

Abstract
FoxA transcription factors play major roles in organ-specific gene expression, regulating, for example, glucagon expression in the pancreas, GLUT2 expression in the liver, and tyrosine hydroxylase expression in dopaminergic neurons. Organ-specific gene regulation by FoxA proteins is achieved through cooperative regulation with a broad array of transcription factors with more limited expression domains. Fork head (Fkh), the sole Drosophila FoxA family member, is required for the development of multiple distinct organs, yet little is known regarding how Fkh regulates tissue-specific gene expression. Here, we characterize Sage, a bHLH transcription factor expressed exclusively in the Drosophila salivary gland (SG). We show that Sage is required for late SG survival and normal tube morphology. We find that many Sage targets, identified by microarray analysis, encode SG-specific secreted cargo, transmembrane proteins, and the enzymes that modify these proteins. We show that both Sage and Fkh are required for the expression of Sage target genes, and that co-expression of Sage and Fkh is sufficient to drive target gene expression in multiple cell types. Sage and Fkh drive expression of the bZip transcription factor Senseless (Sens), which boosts expression of Sage-Fkh targets, and Sage, Fkh and Sens colocalize on SG chromosomes. Importantly, expression of Sage-Fkh target genes appears to simply add to the tissue-specific gene expression programs already established in other cell types, and Sage and Fkh cannot alter the fate of most embryonic cell types even when expressed early and continuously.
Keywords: bHLH proteins, Drosophila, Fork head (Fkh), FoxA, Sage, Salivary gland, Senseless (Sens)
INTRODUCTION
Among the major questions in developmental biology are how cells are specified to form distinct cell types and how tissue-specific programs of gene expression are established and maintained. Key players in this process are the FoxA family of winged-helix transcription factors, originally discovered in mammals because of their roles in activating transcription of liver-specific genes (Lee et al., 2005; Kaestner, 2010). More recently, however, FoxA proteins have been shown to play major roles in regulating gene expression in additional organs, including the pancreas (Gao et al., 2007; Gao et al., 2008), lungs (Wan et al., 2005), midbrain (Ferri et al., 2007; Lin et al., 2009), mammary glands (Bernardo et al., 2010) and prostate (Mirosevich et al., 2005). The FoxA genes function at multiple levels, from the specification of whole organs to the regulation of distinct cell types within these organs to the expression of cell type-specific enzymes (Lee et al., 2005; Ferri et al., 2007; Gao et al., 2008; Gao et al., 2010). Current models suggest that FoxA proteins, through their ability to displace histones, bind to DNA creating an open conformation that allows for subsequent binding of other transcription factors that then provide tissue specificity to gene expression (Cirillo et al., 1998; Zaret, 1999; Cirillo et al., 2002).
Challenges to this model come from studies in breast and prostate cancer cell lines revealing that FoxA proteins bind different enhancers in the two cell types (Lupien et al., 2008). Additional studies in the pancreas have revealed differential binding of FoxA proteins in fetal versus adult pancreatic islet cells (Gao et al., 2008). Beyond the cell type and stage specificity of FoxA binding is the additional complication that FoxA proteins have also been localized to closed chromatin (Eeckhoute et al., 2009). Thus, FoxA proteins might not bind a universal set of targets and FoxA binding might not always result in changes in chromatin conformation. Recent studies reveal that FoxA proteins bind DNA and activate gene expression in a combinatorial manner with other transcription factors. FoxA proteins often exhibit co-occupancy on sites with other transcription factors such as the glucocorticoid (Nitsch and Schütz, 1993), androgen (Gao et al., 2003) and estrogen (Carroll et al., 2005) nuclear receptors. Indeed, gender bias in liver cancer is due to FoxA proteins working either with the estrogen or androgen nuclear receptors to regulate a differential set of downstream target genes (Li et al., 2012).
Simple model systems provide excellent paradigms for understanding mechanisms of gene function in mammals, and the FoxA proteins are no exception. Studies of the sole worm FoxA protein, PHA-4, suggest that PHA-4 binds directly to all genes expressed in the multiple cell types that make up the C. elegans pharynx. The current understanding is that the low concentrations of PHA-4 present at early stages are only sufficient to activate the expression of genes with high-affinity binding sites. The concentrations of PHA-4 that build up over time are eventually high enough to activate genes with lower affinity binding sites, providing a mechanism for the temporal control of gene expression by a single transcription factor (Gaudet and Mango, 2002). Like the vertebrate FoxA proteins, which are expressed in a wide variety of tissues early in development, PHA-4 is also expressed in tissues other than the pharynx, including the intestine, rectum and somatic gonad (see http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=worm&c=Gene&l=pha-4). Even within the pharynx, PHA-4 contributes to the development of several distinct cell types, including muscles, epithelia, marginal cells and glands (Kormish et al., 2010).
The Drosophila salivary gland (SG) provides an excellent model for learning how FoxA proteins function in organ morphogenesis. Much is known regarding the specification of this organ and the sole Drosophila FoxA family member, Fork head (Fkh), plays major roles in its development. SGs are initially specified by the combined activities of the homeotic protein Sex combs reduced (Scr) and its co-factors Extradenticle (Exd) and Homothorax (Hth) (Panzer et al., 1992; Ryoo and Mann, 1999; Henderson and Andrew, 2000). All three factors are essential for SG formation and ectopic expression of Scr, the one spatially limited component, can induce SG cell fates in the subset of ectodermal cells that do not experience activated Dpp signaling (dorsal cells) or express neither Teashirt (Tsh) (parasegments 3-14) or Abdominal B (Abd-B) (parasegment 15) (Panzer et al., 1992; Andrew et al., 1994; Henderson et al., 1999). In the SG secretory cells, Scr and its co-factors activate the expression of several transcription factors, including Fkh, the bZip protein CrebA, the bHLH protein Sage and the SP1-like protein Huckebein (Hkb) (Panzer et al., 1992; Andrew et al., 1994; Andrew et al., 1997; Myat and Andrew, 2000b). Since the expression of Scr and Hth and the nuclear localization of Exd disappear shortly after SGs are specified, the early expressed SG transcription factors play major roles in maintaining and implementing the SG fate decision. Indeed, Fkh is required for many aspects of SG development, including maintaining its own expression and that of at least two other early expressed SG transcription factors: CrebA and Sage (Zhou et al., 2001; Abrams and Andrew, 2005; Abrams et al., 2006). Fkh prevents cell death in SG cells and is required for invagination of the SG primordia to form the initial tubes (Myat and Andrew, 2000a). Fkh also prevents expression of duct genes in the secretory primordia (Kuo et al., 1996; Haberman et al., 2003) and activates and maintains SG expression of Senseless (Sens) (Chandrasekaran and Beckendorf, 2003), a zinc-finger protein expressed in the SG and peripheral nervous system (Nolo et al., 2000; Chandrasekaran and Beckendorf, 2003).
As with all FoxA family proteins, Drosophila Fkh is expressed in many tissues in addition to the SG, including the anterior and posterior midgut, proventriculus, hindgut, Malpighian tubules, hemocytes and a subset of CNS cells (Weigel et al., 1989). How does this one protein have such profound effects on development and gene expression in one organ, yet regulate distinct functions and target genes in the other cell types in which it is expressed? The bHLH factor Sage is exclusively expressed in the SG and is thus an excellent candidate to regulate SG-specific gene expression (Moore et al., 2000). Indeed, Sage has been implicated in the regulation of two prolyl-4-hydroxylase genes that encode SG1 and SG2, proteins that modify cargo being trafficked through the secretory pathway (Abrams et al., 2006). However, because loss-of-function mutations in sage were not previously available, these and other studies of sage function have been limited to either RNAi knockdown or overexpression analysis (Chandrasekaran and Beckendorf, 2003; Li and White, 2003; Abrams et al., 2006).
Here, we create a deletion of sage by homologous recombination and use genome-wide strategies to identify its downstream targets, revealing for the first time what types of proteins are produced by embryonic SGs. We show that both Fkh and Sage are required for the expression of all Sage target genes and that together they can induce ectopic expression of these target genes in multiple additional cell types. We demonstrate that Sage and Fkh colocalize on SG chromosomes, along with their downstream target Sens, and that both proteins are enriched at target gene enhancers. Thus, Drosophila Fkh collaborates with Sage and Sens to achieve SG-specific functions. Importantly, Fkh and Sage are not sufficient to alter cell fate, they simply add to the gene expression programs already established in other cell types. Our findings suggest that, whereas Fkh plays an instrumental role in implementing and maintaining the cell fate decision made by the earlier acting patterning genes, it is not sufficient to establish cell fate, even when partnered with Sage.
MATERIALS AND METHODS
Fly strains
fkh6 has an 11 bp deletion, resulting in a frame-shift after codon seven (Weigel et al., 1989). sensE2 is an amorphic EMS-induced allele with a stop codon in the middle of the ORF (Wang et al., 2010). The CrebA23w protein null allele was generated by P-element excision (Andrew et al., 1997). tub-Gal4 (Lee and Luo, 1999) and en-Gal4 (Weiss et al., 2001) drive expression of UAS constructs in the entire embryo or in epidermal stripes, respectively. Gal4-driven expression of the baculovirus P35 caspase inhibitor blocks programmed cell death (Clem et al., 1991). The fkh6 H99 chromosome contains Df(3L)H99, which deletes the pro-apoptotic genes reaper, hid (Wrinkled - FlyBase) and grim, allowing survival of fkh mutant SG cells (Myat and Andrew, 2000a). UAS-Sage, sage-Gal4 and UAS-Fkh were generated in our laboratory (Abrams et al., 2006; Chung et al., 2009; Maruyama et al., 2011). H. Bellen provided the UAS-Sens flies (Nolo et al., 2000). All homozygous lethal mutations were maintained over the TM3, ftz-lacZ, the TM3, twi-GFP or the TM6B, Ubx-lacZ balancer chromosomes, allowing unambiguous identification and/or isolation of homozygous mutants.
Generation of the sage knockout allele
Null sage alleles were generated by homologous recombination (Gong and Golic, 2003) and verified by PCR (supplementary material Fig. S2). As expected, the mutant lines do not express detectable sage mRNA or protein. The sage knockout does not significantly affect the function of the upstream Aats-trp gene, based on complementation analysis, but does affect the downstream VhaM8.9 gene based on microarray analysis, which showed the transcript levels of this gene to be significantly reduced.
Antibody production, embryo staining and whole-mount in situ hybridization
Antiserum was generated in both rat and guinea pig to the product of the full-length sage ORF subcloned into HindIII and XhoI sites of the pTrcHisA vector (Invitrogen). Recombinant Sage was expressed and purified from E. coli as inclusion bodies and injected into host animals following standard immunization protocols (Covance).
Embryo fixation and antibody staining were performed as described (Reuter and Scott, 1990). Primary antibodies were used at the following dilutions: guinea pig anti-Sage 1:2000, rat anti-Sage 1:1000, mouse anti-Crb [1:100; Drosophila Hybridoma Studies Bank (DHSB)], rabbit anti-Fkh (1:2000; a gift from S. Beckendorf, Berkeley, CA, USA), rat anti-CrebA [1:5000 for HRP, 1:1000 for fluorescence (Andrew et al., 1997)], rabbit anti-CrebA [1:10,000 (Fox et al., 2010)], guinea pig anti-Sens (1:2000 for embryos; a gift from H. Bellen, Baylor College of Medicine, Houston, TX, USA), rat anti-PH4αSG1 [1:15,000 (Abrams et al., 2006)], rabbit anti-PH4αSG2 [1:8000 (Abrams et al., 2006)], rabbit anti-Sas (1:500; a gift from D. Cavener, Penn State University, PA, USA), mouse anti-α-Spectrin (1:2; DSHB), anti-cleaved caspase 3 (1:100; Cell Signaling), mouse anti-Cut (1:50; DSHB) and mouse anti-β-gal (1:5000 for HRP, 1:500 for fluorescence; Promega). Biotin-tagged secondary antibodies were used at 1:500 and staining was amplified using the Vectastain ABC kit (Vector Labs). Fluorescently tagged secondary antibodies (Alexa Fluor 488, 568, 647; Molecular Probes) were used at 1:500. HRP-stained embryos were imaged using Nomarski optics on a Zeiss Axiophot microscope equipped with a Nikon Coolpix 4500 camera. Confocal images were obtained using a Zeiss LSM 510 Meta microscope.
Whole-mount in situ hybridization was performed as described previously (Lehmann and Tautz, 1994).
Chromosome immunostaining
Polytene chromosomes were isolated and fixed for immunostaining as described previously (Andrew and Scott, 1994), except that the second fixation was in 50% glacial acetic acid. Preimmune and immune sera were used at a dilution of 1:200. Secondary fluorescently tagged antibodies (Alexa Fluor 488 or 568; Molecular Probes) were used at 1:100. Chromosomes were also stained with DAPI (1 μg/ml). No antibody staining of chromosomes was detected with either Sage or CrebA preimmune sera.
Microarray analysis
Total RNA was isolated from stage 11 to 17 wild-type embryos and compared with total RNA isolated from stage-matched sageko embryos or tub-Gal4::UAS-Sage embryos by Trizol (Invitrogen) extraction. The Qiagen RNeasy kit was used for RNA cleanup. Total RNA (100 ng) was labeled and amplified using standard Affymetrix protocols. Three samples for each genotype were hybridized to the Drosophila Genome 2.0 Chip. Scanned intensity values were normalized using RMA [Partek software (Irizarry et al., 2003a; Irizarry et al., 2003b)] and statistical analysis was performed using the Spotfire software package (TIBCO). Target genes downregulated in sage null mutants were identified based on a ≥1.5-fold decrease in gene expression with P≤0.05 when compared with Oregon R controls. Target genes upregulated with Sage overexpression were identified based on a ≥1.5-fold increase in gene expression with P≤0.05 compared with Oregon R or tub-Gal4 controls. All data have been deposited at GEO with accession numbers GSE40358 (tub-Gal4::UAS-Sage) and GSE40963 (sage mutant).
ChIP-qPCR
Chromatin immunoprecipitation (ChIP) was performed as described (Fox et al., 2010) except that samples were normalized to Actin 5C (Sage experiments) or RpL32 (Fkh experiments) and that fold change represents the difference between the control and the experimental antiserum (Livak and Schmittgen, 2001). Fold change was calculated using the ΔΔCt method (Livak and Schmittgen, 2001), with the assumption that PCR efficiency was the same for all samples.
RESULTS
sage encodes an SG-specific nuclear bHLH protein required for SG cell survival
Transcripts for the bHLH gene sage were detected exclusively in the SG, beginning at embryonic stage 10 and continuing through embryogenesis, consistent with previous reports (Fig. 1A) (Moore et al., 2000, Abrams et al., 2006). sage mRNA expression is absent in embryos mutant for the homeotic protein encoded by Scr, consistent with the requirement for Scr to form SGs (supplementary material Fig. S2D). Antibodies generated against full-length Sage protein revealed strong nuclear staining in the same spatial and temporal pattern as the sage transcripts (Fig. 1B). Sage protein was also detected in third instar larval SGs (see below) and sage-Gal4-driven UAS-GFP expression was detected in embryonic, larval and adult SGs (data not shown). Thus, Sage is expressed early and continuously in the embryonic, larval and adult SGs.
Fig. 1.
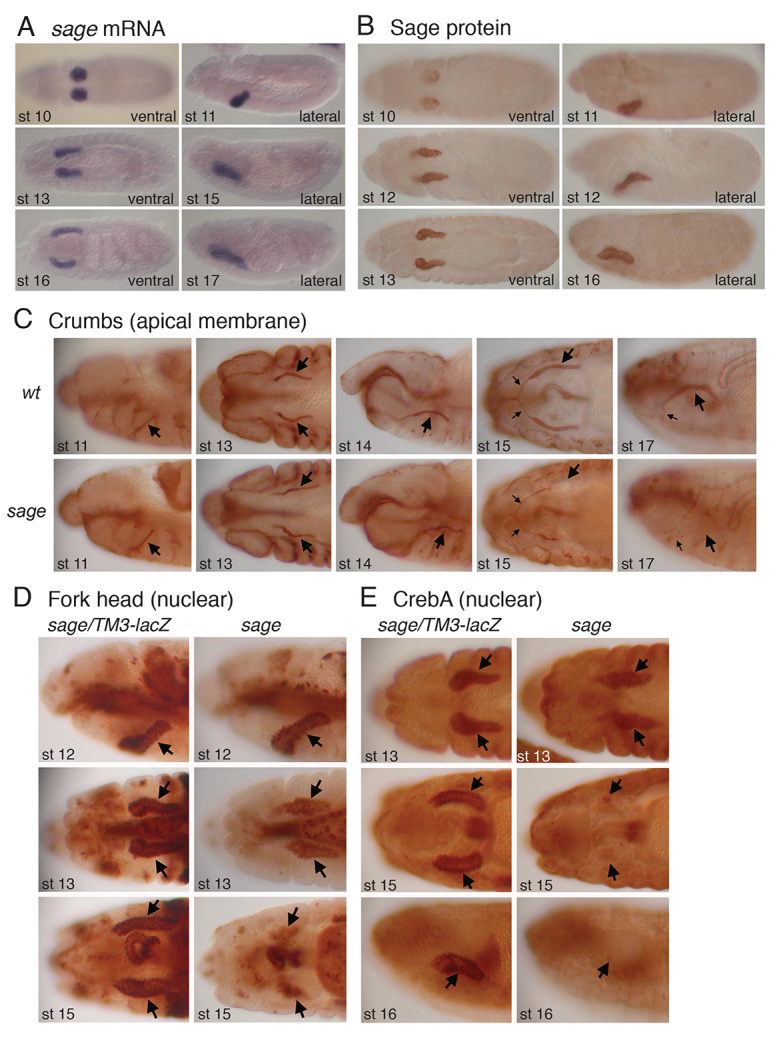
Drosophila Sage is expressed and required in the embryonic salivary gland. (A,B) In situ hybridization of sage mRNA (A) and immunostaining with guinea pig antiserum directed against Sage protein (B) reveal similar patterns in stage 10-16/17 Drosophila embryos. (C) Crb apical staining in stage 11-13 sage mutant salivary glands (SGs) was relatively normal. By embryonic stage 15, Crb staining was not detected in the distal half of the SGs of sage mutants, and by stage 17 Crb was not detected in secretory cells (large arrows), although staining appeared normal in the salivary duct (small arrows). wt, wild type. (D) Fkh nuclear staining was relatively normal in stage 12 and declined in intensity by stage 13. Fkh was detected in far fewer SG cells in stage 15 sage mutants than in heterozygous age-matched siblings (arrows). (E) CrebA nuclear staining was relatively normal in stage 13 sage mutant SGs. Very low CrebA levels were detected at stage 15 and staining was not detectable in SGs by stage 16.
Sage-related proteins are readily identified in other insects, but only distantly related proteins exist in vertebrates. The closest mammalian relatives are neurogenin 1, 2 and 3, mesoderm posterior protein 1 and 2, and pancreas transcription factor 1; homology between these proteins and Sage is limited, however, to the bHLH domain (supplementary material Fig. S1C). A PHYLIP unrooted tree using only this domain of the human versions of these proteins and Drosophila Sage suggests that Sage is equally related to all three proteins (supplementary material Fig. S1B), in contrast to the previous grouping of Sage with only a subset of these proteins (Stevens et al., 2008).
To date, analysis of sage function has been limited to RNAi (Chandrasekaran and Beckendorf, 2003; Li and White, 2003) and overexpression (Abrams et al., 2006) studies. To fully characterize the role of Sage, we created a 4 kb deletion that removed sage as well as a portion of the 3′ ends of the neighboring genes by homologous recombination (referred to herein as sageko or sage mutant) (supplementary material Fig. S2). Examination of homozygous sageko embryos using a variety of membrane and nuclear markers revealed that SG development was relatively normal through embryonic stage 13 (Fig. 1C-E). Marker staining was not detectable, however, in the distal SG cells of some embryonic stage 14 and all embryonic stage 15 sage mutants, and marker staining was almost completely absent in stage 16 and older mutant embryos. Staining of sage mutants with antibodies to the SG-specific endoplasmic reticulum proteins PH4αSG1 (SG1) and PH4αSG2 (SG2) revealed reduced levels of both proteins in early embryos (Fig. 2A; data not shown) (Abrams et al., 2006). SG1 and SG2 staining in late embryos was observed in small puncta in the anterior half of the embryo, suggesting that the SGs were dying and that the cellular debris was being ingested by macrophages, as would occur if the cells were undergoing apoptosis. Indeed, high-magnification images revealed punctate staining of SG1 and SG2 in large macrophage-like cells (Fig. 2B; data not shown).
Fig. 2.

Sage is required for SG viability and normal lumenal morphology. (A) PH4αSG2 endoplasmic reticulum staining in sageko SGs was reduced relative to WT through stage 14 (large arrows), when staining in small puncta in the region of the SG was first visible (small arrows). By embryonic stage 15, most of the PH4αSG2 staining in sage mutants was in small puncta in the region of the SGs (small arrows). (B) Large cells with irregular PH4αSG2 staining (arrows) are likely to be macrophages that have engulfed dying SG cell debris. (C) Cleaved caspase 3 (CC3) staining at stage 15 reveals extensive apoptotic cell death in sageko compared with WT SGs. (D) rpr mRNA levels are upregulated (arrowheads) in stage 14 sageko and sensE2 SGs. (E) Expression of the anti-apoptotic P35 gene in sageko rescues SG cell death associated with sage loss. Arrowheads indicate the secretory portion of the SG. (F) SGs stained with antibodies to the apical membrane protein Stranded at second (Sas), the basolateral protein α-Spectrin and nuclear CrebA reveal lumen irregularities in P35-rescued sageko SGs.
To confirm that SG loss is due specifically to loss of sage and not the adjacent genes also affected in the sageko animals, we expressed the sage ORF in the SGs of sageko/Df(3R)ED5339 embryos using the sage-Gal4 driver. Staining with antibodies to the apical membrane protein Crb or nuclear protein CrebA revealed that SG-specific expression of sage largely rescued the SG lethality and that the rescued SGs had normal morphology. In situ hybridization for Sage downstream target genes (see below) also revealed rescued SG gene expression (supplementary material Fig. S3).
P35-mediated rescue of SG cell survival reveals abnormal lumenal morphologies
To determine whether SGs in sage mutants were dying by apoptosis, we stained wild-type (WT) and sageko embryos with an antibody directed against human cleaved caspase 3 (CC3) (Fan and Bergmann, 2010). Very high levels of CC3 staining were seen in stage 14 and 15 sage mutant SGs, overlapping significantly with CrebA staining (Fig. 2C). Consistent with the apoptotic death observed in SGs at these stages, the proapoptotic genes reaper (rpr) and head involution defective (hid) were expressed to high levels in the SGs of stage 13/14 sage mutants but were undetectable in WT SGs (Fig. 2D; data not shown). To determine whether any late morphological defects are associated with loss of sage, we rescued SG death by expressing the baculovirus anti-apoptotic protein P35 (Sahdev et al., 2010) in sage mutant SGs. P35-rescued SGs stained with antibodies to both membrane and nuclear proteins revealed marked irregularities at the lumenal surface of the rescued SGs as well as reduced lumenal volume (Fig. 2E,F). Thus, Sage is required for late SG viability and normal lumenal morphology.
Similar late SG cell death phenotypes have been described with loss of sens, which encodes a bZip transcription factor related to mammalian Gfi1, and whose SG expression absolutely depends on Fkh (supplementary material Figs S1, S4) (Chandrasekaran and Beckendorf, 2003). Staining of sens null embryos with a variety of SG markers also revealed extensive SG cell death, but the phenotypes were milder; death occurred at later stages and involved fewer cells (data not shown). To examine whether the phenotypes might be linked, we examined Sens and Sage in sage and sens mutant backgrounds. Levels of Sage were not detectably altered in sens mutants, whereas levels of Sens in sage null embryos were slightly reduced, even at early stages (supplementary material Fig. S4).
Sage regulates SG-specific secreted proteins and their modifiers
To gain insight into the role of Sage in the SG, we carried out microarray analyses comparing RNA isolated from age-matched sageko and WT embryos. When compared with WT, 579 genes were downregulated in sage mutants (≥1.5-fold, P≤0.05); however, because sage mutant SGs die by stage 15, potentially affecting the expression of all SG genes, we also isolated RNA from WT embryos expressing Sage protein in all cells using the ubiquitously expressed tub-Gal4 (supplementary material Fig. S2). This analysis revealed 661 genes that were upregulated relative to WT controls and 805 genes that were upregulated relative to tub-Gal4 controls (≥1.5-fold, P≤0.05) (Fig. 3; supplementary material Tables S1-S3). A comparison of the downregulated genes and combined set of upregulated genes revealed 55 overlapping genes. In addition to these 55, we also included nine genes for which expression decreased at least 1.9-fold in the sage knockout for additional studies (supplementary material Table S4). Based on our own findings, as well as the available online expression data for each of these genes, 43 (67%) of them are known to be expressed in embryonic, larval and/or adult SGs. Examination of the gene ontology descriptions assigned to the corresponding proteins revealed that 38 encode either secreted or transmembrane proteins (59%), with an additional ten (∼16%) implicated in the folding and modification of proteins traveling through the secretory pathway. Thus, nearly 75% of the genes that were most highly affected by changes in sage expression encode proteins that travel through the secretory pathway or the proteins that modify them.
Fig. 3.

Microarray analysis of sage loss and overexpression identifies targets regulated by both Sage and Fkh. (A,B) Total RNA (100 ng) was isolated from three individual samples of stage 11-17 embryos for each genotype. Volcano plots show genes that were upregulated (red) or downregulated (blue) at least 1.5-fold (P≤0.05) in sageko embryos (A) or at least 1.5-fold (P≤0.05) in embryos in which Sage is expressed ubiquitously (tub-Gal4::UAS-Sage) (B) relative to WT (left plot) or tub-Gal4 (right plot) controls. (C) The expression of 55 genes was significantly reduced with loss of sage and significantly elevated with sage overexpression (combining comparisons with the two control sets). Using equations for a hypergeometric distribution (Blom, 1989) and Mathematica (Wolfram Research) to calculate the probability that the observed overlap in genes that are downregulated in sage nulls and upregulated with sage overexpression could occur by chance is 0.0018 for the WT control (overlap of 35 or more) and 0.00025 for the tub-Gal4 control sets (overlap of 44 or more). (D) In situ hybridization analysis confirms the regulation of target genes by Sage. Arrows indicate ectopic expression in the proventriculus when UAS-Sage is expressed using tub-Gal4 and boxed regions highlight ectopic expression in the Malpighian tubules and hindgut. (E) SG expression of Sage target genes requires Fkh and, in some cases, also requires Sens.
Sage target genes are also regulated by Fkh
Twelve genes that were significantly downregulated in sageko embryos were examined by in situ hybridization (Fig. 3D; Table 1). In WT embryos, all 12 genes exhibited SG expression beginning during embryonic stage 11 or 12 and continuing through embryogenesis. In sage null embryos, this SG expression either disappeared completely or was significantly diminished (Fig. 3D; Table 1). In embryos expressing Sage protein ubiquitously (tub-Gal4::UAS-Sage), ectopic expression of all 12 genes was observed, but only in a subset of tissues, typically including the hindgut, Malpighian tubules and proventriculus (Fig. 3D; Table 1) (Abrams et al., 2006). Since these other tissues also express Fkh (supplementary material Fig. S4), this finding suggested that Fkh could be required for the SG expression of Sage target genes. Indeed, all of the Sage target genes we tested had reduced SG expression in fkh mutant embryos, with levels typically as low as in surrounding non-SG tissues. Thus, both Sage and Fkh are required for full expression of the SG genes identified by our microarray analysis as Sage dependent (Fig. 3D,E; Table 1).
Table 1.
Expression patterns of 12 Sage target genes in embryos missing or overexpressing SG transcription factors
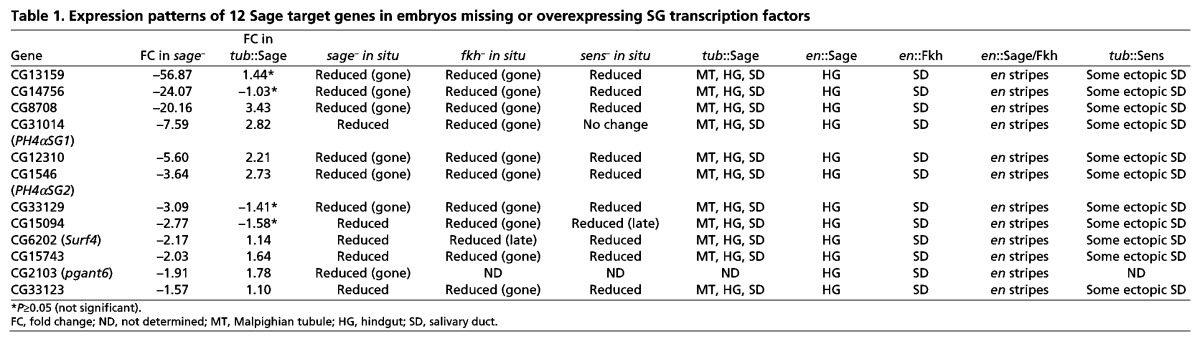
Fkh is required to maintain sage expression and could therefore be indirectly regulating Sage targets through its regulation of sage (Abrams et al., 2006) (supplementary material Fig. S4). To address this possibility, we expressed Sage, Fkh or both proteins in ectodermal stripes using the en-Gal4 driver (supplementary material Fig. S5). With every target gene tested we observed the same changes in expression pattern (Fig. 4A; Table 1). en-Gal4-driven expression of UAS-Sage alone resulted in ectopic expression of Sage target genes in only a subset of hindgut cells. en-Gal4-driven expression of UAS-Fkh alone resulted in ectopic expression of Sage target genes in only a subset of salivary duct cells, presumably because of the role that Fkh plays in establishing the boundary between SG secretory cell and duct cell fates (Kuo et al., 1996; Haberman et al., 2003). en-Gal4-driven expression of both UAS-Sage and UAS-Fkh resulted in stripes of target gene expression in every cell expressing en-Gal4. Importantly, the levels of expression observed in the ectopic stripes were comparable to the levels observed with each gene in the SG. Thus, the combined activities of Fkh and Sage are both necessary and sufficient to achieve wild-type levels of SG-specific target gene expression.
Fig. 4.

Sage and Fkh together are sufficient to drive high-level expression of SG genes. (A) Sage target gene expression in embryos expressing UAS-Sage or UAS-Fkh or both under control of en-Gal4. Black arrows indicate ectopic hindgut expression when UAS-Sage is expressed. White arrows indicate ectopic expression in the salivary duct with expression of UAS-Fkh. (B) Hindgut (HG) and Malpighian tubule (MT) staining of WT and tub-Gal4::UAS-Sage embryos. The white staining in the lower right panel indicates expression of all three markers in Malpighian tubules. (C) Global expression of both Sage and Fkh drives ectopic high-level expression of the SG marker protein SG1 in all ventral cells in early embryos but persistent high-level expression in late embryos occurs only in PS2 and in lateral regions of PS3, cells that also express Scr and its co-factors.
sens is a target of Sage-Fkh
Because loss of sage and loss of sens result in similar phenotypes, we asked whether Sens also regulates Sage-Fkh targets. sens was identified as a Sage target in our microarray analysis (supplementary material Table S4) and Sens shows the same changes in expression as observed with all of the Sage target genes that we examined (supplementary material Fig. S5). Thus, Sens is activated in all cells that express both Sage and Fkh and could also contribute to target gene expression. Indeed, in sens mutants the expression of Sage-Fkh target genes showed a range of effects, from no apparent change to a reduction to levels observed in sage mutants (Fig. 3E). Global expression of Sens on its own had similar, albeit milder, effects as expression of Fkh alone (Table 1; supplementary material Fig. S4), causing ectopic expression of SG-specific genes in salivary duct cells. Thus, Sens appears to boost the expression levels of shared Sage-Fkh SG targets as well as enhancing the Sage-independent Fkh activity that establishes the boundary between secretory gland versus duct fates.
Fkh and Sage do not reprogram cell fate
To examine whether co-expression of Sage and Fkh is sufficient to reprogram cells to an SG fate, we stained tub-Gal4::UAS-Sage embryos with an antibody to the Malpighian tubule (MT) marker Cut (Liu and Jack, 1992). Since Fkh is normally expressed in the hindgut (HG) and MTs, ectopic expression of Sage in these cells leads to ectopic SG gene expression (Fig. 3D; Fig. 4A; Table 1), along with changes in MT and HG morphology (Fig. 4B). If expression of Fkh and Sage causes these organs to adopt an SG fate, we expect that normal tissue-specific markers would disappear. Instead, we find that Cut expression persists in the MTs (Fig. 4B). Thus, Fkh and Sage are not sufficient to reprogram MT cells to become SGs, but instead they upregulate the expression of SG-specific genes. The altered MT and HG morphologies might be explained by the high levels of SG proteins that are being synthesized and, possibly, modified and secreted into the lumen of each tissue.
To determine whether earlier expression of both Fkh and Sage results in additional cells adopting SG fates, we expressed UAS-Fkh with UAS-Sage using the ubiquitous tub-Gal4 driver. SG1 antibody staining revealed ectopic expression in the ventral regions of every segment of the embryo at early stages (Fig. 4C). Interestingly, at later stages, high-level SG1 staining was observed only in parasegment (PS) 2 (where SGs normally form) and in a subset of cells in PS3 - cells that normally also express Scr. These findings are similar to those observed with global expression of Fkh alone, where transient activation of another SG marker was observed in the ventral regions of all segments at early stages but in only PS2 at late stages. Thus, expression of both Fkh and Sage can recruit additional cells to persistently express SG markers, whereas global expression of Fkh alone cannot. However, since the PS3 cells normally express Scr, this finding suggests that although Fkh and Sage can induce transient expression of SG markers they are either not expressed to high enough levels with the tub-Gal4 driver or that additional Scr-Exd-Hth-dependent factors are also necessary to specify and maintain an SG fate.
Sage and Fkh directly co-regulate the expression of SG-specific genes
Sage and Fkh together are necessary and sufficient to upregulate SG gene expression, suggesting that both proteins might bind directly to the enhancers of SG-specific genes to activate tissue-specific expression. Since both Fkh and Sage are also expressed in late stage SGs, where they would presumably continue to activate target gene expression, we first examined whether Fkh and Sage colocalize on polytene chromosomes of SGs isolated from late third instar WT larvae. Indeed, most sites bound by Fkh were also bound by Sage and vice versa (Fig. 5A). This is in contrast to chromosomes stained for CrebA and Fkh or for CrebA and Sage, where only a subset of sites bound by CrebA were bound by Fkh or Sage and vice versa (Fig. 5C,E). We also found a very high level of coincidence in sites bound by Sage and Sens and in sites bound by Fkh and Sens (Fig. 5B,D). As observed with Fkh and Sage, the overlap in the binding sites for Sens and CrebA was more limited (Fig. 5F).
Fig. 5.

Sage, Fkh and Sens colocalize on polytene chromosomes. SG polytene chromosomes were stained with different combinations of antiserum directed against Sage, Fkh, Sens and CrebA to detect the endogenous proteins and with DAPI (blue) to detect DNA. (A) Sage and Fkh. (B) Sage and Sens. (C) CrebA and Sage. (D) Fkh and Sens. (E) Fkh and CrebA. (F) CrebA and Sens. (G) Rat Sage and guinea pig Sage. (H) Rat CrebA and rabbit CrebA. Yellow arrows indicate colocalization.
Finally, we used ChIP to determine whether Fkh and Sage show increased occupancy at the enhancers for a subset of Sage target genes in whole embryos. Primer sets were designed to amplify the regions that contain a high density of putative Sage, Fkh and Sens binding sites (supplementary material Fig. S6). In most cases, Sage and Fkh preferentially pulled down these enhancer regions when compared with control immunoprecipitations performed using either the Sage pre-immune serum or anti-GFP antiserum (Fig. 6).
Fig. 6.
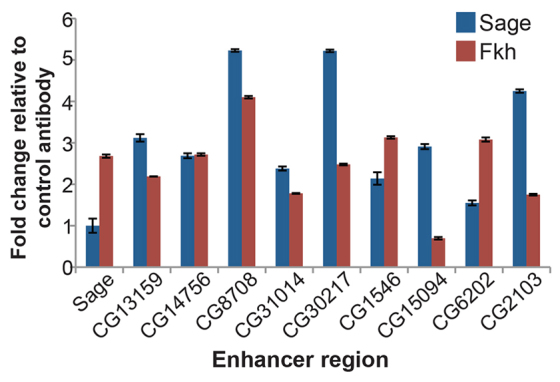
Sage and Fkh bind SG enhancers. ChIP-qPCR reveals that both Fkh and Sage preferentially bind enhancer regions for many of the target genes tested. Error bars indicate s.e.m.
Overall, the genome-wide view of in vivo chromosome binding and the enhancer-specific ChIP data support a model wherein Fkh, Sage and Sens work together to directly regulate SG-specific gene expression.
DISCUSSION
The broadly expressed FoxA proteins are required to establish and maintain tissue-specific gene expression programs, often by partnering with transcriptional co-factors (Kaestner, 2010). Here, we demonstrate that the SG-specific bHLH protein Sage is required for SG viability and regulates a large group of downstream SG target genes, many encoding secreted or transmembrane proteins and their modifiers. We show that Sage target genes also require the FoxA protein Fkh for expression and that Sage and Fkh together are sufficient to drive expression of these target genes to SG equivalent levels in multiple distinct cell types. We demonstrate that Sage, Fkh and the downstream bZip transcription factor Sens colocalize to distinct sites on chromatin, suggesting that all three proteins function to directly regulate a largely overlapping set of SG genes. Indeed, consensus sites for Sage, Fkh and Sens binding are clustered within the enhancers of Sage target genes (supplementary material Fig. S6).
The key to establishing how Fkh achieves tissue specificity was to identify SG-specific gene products. Indeed, this study is the first to reveal what is produced and secreted by the Drosophila embryonic SG. Thirteen of the top 20 genes most affected in expression by loss of sage encode secreted proteins; another three encode enzymes that modify secreted proteins. Many of the Sage target genes are found in clusters, including a cluster of four genes in cytological region 71B that encode secreted glutamate/aspartate-rich proteins (Seds 1-3 and 5), a cluster of three genes in cytological region 79F that encode related mucins (short serine/threonine-rich proteins that are typically highly glycosylated; Smucs 2, 3 and 5), and a cluster of procollagen-proline 4-dioxygenase genes (prolyl-4α-hydroxylases) in cytological region 99F, two of which are expressed exclusively in the SG (PH4αSG1 and PH4αSG2) (Abrams and Andrew, 2002). This clustering of related genes, many of which also show similar expression patterns, suggests that gene duplication events preserved not only the gene coding regions but also the control regions.
Nearly all the identified Sage targets encode terminally differentiated gene products. This finding tells us that the genetic hierarchy controlling SG formation is only a few layers deep (Fig. 7). At the top of the hierarchy are the genes that regulate the decision to form SGs: Scr-Exd-Hth, in the absence of the negative regulators Dpp, Tsh and Abd-B (Panzer et al., 1992; Andrew et al., 1994; Henderson et al., 1999; Henderson and Andrew, 2000). Scr-Exd-Hth activate expression of CrebA, Sage, Fkh and Hkb, which function to maintain and implement the SG fate decision by maintaining their own expression, the expression of each other and of terminally differentiated gene products. CrebA primarily regulates components of the secretory machinery (Abrams and Andrew, 2005; Fox et al., 2010) and the Sage-Fkh-Sens module primarily regulates secreted gene products, their modifiers, and other tissue-specific genes (this study). Interestingly, the expression levels of many of the genes regulated by the Sage-Fkh-Sens module are also affected by loss of CrebA (supplementary material Table S4). We know that CrebA functions to boost expression of the core secretory machinery to the very high levels required in cells that are specialized for secretion, such as the SG and epidermis (Abrams and Andrew, 2005; Fox et al., 2010). This would suggest that, whereas Fkh and Sage provide tissue-specific gene activation, CrebA acts to boost transcription levels, possibly acting directly, based on our polytene chromosome binding data showing some overlap of CrebA sites with those of Sage, Fkh and Sens. Fkh also functions independently of Sage and Sens to control tube morphogenesis (Myat and Andrew, 2000a) and, therefore, must control the expression of many genes that are not targets of Sage and Sens. Our studies suggest that this subset of Fkh target genes directly participates in morphogenesis (Maruyama et al., 2011), again consistent with only a few layers of transcriptional regulation being required to build a functional organ.
Fig. 7.
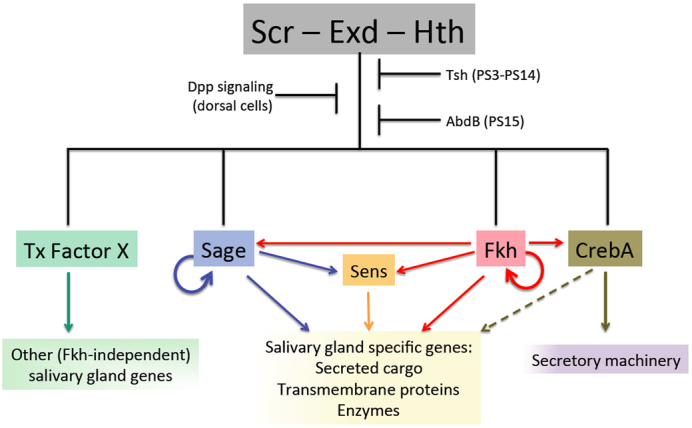
Model for regulation of SG formation. Scr, Exd and Hth regulate SG formation through the activation of a set of transcription factors expressed early and continuously in this tissue: Sage, Fkh and CrebA. Sage and Fkh activate the expression of genes encoding SG-specific secreted cargo proteins, transmembrane proteins and the enzymes that modify proteins that travel through the secretory pathway. Sage and Fkh also activate transcription of sens, which in turn boosts the expression levels of many Sage-Fkh targets. CrebA upregulates expression of the protein components of the secretory machinery to increase overall secretory capacity and contributes, directly or indirectly, to increased expression of many genes encoding SG cargo proteins. Additional uncharacterized Scr-Exd-Hth-dependent transcription factor(s) are proposed to also be required to maintain SG fate and to regulate the many known SG genes whose expression is unaffected by loss of fkh (Maruyama et al., 2011).
High-level SG-specific gene expression requires Fkh, Sage and Sens. These proteins colocalize to multiple sites on SG chromosomes, indicating direct input from all three proteins. How are the enhancers of the SG target genes organized to achieve high-level cell type-specific expression? Three general models of enhancer organization have been proposed. In the ‘enhanceosome’ model, a relatively fixed arrangement of binding sites functions as a scaffold for the cooperative assembly of transcription factors into a complex that directly interacts with the basal transcription machinery, thus controlling genes with only binary states of expression - on or off (Panne, 2008). Developmentally regulated genes, where levels of expression can vary widely in different tissues and at different developmental stages, are thought to be regulated by enhancer elements that recruit one or a small number of transcription factors that act independently and/or redundantly to modulate gene expression levels - the ‘billboard’ model of enhancer organization. In this model, enhancer arrangement is random with respect to spacing and orientation because the enhancers are proposed to assemble and function independently. This model accounts for how some enhancers can function as both activators and repressors, with the basal transcriptional machinery interpreting what is bound and where, thus allowing for more diversity in gene expression patterns (Arnosti and Kulkarni, 2005). The more recent ‘transcription factor collective’ model blends the first two models and suggests that, although a specific group of transcription factors functions cooperatively to regulate gene expression, there is no set ‘grammar’ to dictate the arrangement of binding sites for these transcription factors (Junion et al., 2012).
Our understanding of how tissue-specific gene expression is achieved has largely relied on ChIP-seq to examine transcription factor occupancy at promoter regions, followed by enhancer analysis to look at the minimal elements required for specific expression patterns. Unlike in these other studies, we did not set out to examine regulatory elements in SG enhancers but instead to determine the role of Sage in the SG. We discovered that Sage regulates many SG-specific terminal differentiation genes that also require Fkh, and that together they are sufficient to induce wild-type levels of expression. We also learned that Sens, a downstream target of Sage and Fkh, is necessary to achieve full-level transcriptional activation of many SG genes. Importantly, the arrangement of Fkh, Sage and Sens consensus binding sites within the enhancers of the target genes appears random with respect to position, orientation and distance between sites (supplementary material Fig. S6). Thus, our analysis supports a model in which SG genes are activated by a ‘collective’ of transcription factors - Fkh, Sage, Sens and (perhaps sometimes) CrebA - with the organization of binding sites unlikely to be important based on the random arrangement of consensus binding sites in the SG gene enhancers (supplementary material Fig. S6).
Loss of fkh more profoundly affects the expression of SG genes than loss of sage. We believe this is because in fkh mutants, not only is fkh function lost but also expression of sage, sens and CrebA disappears (Abrams and Andrew, 2005; Abrams et al., 2006). In sage mutants, sage function is lost, Sens levels decline slightly, but Fkh and CrebA levels appear unaffected. Thus, many more components of the SG transcription module are lost in fkh mutants than in sage mutants. Indeed, the compounded effects of fkh loss can explain the difference in SG death phenotypes associated with the loss of each of the three factors - Fkh, Sage and Sens; SG death (and associated expression of the pro-apoptotic regulators rpr and hid) occurs much earlier with loss of fkh than with loss of either sage or sens. Importantly, although Fkh appears capable of inducing ectopic expression of all three SG transcription factors, it can only do so in cells that either also express Sage (CrebA and Sens expression is induced in stripes in en-Gal4::UAS-Sage, UAS-Fkh) or in cells that also express Scr and its co-factors (Sage, CrebA and Sens expression can be induced in duct cells, which also express Scr, in en-Gal4::UAS-Fkh). Importantly, even though Fkh and Sage can induce ectopic expression of every Sage target that we tested, ectopic expression appears to be in addition to the gene expression program already functioning within the different cell types based on the persistent expression of MT markers. Indeed, although expressing both Fkh and Sage in all cells from early stages can persistently drive high-level SG-specific gene expression in additional cells, it is only in cells that also express Scr. Thus, only the Scr-Exd-Hth complex, which initiates the expression of multiple components of the SG collective, is capable of specifying SG fates.
Importantly, this work demonstrates that Fkh, which is expressed and required in many cell types in the Drosophila embryo, achieves SG specificity through its collaboration with the tissue-specific bHLH protein Sage. Parallel relationships between FoxA proteins and Sage-related bHLH proteins are suggested from studies in multiple other systems. For example, neurogenin 2, FoxA1 and FoxA2 have all been implicated in dopaminergic neuron specification and differentiation in mice (Kele et al., 2006; Lin et al., 2009). Similarly, the bHLH protein Ptf1 plays major roles in pancreatic development (Krapp et al., 1996; Krapp et al., 1998) and the FoxA proteins have been implicated in both the development and maintenance of pancreatic islet cells (Gao et al., 2010). Finally, in C. elegans, the HLH-6 protein is required for the expression of pharyngeal-specific secreted mucins (Smit et al., 2008) and the FoxA protein PHA-4 has been implicated in the direct regulation of all genes in the many pharyngeal cell types (Gaudet and Mango, 2002), supporting the possibility that HLH-6 and PHA-4 directly regulate expression of the mucin genes.
Altogether, our findings, combined with studies of the related mammalian and worm proteins in specific cell types, suggest that collaborations between FoxA proteins and tissue-specific bHLH proteins play a major role in achieving tissue specificity for FoxA function and that the origin of these collaborations might be ancient.
Supplementary Material
Acknowledgments
We thank Hugo Bellen for Sens antibodies and UAS-Sens lines; Steve Beckendorf for Fkh antibodies; D. Cavener for Sas antibodies; R. Loganathan for the control Tub-Gal4 microarray data; and members of the D.J.A. laboratory and three anonymous reviewers for critical reading of the manuscript.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health [NIH RO1 DE013899 to D.J.A. and NIH K99 DE021461 to R.M.F.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.092924/-/DC1
References
- Abrams E. W., Andrew D. J. (2002). Prolyl 4-hydroxylase alpha-related proteins in Drosophila melanogaster: tissue-specific embryonic expression of the 99F8-9 cluster. Mech. Dev. 112, 165–171 [DOI] [PubMed] [Google Scholar]
- Abrams E. W., Andrew D. J. (2005). CrebA regulates secretory activity in the Drosophila salivary gland and epidermis. Development 132, 2743–2758 [DOI] [PubMed] [Google Scholar]
- Abrams E. W., Mihoulides W. K., Andrew D. J. (2006). Fork head and Sage maintain a uniform and patent salivary gland lumen through regulation of two downstream target genes, PH4alphaSG1 and PH4alphaSG2. Development 133, 3517–3527 [DOI] [PubMed] [Google Scholar]
- Andrew D. J., Scott M. P. (1994). Immunological methods for mapping protein distributions on polytene chromosomes. Methods Cell Biol. 44, 353–370 [DOI] [PubMed] [Google Scholar]
- Andrew D. J., Horner M. A., Petitt M. G., Smolik S. M., Scott M. P. (1994). Setting limits on homeotic gene function: restraint of Sex combs reduced activity by teashirt and other homeotic genes. EMBO J. 13, 1132–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew D. J., Baig A., Bhanot P., Smolik S. M., Henderson K. D. (1997). The Drosophila dCREB-A gene is required for dorsal/ventral patterning of the larval cuticle. Development 124, 181–193 [DOI] [PubMed] [Google Scholar]
- Arnosti D. N., Kulkarni M. M. (2005). Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J. Cell. Biochem. 94, 890–898 [DOI] [PubMed] [Google Scholar]
- Bernardo G. M., Lozada K. L., Miedler J. D., Harburg G., Hewitt S. C., Mosley J. D., Godwin A. K., Korach K. S., Visvader J. E., Kaestner K. H., et al. (2010). FOXA1 is an essential determinant of ERalpha expression and mammary ductal morphogenesis. Development 137, 2045–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom G. (1989). Probabilities in discrete sample spaces. In Probability and Statistics, Theory and Applications (ed. Feinberg S., Olkin I.). New York, NY: Springer-Verlag; [Google Scholar]
- Carroll J. S., Liu X. S., Brodsky A. S., Li W., Meyer C. A., Szary A. J., Eeckhoute J., Shao W., Hestermann E. V., Geistlinger T. R., et al. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43 [DOI] [PubMed] [Google Scholar]
- Chandrasekaran V., Beckendorf S. K. (2003). senseless is necessary for the survival of embryonic salivary glands in Drosophila. Development 130, 4719–4728 [DOI] [PubMed] [Google Scholar]
- Chung S., Vining M. S., Bradley P. L., Chan C. C., Wharton K. A., Jr, Andrew D. J. (2009). Serrano (sano) functions with the planar cell polarity genes to control tracheal tube length. PLoS Genet. 5, e1000746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo L. A., McPherson C. E., Bossard P., Stevens K., Cherian S., Shim E. Y., Clark K. L., Burley S. K., Zaret K. S. (1998). Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 17, 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo L. A., Lin F. R., Cuesta I., Friedman D., Jarnik M., Zaret K. S. (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289 [DOI] [PubMed] [Google Scholar]
- Clem R. J., Fechheimer M., Miller L. K. (1991). Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254, 1388–1390 [DOI] [PubMed] [Google Scholar]
- Eeckhoute J., Lupien M., Meyer C. A., Verzi M. P., Shivdasani R. A., Liu X. S., Brown M. (2009). Cell-type selective chromatin remodeling defines the active subset of FOXA1-bound enhancers. Genome Res. 19, 372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Bergmann A. (2010). The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 17, 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A. L., Lin W., Mavromatakis Y. E., Wang J. C., Sasaki H., Whitsett J. A., Ang S. L. (2007). Foxa1 and Foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage-dependent manner. Development 134, 2761–2769 [DOI] [PubMed] [Google Scholar]
- Fox R. M., Hanlon C. D., Andrew D. J. (2010). The CrebA/Creb3-like transcription factors are major and direct regulators of secretory capacity. J. Cell Biol. 191, 479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N., Zhang J., Rao M. A., Case T. C., Mirosevich J., Wang Y., Jin R., Gupta A., Rennie P. S., Matusik R. J. (2003). The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 17, 1484–1507 [DOI] [PubMed] [Google Scholar]
- Gao N., White P., Doliba N., Golson M. L., Matschinsky F. M., Kaestner K. H. (2007). Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. 6, 267–279 [DOI] [PubMed] [Google Scholar]
- Gao N., LeLay J., Vatamaniuk M. Z., Rieck S., Friedman J. R., Kaestner K. H. (2008). Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 22, 3435–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N., Le Lay J., Qin W., Doliba N., Schug J., Fox A. J., Smirnova O., Matschinsky F. M., Kaestner K. H. (2010). Foxa1 and Foxa2 maintain the metabolic and secretory features of the mature beta-cell. Mol. Endocrinol. 24, 1594–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet J., Mango S. E. (2002). Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science 295, 821–825 [DOI] [PubMed] [Google Scholar]
- Gong W. J., Golic K. G. (2003). Ends-out, or replacement, gene targeting in Drosophila. Proc. Natl. Acad. Sci. USA 100, 2556–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman A. S., Isaac D. D., Andrew D. J. (2003). Specification of cell fates within the salivary gland primordium. Dev. Biol. 258, 443–453 [DOI] [PubMed] [Google Scholar]
- Henderson K. D., Andrew D. J. (2000). Regulation and function of Scr, exd, and hth in the Drosophila salivary gland. Dev. Biol. 217, 362–374 [DOI] [PubMed] [Google Scholar]
- Henderson K. D., Isaac D. D., Andrew D. J. (1999). Cell fate specification in the Drosophila salivary gland: the integration of homeotic gene function with the DPP signaling cascade. Dev. Biol. 205, 10–21 [DOI] [PubMed] [Google Scholar]
- Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003a). Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. (2003b). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 [DOI] [PubMed] [Google Scholar]
- Junion G., Spivakov M., Girardot C., Braun M., Gustafson E. H., Birney E., Furlong E. E. (2012). A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 [DOI] [PubMed] [Google Scholar]
- Kaestner K. H. (2010). The FoxA factors in organogenesis and differentiation. Curr. Opin. Genet. Dev. 20, 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kele J., Simplicio N., Ferri A. L., Mira H., Guillemot F., Arenas E., Ang S. L. (2006). Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development 133, 495–505 [DOI] [PubMed] [Google Scholar]
- Kormish J. D., Gaudet J., McGhee J. D. (2010). Development of the C. elegans digestive tract. Curr. Opin. Genet. Dev. 20, 346–354 [DOI] [PubMed] [Google Scholar]
- Krapp A., Knöfler M., Frutiger S., Hughes G. J., Hagenbüchle O., Wellauer P. K. (1996). The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. EMBO J. 15, 4317–4329 [PMC free article] [PubMed] [Google Scholar]
- Krapp A., Knöfler M., Ledermann B., Bürki K., Berney C., Zoerkler N., Hagenbüchle O., Wellauer P. K. (1998). The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 12, 3752–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. M., Jones N., Zhou B., Panzer S., Larson V., Beckendorf S. K. (1996). Salivary duct determination in Drosophila: roles of the EGF receptor signalling pathway and the transcription factors fork head and trachealess. Development 122, 1909–1917 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461 [DOI] [PubMed] [Google Scholar]
- Lee C. S., Friedman J. R., Fulmer J. T., Kaestner K. H. (2005). The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–947 [DOI] [PubMed] [Google Scholar]
- Lehmann R., Tautz D. (1994). In situ hybridization to RNA. In Drosophila melanogaster: Practical Uses in Cell and Molecular Biology, Vol. 44 (ed. Goldstein L. S. B., Fyrberg E. A.). San Diego, CA: Academic Press; [Google Scholar]
- Li T. R., White K. P. (2003). Tissue-specific gene expression and ecdysone-regulated genomic networks in Drosophila. Dev. Cell 5, 59–72 [DOI] [PubMed] [Google Scholar]
- Li Z., Tuteja G., Schug J., Kaestner K. H. (2012). Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 148, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Metzakopian E., Mavromatakis Y. E., Gao N., Balaskas N., Sasaki H., Briscoe J., Whitsett J. A., Goulding M., Kaestner K. H., et al. (2009). Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev. Biol. 333, 386–396 [DOI] [PubMed] [Google Scholar]
- Liu S., Jack J. (1992). Regulatory interactions and role in cell type specification of the Malpighian tubules by the cut, Krüppel, and caudal genes of Drosophila. Dev. Biol. 150, 133–143 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- Lupien M., Eeckhoute J., Meyer C. A., Wang Q., Zhang Y., Li W., Carroll J. S., Liu X. S., Brown M. (2008). FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132, 958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R., Grevengoed E., Stempniewicz P., Andrew D. J. (2011). Genome-wide analysis reveals a major role in cell fate maintenance and an unexpected role in endoreduplication for the Drosophila FoxA gene Fork head. PLoS ONE 6, e20901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirosevich J., Gao N., Matusik R. J. (2005). Expression of Foxa transcription factors in the developing and adult murine prostate. Prostate 62, 339–352 [DOI] [PubMed] [Google Scholar]
- Moore A. W., Barbel S., Jan L. Y., Jan Y. N. (2000). A genomewide survey of basic helix-loop-helix factors in Drosophila. Proc. Natl. Acad. Sci. USA 97, 10436–10441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myat M. M., Andrew D. J. (2000a). Fork head prevents apoptosis and promotes cell shape change during formation of the Drosophila salivary glands. Development 127, 4217–4226 [DOI] [PubMed] [Google Scholar]
- Myat M. M., Andrew D. J. (2000b). Organ shape in the Drosophila salivary gland is controlled by regulated, sequential internalization of the primordia. Development 127, 679–691 [DOI] [PubMed] [Google Scholar]
- Nitsch D., Schütz G. (1993). The distal enhancer implicated in the developmental regulation of the tyrosine aminotransferase gene is bound by liver-specific and ubiquitous factors. Mol. Cell. Biol. 13, 4494–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolo R., Abbott L. A., Bellen H. J. (2000). Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell 102, 349–362 [DOI] [PubMed] [Google Scholar]
- Panne D. (2008). The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242 [DOI] [PubMed] [Google Scholar]
- Panzer S., Weigel D., Beckendorf S. K. (1992). Organogenesis in Drosophila melanogaster: embryonic salivary gland determination is controlled by homeotic and dorsoventral patterning genes. Development 114, 49–57 [DOI] [PubMed] [Google Scholar]
- Reuter R., Scott M. P. (1990). Expression and function of the homoeotic genes Antennapedia and Sex combs reduced in the embryonic midgut of Drosophila. Development 109, 289–303 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Mann R. S. (1999). The control of trunk Hox specificity and activity by Extradenticle. Genes Dev. 13, 1704–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahdev S., Saini K. S., Hasnain S. E. (2010). Baculovirus P35 protein: an overview of its applications across multiple therapeutic and biotechnological arenas. Biotechnol. Prog. 26, 301–312 [DOI] [PubMed] [Google Scholar]
- Seshaiah P., Andrew D. J. (1999). WRS-85D: a tryptophanyl-tRNA synthetase expressed to high levels in the developing Drosophila salivary gland. Mol. Biol. Cell 10, 1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit R. B., Schnabel R., Gaudet J. (2008). The HLH-6 transcription factor regulates C. elegans pharyngeal gland development and function. PLoS Genet. 4, e1000222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J. D., Roalson E. H., Skinner M. K. (2008). Phylogenetic and expression analysis of the basic helix-loop-helix transcription factor gene family: genomic approach to cellular differentiation. Differentiation 76, 1006–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H., Dingle S., Xu Y., Besnard V., Kaestner K. H., Ang S. L., Wert S., Stahlman M. T., Whitsett J. A. (2005). Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem. 280, 13809–13816 [DOI] [PubMed] [Google Scholar]
- Wang H., Chattopadhyay A., Li Z., Daines B., Li Y., Gao C., Gibbs R., Zhang K., Chen R. (2010). Rapid identification of heterozygous mutations in Drosophila melanogaster using genomic capture sequencing. Genome Res. 20, 981–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel D., Jürgens G., Küttner F., Seifert E., Jäckle H. (1989). The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 57, 645–658 [DOI] [PubMed] [Google Scholar]
- Weiss J. B., Suyama K. L., Lee H. H., Scott M. P. (2001). Jelly belly: a Drosophila LDL receptor repeat-containing signal required for mesoderm migration and differentiation. Cell 107, 387–398 [DOI] [PubMed] [Google Scholar]
- Zaret K. (1999). Developmental competence of the gut endoderm: genetic potentiation by GATA and HNF3/fork head proteins. Dev. Biol. 209, 1–10 [DOI] [PubMed] [Google Scholar]
- Zhou B., Bagri A., Beckendorf S. K. (2001). Salivary gland determination in Drosophila: a salivary-specific, fork head enhancer integrates spatial pattern and allows fork head autoregulation. Dev. Biol. 237, 54–67 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.