

Abstract
Genomic DNA of 3 patients, born as healthy carriers and developing a late-onset severe transfusion-dependent beta-thalassemia major was studied by high-density genome wide SNP array analysis. A mosaic loss of heterozygosity for almost the entire 11p was found, not attributable to deletions but involving mosaicism for segmental paternal isodisomy of 11p. Mitotic recombination leading to mosaic segmental uniparental isodisomy on chromosome 11p in multiple tissues has been described as a molecular disease mechanism for a subset of sporadic Beckwith-Wiedemann syndrome cases. A similar mechanism also seems to be involved in causing late-onset disease in carriers of recessive mutations in other genes located in 11p, such as late-onset beta-thalassemia major and sickle cell disease. We suggest that the loss of maternally imprinted IGF-2 and H19 genes may account for the selective advantage of hematopoietic cells containing this segmental paternal isodisomy of 11p carrying the β-thalassemia mutation.
Introduction
The beta-thalassemias (β-thalassemias) are a diverse group of disorders of hemoglobin synthesis involving the reduced output of the β-globin chains of adult hemoglobin. More than 200 point mutations that cause reduced gene expression have been described in the β-globin gene.1 Homozygosity or compound heterozygosity generally leads to β-thalassemia major or intermedia from childhood and shows an autosomal recessive Mendelian pattern of inheritance. However, 3 patients born as β-thalassemia minor developed a blood transfusion dependent β-thalassemia major in their third or fourth decade of life. Of 2 patients from whom parents were available, only one parent carried the defective HBB gene. Direct sequencing of DNA isolated from peripheral blood mononuclear cells showed an almost complete homozygosity for the mutant allele in the presence of a reduced wild-type signal in all 3 patients with late-onset β-thalassemia major. To investigate the genomic background of this apparent mosaicism in relationship to late-onset β-globin gene related diseases, we performed a detailed genomic analysis of these 3 independent cases.
Design and Methods
Individuals with late-onset beta-thalassemia
The 3 probands, CG, PF and SSG, were diagnosed as high HbA2 β-thalassemia carriers in infancy. They gradually developed a progressive thalassemia intermedia phenotype and blood transfusion dependency later in life. Propositus 1, a female born in 1978, was diagnosed as a β-thalassemia carrier at eight years of age, with mild microcytic hypochromic anemia and increased HbA2 and HbF levels. She maintained the hematologic phenotype of a healthy carrier until the age of 26 years when her hemoglobin dropped below 8 g/dL and splenomegaly developed. At 30 years of age she developed a severe anemia with jaundice and marked splenomegaly. She subsequently became transfusion-dependent (monthly transfusions). Molecular analysis revealed that her father was a carrier of the β-thalassemia mutation cd76(-C) (HBBc.230delCfs) with the expected mild hematologic phenotype, while her mother was normal (Figure 1A).
Figure 1.

From left to right (A) pedigree of propositus 1; (B) propositus 2; (C) and propositus 3. Propositi 1, 2 and 3 are indicated by arrows. The sequence pattern for the HBBc.230delCfs mutation in propositus 1 inherited from father appears almost homozygous, a small normal pattern is visible indicating mosaicism (B) propositus 2; almost homozygous for the HBBc.118C>T; a small wild-type peak at the mutated site is visible (C) propositus 3; similar to propositus 2, in addition the common polymorphism present in exon 1 at HBBc.9C>T is also mosaic, indicating the presence of a similar small percentage of the normal allele. MLPA analysis excluded allelic loss as a result of a deletion. These results were reproducible in several independent analyses and DNA isolations.
Propositus 2, a male born in 1955, was diagnosed as a β-thalassemia carrier in childhood. He developed severe β-thalassemia intermedia at the more advanced age of 38 years, becoming transfusion dependent at the age of 43 years. The patient was splenectomized at 43 years of age. Molecular analysis revealed the common cd 39 (C→T) mutation (HBBc.118C>T). The patient has 2 sons, both non-carriers, and one daughter who is a β-thalassemia carrier. The parents of this patient have died and no medical history is available (Figure 1B).
Propositus 3 is a male born in 1951, diagnosed as a β-thalassemia carrier in childhood. He developed severe thalassemia intermedia and splenomegaly at the more advanced age of 46 years, becoming transfusion dependent at the age of 51 years. The patient was splenectomized at 51 years of age. The family history revealed a β-thalassemia trait in his father, due to the common cd 39 (C→T) mutation (HBBc.118C>T), while the mother was normal (Figure 1C).
The material for analysis is archived DNA isolated from blood leukocytes and saliva. The patients were not available for collection of other samples. The patients and their parents gave written informed consent. The analysis for the diagnostics for hemoglobinopathies has been approved by the local Ethics Committee.
Hematologic analysis
The patients were underwent clinical, hematologic and molecular study over three decades. The hematologic data were obtained on different standard counters. HbA2 and HbF levels were measured on different automatic high performance liquid chromatographic equipment (HPLC).2 The hematologic analysis was made before transfusion.
DNA isolation and analysis
Genomic DNA was isolated from peripheral leukocytes using the Puregene DNA Purification System (Gentra Systems, Minneapolis, USA). DNA was extracted from buccal mucose using the OrageneTM DNA collection kit (DNA Genotek, Inc., Ottawa, Ontario, Canada) according to the manufacturer’s instructions. Direct DNA sequencing was performed as previously described.3 Molecular analysis for common deletions and the α-triplication was performed using multiplex PCR.4 Multiplex Ligation-dependent Probe Amplification (MLPA) was performed as previously described.5
Illumina SNP bead array analysis
To determine on a genomic scale the extent of homozygosity and the parental source of uniparental segmental isodisomy, we hybridized 200 ng of extracted DNA to Illumina OmniExpress human 730K SNP Bead arrays (Illumina, San Diego, CA, USA). The samples were processed in 3 separate experiments using the automated protocol on an EVO150 Tecan robot for Illumina BeadChips (Tecan Group Ltd., Männedorf, Switzerland) at a dedicated facility at the Leiden Genome Technology Center at the Leiden University Medical Center. Intensity data were imported to Illumina GenomeStudio v2010.3 software with Genotyping module 1.8.4. The 733202 SNPs on this array have a respective mean and median spacing of 4 kb and 2 kb across the genome and on chromosome 11, with 36980 SNPs on chromosome 11. SNPs were clustered by the GenTrain 1.0 calling algorithm and the Illumina provided cluster file (HumanOmniExpress-12v1_A.egt). SNP positions are from genome build 36 (hg18).
Chromosomal-scale LoH analysis
To identify the extent of the loss of heterozygosity (LoH), we analyzed the β-allele frequency (Baf) of each SNP. The β-allele frequency is calculated by the linear interpolation of the distance from the canonical 0 (aa), 0.5 (ab), or 1(bb) allele frequencies for the b allele. The absolute medians of the canonical frequencies, and the size of the cluster around it, are taken from the Illumina provided cluster file that describes allelic ratios for this array in a diverse set of 200 HapMap samples. In Figure 2A, Baf was plotted for each SNP in physical order on chromosome 11 to visualize the extent of near-homozygosity.
Figure 2.

(A) β allele frequencies (Baf) for all SNPs on chromosome 11 are shown (value for genotype AA: 0, AB: 0.5, and BB 1.0). From the top the results are shown of the father of propositus 1, carrier of β cd39, the propositus 1 having the segmental paternal uniparental isodisomy of approx. 47.7Mb in an estimated mosaicism of 65%, and the mother of propositus 1, who is completely normal. Propositus 3 and 2 show a similar segmental isodisomy as propositus 1 of almost the entire short arm of chromosome 11 (approx. 47.2 and 48.9 Mb, respectively), with different percentage of mosaicism (70% and 80%, respectively). Patients showed normal SNP intensity values across 11p, precluding the possibility that deletion events cause the deviating Baf pattern. (B) The Identity-By-State (IBS) per SNP between propositus 1 and each parent plotted by physical position on chromosome 11. The number of SNPs per IBS value is indicated on the right. The comparison with the father (bottom) shows the expected parent-offspring IBS pattern (two IBS 0 SNPs are genotype errors); the comparison with the mother (top) shows 207 unexpected SNPs in IBS 0, caused by the absence of maternal alleles for those SNPs due to paternal segmental UPID.
The Chromosome 11 ideogram is taken from David Adler, Dept. of Pathology at the University of Washington, USA. The Identity By State (IBS) per SNP between parent-offspring pairs on a chromosomal scale (Figure 2B) has been calculated using the SNPduo program6 (http://pevsnerlab.kennedykrieger.org/SNPduo.)
Results and Discussion
Direct sequencing of the entire β-globin gene in the 3 propositi showed repeatedly and on different DNA samples a near to complete homozygous pattern for the mutant allele with only a minor presence of the wild-type signal, indicative for a cellular mosaicism (Figure 1). MLPA analysis revealed normal patterns excluding deletions (data not shown). As a next step, we used the Illumina OmniExpress-12_v1 human 730K SNP array to determine the degree of mosaicism and chromosomes involved. The absence of a deletion, concluded from MLPA, was confirmed by the Illumina data showing normal signal intensities for the complete genome of all 3 patients. Remarkably, a high amount of near-homozygous calls is seen for almost the entire short arm of chromosome 11 in all 3 patients at least 47 Mb in length (Figure 2), indicative for segmental uniparental isodisomy (segmental UPID) of 11p, the segment containing the β-globin gene cluster at 11p15.5.7 The percentage of mosaicism is estimated and differs between patients (Figure 2A).
For propositus 1, both parents were available for blood collection, which allowed us to determine the parental origin of the isodisomic segment by array analysis. We compared the Identity By State (IBS) for each SNP between each parent and the patient across the genome using SNPduo.6 IBS is defined as the number of alleles shared (0, 1, or 2) between individuals, without knowledge of descent. Segmental UPID is visible as the presence of IBS 0 SNPs in the parent-offspring comparison with the non-affected parent, as both alleles containing these SNPs are inherited from the affected parent. The results confirm the paternal origin of the segment showing isodisomy for the majority of the cells used for DNA isolation, carrying the paternal β-thalassemia mutation in propositus 1 (Figure 2B). By plotting a histogram of the β-allele frequency for all SNPs along the short arm of chromosome 11 that is near-homozygous for all patients, we estimated the percentage of mosaicism (Figure 2A). For propositus 2 (80%) and 3 (70%), there was no difference in the percentage of mosaicism between DNA isolated from blood or saliva. Furthermore, there seems to be no correlation between percentage of mosaicism found at the age of onset, as propositus 1 (65%) has the lowest percentage of mosaicism, but the earliest age of onset at the age of 26 years, while propositus 3 has the highest percentage and remained asymptomatic until the age of 46 years.
Table 1.
Hematological parameters and clinical description.
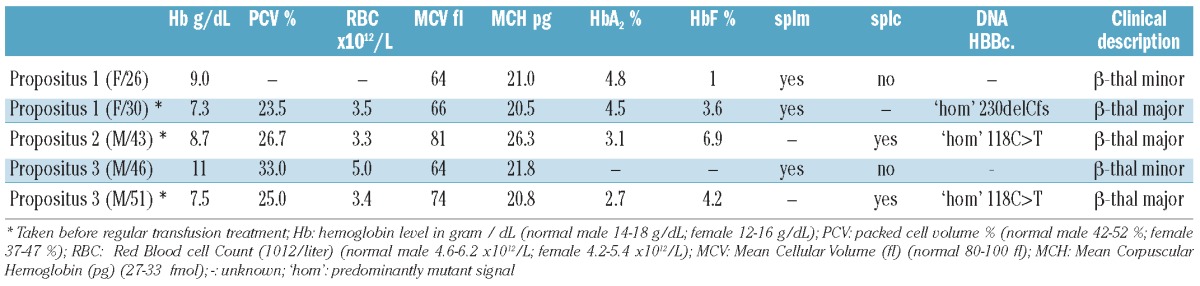
Late-onset β-thalassemia major in a carrier of a single β-globin gene mutation is extremely rare. Beta-thalassemia intermedia patients with a mitotic LoH in the hematopoietic lineage due to a deletion containing the unaffected β-globin gene has been reported by Badens et al. and Galanello et al.8,9 Somatic deletions giving rise to loss of heterozygosity have been discovered and studied in the context of carcinogenesis.10,11 However, as MLPA and Illumina SNP array analysis showed no deletion involving the β-globin gene or neighboring region in the DNA isolated from blood of any of these independent patients, another molecular cause was expected. A previously reported case of late-onset β-thalassemia by Chang et al.,12 and a sickle cell disease case reported by Swensen et al.,13 involve LoH due to segmental isodisomy of chromosome 11p. The striking resemblance between these 2 cases and the 3 presented here uncovers a common molecular cause for this remarkable phenomenon. It is the first time that individuals showing late-onset β-thalassemia have been studied in such molecular detail to allow comparison amongst each other and with previously reported cases in literature. The use of high-density SNP array technology of Illumina revealed the largest segmental UPID ever associated with late-onset β-thalassemia covering a region of up to 45 Mb. The Illumina data in SNP duo software allowed the paternal origin by IBS to be determined.
RNA analysis from cultured erythroid precursor cells, globin chain synthesis and comparison between DNA isolated from blood and hair follicles or fibroblasts to confirm the original carrier status in tissues other than hematopoietic tissue has not been performed, as only archived DNA samples were available. However, this will be the subject of future study. On the other hand, if the segmental UPID had been constitutional, the patients would have been blood transfusion-dependent from six months after birth, following the hemoglobin switch from γ- to β-globin gene expression. In contrast they became transfusion-dependent much later in life (3rd and 4th decade), suggesting they were born as healthy carriers, as is clinically evident.
To explain the late-onset blood transfusion dependency, Chang et al.12 suggested a growth advantage of the mutant hematopoietic cells was responsible. The loss of the maternally-active growth suppressor gene H19 and the presence of the duplicated paternally-active growth-enhancer gene IGF-2 on 11p, as seen in 20% of Beckwith-Wiedemann patients showing UPD of 11p, promotes cellular growth and predisposes to tumor development when experimentally over-expressed in vivo.14,15 If a somatic mutation that causes paternal UPID occurred in the patient’s hematopoietic tissue, the number of cells that become homozygous for the paternal β-thalassemia mutation may outgrow the heterozygous hematopoietic cells during life, explaining the late-onset blood transfusion dependency.
Segmental UPID may arise as the consequence of a postzygotic somatic recombination, between the maternal and paternal homolog, in which the UPID segment lies distally, the rest of the chromosome being biparental. When this happens early in embryogenesis in one cell of the early conceptus, this can be the source of segmentally UPID tissue in a part of the conceptus. Alternatively, this may happen as a mitotic event in hematopoietic progenitor cells, giving a proliferative advantage to segmental UPID cells.7,14,16–19 Interestingly, mosaic segmental paternal isodisomy involving a locus at 11p is responsible for approximately 20% of Beckwith-Wiedemann syndrome (BWS).13,16 There is only one reported case of BWS and β-thalassemia major.20 However, this was in a new born who, in contrast to the patients described here, had congenital BWS and β-thalassemia major from birth, the father being a β-thalassemia carrier. None of the 3 late-onset β-thalassemia major cases presented here showed any clinical signs of BWS.
Mitotic recombination with segmental UPID may play a role in explaining sporadic genetic diseases without a clear Mendelian inheritance, a role that has been frequently ignored.21 Depending on the mutant allele and the affected tissue, this may have an effect on developing disease. In the case of late onset β-thalassemia major, the identification of this mechanism also introduces important consequences for estimating the genetic risk in counseling for thalassemia major to couples who were previously thought to be without an elevated genetic risk.
Acknowledgments
The authors want to thank Prof. André Uiterlinden for making use of the facilities in the EMC in Rotterdam, The Netherlands, for scanning the Illumina BeadChip arrays and the Leiden Genome Technology Center (LGTC) for helpful assistance.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial and other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Weatherall DJ, Clegg JB. The Thalassemia Syndromes, 4thedn. Cambridge, MA: Blackwell Science, 2001 [Google Scholar]
- 2.van DP, Lenters E, Bakker-Verweij M, de KM, Baylan U, Harteveld CL, et al. Evaluating five dedicated automatic devices for haemoglobinopathy diagnostics in multi-ethnic populations. Int J Lab Hematol. 2009;31(5):484–95 [DOI] [PubMed] [Google Scholar]
- 3.Traeger-Synodinos J, Harteveld CL. Disease services: Haemoglobinopathies. 2010. Totowa, New Jersey: Humana Press [Google Scholar]
- 4.Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of α-thalassaemia deletions and α-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108(2):295–9 [DOI] [PubMed] [Google Scholar]
- 5.Harteveld CL, Voskamp A, Phylipsen M, Akkermans N, den Dunnen JT, White SJ, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing α- and β-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet. 2005;42(12):922–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberson ED, Pevsner J. Visualization of shared genomic regions and meiotic recombination in high-density SNP data. PLoS ONE. 2009;4(8):e6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner R.J., Sutherland G.R. Chromosome abnormalities and genetic counseling. New York: Oxford University Press; 2004 [Google Scholar]
- 8.Galanello R, Perseu L, Perra C, Maccioni L, Barella S, Longinotti M, et al. Somatic deletion of the normal β-globin gene leading to thalassaemia intermedia in heterozygous β-thalassaemic patients. Br J Haematol. 2004;127(5):604–6 [DOI] [PubMed] [Google Scholar]
- 9.Badens C, Mattei MG, Imbert AM, Lapoumeroulie C, Martini N, Michel G, et al. A novel mechanism for thalassaemia intermedia. Lancet. 2002. J;359(9301):132–3 [DOI] [PubMed] [Google Scholar]
- 10.Grundy P, Telzerow P, Paterson MC, Haber D, Berman B, Li F, et al. Chromosome 11 uniparental isodisomy predisposing to embryonal neoplasms. Lancet. 1991;338 (8774):1079–80 [DOI] [PubMed] [Google Scholar]
- 11.Shuman C, Smith AC, Steele L, Ray PN, Clericuzio C, Zackai E, et al. Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet A. 2006;140(14):1497–503 [DOI] [PubMed] [Google Scholar]
- 12.Chang JG, Tsai WC, Chong IW, Chang CS, Lin CC, Liu TC. {β}-thalassemia major evolution from {β}-thalassemia minor is associated with paternal uniparental isodisomy of chromosome 11p15. Haematologica. 2008;93(6):913–6 [DOI] [PubMed] [Google Scholar]
- 13.Swensen JJ, Agarwal AM, Esquilin JM, Swierczek S, Perumbeti A, Hussey D, et al. Sickle cell disease resulting from uniparental disomy in a child who inherited sickle cell trait. Blood. 2010;116(15):2822–5 [DOI] [PubMed] [Google Scholar]
- 14.Dutly F, Baumer A, Kayserili H, Yuksel-Apak M, Zerova T, Hebisch G, et al. Seven cases of Wiedmann-Beckwith syndrome, including the first reported case of mosaic paternal isodisomy along the whole chromosome 11. Am J Med Genet. 1998;79 (5):347–53 [DOI] [PubMed] [Google Scholar]
- 15.Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J. 1994;13(14):3286–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper WN, Curley R, Macdonald F, Maher ER. Mitotic recombination and uniparental disomy in Beckwith-Wiedemann syndrome. Genomics. 2007;89(5):613–7 [DOI] [PubMed] [Google Scholar]
- 17.Henry I, Puech A, Riesewijk A, Ahnine L, Mannens M, Beldjord C, et al. Somatic mosaicism for partial paternal isodisomy in Wiedemann-Beckwith syndrome: a post-fertilization event. Eur J Hum Genet. 1993; 1(1):19–29 [DOI] [PubMed] [Google Scholar]
- 18.Slatter RE, Elliott M, Welham K, Carrera M, Schofield PN, Barton DE, et al. Mosaic uniparental disomy in Beckwith-Wiedemann syndrome. J Med Genet. 1994;31(10):749–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010; 154C(3):343–54 [DOI] [PubMed] [Google Scholar]
- 20.Beldjord C, Henry I, Bennani C, Vanhaeke D, Labie D. Uniparental disomy: a novel mechanism for thalassemia major. Blood. 1992;80(1):287–9 [PubMed] [Google Scholar]
- 21.Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330(6000):94–7 [DOI] [PMC free article] [PubMed] [Google Scholar]