fan MMY, Raymond LA. N-Methyl-d-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog. Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer’s disease. Am. J. Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 45.Plaitakis A. Glutamate dysfunction and selective motor neuron degeneration inamyotrophic lateral sclerosis: A hypothesis. Ann. Neurol. 1990;28:328. doi: 10.1002/ana.410280103. [DOI] [PubMed] [Google Scholar]
- 46.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 47.Rothstein J. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin. Neurosci. 1996;3:348–359. [PubMed] [Google Scholar]
- 48.Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monaghan DT, Irvine MW, Costa BM, Fang G, Jane DE. Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochem. Int. 2012;61:581–592. doi: 10.1016/j.neuint.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans R, Francis A, Jones A, Smith D, Watkins J. The effects of a series of omega-phosphonic alpha-carboxylic amino acids on electrically evoked and excitant amino acid-induced responses in isolated spinal cord preparations. Br. J. Pharmacol. 1982;75:65–75. doi: 10.1111/j.1476-5381.1982.tb08758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Löschmann PA, Lange K, Kunow M, Rettig KJ, Jähnig P, Honore T, Turski L, Wachtel H, Jenner P, Marsden C. Synergism of the AMPA-antagonist NBQX and the NMDA-antagonist CPP with L-dopa in models of Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 1991;3:203–213. doi: 10.1007/BF02259538. [DOI] [PubMed] [Google Scholar]
- 53.Vender JR, Nair SN, Lehmann JC. NMDA receptors: the first decade. Expert Opin. Investig. Drugs. 1994;3:341–354. [Google Scholar]
- 54.Bräuner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. Ligands for glutamate receptors: design and therapeutic prospects. J. Med. Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 55.Lehmann JC, Wood PL. 7-Chlorokynurenate prevents NMDA-induced and kainate-induced striatal lesions. Brain Res. 1993;620:1–6. doi: 10.1016/0006-8993(93)90263-m. [DOI] [PubMed] [Google Scholar]
- 56.Wong E, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc. Natl. Acad. Sci. U. S. A. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marin C, Papa S, Engber TM, Bonastre M, Tolosa E, Chase T. MK-801 prevents levodopa-induced motor response alterations in parkinsonian rats. Brain Res. 1996;736:202–205. doi: 10.1016/0006-8993(96)00693-2. [DOI] [PubMed] [Google Scholar]
- 58.Chen HSV, Lipton SA. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 59.Crossman A, Peggs D, Boyce S, Luquin M, Sambrook M. Effect of the NMDA antagonist MK-801 on MPTP-induced parkinsonism in the monkey. Neuropharmacol. 1989;28:1271–1273. doi: 10.1016/0028-3908(89)90221-9. [DOI] [PubMed] [Google Scholar]
- 60.Blanpied TA, Clarke RJ, Johnson JW. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005;25:3312–3322. doi: 10.1523/JNEUROSCI.4262-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uitti R, Rajput A, Ahlskog J, Offord K, Schroeder D, Ho M, Prasad M, Rajput A, Basran P. Amantadine treatment is an independent predictor of improved survival in Parkinson’s disease. Neurology. 1996;46:1551–1556. doi: 10.1212/wnl.46.6.1551. [DOI] [PubMed] [Google Scholar]
- 62.Metman VL, Dotto DP, Natte R, Van den Munckhof P, Chase T. Dextromethorphan improves levodopa-induced dyskinesias in Parkinson’s disease. Neurology. 1998;51:203–206. doi: 10.1212/wnl.51.1.203. [DOI] [PubMed] [Google Scholar]
- 63.Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch. Neurol. 1999;56:1383–1386. doi: 10.1001/archneur.56.11.1383. [DOI] [PubMed] [Google Scholar]
- 64.Crosby N, Deane K, Clarke C. Amantadine for dyskinesia in Parkinson’s disease. Cochrane Database Syst. Rev. 2003;2:1–14. doi: 10.1002/14651858.CD003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shoulson I, Penney J, McDermott M, Schwid S, Kayson E, Chase T, Fahn S, Greenamyre J, Lang A, Siderowf A. A randomized, controlled trial of remacemide for motor fluctuations in Parkinson’s disease. Neurology. 2001;56:455–462. doi: 10.1212/wnl.56.4.455. [DOI] [PubMed] [Google Scholar]
- 66.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 67.Ferris SH. Evaluation of memantine for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2003;4:2305–2313. doi: 10.1517/14656566.4.12.2305. [DOI] [PubMed] [Google Scholar]
- 68.Herrmann N, Li A, Lanctoot K. Memantine in dementia: a review of the current evidence. Expert Opin. Pharmacother. 2011;12:787–800. doi: 10.1517/14656566.2011.558006. [DOI] [PubMed] [Google Scholar]
- 69.Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat. Rev. Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- 70.Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 71.Tahirovic YA, Geballe M, Gruszecka-Kowalik E, Myers SJ, Lyuboslavsky P, Le P, French A, Irier H, Choi W, Easterling K. Enantiomeric Propanolamines as selective N-Methyl-d-aspartate 2B Receptor Antagonists†. J. Med. Chem. 2008;51:5506–5521. doi: 10.1021/jm8002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mosley CA, Myers SJ, Murray EE, Santangelo R, Tahirovic YA, Kurtkaya N, Mullasseril P, Yuan H, Lyuboslavsky P, Le P. Synthesis, structural activity-relationships, and biological evaluation of novel amide-based allosteric binding site antagonists in NR1A/NR2B N-methyl-d-aspartate receptors. Bioorg. Med. Chem. 2009;17:6463–6480. doi: 10.1016/j.bmc.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fischer G, Mutel V, Trube G, Malherbe P, Kew J, Mohacsi E, Heitz M, Kemp J. Ro 25–6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. Characterization in vitro. J. Pharmacol. Exp. Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- 74.Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br. J. Pharmacol. 1997;122:809–812. doi: 10.1038/sj.bjp.0701445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M, Jamerson B, Menniti FS, Landen JW. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and parkinsonism. Movement Disorders. 2008;23:1860–1866. doi: 10.1002/mds.22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J. Med. Chem. 2010:5476–5490. doi: 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hansen KB, Traynelis SF. Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN-D-containing NMDA receptors. J. Neurosci. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Skifter DA, Jane DE, Monaghan DT. A novel family of negative and positive allosteric modulators of NMDA receptors. J. Pharmacol. Exp. Ther. 2010;335:614–621. doi: 10.1124/jpet.110.174144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC. Mechanism for Noncompetitive Inhibition by Novel GluN2C/D N-Methyl-d-aspartate Receptor Subunit-Selective Modulators. Mol. Pharmacol. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L. Identification and characterization of novel NMDA receptor antagonists selective for NR2A-over NR2B-containing receptors. J. Pharmacol. Exp. Ther. 2010;335:636–644. doi: 10.1124/jpet.110.172544. [DOI] [PubMed] [Google Scholar]
- 81.Retchless BS, Gao W, Johnson JW. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat. Neurosci. 2012;15:406–413. doi: 10.1038/nn.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J. Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Xia P, Chen HV, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levine MS, Cepeda C, André VM. Location, location, location: contrasting roles of synaptic and extrasynaptic NMDA receptors in Huntington’s disease. Neuron. 2010;65:145–147. doi: 10.1016/j.neuron.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. The origins of oxidant stress in Parkinson’s disease and therapeutic strategies. Antioxidants Redox Signaling. 2011;14:1289–1301. doi: 10.1089/ars.2010.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Youdim M, Buccafusco J. CNS Targets for multi-functional drugs in the treatment of Alzheimer’s and Parkinson’s diseases. J. Neural Transm. 2005;112:519–537. doi: 10.1007/s00702-004-0214-z. [DOI] [PubMed] [Google Scholar]
- 90.Youdim MBH, Geldenhuys WJ, Van der Schyf CJ. Why should we use multifunctional neuroprotective and neurorestorative drugs for Parkinson’s disease? Parkinsonism Related Disorders. 2007;13:S281–S291. doi: 10.1016/S1353-8020(08)70017-8. [DOI] [PubMed] [Google Scholar]
- 91.Geldenhuys WJ, Youdim MBH, Carroll RT, Van der Schyf CJ. The emergence of designed multiple ligands for neurodegenerative disorders. Prog. Neurobiol. 2011:347–359. doi: 10.1016/j.pneurobio.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 92.Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol. Neurodegener. 2008;3:15. doi: 10.1186/1750-1326-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jones RW. Dimebon disappointment. Alzheimers Res. Ther. 2010;2:25. doi: 10.1186/alzrt49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lermontova N, Redkozubov A, Shevtsova E, Serkova T, Kireeva E, Bachurin S. Dimebon and tacrine inhibit neurotoxic action of β-amyloid in culture and block L-type Ca2+ channels. Bull. Exper. Biol. Med. 2001;132:1079–1083. doi: 10.1023/a:1017972709652. [DOI] [PubMed] [Google Scholar]
- 95.Grigor’ev V, Dranyi O, Bachurin S. Comparative study of action mechanisms of dimebon and memantine on AMPA-and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull. Exper. Biol. Med. 2003;136:474–477. doi: 10.1023/b:bebm.0000017097.75818.14. [DOI] [PubMed] [Google Scholar]
- 96.Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson’s disease. Functional Neurol. 2010;25:65–71. [PubMed] [Google Scholar]
- 97.Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson’s disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem. Pharmacol. 2006;72:1197–1206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 98.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behavioural Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 99.X Sureda F, Junyent F, Verdaguer E, Auladell C, Pelegri C, Vilaplana J, Folch J, Maria Canudas A, Beas Zarate C, Pallas M. Antiapoptotic drugs: a therapeutic strategy for the prevention of neurodegenerative diseases. Curr. Pharm. Des. 2011;17:230–245. doi: 10.2174/138161211795049732. [DOI] [PubMed] [Google Scholar]
- 100.Malgouris C, Daniel M, Doble A. Neuroprotective effects of riluzole on N-methyl-D-aspartate-or veratridine-induced neurotoxicity in rat hippocampal slices. Neuros. Lett. 1994;177:95–99. doi: 10.1016/0304-3940(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 101.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:233S–241S. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 102.Bryson HM, Fulton B, Benfield P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs. 1996;52:549–563. doi: 10.2165/00003495-199652040-00010. [DOI] [PubMed] [Google Scholar]
- 103.Jankovic J, Hunter C. A double-blind, placebo-controlled and longitudinal study of riluzole in early Parkinson’s disease. Parkinsonism Related Disorders. 2002;8:271–276. doi: 10.1016/s1353-8020(01)00040-2. [DOI] [PubMed] [Google Scholar]
- 104.Landwehrmeyer GB, Dubois B, de Yébenes JG, Kremer B, Gaus W, Kraus PH, Przuntek H, Dib M, Doble A, Fischer W. Riluzole in Huntington’s disease: a 3-year, randomized controlled study. Ann. Neurol. 2007;62:262–272. doi: 10.1002/ana.21181. [DOI] [PubMed] [Google Scholar]
- 105.Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nicoletti F, Bruno V, Copani A, Casabona G, Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 107.Caraci F, Battaglia G, Sortino MA, Spampinato S, Molinaro G, Copani A, Nicoletti F, Bruno V. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem. Int. 2012;61:559–565. doi: 10.1016/j.neuint.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 108.Gregory KJ, Dong EN, Meiler J, Conn PJ. Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacol. 2011;60:66–81. doi: 10.1016/j.neuropharm.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Striggow F, Ehrlich BE. Ligand-gated calcium channels inside and out. Curr. Opin. Cell. Biol. 1996;8:490–495. doi: 10.1016/s0955-0674(96)80025-1. [DOI] [PubMed] [Google Scholar]
- 110.Zamponi GW. Voltage-Gated Calcium Channels. Kluwer Academic; New York: 2005. [Google Scholar]
- 111.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 112.Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev. 2003;55:579–581. doi: 10.1124/pr.55.4.8. [DOI] [PubMed] [Google Scholar]
- 113.Eisenberg MJ, Brox A, Bestawros AN. Calcium channel blockers: an update. Am. J. Med. 2004;116:35–43. doi: 10.1016/j.amjmed.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 114.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem. Soc. Trans. 2006;34:903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 115.Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J. Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Epstein BJ, Vogel K, Palmer BF. Dihydropyridine calcium channel antagonists in the management of hypertension. Drugs. 2007;67:1309–1327. doi: 10.2165/00003495-200767090-00005. [DOI] [PubMed] [Google Scholar]
- 117.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 118.Brevetti G, Bonaduce D, Breglio R, Perna S, Simonelli P, Marconi R, Campanella G. Parkinson’s disease and hypotension: 24-hour blood pressure recording in ambulant patients. Clin. Cardiol. 1990;13:474–478. doi: 10.1002/clc.4960130709. [DOI] [PubMed] [Google Scholar]
- 119.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 120.Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang CC, Cao S, Kang S, Kai L, Tian X, Pandey P, Dunne SF, Luan CH, Surmeier DJ, Silverman RB. Antagonism of 4-substituted 1,4-dihydropyridine-3,5-dicarboxylates toward voltage-dependent L-type Ca2+ channels Ca V 1.3 and Ca V 1.2. Bioorg. Med. Chem. 2010;18:3147–3158. doi: 10.1016/j.bmc.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 122.Sorkin EM, Clissold SP, Brogden R. N. Nifedipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy, in ischaemic heart disease, hypertension and related cardiovascular disorders. Drugs. 1985;30:182–274. doi: 10.2165/00003495-198530030-00002. [DOI] [PubMed] [Google Scholar]
- 123.Xia G, Benmohamed R, Kim J, Arvanites AC, Morimoto RI, Ferrante RJ, Kirsch DR, Silverman RB. Pyrimidine-2,4,6-trione derivatives and their inhibition of mutant SOD1-dependent protein aggregation. Toward a treatment for amyotrophic lateral sclerosis. J. Med. Chem. 2011;54:2409–2421. doi: 10.1021/jm101549k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, Silverman RB. Ca(V)1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012;3:1146. doi: 10.1038/ncomms2149. [DOI] [PubMed] [Google Scholar]
- 125.Nakamura T, Lipton SA. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 2009;14:455–468. doi: 10.1007/s10495-008-0301-y. [DOI] [PubMed] [Google Scholar]
- 126.Chung KKK, David KK. Emerging roles of nitric oxide in neurodegeneration. Nitric Oxide. 2010;22:290–295. doi: 10.1016/j.niox.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 127.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 128.Chung KKK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 129.Yao DD, Gu ZZ, Nakamura TT, Shi Z-QZ, Ma YY, Gaston BB, Palmer LAL, Rockenstein EME, Zhang ZZ, Masliah EE, Uehara TT, Lipton SAS. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog. Brain. Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 131.Martínez-Ruiz A, Villanueva L, González de Orduña C, López-Ferrer D, Higueras MA, Tarín C, Rodríguez-Crespo I, Vázquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tsang AHK, Chung KKK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 133.Daff S. NO synthase: structures and mechanisms. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 134.Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Selective Neuronal Nitric Oxide Synthase Inhibitors. Curr. Top. Med. Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]
- 135.Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–8517. doi: 10.1021/bi00084a017. [DOI] [PubMed] [Google Scholar]
- 136.Yamamoto K, Shimamura K, Sekiguchi F, Sunano S. Effects of NG-nitro-L-arginine on the blood pressure of spontaneously hypertensive rats with different degrees of hypertension. Clin. Exper. Hypertension. 2001;23:533–544. doi: 10.1081/ceh-100106824. [DOI] [PubMed] [Google Scholar]
- 137.Silverman RB, Huang H, Marletta MA, Martásek P. Selective Inhibition of Neuronal Nitric Oxide Synthase by Nω-Nitroarginine- and Phenylalanine-Containing Dipeptides and Dipeptide Esters. J. Med. Chem. 1997;40:2813–2817. doi: 10.1021/jm970200u. [DOI] [PubMed] [Google Scholar]
- 138.Huang H, Martásek P, Roman LJ, Masters BSS, Silverman RB. Nω-Nitroarginine-Containing Dipeptide Amides. Potent and Highly Selective Inhibitors of Neuronal Nitric Oxide Synthase. J. Med. Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 139.Huang H, Martásek P, Roman LJ, Silverman RB. Synthesis and evaluation of peptidomimetics as selective inhibitors and active site probes of nitric oxide synthases. J. Med. Chem. 2000;43:2938–2945. doi: 10.1021/jm000127z. [DOI] [PubMed] [Google Scholar]
- 140.Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and selective conformationally restricted neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 141.Hah J-M, Roman LJ, Martásek P, Silverman RB. Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N'-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 142.Hah J-M, Martásek P, Roman LJ, Silverman RB. Aromatic reduced amide bond peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- 143.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 144.Raman CS, Li H, Martásek P, Král V, Masters BSS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 145.Ji H, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Discovery of highly potent and selective inhibitors of neuronal nitric oxide synthase by fragment hopping. J. Med. Chem. 2009;52:779–797. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal Pharmacophoric Elements and Fragment Hopping, an Approach Directed at Molecular Diversity and Isozyme Selectivity. Design of Selective Neuronal Nitric Oxide Synthase Inhibitors. J. Am. Chem. Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann. Neurol. 2009;65:209–217. doi: 10.1002/ana.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of a cerebral palsy phenotype in an animal model. J. Am. Chem. Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ji H, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Exploration of the active site of neuronal nitric oxide synthase by the design and synthesis of pyrrolidinomethyl 2-aminopyridine derivatives. J. Med. Chem. 2010;53:7804–7824. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xue F, Fang J, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Symmetric Double-Headed Aminopyridines, a Novel Strategy for Potent and Membrane-Permeable Inhibitors of Neuronal Nitric Oxide Synthase. J. Med. Chem. 2011;54:2039–2048. doi: 10.1021/jm101071n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Faraci WS, Nagel AA, Verdries KA, Vincent LA, Xu H, Nichols LE, Labasi JM, Salter ED, Pettipher ER. 2-Amino-4-methylpyridine as a potent inhibitor of inducible NO synthase activity in vitro and in vivo. Br. J. Pharmacol. 1996;119:1101–1108. doi: 10.1111/j.1476-5381.1996.tb16010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hagmann WK, Caldwell CG, Chen P, Durette PL, Esser CK, Lanza TJ, Kopka IE, Guthikonda R, Shah SK, MacCoss M, Chabin RM, Fletcher D, Grant SK, Green BG, Humes JL, Kelly TM, Luell S, Meurer R, Moore V, Pacholok SG, Pavia T, Williams HR, Wong KK. Substituted 2-aminopyridines as inhibitors of nitric oxide synthases. Bioorg. Med. Chem. Lett. 2000;10:1975–1978. doi: 10.1016/s0960-894x(00)00389-9. [DOI] [PubMed] [Google Scholar]
- 153.Lowe JA, Qian W, Volkmann RA, Heck S, Nowakowski J, Nelson R, Nolan C, Liston D, Ward K, Zorn S, Johnson C, Vanase M, Faraci WS, Verdries KA, Baxter J, Doran S, Sanders M, Ashton M, Whittle P, Stefaniak M. A new class of selective and potent inhibitors of neuronal nitric oxide synthase. Bioorg. Med. Chem. Lett. 1999;9:2569–2572. doi: 10.1016/s0960-894x(99)00432-1. [DOI] [PubMed] [Google Scholar]
- 154.Lowe JA, Qian W, Drozda SE, Volkmann RA, Nason D, Nelson RB, Nolan C, Liston D, Ward K, Faraci S, Verdries K, Seymour P, Majchrzak M, Villalobos A, White WF. Structure-activity relationships of potent, selective inhibitors of neuronal nitric oxide synthase based on the 6-phenyl-2-aminopyridine structure. J. Med. Chem. 2004;47:1575–1586. doi: 10.1021/jm030519g. [DOI] [PubMed] [Google Scholar]
- 155.McCall TB, Feelisch M, Palmer RM, Moncada S. Identification of N-iminoethyl-L-ornithine as an irreversible inhibitor of nitric oxide synthase in phagocytic cells. Br. J. Pharmacol. 1991;102:234–238. doi: 10.1111/j.1476-5381.1991.tb12159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Collins JL, Shearer BG, Oplinger JA, Lee S, Garvey EP, Salter M, Duffy C, Burnette TC, Furfine ES. N-Phenylamidines as selective inhibitors of human neuronal nitric oxide synthase: structure-activity studies and demonstration of in vivo activity. J. Med. Chem. 1998;41:2858–2871. doi: 10.1021/jm980072p. [DOI] [PubMed] [Google Scholar]
- 157.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 158.Maccallini C, Patruno A, Lannutti F, Ammazzalorso A, De Filippis B, Fantacuzzi M, Franceschelli S, Giampietro L, Masella S, Felaco M, Re N, Amoroso R. N-Substituted acetamidines and 2-methylimidazole derivatives as selective inhibitors of neuronal nitric oxide synthase. Bioorg. Med. Chem. Lett. 2010;20:6495–6499. doi: 10.1016/j.bmcl.2010.09.059. [DOI] [PubMed] [Google Scholar]
- 159.Reif DW, McCarthy DJ, Cregan E, Macdonald JE. Discovery and development of neuronal nitric oxide synthase inhibitors. Free Radic. Biol. Med. 2000;28:1470–1477. doi: 10.1016/s0891-5849(00)00250-1. [DOI] [PubMed] [Google Scholar]
- 160.O’Neill MJ, Murray TK, McCarty DR, Hicks CA, Dell CP, Patrick KE, Ward MA, Osborne DJ, Wiernicki TR, Roman CR, Lodge D, Fleisch JH, Singh J. ARL 17477, a selective nitric oxide synthase inhibitor, with neuroprotective effects in animal models of global and focal cerebral ischaemia. Brain Res. 2000;871:234–244. doi: 10.1016/s0006-8993(00)02471-9. [DOI] [PubMed] [Google Scholar]
- 161.Fedorov R, Vasan R, Ghosh DK, Schlichting I. Structures of nitric oxide synthase isoforms complexed with the inhibitor AR-R17477 suggest a rational basis for specificity and inhibitor design. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5892–5897. doi: 10.1073/pnas.0306588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Renton P, Green B, Maddaford SP, Rakhit S, Andrews JS. Novel Dual Action Neuronal Nitric Oxide Synthase Inhibitors with μ-Opioid Agonist Activity. ACS Med. Chem. Lett. 2012;3:227–231. doi: 10.1021/ml200268w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Annedi SC, Ramnauth J, Maddaford SP, Renton P, Rakhit S, Mladenova G, Dove P, Silverman S, Andrews JS, Felice MD, Porreca F. Discovery of cis-N-(1-(4-(Methylamino)cyclohexyl)indolin-6-yl)thiophene-2-carboximidamide: A 1,6-Disubstituted Indoline Derivative as a Highly Selective Inhibitor of Human Neuronal Nitric Oxide Synthase (nNOS) without Any Cardiovascular Liabilities. J. Med. Chem. 2012;55:943–955. doi: 10.1021/jm201564u. [DOI] [PubMed] [Google Scholar]
- 164.Maddaford S, Annedi SC, Ramnauth J, Rakhit S. Advancements in the Development of Nitric Oxide Synthase Inhibitors. Annual Rep. Med. Chem. 2009;44:27–50. [Google Scholar]
- 165.Serafim RA, Primi MC, Trossini GH, Ferreira EI. Nitric oxide: state of the art in drug design. Curr. Med. Chem. 2012;19:386–405. doi: 10.2174/092986712803414321. [DOI] [PubMed] [Google Scholar]
- 166.Payne JE, Bonnefous C, Symons KT, Nguyen PM, Sablad M, Rozenkrants N, Zhang Y, Wang L, Yazdani N, Shiau AK, Noble SA, Rix P, Rao TS, Hassig CA, Smith ND. Discovery of Dual Inducible/Neuronal Nitric Oxide Synthase (iNOS/nNOS) Inhibitor Development Candidate 4-(2-Cyclobutyl-1H-imidazo[4,5-b]pyrazin-1-yl)methyl)-7,8-difluoroquinolin-2(1H)-one (KD7332) Part 2: Identification of a Novel, Potent, and Selective Series of Benzimidazole-Quinolinone iNOS/nNOS Dimerization Inhibitors That Are Orally Active in Pain Models. J. Med. Chem. 2010;53:7739–7755. doi: 10.1021/jm100828n. [DOI] [PubMed] [Google Scholar]
- 167.Matter H, Kumar HSA, Fedorov R, Frey A, Kotsonis P, Hartmann E, Fröhlich LG, Reif A, Pfleiderer W, Scheurer P, Ghosh DK, Schlichting I, Schmidt HHHW. Structural analysis of isoform-specific inhibitors targeting the tetrahydrobiopterin binding site of human nitric oxide synthases. J. Med. Chem. 2005;48:4783–4792. doi: 10.1021/jm050007x. [DOI] [PubMed] [Google Scholar]
- 168.Behl C. Alzheimer’s disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 169.Christen Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000;71:621S–629S. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- 170.Harman D. A hypothesis on the pathogenesis of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996;786:152–168. doi: 10.1111/j.1749-6632.1996.tb39059.x. [DOI] [PubMed] [Google Scholar]
- 171.Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA. Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol. Aging. 2001;22:131–146. doi: 10.1016/s0197-4580(00)00211-6. [DOI] [PubMed] [Google Scholar]
- 172.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Prasad KN, Hovland AR, Cole WC, Prasad KC, Nahreini P, Edwards-Prasad J, Andreatta CP. Multiple antioxidants in the prevention and treatment of Alzheimer disease: analysis of biologic rationale. Clin. Neuropharmacol. 2000;23:2–13. doi: 10.1097/00002826-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 174.Beal MF. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001;2:325–334. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- 175.Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 176.Foley P, Riederer P. Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson’s disease. J. Neurol. 2000;247(Suppl 2):II8–294. doi: 10.1007/pl00007766. [DOI] [PubMed] [Google Scholar]
- 177.Gerlach M, Riederer P. Animal models of Parkinson’s disease: an empirical comparison with the phenomenology of the disease in man. J. Neural. Transm. 1996;103:987–1041. doi: 10.1007/BF01291788. [DOI] [PubMed] [Google Scholar]
- 178.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, Kasarskis E, Mattson MP. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- 180.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr. Med. Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 181.Grodstein F, Chen J, Willett WC. High-dose antioxidant supplements and cognitive function in community-dwelling elderly women. Am. J. Clin. Nutr. 2003;77:975–984. doi: 10.1093/ajcn/77.4.975. [DOI] [PubMed] [Google Scholar]
- 182.Laurin D, Masaki KH, Foley DJ, White LR, Launer LJ. Midlife dietary intake of antioxidants and risk of late-life incident dementia: the Honolulu-Asia Aging Study. Am. J. Epidemiol. 2004;159:959–967. doi: 10.1093/aje/kwh124. [DOI] [PubMed] [Google Scholar]
- 183.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 184.Hayden KM, Welsh-Bohmer KA, Wengreen HJ, Zandi PP, Lyketsos CG, Breitner JC. Risk of mortality with vitamin E supplements: the Cache County study. Am. J. Med. 2007;120:180–184. doi: 10.1016/j.amjmed.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 185.Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch. Neurol. 2003;60:203–208. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 186.Sinha K, Chaudhary G, Gupta YK. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci. 2002;71:655–665. doi: 10.1016/s0024-3205(02)01691-0. [DOI] [PubMed] [Google Scholar]
- 187.The Parkinson Study Group Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 188.Kieburtz K, McDermott M, Como P, Growdon J, Brady J, Carter J, Huber S, Kanigan B, Landow E, Rudolph A, Saint-Cyr J, Stern Y, Tennis M, Thelen J, Soulson I, Parkinson Study Group The effect of deprenyl and tocopherol on cognitive performance in early untreated Parkinson’s disease. Neurology. 1994;44:1756–1759. doi: 10.1212/wnl.44.9.1756. Parkinson Study Group. [DOI] [PubMed] [Google Scholar]
- 189.Mihatsch W, Russ H, Gerlach M, Riederer P, Przuntek H. Treatment with antioxidants does not prevent loss of dopamine in the striatum of MPTP-treated common marmosets: preliminary observations. J. Neural. Transm. Park. Dis. Dement. Sect. 1991;3:73–78. doi: 10.1007/BF02251138. [DOI] [PubMed] [Google Scholar]
- 190.Perry TL, Yong VW, Hansen S, Jones K, Bergeron C, Foulks JG, Wright JM. Alpha-tocopherol and beta-carotene do not protect marmosets against the dopaminergic neurotoxicity of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurol. Sci. 1987;81:321–331. doi: 10.1016/0022-510x(87)90106-7. [DOI] [PubMed] [Google Scholar]
- 191.Peyser CE, Folstein M, Chase GA, Starkstein S, Brandt J, Cockrell JR, Bylsma F, Coyle JT, McHugh PR, Folstein SE. Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry. 1995;152:1771–1775. doi: 10.1176/ajp.152.12.1771. [DOI] [PubMed] [Google Scholar]
- 192.Desnuelle C, Dib M, Garrel C, Favier A, ALS riluzole-tocopherol Study Group A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2001;2:9–18. doi: 10.1080/146608201300079364. [DOI] [PubMed] [Google Scholar]
- 193.Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- 194.Asthana S, Baker LD, Craft S, Stanczyk FZ, Veith RC, Raskind MA, Plymate SR. High-dose estradiol improves cognition for women with AD: results of a randomized study. Neurology. 2001;57:605–612. doi: 10.1212/wnl.57.4.605. [DOI] [PubMed] [Google Scholar]
- 195.Carlson MC, Zandi PP, Plassman BL, Tschanz JT, Welsh-Bohmer KA, Steffens DC, Bastian LA, Mehta KM, Breitner JC. Hormone replacement therapy and reduced cognitive decline in older women: the Cache County Study. Neurology. 2001;57:2210–2216. doi: 10.1212/wnl.57.12.2210. [DOI] [PubMed] [Google Scholar]
- 196.Henderson VW, Paganini-Hill A, Miller BL, Elble RJ, Reyes PF, Shoupe D, McCleary CA, Klein RA, Hake AM, Farlow MR. Estrogen for Alzheimer’s disease in women: randomized, double-blind, placebo-controlled trial. Neurology. 2000;54:295–301. doi: 10.1212/wnl.54.2.295. [DOI] [PubMed] [Google Scholar]
- 197.Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer’s Disease Cooperative Study. J. Am. Med. Assoc. 2000;283:1007–1015. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- 198.Wang PN, Liao SQ, Liu RS, Liu CY, Chao HT, Lu SR, Yu HY, Wang SJ, Liu HC. Effects of estrogen on cognition, mood, and cerebral blood flow in AD: a controlled study. Neurology. 2000;54:2061–2066. doi: 10.1212/wnl.54.11.2061. [DOI] [PubMed] [Google Scholar]
- 199.Blanchet PJ, Fang J, Hyland K, Arnold LA, Mouradian MM, Chase TN. Short-term effects of high-dose 17beta-estradiol in postmenopausal PD patients: a crossover study. Neurology. 1999;53:91–95. doi: 10.1212/wnl.53.1.91. [DOI] [PubMed] [Google Scholar]
- 200.Fernandez HH, Lapane KL. Estrogen use among nursing home residents with a diagnosis of Parkinson’s disease. Mov. Disord. 2000;15:1119–1124. doi: 10.1002/1531-8257(200011)15:6<1119::aid-mds1009>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 201.Tsang KL, Ho SL, Lo SK. Estrogen improves motor disability in parkinsonian postmenopausal women with motor fluctuations. Neurology. 2000;54:2292–2298. doi: 10.1212/wnl.54.12.2292. [DOI] [PubMed] [Google Scholar]
- 202.Baldereschi M, Di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, Inzitari D. Parkinson’s disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology. 2000;55:1358–1363. doi: 10.1212/wnl.55.9.1358. [DOI] [PubMed] [Google Scholar]
- 203.Mayeux R, Marder K, Cote LJ, Denaro J, Hemenegildo N, Mejia H, Tang MX, Lantigua R, Wilder D, Gurland B, Hauser A. The frequency of idiopathic Parkinson’s disease by age, ethnic group, and sex in northern Manhattan, 1988-1993. Am. J. Epidemiol. 1995;142:820–827. doi: 10.1093/oxfordjournals.aje.a117721. [DOI] [PubMed] [Google Scholar]
- 204.Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009;41:40–59. doi: 10.1016/j.biocel.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Richard T, Pawlus AD, Iglesias ML, Pedrot E, Waffo-Teguo P, Merillon JM, Monti JP. Neuroprotective properties of resveratrol and derivatives. Ann. N. Y. Acad. Sci. 2011;1215:103–108. doi: 10.1111/j.1749-6632.2010.05865.x. [DOI] [PubMed] [Google Scholar]
- 206.Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J. Nutrition. Biochem. 2002;13:572–584. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- 207.Pietta PG. Flavonoids as antioxidants. J. Nat. Prod. 2000;63:1035–1042. doi: 10.1021/np9904509. [DOI] [PubMed] [Google Scholar]
- 208.Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- 209.Owuor ED, Kong AN. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002;64:765–770. doi: 10.1016/s0006-2952(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 210.Filomeni G, Rotilio G, Ciriolo MR. Cell signalling and the glutathione redox system. Biochem. Pharmacol. 2002;64:1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 211.Paolicchi A, Dominici S, Pieri L, Maellaro E, Pompella A. Glutathione catabolism as a signaling mechanism. Biochem. Pharmacol. 2002;64:1027–1035. doi: 10.1016/s0006-2952(02)01173-5. [DOI] [PubMed] [Google Scholar]
- 212.Fridovich I. Superoxide anion radical (O22.), superoxide dismutases, and related matters. J. Biol. Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 213.Masumoto H, Sies H. The reaction of 2-(methylseleno)benzanilide with peroxynitrite. Chem. Res. Toxicol. 1996;9:1057–1062. doi: 10.1021/tx9600560. [DOI] [PubMed] [Google Scholar]
- 214.Parnham M, Sies H. Ebselen: prospective therapy for cerebral ischaemia. Expert Opin. Investig. Drugs. 2000;9:607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- 215.Moussaoui S, Obinu MC, Daniel N, Reibaud M, Blanchard V, Imperato A. The antioxidant ebselen prevents neurotoxicity and clinical symptoms in a primate model of Parkinson’s disease. Exp. Neurol. 2000;166:235–245. doi: 10.1006/exnr.2000.7516. [DOI] [PubMed] [Google Scholar]
- 216.Aitken JB, Lay PA, Duong TT, Aran R, Witting PK, Harris HH, Lai B, Vogt S, Giles GI. Synchrotron radiation induced X-ray emission studies of the antioxidant mechanism of the organoselenium drug ebselen. J. Biol. Inorg. Chem. 2012;17:589–598. doi: 10.1007/s00775-012-0879-y. [DOI] [PubMed] [Google Scholar]
- 217.d’Alessio P, Moutet M, Coudrier E, Darquenne S, Chaudiere J. ICAM-1 and VCAM-1 expression induced by TNF-alpha are inhibited by a glutathione peroxidase mimic. Free Radic. Biol. Med. 1998;24:979–987. doi: 10.1016/s0891-5849(97)00396-1. [DOI] [PubMed] [Google Scholar]
- 218.Moutet M, d’Alessio P, Malette P, Devaux V, Chaudiere J. Glutathione peroxidase mimics prevent TNFalpha- and neutrophil-induced endothelial alterations. Free Radic. Biol. Med. 1998;25:270–281. doi: 10.1016/s0891-5849(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 219.Batinic-Haberle I, Benov L, Spasojevic I, Fridovich I. The ortho effect makes manganese(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J. Biol. Chem. 1998;273:24521–24528. doi: 10.1074/jbc.273.38.24521. [DOI] [PubMed] [Google Scholar]
- 220.Bowler RP, Sheng H, Enghild JJ, Pearlstein RD, Warner DS, Crapo JD. A catalytic antioxidant (AEOL 10150) attenuates expression of inflammatory genes in stroke. Free Radic. Biol. Med. 2002;33:1141–1152. doi: 10.1016/s0891-5849(02)01008-0. [DOI] [PubMed] [Google Scholar]
- 221.Patel M, Day BJ. Metalloporphyrin class of therapeutic catalytic antioxidants. Trends Pharmacol. Sci. 1999;20:359–364. doi: 10.1016/s0165-6147(99)01336-x. [DOI] [PubMed] [Google Scholar]
- 222.Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J. Biol. Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- 223.Peng J, Stevenson FF, Doctrow SR, Andersen JK. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. J. Biol. Chem. 2005;280:29194–29198. doi: 10.1074/jbc.M500984200. [DOI] [PubMed] [Google Scholar]
- 224.Young AJ, Lowe GM. Antioxidant and prooxidant properties of carotenoids. Arch. Biochem. Biophys. 2001;385:20–27. doi: 10.1006/abbi.2000.2149. [DOI] [PubMed] [Google Scholar]
- 225.Di Mascio P, Kaiser S, Sies H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch. Biochem. Biophys. 1989;274:532–538. doi: 10.1016/0003-9861(89)90467-0. [DOI] [PubMed] [Google Scholar]
- 226.Burton GW, Ingold KU. beta-Carotene: an unusual type of lipid antioxidant. Science. 1984;224:569–573. doi: 10.1126/science.6710156. [DOI] [PubMed] [Google Scholar]
- 227.Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 2003;100:2901–2905. doi: 10.1073/pnas.0437937100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Goodman AB. Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol. 2006;209:598–603. doi: 10.1002/jcp.20784. [DOI] [PubMed] [Google Scholar]
- 229.Ding Y, Qiao A, Wang Z, Goodwin JS, Lee ES, Block ML, Allsbrook M, McDonald MP, Fan GH. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J. Neurosci. 2008;28:11622–11634. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mann KG. Biochemistry and physiology of blood coagulation. Thromb. Haemost. 1999;82:165–174. [PubMed] [Google Scholar]
- 231.Price PA. Role of vitamin-K-dependent proteins in bone metabolism. Annu. Rev. Nutr. 1988;8:565–583. doi: 10.1146/annurev.nu.08.070188.003025. [DOI] [PubMed] [Google Scholar]
- 232.Allison AC. The possible role of vitamin K deficiency in the pathogenesis of Alzheimer’s disease and in augmenting brain damage associated with cardiovascular disease. Med. Hypotheses. 2001;57:151–155. doi: 10.1054/mehy.2001.1307. [DOI] [PubMed] [Google Scholar]
- 233.Beal MF, Shults CW. Effects of Coenzyme Q10 in Huntington’s disease and early Parkinson’s disease. Biofactors. 2003;18:153–161. doi: 10.1002/biof.5520180218. [DOI] [PubMed] [Google Scholar]
- 234.Bhat V, Weiner WJ. Parkinson’s disease. Diagnosis and the initiation of therapy. Minerva Med. 2005;96:145–154. [PubMed] [Google Scholar]
- 235.Yang X, Yang Y, Li G, Wang J, Yang ES. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J. Mol. Neurosci. 2008;34:165–171. doi: 10.1007/s12031-007-9033-7. [DOI] [PubMed] [Google Scholar]
- 236.Yang X, Dai G, Li G, Yang ES. Coenzyme Q10 reduces beta-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010;41:110–113. doi: 10.1007/s12031-009-9297-1. [DOI] [PubMed] [Google Scholar]
- 237.Skulachev VP. Mitochondria-targeted antioxidants as promising drugs for treatment of age-related brain diseases. J. Alzheimers Dis. 2012;28:283–289. doi: 10.3233/JAD-2011-111391. [DOI] [PubMed] [Google Scholar]
- 238.Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm. Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- 239.Floyd RA, Hensley K. Nitrone inhibition of age-associated oxidative damage. Ann. N. Y. Acad. Sci. 2000;899:222–237. doi: 10.1111/j.1749-6632.2000.tb06189.x. [DOI] [PubMed] [Google Scholar]
- 240.Floyd RA, Hensley K, Bing G. Evidence for enhanced neuro-inflammatory processes in neurodegenerative diseases and the action of nitrones as potential therapeutics. J. Neural. Transm. Suppl. 2000:387–414. doi: 10.1007/978-3-7091-6301-6_28. [DOI] [PubMed] [Google Scholar]
- 241.Das A, Gopalakrishnan B, Voss OH, Doseff AI, Villamena FA. Inhibition of ROS-induced apoptosis in endothelial cells by nitrone spin traps via induction of phase II enzymes and suppression of mitochondria-dependent pro-apoptotic signaling. Biochem. Pharmacol. 2012;84:486–497. doi: 10.1016/j.bcp.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Floyd RA, Hensley K, Jaffery F, Maidt L, Robinson K, Pye Q, Stewart C. Increased oxidative stress brought on by pro-inflammatory cytokines in neurodegenerative processes and the protective role of nitrone-based free radical traps. Life Sci. 1999;65:1893–1899. doi: 10.1016/s0024-3205(99)00443-9. [DOI] [PubMed] [Google Scholar]
- 243.Floyd RA, Hensley K, Forster MJ, Kelleher-Andersson JA, Wood PL. Nitrones, their value as therapeutics and probes to understand aging. Mech. Ageing Dev. 2002;123:1021–1031. doi: 10.1016/s0047-6374(01)00385-2. [DOI] [PubMed] [Google Scholar]
- 244.Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 245.Klivenyi P, Matthews RT, Wermer M, Yang L, MacGarvey U, Becker DA, Natero R, Beal MF. Azulenyl nitrone spin traps protect against MPTP neurotoxicity. Exp. Neurol. 1998;152:163–166. doi: 10.1006/exnr.1998.6824. [DOI] [PubMed] [Google Scholar]
- 246.Yang L, Calingasan NY, Chen J, Ley JJ, Becker DA, Beal MF. A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicities. Exp. Neurol. 2005;191:86–93. doi: 10.1016/j.expneurol.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 247.Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, Chen S, Corpe C, Dutta A, Dutta SK, Levine M. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003;22:18–35. doi: 10.1080/07315724.2003.10719272. [DOI] [PubMed] [Google Scholar]
- 248.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Dringen R, Hamprecht B. N-acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neurosci. Lett. 1999;259:79–82. doi: 10.1016/s0304-3940(98)00894-5. [DOI] [PubMed] [Google Scholar]
- 250.Cudkowicz ME, Sexton PM, Ellis T, Hayden DL, Gwilt PR, Whalen J, Brown RH., Jr. The pharmacokinetics and pharmaco-dynamics of Procysteine in amyotrophic lateral sclerosis. Neurology. 1999;52:1492–1494. doi: 10.1212/wnl.52.7.1492. [DOI] [PubMed] [Google Scholar]
- 251.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74:1526–1539. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- 253.Magesh S, Chen Y, Hu LQ. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012;32:687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J, Johnson JA. The Nrf2/ARE Pathway as a Potential Therapeutic Target in Neurodegenerative Disease. Antioxidants Redox Signaling. 2009;11:497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Jung KA, Kwak MK. The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules. 2010;15:7266–7291. doi: 10.3390/molecules15107266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75(NTR)-dependent motor neuron apoptosis. J. Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 257.Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J, Shaw PJ. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–1706. doi: 10.1093/brain/awh503. [DOI] [PubMed] [Google Scholar]
- 258.Trinh K, Moore K, Wes PD, Muchowski PJ, Dey J, Andrews L, Pallanck LJ. Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson’s disease. J. Neurosci. 2008;28:465–472. doi: 10.1523/JNEUROSCI.4778-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exper. Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 262.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 263.Bartolini M, Andrisano V. Strategies for the inhibition of protein aggregation in human diseases. ChemBioChem. 2010;11:1018–1035. doi: 10.1002/cbic.200900666. [DOI] [PubMed] [Google Scholar]
- 264.Schneider A, Mandelkow E. Tau based treatment strategies in neurodegenerative diseases. Neurother. 2008;5:443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Shelton SB, Johnson GV. Cyclin-dependent kinase-5 in neurodegeneration. J. Neurochem. 2004;88:1313–1326. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- 266.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 267.Meijer L, Thunnissen AMWH, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkow E, Kim SH, Pettit GR. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 268.Palomo V, Perez DI, Gil C, Martinez A. The potential role of glycogen synthase kinase 3 inhibitors as amyotrophic lateral sclerosis pharmacological therapy. Curr. Med. Chem. 2011;18:3028–3034. doi: 10.2174/092986711796391697. [DOI] [PubMed] [Google Scholar]
- 269.Chen PC, Gaisina IN, El-Khodor BF, Ramboz S, Makhortova NR, Rubin LL, Kozikowski AP. Identification of a Maleimide-Based Glycogen Synthase Kinase-3 (GSK23) Inhibitor, BIP-135, That Prolongs the Median Survival Time of Delta 7 SMA KO Mouse Model of Spinal Muscular Atrophy. ACS Chem. Neurosci. 2012;3:5–11. doi: 10.1021/cn200085z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Koh SH, Kim Y, Kim HY, Hwang S, Lee CH, Kim SH. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007;205:336–346. doi: 10.1016/j.expneurol.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 271.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ballatore C, Brunden KR, Trojanowski JQ, Lee VM, Smith AB, 3rd, Huryn DM. Modulation of protein-protein interactions as a therapeutic strategy for the treatment of neurodegenerative tauopathies. Curr. Top. Med. Chem. 2011;11:317–330. doi: 10.2174/156802611794072605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U. S. A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ballatore C, Crowe A, Piscitelli F, James M, Lou K, Rossidivito G, Yao Y, Trojanowski JQ, Lee VM, Brunden KR, Smith AB., 3rd Aminothienopyridazine inhibitors of tau aggregation: evaluation of structure-activity relationship leads to selection of candidates with desirable in vivo properties. Bioorg. Med. Chem. 2012;20:4451–4461. doi: 10.1016/j.bmc.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Messing L, Decker JM, Joseph M, Mandelkow E, Mandelkow EM. Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol. Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.10.024. in press, doi: 10.1016/j.neurobiolaging.2012.1010.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ghosh AK, Brindisi M, Tang J. Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012;120:71–83. doi: 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, Monk SA, Mathes BM, Mergott DJ, Watson BM, Stout SL, Timm DE, Smith Labell E, Gonzales CR, Nakano M, Jhee SS, Yen M, Ereshefsky L, Lindstrom TD, Calligaro DO, Cocke PJ, Greg Hall D, Friedrich S, Citron M, Audia JE. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J. Neurosci. 2011;31:16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.D’Onofrio G, Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Paroni G, Cascavilla L, Seripa D, Pilotto A. Advances in the identification of γ-secretase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Discov. 2012;7:19–37. doi: 10.1517/17460441.2012.645534. [DOI] [PubMed] [Google Scholar]
- 279.Gillman KW, Starrett JE, Parker MF, Xie K, Bronson JJ, Marcin LR, McElhone KE, Bergstrom CP, Mate RA, Williams R, Meredith JE, Burton CR, Barten DM, Toyn JH, Roberts SB, Lentz KA, Houston JG, Zaczek R, Albright CF, Decicco CP, Macor JE, Olson RE. Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable gamma-Secretase Inhibitor. ACS Med. Chem. Lett. 2010;1:120–124. doi: 10.1021/ml1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Panza F, Solfrizzi V, Frisardi V, Capurso C, D’Introno A, Colacicco AM, Vendemiale G, Capurso A, Imbimbo BP. Disease-modifying approach to the treatment of Alzheimer’s disease: from alpha-secretase activators to gamma-secretase inhibitors and modulators. Drugs Aging. 2009;26:537–555. doi: 10.2165/11315770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 281.Khan TK, Nelson TJ, Verma VA, Wender PA, Alkon DL. A cellular model of Alzheimer’s disease therapeutic efficacy: PKC activation reverses Abeta-induced biomarker abnormality on cultured fibroblasts. Neurobiol. Dis. 2009;34:332–339. doi: 10.1016/j.nbd.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Wender PA, Baryza JL, Brenner SE, DeChristopher BA, Loy BA, Schrier AJ, Verma VA. Design, synthesis, and evaluation of potent bryostatin analogs that modulate PKC translocation selectivity. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6721–6726. doi: 10.1073/pnas.1015270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Simons M, Keller P, Dichgans J, Schulz JB. Cholesterol and Alzheimer’s disease: is there a link? Neurology. 2001;57:1089–1093. doi: 10.1212/wnl.57.6.1089. [DOI] [PubMed] [Google Scholar]
- 284.Simons M, Schwarzler F, Lutjohann D, von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002;52:346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 285.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat. Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 286.Roy A, Pahan K. Prospects of statins in Parkinson disease. Neuroscientist. 2011;17:244–255. doi: 10.1177/1073858410385006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Valenza M, Cattaneo E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011;34:474–486. doi: 10.1016/j.tins.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 288.Bate C, Tayebi M, Williams A. Sequestration of free cholesterol in cell membranes by prions correlates with cytoplasmic phospholipase A-activation. BMC Biol. 2008;6:8. doi: 10.1186/1741-7007-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Tobert JA. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 290.Arawaka S, Machiya Y, Kato T. Heat shock proteins as suppressors of accumulation of toxic prefibrillar intermediates and misfolded proteins in neurodegenerative diseases. Curr. Pharma. Biotechnol. 2010;11:158–166. doi: 10.2174/138920110790909713. [DOI] [PubMed] [Google Scholar]
- 291.Wyttenbach A, Arrigo AP. The role of heat shock proteins during neurodegeneration in Alzheimer’s, Parkinson’s and Huntington’s Disease. In: Richter-Landsberg C, editor. Heat Shock Proteins in Neural Cells. Springer and R. G. Landes; Georgetown, USA: 2009. pp. 1–5. [Google Scholar]
- 292.Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Luo W, Sun W, Tadone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Trott A, West JD, Klaic L, Westerheide SD, Silverman RB, Morimoto RI, Morano KA. Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell. 2008;19:1104–1112. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Klaic L, Trippier PC, Mishra RK, Morimoto RI, Silverman RB. Remarkable Stereospecific Conjugate Additions to the Hsp90 Inhibitor Celastrol. J. Am. Chem. Soc. 2011;133:19634–19637. doi: 10.1021/ja208359a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- 297.Evans CG, Chang L, Gestwicki JE. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target. J. Med. Chem. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 299.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat. Rev. Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 300.Casarejos MJ, Solano RM, Gomez A, Perucho J, de Yebenes JG, Mena MA. The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem. Int. 2011;58:512–520. doi: 10.1016/j.neuint.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 301.Dennissen FJ, Kholod N, van Leeuwen FW. The ubiquitin proteasome system in neurodegenerative diseases: Culprit, accomplice or victim? Prog. Neurobiol. 2012;96:190–207. doi: 10.1016/j.pneurobio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 302.Luo G-R, Chen S, Le W-D. Are heat shock proteins therapeutic target for Parkinson’s Disease. Int. J. Biol. Sci. 2007;3:20–26. doi: 10.7150/ijbs.3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Zhao JH, Liu HL, Lin HY, Huang CH, Fang HW, Chen SS, Ho Y, Tsai WB, Chen WY. Chemical chaperone and inhibitor discovery: potential treatments for protein conformational diseases. Perspect. Med. Chem. 2008;1:39–48. doi: 10.4137/pmc.s212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ali YO, Kitay BM, Zhai RG. Dealing with Misfolded Proteins: Examining the Neuroprotective Role of Molecular Chaperones in Neurodegeneration. Molecules. 2010;15:6859–6887. doi: 10.3390/molecules15106859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Cuajungco MP, Faget KY, Huang X, Tanzi RE, Bush AI. Metal Chelation as a Potential Therapy for Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2000;920:292–304. doi: 10.1111/j.1749-6632.2000.tb06938.x. [DOI] [PubMed] [Google Scholar]
- 306.Wang X, Wang X, Zhang C, Jiao Y, Guo Z. Inhibitory action of macrocyclic platiniferous chelators on metal-induced Ab aggregation. Chem. Sci. 2012;3:1304–1312. [Google Scholar]
- 307.Wisniewski T, Sadowski M. Preventing beta-amyloid fibrillization and deposition: beta-sheet breakers and pathological chaperone inhibitors. BMC Neurosci. 2008;9(Suppl 2):S5. doi: 10.1186/1471-2202-9-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Simons LJ, Caprathe BW, Callahan M, Graham JM, Kimura T, Lai Y, LeVine H, 3rd, Lipinski W, Sakkab AT, Tasaki Y, Walker LC, Yasunaga T, Ye Y, Zhuang N, Augelli-Szafran CE. The synthesis and structure-activity relationship of substituted N-phenyl anthranilic acid analogs as amyloid aggregation inhibitors. Bioorg. Med. Chem. Lett. 2009;19:654–657. doi: 10.1016/j.bmcl.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 309.Heiser V, Engernann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, Nordhoff E, Lurz R, Schugardt N, Rautenberg S, Herhaus C, Barnickel G, Bottcher H, Lehrach H, Wanker EE. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington’s disease by using an automated filter retardation assay. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16400–16406. doi: 10.1073/pnas.182426599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Geldenhuys WJ, Van der Schyf CJ. Designing drugs with multi-target activity: the next step in the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2013;8:115–129. doi: 10.1517/17460441.2013.744746. [DOI] [PubMed] [Google Scholar]
- 311.Rizzo S, Tarozzi A, Bartolini M, Da Costa G, Bisi A, Gobbi S, Belluti F, Ligresti A, Allara M, Monti JP, Andrisano V, Di Marzo V, Hrelia P, Rampa A. 2-Arylbenzofuran-based molecules as multipotent Alzheimer’s disease modifying agents. Eur. J. Med. Chem. 2012;58:519–532. doi: 10.1016/j.ejmech.2012.10.045. [DOI] [PubMed] [Google Scholar]
- 312.Sorrenti V, Salerno L, Di Giacomo C, Acquaviva R, Siracusa MA, Vanella A. Imidazole derivatives as antioxidants and selective inhibitors of nNOS. Nitric Oxide. 2006;14:45–50. doi: 10.1016/j.niox.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 313.Galdeano C, Viayna E, Sola I, Formosa X, Camps P, Badia A, Clos MV, Relat J, Ratia M, Bartolini M, Mancini F, Andrisano V, Salmona M, Minguillon C, Gonzalez-Munoz GC, Rodriguez-Franco MI. Bidon-Chanal, A.; Luque, F. J.; Munoz-Torrero, D. Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer’s and prion diseases. J. Med. Chem. 2012;55:661–669. doi: 10.1021/jm200840c. [DOI] [PubMed] [Google Scholar]