

Abstract
Halogenated organic matter buried in marine subsurface sediment may serve as a source of electron acceptors for anaerobic respiration of subseafloor microbes. Detection of a diverse array of reductive dehalogenase-homologous (rdhA) genes suggests that subseafloor organohalide-respiring microbial communities may play significant ecological roles in the biogeochemical carbon and halogen cycle in the subseafloor biosphere. We report here the spatial distribution of dehalogenation activity in the Nankai Trough plate-subduction zone of the northwest Pacific off the Kii Peninsula of Japan. Incubation experiments with slurries of sediment collected at various depths and locations showed that degradation of several organohalides tested only occurred in the shallow sedimentary basin, down to 4.7 metres below the seafloor, despite detection of rdhA in the deeper sediments. We studied the phylogenetic diversity of the metabolically active microbes in positive enrichment cultures by extracting RNA, and found that Desulfuromonadales bacteria predominate. In addition, for the isolation of genes involved in the dehalogenation reaction, we performed a substrate-induced gene expression screening on DNA extracted from the enrichment cultures. Diverse DNA fragments were obtained and some of them showed best BLAST hit to known organohalide respirers such as Dehalococcoides, whereas no functionally known dehalogenation-related genes such as rdhA were found, indicating the need to improve the molecular approach to assess functional genes for organohalide respiration.
Keywords: reductive dehalogenation, reductive dehalogenase-homologous gene, substrate-induced gene expression, subseafloor biosphere, biogeochemical carbon and halogen cycle
1. Introduction
Numerous previous microbiological studies demonstrate that microbes play important ecological roles in halogen cycles on our planet. Reductive dehalogenation mediated by organohalide-respiring microorganisms has been used in the bioremediation of soil and groundwater contaminated with compounds such as tetrachloroethene (PCE) and trichloroethene (TCE) [1–3]. Many organohalides are used in industrial processes (e.g. PCE is widely used in the dry cleaning industry), and improper disposal of these chemicals has caused serious environmental pollutions. There are also many naturally produced organohalides. Gribble [4] reported that at least 4714 organohalides have been identified from biotic and abiotic natural sources [4]. For example, both PCE and TCE may also enter the environment through production by various marine algae and as a result of volcanic emissions. In this context, microbial dehalogenation is believed to have played an ecological role in the natural halogen cycle, having arisen through microbial utilization of naturally produced organohalides before anthropogenic release of these compounds into the environment [3,5,6].
The marine aquatic environment is the primary source of naturally occurring organohalides [4]. Microbial reductive dehalogenation has been experimentally demonstrated in both marine and estuarine environments [7]. King [8] was the first to report microbial dehalogenation of 2,4-dibromophenol produced by animal activity in coastal sediments. Dehalogenation activity and the reductive dehalogenase-homologous (rdhA) genes, the key functional genes necessary for organohalide respiration, have also been detected in the microbial community associated with bromophenol-producing marine sponge Aplysina aerophoba [5]. Brominated compounds actually serve as growth supporting electron acceptors. Recently, for example, Dehalococcoides mccartyi strain CBDB1 was shown to have an even wider dehalogenation potential for brominated benzenes than for chlorinated benzenes [9]. The molecular ecological and biochemical studies indicate the significance of enzymatic dehalogenation reactions in marine halogen cycles.
The organohalide-respiring bacteria isolated from marine and estuarine environments to date are members of the genera Desulfomonile, Dehalobium and Dehalococcoides. Desulfomonile limimaris strains DCB-M and DCB-F capable of dechlorinating 3-chlorobenzoate to benzoate were isolated from marine sediments in Florida [10]. Members of the genera Dehalobium and Dehalococcoides belong to the class Dehalococcoidetes (subphylum II) within the phylum Chloroflexi [11–13]. Dehalobium chlorocoercia strain DF-1, isolated from Charleston harbour in South Carolina, is capable of respiring polychlorinated biphenyls [14,15]. Dehalococcoides mccartyi strain MB, isolated from San Francisco Bay, dechlorinates PCE to trans-1,2-dichloroethene (trans-DCE) [12,16]. All of the organohalide-respiring members of the phylum Chloroflexi isolated so far are obligate organohalide respirers. A number of molecular ecological studies have shown that organohalide-respiring Chloroflexi may play significant roles in natural dehalogenation processes in estuarine and tidal flat sediments [1,17–20].
Previous molecular ecological studies of deep subseafloor sedimentary habitats revealed that 16S rRNA genes derived from potential dehalogenating bacteria within the Chloroflexi and Deltaproteobacteria are widely distributed in organic-rich marine subsurface sediments on the continental margins [1,21,22]. PCR amplification and sequencing of the rdhA genes showed that phylogenetically diverse rdhA genes are widely distributed in the subseafloor sediments of the Pacific Ocean, such as those located southeast of Peru, in the eastern equatorial Pacific, the Juan de Fuca Ridge flank off Oregon, and in the northwest Pacific off the Shimokita Peninsula and Nankai Trough, down to as far as 358 m below the seafloor (mbsf) (figure 2; [23]). Dehalogenation of 2,4,6-tribromophenol (2,4,6-TBP) and TCE has been detected in samples of sediment collected from the Nankai Trough, suggesting that organohalides may serve as possible electron acceptors for deep subseafloor microbes [23].
Figure 2.

Distribution of the rdhA genes in the subseafloor sediments of the Pacific Ocean. The data was summarized from table 1 and our previous study [23]. The data from our previous study [23] are indicated by asterisks. Filled circles indicate that PCR product of expected size was amplified, whereas open circles indicate that PCR product of expected size was not amplified. The red circles indicate that the amplified rdhA genes were further confirmed by sequencing.
In this study, we examined the spatial distribution of dehalogenation activity in the forearc basin and accretionary wedge of the Nankai Trough using 10 organohalides as test compounds. Sediment samples were collected by drilling from six sites during the Integrated Ocean Drilling Program (IODP) expeditions 315 and 316. Given the significant fraction of functionally uncharacterized genes in subseafloor sedimentary habitats [24,25], we also performed a screening method, designated as ‘substrate-induced gene expression (SIGEX) screening’, to the cultures showing dehalogenating activities for identification of genes involved in the dehalogenation activity. The SIGEX was a method developed for isolating catabolic genes from metagenomes using the substrate-dependent gene-induction assay [26,27]. Although we could not retrieve any dehalogenation-related genes such as rdhA in this study, we discuss the result of SIGEX screening as applicable information for studying the samples that have low microbial metabolic activities such as deep marine subsurface microbes [28,29].
2. Material and methods
(a). Sample collection and cultivation conditions
The site location, water depth and sediment depth of the core samples collected for use in this study are summarized in table 1. Core samples were obtained using the deep-sea drilling vessel Chikyu during the IODP expeditions 315 and 316. Site C0002 is located in the Kumano forearc basin, whereas sites C0001, C0004, C0006, C0007 and C0008 are located in the slope and toe of the accretionary wedge associated with seismogenic faults (figure 1), and hence the distances from land and water depths for these sites are far greater than for the Kumano basin site (table 1). All sediment core samples were collected and processed as the whole round by shipboard microbiologists, immediately placed in an anaerobic glove box where they were placed into oxygen-impermeable bags with AnaeroPack oxygen-removers (Mitsubishi Gas Chemical, Japan), and then stored at 4°C until use in experiments.
Table 1.
Characteristics of sampling sites and results of PCR amplification of dehalogenase-homologous gene (rdh). +, PCR product of expected size was amplified; −, PCR product of expected size was not amplified; n.t., not tested. The data of PCR-detection of rdh in site C0002 were used from our previous study [23].
| sampling site (expedition name) | water depth (m) | hole | core section | sediment depth (mbsf) | rdh amplification |
|---|---|---|---|---|---|
| site C0002 (IODP expedition 315) | 1937.1 | D | 1H-3 | 1.9 | + |
| D | 1H-6 | 4.7 | − | ||
| D | 2H-4 | 9.2 | + | ||
| D | 2H-8 | 13.4 | − | ||
| D | 3H-5 | 20.2 | + | ||
| D | 4H-5 | 30.0 | − | ||
| D | 8H-3 | 66.6 | + | ||
| D | 16H-4 | 155.4 | − | ||
| site C0001 (IODP expedition 315) | 2198.0 | E | 1H-2 | 0.7 | + |
| E | 2H-2 | 6.1 | − | ||
| E | 2H-6 | 10.8 | − | ||
| E | 4H-5 | 28.1 | − | ||
| E | 6H-7 | 50.3 | − | ||
| E | 8H-6 | 66.3 | − | ||
| E | 10H-2 | 82.0 | − | ||
| E | 12H-3 | 102.2 | − | ||
| site C0004 (IODP expedition 316) | 2627.0 | C | 1H-1 | 0.8 | + |
| C | 1H-4 | 4.0 | − | ||
| C | 2H-4 | 10.4 | − | ||
| C | 3H-4 | 19.7 | − | ||
| C | 5H-2 | 36.6 | + | ||
| C | 7H-3 | 57.7 | − | ||
| C | 9H-2 | 75.0 | − | ||
| C | 11H-1 | 86.3 | − | ||
| site C0008 (IODP expedition 316) | 2797.0 | B | 1H-3 | 3.1 | + |
| C | 1H-4 | 3.1 | − | ||
| B | 1H-7 | 7.3 | − | ||
| C | 2H-3 | 7.5 | − | ||
| C | 2H-5 | 9.7 | − | ||
| C | 3H-2 | 16.6 | − | ||
| C | 5H-3 | 36.5 | − | ||
| C | 13H-6 | 93.7 | n.t. | ||
| site C0006 (IODP expedition 316) | 3880.5 | C | 1H-1 | 0.7 | + |
| E | 1H-3 | 2.6 | − | ||
| D | 1H-4 | 3.8 | − | ||
| C | 1H-5 | 4.7 | − | ||
| E | 2H-6 | 11.1 | − | ||
| E | 3H-5 | 19.2 | − | ||
| E | 5H-2 | 35.6 | − | ||
| E | 7H-7 | 46.5 | − | ||
| site C0007 (IODP expedition 316) | 4081.0 | A | 1H-3 | 2.3 | − |
| B | 1H-1 | 4.3 | − | ||
| B | 1H-5 | 8.2 | − | ||
| C | 1H-3 | 15.0 | + | ||
| C | 3H-1 | 30.2 | + | ||
| C | 16H-1 | 148.8 | − |
Figure 1.

Geographical setting of the Nankai Trough plate-subduction zone and location of sites C0001, C0002, C0004, C0006, C0007 and C0008 during the IODP Expeditions 315 and 316. The red line in the inlet indicates the transect line.
Sediment slurries for incubation experiments were prepared in the laboratory on land as described previously [23]. Briefly, the innermost sediment of the whole-round core was collected with sterilized spatulas in an anaerobic glove box. The sediment samples were slurried in anaerobic artificial seawater medium containing a trace element solution and a vitamin solution with a modified concentration of vitamin B12 [31,32]. Next, slurry samples were spiked with or without one of the following organohalides to a final concentration of approximately 100 μM: 2,4,6-TBP, 2,4,6-trichlorophenol (2,4,6-TCP), 2,4,6-triiodophenol (2,4,6-TIP), TCE, cis-DCE, trans-DCE, carbon tetrachloride, chloroform, carbon dichloride or dibromomethane. The spiked samples were statically incubated at 15°C in the dark.
(b). Measurement of dehalogenation activity
During the slurry incubation, chlorophenols, iodophenols, bromophenols and phenol were quantified using high-performance liquid chromatography with UV detection (Shimadzu, Japan). Compounds were resolved on a TSKgel ODS-100Z C18 column (Tosoh, Japan), as described previously [23]. Chromatographic peaks were identified and quantified based on retention time and peak area as compared with chromatograms of the following standards: 2,4,6-TBP, 2,6-dibromophenol, 2,4-dibromophenol, 4-bromophenol (4-BP), 2-bromophenol, 2,4,6-TIP, 2,4,6-TCP and phenol. Halogenated methanes and ethenes were quantified on a gas chromatograph (7890A GC System, Agilent Technologies, Santa Clara, CA, USA) equipped with a DB-624 column (J&W Scientific, Folsom, CA, USA) and flame ionization detector.
(c). Analysis of the diversity of metabolically active bacteria in enrichment cultures
After complete dehalogenation was observed, sediment slurry samples were preserved at −80°C with RNAlater (Ambion, Austin, TX, USA). A 1.6 ml volume of each frozen sample was homogenized using a freezer mill (SPEX CertiPrep 6850, Metuchen, NJ, USA) for eight cycles of 1 min homogenization at 15-impact frequency per second. The powdered slurry was then transferred to a new tube and dissolved in 2 ml of TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Total RNA was extracted using a TRIzol Plus RNA purification kit (Invitrogen) according to the manufacturer's protocol. The total RNA fraction was eluted with 100 μl of water treated with diethylpyrocarbonate. Because reverse transcription–PCR (RT-PCR) amplification using these samples as templates was unsuccessful, the samples were purified further and then concentrated to 30 μl using Agencourt RNAClean (Beckman Coulter, Brea, CA, USA). The extracted RNA was treated with DNase I (TURBO DNA-free Kit, Ambion) and the absence of contaminating DNA was confirmed through a PCR amplification test using LA Taq polymerase (Takara, Japan). The 16S rRNA gene was amplified using the Superscript III One-Step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen) using Bac27F and U1490R as primers [33]. The amplicons were cloned into pCR2.1 TOPO vector (Invitrogen) and sequenced using an ABI3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences with a 97% identity were tentatively assigned to the same phylotype. The 16S rRNA gene sequences were compared with data deposited in DDBJ/EMBL/GenBank using BLASTN analysis. A phylogenetic tree for the resulting sequences was constructed according to the neighbor-joining method using MEGA software, v. 4.0 [34].
(d). Detection of rdhA genes
Bulk environmental DNA was extracted from each core sample using a PowerMax Soil DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA). The extracted DNA was purified using a MagExtractor DNA purification kit (Toyobo, Japan) according to the manufacturer's instructions. Because the amount of DNA extracted was too low to conduct molecular analyses, multiple displacement amplification (MDA) was performed using an illustra GenomiPhi HY kit (GE Healthcare, UK) according to the manufacturer's protocol. Briefly, the extracted DNA was heat denatured at 95°C for 3 min, cooled on ice, then incubated with phi29 polymerase, random hexamers and SYBR Green I (0.05× in final concentration) at 30°C for 2 h while monitoring its amplification with real-time PCR (StepOnePlus, Life Technologies). The amplification products were then treated at 65°C for 10 min to inactivate enzyme. The rdhA genes were detected by PCR using Ex Taq polymerase (Takara, Kyoto, Japan), and RRF2 and B1R as primers, as described previously [23,35].
(e). Determination of cell density
The density of microbial cells in sediment core samples was determined using a fluorescent image-based microscopic assay with SYBR Green I staining, as described previously [36,37].
(f). Screening of genes involved in organohalides
The SIGEX technique was used to screen for isolation of catabolic genes for organohalides in incubation slurries that were positive for dehalogenation activity, according to previously published procedures with some modifications [26,27]. Bulk environmental DNA was extracted from the incubation slurry using a PowerMax Soil DNA isolation kit (MO BIO Laboratories) from the enrichment cultures that were positive for dehalogenation activity to 2,4,6-TBP, 2,4,6-TIP and TCE. The extracted DNA was purified using a MagExtractor DNA purification kit (Toyobo) according to the manufacturer's instructions. To obtain a sufficient amount of DNA to construct the SIGEX library, extracted DNA was amplified by MDA using illustra GenomiPhi HY Kit (GE Healthcare) as described above. After the purification using Montage PCR centrifugal filter device (Millipore, Billerica, MA, USA), the MDA-amplified, hyperbranched DNA products were then treated with phi29 polymerase without adding any primer to reduce branching junctions. To remove junctions and 3′ single-stranded overhangs, the products were treated with S1 nuclease. The nicks created by S1 nuclease treatment were resolved by nick translation with DNA polymerase I (Takara) [38]. The debranched DNA was A-tailed with TAKARA Taq DNA polymerase (Takara) and used as insertion DNA fragments. The modified promoter trap vector pK18GreenTIR was constructed according to the method by Uchiyama et al. [26] by using pK18 [39] as starting vector and pGreen TIR [40] as a source of gfp gene. The pK18GreenTIR vector was then attached to topoisomerase I by Invitrogen pK18GreenTIR-TOPO. Prepared DNA fragments were inserted into pK18GreenTIR-TOPO following the manufacturer's instructions and electropolated into Escherichia coli MegaX DH10B T1R cells (Invitrogen). After a 1 h incubation of electroporated cells, a small portion of the cells was plated onto a kanamycin and isopropylthiogalactoside (IPTG)-containing Luria-Bertani (LB) agar plate to check the cloning efficiency. This pre-master library in liquid culture was stored at −80°C with 15 per cent glycerol. From the pre-master library, fluorescence-negative clones were sorted by an Epics ALTRA flow cytometer (Beckman Coulter). The number of sorted cells was three times greater than the initial number of the clones. The sorted cells were then incubated in kanamycin-containing LB media and then stored at −80°C with 15 per cent glycerol as master library.
The cells in the master library were grown in dLB medium (1 g Bacto-tryptone, 0.5 g bacto yeast extract, 1 g NaCl and 2 g maltose per litre) until the OD600 reached 0.6–0.8, then substrate for transcriptional induction, including 2,4,6-TBP or 2,4,6-TCP individually or a mixture consisting of 2,4,6-TBP, 2,4,6-TCP and 2,4,6-TIP, at a concentration of 50 μM was added and incubated overnight to ensure gene fragment induction. The fluorescence positive clones were sorted by the Epics ALTRA flow cytometer and plated on the LB plate with kanamycin. The selected clones were grown in a liquid medium with or without induction substrate and the induction of the fluorescence by the substrate was confirmed by the Epics ALTRA flow cytometer or a BD Accuri C6 flow cytometer (Becton, Dickinson and Company, NJ, USA) (see the electronic supplementary material, figure S1). The gene fragments from the induction-positive clones were sequenced using an ABI3130xl Genetic Analyzer (Applied Biosystems). The obtained sequence data was base-called and assembled with Phred, Phrap and Consed software [30,41]. Potential open reading frames (ORFs) were identified using NCBI ORF Finder and analysed using the NCBI programs BLASTN and BLASTP, and Pfam.
(g). Nucleotide sequence accession numbers
The sequences reported in this study have been deposited in the DDBJ/EMBL/GenBank databases under accession numbers AB716277–AB716322.
3. Results
(a). Spatial distribution of dehalogenation activity in a transect line of the Nankai Trough accretionary wedge
Dehalogenation activity was screened in samples collected from six different drilling sites (i.e. C0002, C0001, C0004, C0008, C0006 and C0007) along a transect line of the Nankai Trough plate-subduction zone (figure 1 and table 2). Sediment samples collected from several different depths listed in table 1 were mixed with culture medium and spiked with 1 of 10 organohalides, which included halogenated phenols, ethenes and methanes (listed in table 2). A previous pilot study conducted by our laboratory demonstrated the debromination of 2,4,6-TBP to phenol and the dechlorination of TCE to cis-DCE in samples collected from site C0002 [23]. In the present study, we also detected the deiodination of 2,4,6-TIP to phenol in samples collected from site C0002 and the debromination of 2,4,6-TBP and deiodination of 2,4,6-TIP to phenol in samples collected from site C0001. The site C0002 is located in the forearc basin, whereas site C0001 is located in the slope apron. Even after 200 days of incubation, no dehalogenation activity was observed in samples collected from sites C0004, C0006, C0007 and C0008, which are located along the slope of the accretionary wedge (figure 1).
Table 2.
Dehalogenation activity in marine subsurface sediments collected from the Nankai Trough subduction zone. +, Dehalogenation activity was detected; −, dechlorination activity was not detected.
| halogenated compounds | C0002 | C0001 | C0004 | C0008 | C0006 | C0007 |
|---|---|---|---|---|---|---|
| 2,4,6-trichlorophenol | − | − | − | − | − | − |
| 2,4,6-tribromophenol | + (1.2)a | + (1.7)a | − | − | − | − |
| 2,4,6-triiodophenol | + (0.62)a | + (0.44)a | − | − | − | − |
| trichloroethene | + (0.049)a | − | − | − | − | − |
| cis-1,2-dichloroethene | − | − | − | − | − | − |
| trans-1,2-dichloroethene | − | − | − | − | − | − |
| carbon tetrachloride | − | − | − | − | − | − |
| chloroform | − | − | − | − | − | − |
| carbon dichloride | − | − | − | − | − | − |
| dibromomethane | − | − | − | − | − | − |
aThe maximum reduction rate of each organohalide was calculated (μM per 1 cm3 of sediment per day).
The vertical distribution of 2,4,6-TBP debromination activity was further investigated using sediment samples collected from site C0002, because rdhA genes were most frequently detected in these samples (table 1 and figure 2) [23]. Slurries of sediment samples collected at depths of 1.9, 4.7, 9.2, 13.4, 20.2, 30.0, 66.6 and 155.4 mbsf were spiked with 2,4,6-TBP. Debromination was detected only in the shallow subsurface samples collected at 1.9 and 4.7 mbsf. Production of 4-BP and phenol were detected in the sample of sediment from 1.9 mbsf, whereas a lower concentration of 4-BP and no phenol were detected in the sample from 4.7 mbsf (figure 3a,b).
Figure 3.

Dehalogenation of 2,4,6-TBP in sediment from site C0002 collected at a depth of (a) 1.9 mbsf and (b) 4.7 mbsf and (c) microbial cell density in samples collected at site C0002. The cell densities at depths of 1.9 mbsf and 4.7 mbsf are noted by red circles. (a,b) Red circle, 2,4,6-TBP; green circle, 4-BP; blue circle, phenol.
The cell density did not seem to be a limiting factor for dehalogenation. As shown in figure 3c, while we had highest cell density in the sample at 1.9 mbsf, in which we detected debromination activity, the other sample from 4.7 mbsf (1.76 × 106 cells cm−3) was smaller in cell density than the samples from 13.4 and 155.4 mbsf (8.42 × 106 and 5.45 × 106 cells cm−3, respectively).
(b). Distribution of rdhA-homologous genes in the Nankai Trough plate-subduction zone
To evaluate the potential for organohalide respiration in the Nankai Trough, we investigated the amplifiability of rdhA genes using PCR (table 1 and figure 2). We amplified rdhA genes in samples collected from all sites in the Nankai Trough, even from those in which no dehalogenation activity was detected (i.e. rdhA genes were detected in sediment collected from depths of 1.9, 9.2, 20.2 and 66.6 mbsf at site C0002, which did not correlate with the vertical distribution of 2,4,6-TBP debromination activity as described above).
(c). Diversity of the metabolically active microbial community during slurry incubation with organohalides
Following the incubation of slurry samples of sediment collected from site C0001 with and without 2,4,6-TBP and 2,4,6-TIP, the microbial community structure was investigated by sequencing the 16S rRNA genes. Cultures spiked with either 2,4,6-TBP or 2,4,6-TIP showed a significant change in community structure compared with the no-addition control. In cultures spiked with 2,4,6-TBP or 2,4,6-TIP, the community was only composed of Deltaproteobacteria, whereas the control culture included Gammaproteobacteria, Firmicutes and Bacteroidetes (table 3 and figure 4a). The predominant Deltaproteobacteria 16S rRNA gene sequences matched to the order Desulfuromonadales with high confidence, and these organisms were phylogenetically classified into the Desulfuromusa cluster (figure 5).
Table 3.
Results of BLASTN and RDP classifier of representative sequences obtained by clone analysis of the 16S rRNA gene.
| culture condition | representative clone name | no. of clone grouped into same phylotype | accession no. of closest sequence | name of closest sequence | % identities (query bp/target bp) | isolation source of closest sequence | taxonomic classification by RDP classifier | accession no. |
|---|---|---|---|---|---|---|---|---|
| no addition | C0001_1 | 20 | FM863760.1 | R65SW | 97% (710/733) | Rimicaris exoculata gut | Deltaproteobacteria | AB716277 |
| C0001_2 | 1 | GU302447.1 | Out0bac29 | 92% (652/711) | marine sediments (900 m water depth, 0–24 m sediment depth) from Mississippi Canyon 118, northern slope of the Gulf of Mexico | Bacteroidetes | AB716278 | |
| C0001_3 | 1 | AJ532694.1 | Gitt-KF-166 | 97% (546/565) | uranium mill tailings | Gammaproteobacteria | AB716279 | |
| C0001_4 | 2 | EU050842.1 | ss1_B_06_70 | 98% (734/746) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716280 | |
| C0001_5 | 2 | FJ716952.1 | D10_10.2_2 | 97% (752/775) | marine sediment from Cullercoats, Northumberland, United Kingdom used in the laboratory to culture Arenicola marina (lugworm) | Bacteroidetes | AB716281 | |
| C0001_6 | 1 | AB634592.1 | Thiomicrospira sp. V2501 | 98% (709/720) | — | Gammaproteobacteria | AB716282 | |
| C0001_7 | 3 | AJ532694.1 | Gitt-KF-166 | 99% (701/706) | uranium mill tailings | Gammaproteobacteria | AB716283 | |
| C0001_8 | 1 | FJ716931.1 | B3_10.2_2 | 96% (683/708) | marine sediment from Cullercoats, Northumberland, United Kingdom used in the laboratory to culture Arenicola marina (lugworm) | Bacteroidetes | AB716284 | |
| C0001_9 | 1 | AJ532694.1 | Gitt-KF-166 | 98% (521/530) | uranium mill tailings | Gammaproteobacteria | AB716285 | |
| C0001_10 | 5 | GQ259272.1 | sediment_deep7 | 97% (724/746) | deep sediment | Bacteroidetes | AB716286 | |
| C0001_11 | 4 | GU302447.1 | Out0bac29 | 99% (751/753) | marine sediments (900 m water depth, 0–24 m sediment depth) from Mississippi Canyon 118, northern slope of the Gulf of Mexico | Bacteroidetes | AB716287 | |
| C0001_12 | 1 | AB305559.1 | p816_b_1.24 | 95% (736/774) | PCR-derived sequence from hydrothermal sediments | Bacteroidetes | AB716288 | |
| C0001_13 | 1 | FJ264785.1 | so4B5 | 99% (692/701) | methane seep sediment | Firmicutes | AB716289 | |
| C0001_14 | 1 | EU287256.1 | S11-73 | 97% (747/767) | arctic surface sediment | Bacteroidetes | AB716290 | |
| 2,4,6-TBP | C0001_TBP1 | 14 | EU050842.1 | ss1_B_06_70 | 99% (733/744) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716291 |
| C0001_TBP2 | 5 | EU050842.1 | ss1_B_06_70 | 99% (698/705) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716292 | |
| C0001_TBP3 | 13 | FM863760.1 | R65SW | 97% (707/730) | Rimicaris exoculata gut | Deltaproteobacteria | AB716293 | |
| C0001_TBP4 | 4 | EU050842.1 | ss1_B_06_70 | 98% (698/710) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716294 | |
| C0001_TBP5 | 1 | EU050842.1 | ss1_B_06_70 | 95% (691/729) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716295 | |
| 2,4,6-TIP | C0001_TIP1 | 13 | FM863760.1 | R65SW | 97% (678/699) | Rimicaris exoculata gut | Deltaproteobacteria | AB716296 |
| C0001_TIP2 | 20 | EU050842.1 | ss1_B_06_70 | 99% (687/694) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716297 | |
| C0001_TIP3 | 1 | JF747621.1 | MT5B102 | 95% (750/790) | Manantial del Toro hypersaline groundwater | Deltaproteobacteria | AB716298 | |
| C0001_TIP4 | 1 | EU050842.1 | ss1_B_06_70 | 99% (750/757) | sediment from the Kings Bay, Svalbard, Arctic | Deltaproteobacteria | AB716299 |
Figure 4.

Diversity of clones obtained from subseafloor sediments. (a) Bacterial groups in the 16S rRNA gene clone libraries constructed from site C0001 core sediments amended with or without 2,4,6-TBP or 2,4,6-TIP. (b) Microbial groups in the clones obtained by SIGEX analysis using 2,4,6-TBP or 2,4,6-TCP as an induction substrate.
Figure 5.

Phylogenetic tree showing 16S rRNA clones obtained from 2,4,6-TBP- and 2,4,6-TIP-dehalogenating cultures. Clones denoted in bold were representative sequences obtained in this study. The functionally known organohalide-respiring bacteria are indicated by asterisks. The Geobacter, Desulfuromonas and Desulfuromusa clusters were grouped according a previous report [42]. Bootstrap values are represented as percentages determined from 1000 trials. Scale bar represents 5% estimated sequence divergence.
(d). Phylogenetic diversity of the clones obtained by substrate-induced gene expression
We used the SIGEX method to identify genes involved in the reductive dehalogenation. SIGEX is the method developed to screen catabolic genes based on knowledge that catabolic gene expression is generally induced by relevant compounds and, in many cases, controlled by regulatory elements situated proximate to catabolic genes [26,27]. In this study, we constructed a SIGEX library using sediment slurries that dechlorinate 2,4,6-TBP and 2,4,6-TIP and TCE. The vector we used in this study (pK18GreenTIR-TOPO) contains a lac promoter upstream of the insertion site. After induction with IPTG and following sorting of fluorescent-negative cells, we could selectively obtained the clone that has an inserted DNA fragment and is negative for GFP fluorescence without induction substrates, and used it as a master library for induction. The constructed master libraries contained a total of ca 300 000 clones. Then, the cells that fluoresce after the induction with the substrates were sorted from the master library as induction-positive clones.
A total of 42 GFP-positive clones were screened from the SIGEX library when 2,4,6-TBP, 2,4,6-TCP and 2,4,6-TIP were used as induction substrates. We obtained clones from all the induction conditions, and interestingly we could obtain 2,4,6-TCP induction-positive clones while no dechlorination activity was detected in the incubation experiment, as shown in table 2. Phylogenic affiliations of DNA fragments in 23 clones were estimated based on taxa of BLASTN matched sequences (table 4; [43]). Although analysis of 16S rRNA gene sequences showed that Deltaproteobacteria dominated the dehalogenating cultures (table 3 and figure 4a), the clones obtained by SIGEX analysis indicated that there were phylogenetically more diverse DNA fragments. A BLASTN-based estimation of the taxa of DNA fragments indicated the presence of sequences showing similarities to those in Deltaproteobacteria, Gammaproteobacteria, Dehalococcoidetes and Clostridiales as well as members of the Methanomicrobia methanogenic Archaea class (table 4 and figure 4b). Several clone sequences did not show any significant similarity to those contained in DDBJ/EMBL/GenBank, and thus their phylotypes could not be predicted.
Table 4.
Results of BLASTN analysis of representative clones obtained using SIGEX.
| clone namea | accession no. of closest sequence | closest sequence by BLASTN | % identities (query bp/target bp) | e-value | BLASTN-based classification | accession no. |
|---|---|---|---|---|---|---|
| TBP1 | CP000300.1 | Methanococcoides burtonii DSM 6242 | 90% (776/865) | 0 | Methanomicrobia | AB716300 |
| TBP2 | CP000300.1 | M. burtonii DSM 6242 | 84% (96/114) | 2.00 × 10−23 | Methanomicrobia | AB716301 |
| TBP3 | CP000109.2 | Thiomicrospira crunogena XCL-2 | 76% (239/315) | 2.00 × 10−53 | Gammaproteobacteria | AB716302 |
| TBP4 | — | — | — | — | — | AB716303 |
| TBP5 | — | — | — | — | — | AB716304 |
| TBP6 | CP003171.1 | Oceanimonas sp. GK1 | 69% (641/923) | 8.00 × 10−97 | Gammaproteobacteria | AB716305 |
| TBP7 | CP003083.1| | Methanolobuspsychrophilus R15 | 81% (419/520) | 5.00 × 10−131 | Methanomicrobia | AB716306 |
| TBP8 | — | — | — | — | — | AB716307 |
| TCP1 | CP001924.1 | Dehalococcoides sp. GT | 70% (225/321) | 4.00 × 10−31 | Dehalococcoidetes | AB716308 |
| TCP2 | CP002585.1 | Pseudomonas brassicacearum ssp. brassicacearum NFM421 | 68% (346/511) | 2.00 × 10−34 | Gammaproteobacteria | AB716309 |
| TCP3 | CP000300.1 | M. burtonii DSM 6242 | 87% (413/476) | 8.00 × 10−160 | Methanomicrobia | AB716310 |
| TCP4 | CP000821.1 | Shewanella sediminis HAW-EB3 | 73% (288/397) | 6.00 × 10−54 | Gammaproteobacteria | AB716311 |
| TCP5 | — | — | — | — | — | AB716312 |
| TCP6 | — | — | — | — | — | AB716313 |
| TCP7 | CP003869.1 | Dehalobacter sp. DCA | 63% (817/1295) | 3.00 × 10−32 | Clostridiales | AB716314 |
| TCP8 | CP000300.1 | M. burtonii DSM 6242 | 85% (1240/1467) | 0 | Methanomicrobia | AB716315 |
| TCP9 | — | — | — | — | — | AB716316 |
| TCP10 | — | — | — | — | — | AB716317 |
| TCP11 | — | — | — | — | — | AB716318 |
| TBP/TCP1 | — | — | — | — | — | AB716319 |
| TBP/TCP2 | CP000300.1 | M. burtonii DSM 6242 | 89% (1314/1480) | 0 | Methanomicrobia | AB716320 |
| THP mix1 | AP011177.1 | Shewanella violacea DSS12 | 71% (2041/2886) | 0 | Gammaproteobacteria | AB716321 |
| THP mix2 | CP002607.1 | Aeromonas veronii B565 | 76% (102/134) | 2.00 × 10−16 | Gammaproteobacteria | AB716322 |
aTBP, 2,4,6-TBP was used as induction substrate; TCP, 2,4,6-TCP was used as induction substrate; TBP/TCP, 2,4,6-TBP or 2,4,6-TCP were used as induction substrates; THP mix, 2,4,6-TBP, 2,4,6-TCP and 2,4,6-TIP were used as induction substrates.
(e). Putative functions of the open reading frames identified using substrate-induced gene expression
Because transcriptional factor is necessary for the function of SIGEX screening [26], we looked for the putative functional genes involved in transcriptional regulation using BLASTP (table 5 and figure 6). We found clones that showed similarities to HxlR family protein from Methanococcoides (clone TCP3), and ArsR family protein from Shewanella (clone THP mix2). Also several ORFs showed significant similarity to those from the physiologically and functionally well-characterized organohalide-respiring bacteria such as Dehalococcoides, Dehalobacter, Desulfitobacterium and Geobacter (see the electronic supplementary material, table S1). However, most of the ORFs representing genes encoding proteins other than transcriptional factors were apparently unrelated to organohalide metabolism (e.g. proteins involved in flagellum biogenesis, phage/plasmid primase, endonuclease, site-specific recombinase, antitoxin, dihydroototase, NAD-dependent epimerase dehydratase, 4Fe–4S ferredoxin, serine protease inhibitor, copper-resistance protein transporter and epimerase; electronic supplementary material, table S1). A single ORF involved in the degradation of organohalides was similar to a haloacid dehalogenase (HAD) superfamily hydrolase from Geobacter (clone TCP6; electronic supplementary material, table S1). This ORF showed significant Pfam-A-match to the HAD_2 family domain (e-value = 5.6 × 10−13), although it showed greater similarity to phosphatase in BLASTP search.
Table 5.
Results of BLASTP analysis of ORFs encoded on clones obtained by SIGEX.
| clone no. (ORF no.) | length (aa) | gene product (host organism) | length (aa) | % identities (aa) (query aa/target aa) | e-value | accession no. |
|---|---|---|---|---|---|---|
| TCP3 (1) | 353 | permease (Shewanella pealeana ATCC 700345) | 356 | 47% (164/350) | 1.0 × 10−110 | YP_001503275 |
| TCP3 (2) | 64 | hypothetical protein Dole_2888 (Desulfococcus oleovorans Hxd3) | 112 | 58% (35/60) | 8.0 × 10−17 | YP_001530768 |
| TCP3 (3) | 111 | transporter (Photobacterium damselae subsp. damselae CIP) | 357 | 64% (65/101) | 2.0 × 10−36 | ZP_06155623 |
| TCP3 (4) | 104 | regulatory protein ArsR (Pseudoalteromonas rubra ATCC 29570) | 119 | 62% (63/102) | 4.0 × 10−37 | ZP_10294511 |
| TCP3 (5) | 62 | — | — | — | — | — |
| TCP3 (6) | 77 | — | — | — | — | — |
| THP mix2 (1) | 125 | HxlR family transcriptional regulator (Methanococcoides burtonii DSM 6242) | 125 | 84% (105/125) | 3.0 × 10−69 | YP_565113 |
| THP mix2 (2) | 197 | serine protease inhibitor (Methanosarcina mazei Go1) | 427 | 58% (101/173) | 2.0 × 10−66 | NP_634699 |
Figure 6.
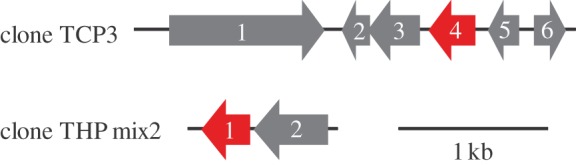
ORF organization around the putative transcriptional factor gene cluster of clones TCP3 and THP mix2. The putative transcriptional factor genes are indicated by red. The numbers corresponds to the ORF numbers in table 5.
4. Discussion
Because subseafloor sedimentary microbes play a significant role in the biogeochemical carbon cycle through anaerobic degradation of buried organic matter, halogenated organic compounds may support microbial activity by serving as electron acceptors. Indeed, our previous study of various coastal locations in the Pacific Ocean revealed the presence of a diverse array of rdhA genes in ocean sediment, a result that was consistent with the detection of a diverse assortment of 16S rRNA genes within the Chloroflexi (figure 2). Given the ecological significance of microbial dehalogenation in the subseafloor environment, we further investigated the spatial distribution of dehalogenation activity using drill-cored samples obtained from the Nankai Trough, which is one of the most geologically active plate-subduction zones in the world, and is associated with the occurrence of devastating earthquakes and tsunamis [44,45]. The Nankai Trough is a geologically complex environment. Site C0002 is located in the Kumano basin of the Nankai Trough, and the sediment core samples collected at those sites for use in this study are well-stratified and characterized by relatively low structural disturbance. Sites C0001, C0004, C0006, C0007 and C0008 are located along the slope to toe of the Nankai Trough accretionary wedge, where the structures are mainly composed of geologically old accretionary prism.
In our vertical investigation of debromination activity in samples collected at 1.9, 4.7, 9.2, 13.4, 20.2, 30.0, 66.6 and 155.4 mbsf from site C0002, we detected debromination only in samples collected at 1.9 and 4.7 mbsf, despite the fact that PCR analyses indicated that rdhA genes are well-distributed in the deeper sediments (e.g. 9.2, 20.2 and 66.6 mbsf). The deepest site C0002 sediment sample from which rdhA genes were detected (66.6 mbsf) was formed at least ca 500 000 years ago, during the Quaternary period [46]. Some conceivable explanations for our inability to detect debromination in the deeper sediments could be: (i) the cultivation conditions used might not be optimal for deep subseafloor dehalogenating microbes (e.g. we have many choices of organohalide substrates), (ii) the metabolic processes at depth may be very slow, thus requiring more sensitive methods (e.g. radiotracer incubation experiments) than those used in this study, and (iii) the PCR-based detection of rdhA might be the result of unspecific amplification. In a similar fashion, we detected no dehalogenation of the test 10 organohalides in sediment samples collected from sites C0004, C0006, C0007 and C0008, where PCR products of the expected sizes of rdhA genes were detected as well in our previous study [23]. In the results of incubation experiments, it was worth noting that the deiodination of 2,4,6-TIP detected in the present study in sediments from sites C0001 and C0002 is the first report of microbial deiodination of 2,4,6-TIP.
Clonal analysis of 16S rRNA genes indicated that members of the Desulfuromusa cluster within the order Desulfuromonadales were the dominant bacteria in the 2,4,6-TBP- and 2,4,6-TIP-dehalogenating cultures prepared from site C0001. This taxonomic order of bacteria includes various anaerobic respirers that use sulfur, nitrate, Fe (III), Mn (IV) and organochlorines, although it is not always true that they are organohalide-respiring bacteria. To date, organohalide-respiring bacteria from the Desulfuromonadales belonging to the genera Geobacter and Desulfuromonas have been isolated from terrestrial environments. For example, Geobacter thiogenes K1 (formerly Trichlorobacter thiogenes K1) is able to grow with trichloroacetate serving as an electron acceptor [47–49], and Geobacter lovleyi SZ dechlorinates PCE to cis-DCE [50]. Desulfuromonas chloroethenica TT4B and Desulfuromonas michiganensis strains BB1 and BRS1 are able to respire with PCE and TCE and convert them to cis-DCE [51,52].
We applied the SIGEX screening to obtain genes involved in the reductive dehalogenation reaction in the dehalogenating cultures. This was for the reason that functional screening of rdhA seems to be difficult because functional expression of rdhA genes in E. coli has not been successful [53,54]. On the other hand, the utility of an in vivo transcription assay of the transcriptional regulator CprK in E. coli has been proved [55], indicating that it might be possible to explore genes involved in organohalide respiration using SIGEX transcriptional screening. Contrary to the results of the 16S rRNA gene sequence analysis, which indicated that Desulfuromonadales species predominated in the dehalogenation-positive enrichment cultures, SIGEX suggested the presence of a phylogenetically diverse assortment of clones putatively belong to the Gammaproteobacteria, Deltaproteobacteria, Dehalococcoidetes, Deferribacteres and Methanomicrobia. Interestingly, SIGEX also retrieved clones that showed similarity to the gene of methanogenic Archaea, Methanomicrobia. No organohalide-respiring member of the Archaea has been identified to date, but Archaea are known to be capable of co-metabolic dehalogenation, implying that Archaea may have an organohalide-response system. For example, it has been reported that Methanosarcina sp. strain DCM dechlorinates PCE to TCE [56], potentially mediated in a cometabolic reaction by cofactor F430 containing vitamin B12 [57].
However, importantly, we could found only two putative transcriptional regulator encoding clones among the 23 representative clones obtained by SIGEX screening. This result appears to contradict the fact that SIGEX is based on GFP expression by transcriptional regulation with induction substrates [26]. We hypothesized that the expressed gene fragments obtained using SIGEX might encode novel transcriptional factors because the environmental gene pools from subseafloor sediments contain significant proportions of unannotated ORFs, and most of the metagenomic sequences of subseafloor microbial communities are distinct from those associated with surface environments [24,25]. Also there is another concern on the false positives in the SIGEX procedure. A stress or other response system in the host E. coli might affect the expression level of GFP in clones where a putative transcriptional factor that encodes genes was not found. In addition, we could not find any dehalogenation-related genes near the putative transcriptional factor genes retrieved in our SIGEX survey, most likely because the average length of the DNA library was relatively short (i.e. ca 1 and 2.9 kb). Those above are the current limitation of our SIGEX approach. We are trying to address this issue by constructing a library with longer DNA fragments by using a number of the environmental samples. These efforts will make the SIGEX approach more effective and reliable in future.
5. Conclusion and perspectives
Dehalogenating microorganisms may play significant ecological and biogeochemical roles in the carbon and halogen cycles within the deep, dark and old subseafloor sedimentary environment. In such a geological habitat, the metabolic processes of most cells are extremely slow, leading to an expected generation time ranging from hundreds to thousands of years [28,29]. These microbial metabolic activities are considered to be strongly associated with the availability of electron acceptors, which may include halogenated organic matter in the sediment, and may be involved in the mineralization and thermogenic degradation of deeply buried organic matter. In this study, we detected dehalogenation activity only in relatively shallow (approx. 4.7 mbsf) sediment samples collected from the stratified sedimentary basin, although the rdhA genes were found in the deep and old accretionary prism. In addition, we performed the gene expression-based metagenomic survey called SIGEX for exploring organohalide respiration-related genes from the largely functionally ‘unknown’ gene pools in the subseafloor biosphere [24,25]. To obtain a better understanding of the ecological importance of microbial dehalogenation in deep marine sediments, it is clear that we should use more sensitive biogeochemical approaches to assess the activity (e.g. stable and/or radioactive isotopic tracers). In addition, an effective metagenomic screening assay customized for the microbes having a quite low metabolic activity should also be used for the deep marine subsurface microbes. We are currently focusing on studies using such techniques.
Acknowledgements
The authors are grateful to the shipboard scientific parties and crews of IODP expeditions 315 and 316. We also thank Satoko Tanaka, Sae Fukunaga and Noriaki Masui for technical assistance. This study was supported in part by the Strategic Fund for Strengthening Leading-Edge Research and Development (to JAMSTEC) and the Funding Programme for Next Generation World-Leading Researchers (to F.I.) by the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). This study was also supported by a JAMSTEC Multidisciplinary Research Promotion Award (to Y.M.), a Grant-in-Aid for Challenging Exploratory Research (no. 24651018, to Y.M.), and a Grant-in-Aid for Young Scientists (B) (no. 21780085, to T.F.). Support for A.H.K. was provided by a grant from the Academy of Finland (no. 122394), a Finnish Funding Agency for Technology and Innovation grant (no. 40149/07), an Osk Huttunen's Foundation grant, and a Finnish Cultural Foundation grant.
References
- 1.Adrian L. 2009. ERC-group microflex: microbiology of Dehalococcoides-like Chloroflexi. Rev. Environ. Sci. Biotechnol. 8, 225–229 10.1007/s11157-009-9166-y (doi:10.1007/s11157-009-9166-y) [DOI] [Google Scholar]
- 2.Löffler FE, Edwards EA. 2006. Harnessing microbial activities for environmental cleanup. Curr. Opin. Biotechnol. 17, 274–284 10.1016/j.copbio.2006.05.001 (doi:10.1016/j.copbio.2006.05.001) [DOI] [PubMed] [Google Scholar]
- 3.Smidt H, de Vos WM. 2004. Anaerobic microbial dehalogenation. Annu. Rev. Microbiol. 58, 43–73 10.1146/annurev.micro.58.030603.123600 (doi:10.1146/annurev.micro.58.030603.123600) [DOI] [PubMed] [Google Scholar]
- 4.Gribble GW. 2010. Naturally occurring organohalogen compounds—a comprehensive update, 1st edn Vienna, Austria: Springer-Verlag [Google Scholar]
- 5.Ahn YB, Rhee SK, Fennell DE, Kerkhof LJ, Hentschel U, Häggblom MM. 2003. Reductive dehalogenation of brominated phenolic compounds by microorganisms associated with the marine sponge Aplysina aerophoba. Appl. Environ. Microbiol. 69, 4159–4166 10.1128/AEM.69.7.4159-4166.2003 (doi:10.1128/AEM.69.7.4159-4166.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krzmarzick MJ, Crary BB, Harding JJ, Oyerinde OO, Leri AC, Myneni SC, Novak PJ. 2012. Natural niche for organohalide-respiring Chloroflexi. Appl. Environ. Microbiol. 78, 393–401 10.1128/AEM.06510-11 (doi:10.1128/AEM.06510-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Häggblom MM, Ahn YB, Fennell DE, Kerkhof LJ, Rhee SK. 2003. Anaerobic dehalogenation of organohalide contaminants in the marine environment. Adv. Appl. Microbiol. 53, 61–84 10.1016/S0065-2164(03)53002-7 (doi:10.1016/S0065-2164(03)53002-7) [DOI] [PubMed] [Google Scholar]
- 8.King GM. 1988. Dehalogenation in marine sediments containing natural sources of halophenols. Appl. Environ. Microbiol. 54, 3079–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner A, Cooper M, Ferdi S, Seifert J, Adrian L. 2012. Growth of Dehalococcoides mccartyi strain CBDB1 by reductive dehalogenation of brominated benzenes to benzene. Environ. Sci. Technol. 46, 8960–8968. 10.1021/es3003519 (doi:10.1021/es3003519) [DOI] [PubMed] [Google Scholar]
- 10.Sun B, Cole JR, Tiedje JM. 2001. Desulfomonile limimaris sp. nov., an anaerobic dehalogenating bacterium from marine sediments. Int. J. Syst. Evol. Microbiol. 51, 365–371 [DOI] [PubMed] [Google Scholar]
- 11.Hugenholtz P, Stackebrandt E. 2004. Reclassification of Sphaerobacter thermophilus from the subclass Sphaerobacteridae in the phylum Actinobacteria to the class Thermomicrobia (emended description) in the phylum Chloroflexi (emended description). Int. J. Syst. Evol. Microbiol. 54, 2049–2051 10.1099/ijs.0.03028-0 (doi:10.1099/ijs.0.03028-0) [DOI] [PubMed] [Google Scholar]
- 12.Löffler FE, et al. In press. Dehalococcoides mccartyi gen. nov., sp. nov., obligate organohalide-respiring anaerobic bacteria, relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidetes classis nov., within the phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 10.1099/ijs.0.034926-0 (doi:10.1099/ijs.0.034926-0) [DOI] [PubMed] [Google Scholar]
- 13.Sekiguchi Y, Yamada T, Hanada S, Ohashi A, Harada H, Kamagata Y. 2003. Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain Bacteria at the subphylum level. Int. J. Syst. Evol. Microbiol. 53, 1843–1851 10.1099/ijs.0.02699-0 (doi:10.1099/ijs.0.02699-0) [DOI] [PubMed] [Google Scholar]
- 14.May HD, Miller GS, Kjellerup BV, Sowers KR. 2008. Dehalorespiration with polychlorinated biphenyls by an anaerobic ultramicrobacterium. Appl. Environ. Microbiol. 74, 2089–2094 10.1128/AEM.01450-07 (doi:10.1128/AEM.01450-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Q, Watts JE, Sowers KR, May HD. 2002. Identification of a bacterium that specifically catalyzes the reductive dechlorination of polychlorinated biphenyls with doubly flanked chlorines. Appl. Environ. Microbiol. 68, 807–812 10.1128/AEM.68.2.807-812.2002 (doi:10.1128/AEM.68.2.807-812.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng D, He J. 2009. Isolation and characterization of ‘Dehalococcoides’ sp. strain MB, which dechlorinates tetrachloroethene to trans-1,2-dichloroethene. Appl. Environ. Microbiol. 75, 5910–5918 10.1128/AEM.00767-09 (doi:10.1128/AEM.00767-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cutter LA, Watts JE, Sowers KR, May HD. 2001. Identification of a microorganism that links its growth to the reductive dechlorination of 2,3,5,6-chlorobiphenyl. Environ. Microbiol. 3, 699–709 10.1046/j.1462-2920.2001.00246.x (doi:10.1046/j.1462-2920.2001.00246.x) [DOI] [PubMed] [Google Scholar]
- 18.Kittelmann S, Friedrich MW. 2008. Novel uncultured Chloroflexi dechlorinate perchloroethene to trans-dichloroethene in tidal flat sediments. Environ. Microbiol. 10, 1557–1570 10.1111/j.1462-2920.2008.01571.x (doi:10.1111/j.1462-2920.2008.01571.x) [DOI] [PubMed] [Google Scholar]
- 19.Watts JE, Fagervold SK, May HD, Sowers KR. 2005. A PCR-based specific assay reveals a population of bacteria within the Chloroflexi associated with the reductive dehalogenation of polychlorinated biphenyls. Microbiology 151, 2039–2046 10.1099/mic.0.27819-0 (doi:10.1099/mic.0.27819-0) [DOI] [PubMed] [Google Scholar]
- 20.Zanaroli G, Balloi A, Negroni A, Borruso L, Daffonchio D, Fava F. 2012. A Chloroflexi bacterium dechlorinates polychlorinated biphenyls in marine sediments under in situ-like biogeochemical conditions. J. Hazard Mater. 209–210, 449–457 10.1016/j.jhazmat.2012.01.042 (doi:10.1016/j.jhazmat.2012.01.042) [DOI] [PubMed] [Google Scholar]
- 21.Inagaki F, et al. 2006. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl Acad. Sci. USA 103, 2815–2820 10.1073/pnas.0511033103 (doi:10.1073/pnas.0511033103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inagaki F, Suzuki M, Takai K, Oida H, Sakamoto T, Aoki K, Nealson KH, Horikoshi K. 2003. Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Appl. Environ. Microbiol. 69, 7224–7235 10.1128/AEM.69.12.7224-7235.2003 (doi:10.1128/AEM.69.12.7224-7235.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Futagami T, Morono Y, Terada T, Kaksonen AH, Inagaki F. 2009. Dehalogenation activities and distribution of reductive dehalogenase homologous genes in marine subsurface sediments. Appl. Environ. Microbiol. 75, 6905–6909 10.1128/AEM.01124-09 (doi:10.1128/AEM.01124-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH. 2008. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl Acad. Sci. USA 105, 10 583–10 588 10.1073/pnas.0709942105 (doi:10.1073/pnas.0709942105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biddle JF, White JR, Teske AP, House CH. 2011. Metagenomics of the subsurface Brazos-Trinity Basin (IODP site 1320): comparison with other sediment and pyrosequenced metagenomes. ISME J. 5, 1038–1047 10.1038/ismej.2010.199 (doi:10.1038/ismej.2010.199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uchiyama T, Abe T, Ikemura T, Watanabe K. 2005. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 23, 88–93 10.1038/nbt1048 (doi:10.1038/nbt1048) [DOI] [PubMed] [Google Scholar]
- 27.Uchiyama T, Watanabe K. 2008. Substrate-induced gene expression (SIGEX) screening of metagenome libraries. Nat. Protocol 3, 1202–1212 10.1038/nprot.2008.96 (doi:10.1038/nprot.2008.96) [DOI] [PubMed] [Google Scholar]
- 28.Jørgensen BB. 2011. Deep subseafloor microbial cells on physiological standby. Proc. Natl Acad. Sci. USA 108, 18 193–18 194 10.1073/pnas.1115421108 (doi:10.1073/pnas.1115421108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morono Y, Terada T, Nishizawa M, Ito M, Hillion F, Takahata N, Sano Y, Inagaki F. 2011. Carbon and nitrogen assimilation in deep subseafloor microbial cells. Proc. Natl Acad. Sci. USA 108, 18 295–18 300 10.1073/pnas.1107763108 (doi:10.1073/pnas.1107763108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewing B, Hillier L, Wendl M, Green P. 1998. Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 10.1101/gr.8.3.175 (doi:10.1101/gr.8.3.175) [DOI] [PubMed] [Google Scholar]
- 31.Löffler FE, Sanford RA, Ritalahti KM. 2005. Enrichment, cultivation, and detection of reductively dechlorinating bacteria. Methods Enzymol. 397, 77–111 10.1016/S0076-6879(05)97005-5 (doi:10.1016/S0076-6879(05)97005-5) [DOI] [PubMed] [Google Scholar]
- 32.Wolin EA, Wolin MJ, Wolfe RS. 1963. Formation of methane by bacterial extracts. J. Biol. Chem. 238, 2882–2886 [PubMed] [Google Scholar]
- 33.Lane DJ. 1991. 16S/23S rRNA sequencing. In Nucleic acid techniques in bacterial systematics (eds Stackebrandt E, Goodfellow M.), pp. 115–175 New York, NY: John Wiley and Sons [Google Scholar]
- 34.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 10.1093/molbev/msm092 (doi:10.1093/molbev/msm092) [DOI] [PubMed] [Google Scholar]
- 35.Krajmalnik-Brown R, Holscher T, Thomson IN, Saunders FM, Ritalahti KM, Löffler FE. 2004. Genetic identification of a putative vinyl chloride reductase in Dehalococcoides sp. strain BAV1. Appl. Environ. Microbiol. 70, 6347–6351 10.1128/AEM.70.10.6347-6351.2004 (doi:10.1128/AEM.70.10.6347-6351.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morono Y, Inagaki F. 2010. Automatic slide-loader fluorescence microscope system for high-throughput discriminative enumeration of subseafloor life. Sci. Drill. 9, 32–36 10.2204/iodp.sd.9.05.2010 (doi:10.2204/iodp.sd.9.05.2010) [DOI] [Google Scholar]
- 37.Morono Y, Terada T, Masui N, Inagaki F. 2009. Discriminative detection and enumeration of microbial life in marine subsurface sediments. ISME J. 3, 503–511 10.1038/ismej.2009.1 (doi:10.1038/ismej.2009.1) [DOI] [PubMed] [Google Scholar]
- 38.Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, Church GM. 2006. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 24, 680–686 10.1038/nbt1214 (doi:10.1038/nbt1214) [DOI] [PubMed] [Google Scholar]
- 39.Pridmore RD. 1987. New and versatile cloning vectors with kanamycin-resistance marker. Gene 56, 309–312 10.1016/0378-1119(87)90149-1 (doi:10.1016/0378-1119(87)90149-1) [DOI] [PubMed] [Google Scholar]
- 40.Miller WG, Lindow SE. 1997. An improved GFP cloning cassette designed for prokaryotic transcriptional fusions. Gene 191, 149–153 10.1016/S0378-1119(97)00051-6 (doi:10.1016/S0378-1119(97)00051-6) [DOI] [PubMed] [Google Scholar]
- 41.Ewing B, Green P. 1998. Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 10.1101/gr.8.3.186 (doi:10.1101/gr.8.3.186) [DOI] [PubMed] [Google Scholar]
- 42.Holmes DE, Nevin KP, Lovley DR. 2004. Comparison of 16S rRNA, nifD, recA, gyrB, rpoB and fusA genes within the family Geobacteraceae fam. nov. Int. J. Syst. Evol. Microbiol. 54, 1591–1599 10.1099/ijs.0.02958-0 (doi:10.1099/ijs.0.02958-0) [DOI] [PubMed] [Google Scholar]
- 43.Brady A, Salzberg SL. 2009. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods 6, 673–676 10.1038/nmeth.1358 (doi:10.1038/nmeth.1358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riedinger N, Brunner B, Formolo MJ, Solomon E, Kasten S, Strasser M, Ferdelman TG. 2010. Oxidative sulfur cycling in the deep biosphere of the Nankai Trough, Japan. Geology 38, 851–854 10.1130/G31085.1 (doi:10.1130/G31085.1) [DOI] [Google Scholar]
- 45.Strasser M, et al. 2009. Origin and evolution of a splay fault in the Nankai accretionary wedge. Nat. Geosci. 2, 648–652 10.1038/ngeo609 (doi:10.1038/ngeo609) [DOI] [Google Scholar]
- 46.Expedition 315 Scientists 2009. Expedition 315 site C0002. In Proc. IODP vol. 314/315/316 (eds Kinoshita M, Tobin H, Ashi J, Kimura G, Lallemant S, Screaton EJ, Curewitz D, Masago H, Moe KT, the Expedition 314/315/316 Scientists). Washington, DC: IODP Management International, Inc [Google Scholar]
- 47.De Wever H, Cole JR, Fettig MR, Hogan DA, Tiedje JM. 2000. Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen. nov., sp. nov. Appl. Environ. Microbiol. 66, 2297–2301 10.1128/AEM.66.6.2297-2301.2000 (doi:10.1128/AEM.66.6.2297-2301.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nevin KP, Holmes DE, Woodard TL, Covalla SF, Lovley DR. 2007. Reclassification of Trichlorobacter thiogenes as Geobacter thiogenes comb. nov. Int. J. Syst. Evol. Microbiol. 57, 463–466 10.1099/ijs.0.63408-0 (doi:10.1099/ijs.0.63408-0) [DOI] [PubMed] [Google Scholar]
- 49.Snoeyenbos-West O, Van Praagh CG, Lovley DR. 2001. Trichlorobacter thiogenes should be renamed as a Geobacter species. Appl. Environ. Microbiol. 67, 1020–1022 10.1128/AEM.67.2.1020-1022.2001 (doi:10.1128/AEM.67.2.1020-1022.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sung Y, Fletcher KE, Ritalahti KM, Apkarian RP, Ramos-Hernandez N, Sanford RA, Mesbah NM, Löffler FE. 2006. Geobacter lovleyi sp. nov. strain SZ, a novel metal-reducing and tetrachloroethene-dechlorinating bacterium. Appl. Environ. Microbiol. 72, 2775–2782 10.1128/AEM.72.4.2775-2782.2006 (doi:10.1128/AEM.72.4.2775-2782.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krumholz LR. 1997. Desulfuromonas chloroethenica sp. nov. uses tetrachloroethylene and trichloroethylene as electron acceptors. Int. J. Syst. Evol. Microbiol. 47, 1262–1263 10.1099/00207713-47-4-1262 (doi:10.1099/00207713-47-4-1262) [DOI] [Google Scholar]
- 52.Sung Y, Ritalahti KM, Sanford RA, Urbance JW, Flynn SJ, Tiedje JM, Löffler FE. 2003. Characterization of two tetrachloroethene-reducing, acetate-oxidizing anaerobic bacteria and their description as Desulfuromonas michiganensis sp. nov. Appl. Environ. Microbiol. 69, 2964–2974 10.1128/AEM.69.5.2964-2974.2003 (doi:10.1128/AEM.69.5.2964-2974.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neumann A, Wohlfarth G, Diekert G. 1998. Tetrachloroethene dehalogenase from Dehalospirillum multivorans: cloning, sequencing of the encoding genes, and expression of the pceA gene in Escherichia coli. J. Bacteriol. 180, 4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suyama A, Yamashita M, Yoshino S, Furukawa K. 2002. Molecular characterization of the PceA reductive dehalogenase of Desulfitobacterium sp. strain Y51. J. Bacteriol. 184, 3419–3425 10.1128/JB.184.13.3419-3425.2002 (doi:10.1128/JB.184.13.3419-3425.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gábor K, Hailesellasse Sene K, Smidt H, de Vos WM, van der Oost J. 2008. Divergent roles of CprK paralogues from Desulfitobacterium hafniense in activating gene expression. Microbiology 154, 3686–3696 10.1099/mic.0.2008/021584-0 (doi:10.1099/mic.0.2008/021584-0) [DOI] [PubMed] [Google Scholar]
- 56.Fathepure BZ, Boyd SA. 1988. Dependence of tetrachloroethylene dechlorination on methanogenic substrate consumption by Methanosarcina sp. strain DCM. Appl. Environ. Microbiol. 54, 2976–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gantzer CJ, Wackett LP. 1991. Reductive dechlorination catalyzed by bacterial transition-metal coenzymes. Environ. Sci. Technol. 25, 715–721 10.1021/es00016a017 (doi:10.1021/es00016a017) [DOI] [Google Scholar]