

Abstract
Steroid receptor coactivator 3 (SRC-3) is a multifunctional protein that plays an important role in regulation of bacterial lipopolysaccharide-induced inflammation. However, its involvement in host defense against bacterial infection remains unclear. Herein we used SRC-3 knockout mice to assess the role of SRC-3 in antibacterial defense in E. coli-induced septic peritonitis. After E. coli bacteria were injected intraperitoneally, SRC-3 deficient mice exhibited excessive local and systemic inflammatory responses and more severe bacterial burdens, leading to a significantly higher mortality compared with wild-type mice. Peritoneal macrophages of SRC-3 deficient mice showed a decrease in bacterial phagocytosis in culture and an increase in apoptosis, which was consistent with the defective bacterial clearance observed in SRC-3-deficient mice. Accordingly, SRC-3 null macrophages expressed much lower levels of the scavenger receptor A, the antioxidant enzyme catalase, and anti-apoptotic gene Bcl-2. Collectively, our data demonstrate that SRC-3 is important in not only modulating the local and systemic inflammation but also intensifying bacterial clearance, which highlights a pivotal role of SRC-3 in the host defense system against bacterial infection.
Introduction
Severe sepsis is a common, life-threatening infection resulting from a range of causative pathogens (1). The prevailing concept of the pathogenesis of sepsis is that mortality is a consequence of an uncontrolled hyperinflammatory response of the host mediated by proinflammatory cytokines (2, 3). Indeed, inhibition of proinflammatory cytokines such as IL-1β and TNF-α improves organ function and survival in animal models of sepsis (4, 5). However, deficiency in another proinflammatory cytokine IL-6 resulted in accumulation of live bacteria and increased mortality of mice infected by Escherichia coli (E. coli) (6). In addition, phagocytic cells and complements also play crucial roles in prevention of sepsis by clearing bacteria (7, 8). Therefore, precise control of proinflammatory cytokines and effective elimination of bacteria are crucial for host survival from infection.
Steroid receptor coactivator-3 (SRC-3/AIB1/ACTR/pCIP/RAC3/TRAM-1)3 is a member of the p160 coactivator family that also includes SRC-1 and SRC-2, which interacts with nuclear receptors and other transcription factors to enhance their effects on target gene transcription (9–13). SRC-3 was initially found to be amplified and overexpressed in breast cancer (9). Later studies demonstrated that SRC-3 played an important role in many developmental, physiological and pathologic events, such as cell growth, oncogenesis (14–18), cancer metastasis (19), differentiation (20) and energy homeostasis (21). We recently reported that SRC-3 deficient mice are markedly more susceptible to lipopolysaccharide (LPS)-induced endotoxic shock due to an increased production of proinflammatory cytokines including TNF-α, IL-1β, and IL-6 (22); in addition, SRC-3 is involved in the regulation of proinflammatory cytokine expression through suppressing cytokine translation from mRNA (22). These results indicate that SRC-3 has an essential function of suppressing LPS-induced inflammatory response. However, the biological role of SRC-3 in host defense against bacterial infection remains unclear.
Peritonitis is the second most common cause of sepsis (5) and a major abdominal emergency causing high rate of morbidity and mortality (23). Although different bacteria have been found responsible for peritonitis, E. coli remains one of the most common pathogens in intraperitoneal infection (23). In the present study, we used SRC-3 knockout mice to determine the role of SRC-3 in antibacterial defense in E. coli-induced septic peritonitis. We found that SRC-3 deficient mice were more susceptible to peritonitis-induced lethality caused by excessive local and systemic inflammatory responses as well as bacterial outgrowth and disseminations. Our findings demonstrate that SRC-3 plays a pivotal protective role in the host defense against E. coli-induced peritonitis by enhancing bacterial clearance and modulating inflammatory responses.
Materials and Methods
Mice
SRC-3 deficient mice (SRC-3−/−) were generated as described previously (24). Wild-type littermates served as control mice. The average body weight of SRC-3−/− mice was about 7.8% less than that of wild-type mice. Female mice (6–8 weeks of age) were used in all experiments. Animal experiments were approved by the Laboratory Animal Center of Xiamen University.
Cell culture and transfection
The RAW 264.7 murine macrophages were grown in DMEM supplemented with 10% FBS. All plasmid were transfected by using Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer’s instructions. The SRC-3 siRNA and control siRNA were purchased from Dharmacon and transfected into cells using TransIT-TKO transfection reagent (Mirus, Madison, WI).
Bacterial strain and infection of mice
E. coli (ATCC25922) was inoculated into tryptic soy broth (Oxoid, Basingstoke, Hampshire, UK) and incubated in a shaker incubator for 10 h at 37 °C. Bacteria were washed twice with PBS before infection of mice. Mice were intraperitoneally (i.p.) injected with 1 × 107 CFU of E. coli. An initial estimate of E. coli numbers was performed by densitometry. Serial dilution was performed, and inoculum size was established by enumeration of CFU on Luria-Bertani (LB) plates after overnight incubation at 37 °C.
Chemical analyses and measurement of cytokines
AST (Aspartate aminotransferase), ALT (Alanine aminotransferase), BUN (Blood urea nitrogen) were determined by commercially available kits (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions. The concentrations of TNF-α, IL-1β, IL-6, MCP-1, MIP-2 and KC in either plasma or peritoneal fluid were determined using ELISA kits (eBiosciences, San Diego, CA) according to the manufacturer’s instructions.
Isolation of peritoneal macrophages
Isolation of peritoneal macrophages from wild-type and SRC-3−/− mice were performed as described previously (22). Three days before isolation, wild-type and SRC-3−/− mice were injected i.p. with 2 ml of 4% thioglycollate medium (Sigma, St. Louis, MO) to elicit macrophages. Peritoneal macrophages were collected by peritoneal lavage with 10 ml of cold PBS. Cells were incubated in DMEM supplemented with 10% FBS at 37 °C for 2 h and then washed with PBS to eliminate non-adherent cells. Adherent cells were used as peritoneal macrophages.
Phagocytosis of peritoneal macrophages
E. coli strain ATCC25922 and Staphyloccocus aureus strain CMCC(B)26003 were labeled with FITC (Amresco, Cleveland, Ohio) according to Saresella et al (25) (hereafter designated as E. coli-FITC or S. aureus-FITC). For phagocytosis, the bacteria were opsonized with sera obtained from syngeneic mice at 37 °C for 30 min. Peritoneal macrophages were seeded in six-well plates (1×106 cells/well) for 2 h, and subsequently co-cultured with E. coli-FITC or S. aureus-FITC at 37 °C for 1 h. For phagocytosis of fluospheres, fluospheres suspension (F8827, Molecular probes) was redispersed by means of sonicator and mixed with macrophages at 37 °C for 1 h. After 1 h, the plates were covered in a dark container on ice and cells were washed extensively with PBS to remove extracellular bacteria. Then, the fluorescence that resulted from bacteria that were outside of the cells or sticking to the surface of the cells was quenched by trypan blue, and bacterial uptake was analyzed by flow cytometry (EPICS XL, Backman Coulter).
Bactericidal assay
Peritoneal macrophages were plated on a 24-well plate at 5 × 105. After 2 h, E. coli at a multiplicity of infection (m.o.i.) of 1 in RPMI 1640 supplemented with 10% syngeneic mouse serum without antibiotics was added. At the 30 min time point, each well was washed in PBS to remove extracellular bacteria and 1 ml of 0.05% Triton X-100 per well was added in the first three wells to lyse cells, and then the cell lysates were collected and designated as t = 0. Other wells were incubated with RPMI 1640 supplemented with 10% FBS and 5 μg/ml gentamycin for 2 h and then washed with PBS and lysed with 0.05% Triton X-100 (26). Serial dilutions of the cell lysates were plated on the LB plates, and bacterial counts were enumerated after 24 h. Bactericidal activity was expressed as the percentage of killed bacteria in relation to t = 0 (percent killing=1- CFU of E. coli at 2 h/CFU of E. coli at time 0 x100%)
Acute phase serum assay
Acute phase serum assay was performed as described (27). Briefly, mice were injected i.p. with heat-killed E. coli, and after 16 h the mice were sacrificed and bled to collect sera. The sera were regarded as acute phase serum. For in vitro growth measurements of E. coli, 5 × 103 CFU of E. coli were grown in RPMI 1640 supplemented with 20% acute phase serum or normal mouse serum at 37 °C, and the number of viable bacteria in the supernatant at the indicated time was determined by plating serial dilutions onto LB plates.
Apoptosis assay
Peritoneal macrophages were plated on a 6-well plate at 2 × 106 and infected with E. coli at an m.o.i. of 100 in antibiotic-free DMEM containing 10% FBS for 2 h as described (28), and medium was then removed and replaced with DMEM plus 10% FBS containing 100 μg/ml gentamycin. After 6 h, macrophage apoptosis was quantified by flow cytometry using sub-G0-G1 staining nuclei or annexin-V staining. For analysis of sub-G0-G1 phase cells, cells were collected, fixed overnight in 70% ethanol, and stained by incubation in PBS containing 50 μg/ml propidium iodide (PI) for 30 min. For annexin V staining, cells were stained by annexin-V-FLUOS Staining Kit (Roche, Penzberg, Germany) according to the manufacturer’s instructions.
Determination of reactive oxygen species (ROS)
Intracellular H2O2 was measured by 2′, 7′-dichlorofluorescein (DCF) fluorescence (29). In brief, Peritoneal macrophages were incubated with 10 μM 2′, 7′-dichlorofluorescein diacetate (DCFH-DA, Fluka), which is taken up and oxidized to the fluorescent DCF by intracellular H2O2 at 37 °C for 30 min. After that, the cells were immediately measured by flow cytometry.
Catalase activity assay
The activity of catalase was measured according to Sinha et al (30). Briefly, dichromate/acetic acid was prepared by mixing a 5% solution of potassium dichromate with acetic acid (1:3 by volume). The assay mixture containing 2 ml of H2O2 (400 μmoles) and 2.5 ml of PBS was rapidly mixed with 0.5 ml of cell sample, and the reaction was run for 2 min, then 1 ml portion of the reaction mixture was withdrawn and blown into 2 ml of dichromate/acetic acid for measuring the hydrogen peroxide contents. The catalase activity was calculated according to the content of H2O2 remained.
Quantitative real-time PCR
Total RNA was prepared from the cells using Trizol reagent (Invitrogen, Carlsbad, CA). One microgram of total RNA was converted to cDNA by SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR reactions were performed using SYBR Premix ExTaq (TaKaRa, Dalian, China). Relative quantification was achieved by normalization to endogenous TBP. The primers used for real-time PCR were listed in Supplementary Table I.
Luciferase reporter assay
The −494 to +68 fragment of the catalase promoter was inserted into the pGL3-basic vector to generate the catalase reporter plasmid according to the method reported previously (31). The pCR3.1-Rluc vector that constitutively expresses Renilla luciferase was used to normalize transfection efficiency. Luciferase activity was assayed using a dual luciferase reporter assay system (Promega, Madison, WI).
Chromatin immunoprecipitation (ChIP) assay
RAW264.7 cells were used for ChIP and processed according to the method described by abcam. The following primers were used: catalase promoter sense strand 5′-ATTGGACCCTGAGCTGTGAC-3′, antisense strand 5′-GACTTCAGGCTCCACCAATC -3. Anti-SRC-3 antibody was purchased from Santa Cruz (Santa Cruz, CA).
Determination of SR-A expression by flow cytometry
SR-A protein on macrophages was stained with SR-A antibody (Santa Cruz, CA). Then, secondary FITC-labeled antibody F(ab)2 goat anti rat IgG (Pierce, Rockford, IL) was added and the expression was analyzed by flow cytometry.
Statistical analysis
The Kaplan-Meier and log-rank methods were used to analyze mortality. Data were collected from several independent experiments, with triplicates per experiment. All data were expressed as mean + SD. Statistically significant effects were examined using 2-tailed student’s t-test in SPSS 11.0 for Windows.
Results
SRC-3−/− mice are ultra-susceptible to E. coli-induced peritonitis
To determine whether SRC-3 plays roles in antibacterial defense in E. coli-induced septic peritonitis, we first examined the survival results of SRC-3−/− and wild-type mice in E. coli-induced septic peritonitis. After infected with 1 × 107 CFU of E. coli, SRC-3−/− mice displayed more severe symptoms of sepsis such as a crouched position, shivering and ruffled fur as compared to wilt-type controls. SRC-3−/− mice showed a significantly accelerated lethality compared with wild-type mice. Within 24 h, 42% (5 of 12) of SRC-3−/− mice died while no (0 of 12) wild-type mice died at this time point. All SRC-3−/− mice (12 of 12) died within 48 h post-infection, but 58% of the wild-type controls (7 of 12) still survived and recovered by the end of experiment (Fig. 1A). Therefore, SRC-3−/− mice are hypersensitive to E. coli infection compared with wild-type mice.
FIGURE 1.
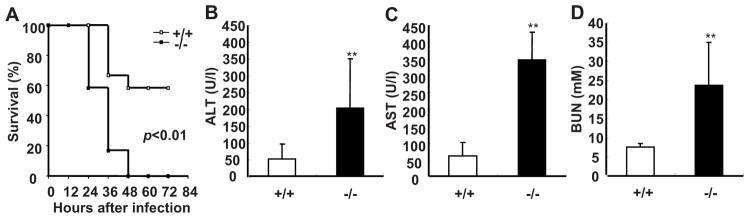
SRC-3−/− mice are markedly susceptible to E. coli-induced peritonitis. A, SRC-3−/− mice exhibited the increased mortality during E. coli-induced peritonitis. After infection with E. coli, mice were observed every 12 h for 3 d. Each group consisted of 12 mice. B–D, SRC-3−/− mice exhibited more severe multiple organ damage compared to wild-type mice. Liver injury was reflected by measurement of ALT and AST in plasma of SRC-3−/− and wild-type mice at 20 h post-infection (B and C). Kidney damage was assessed by determining the level of BUN in plasma of both mice at 20 h post-infection (D). Data are the means + SD of five mice per time point. ** p<0.01.
Multiple organ damage occurs frequently during sepsis (32). Therefore, we examined whether the liver and kidney were damaged upon E. coli infection. The levels of AST and ALT (indicators of liver damage) as well as BUN (an indicator of kidney damage) in plasma of SRC-3−/− mice were significantly higher than that of wild-type mice after infection (Fig. 1, B–D). Histopathological examination further showed that the liver and kidney of SRC-3−/− mice had more necrotic areas (Supplementary Fig. 1). Thus, consistent with more lethality in SRC-3−/− mice, the degree of multiple organ damages in SRC-3−/− mice is much more severe than that in wild-type mice after E. coli infection.
SRC-3−/− mice produce more proinflammatory cytokines than wild-type mice during E. coli-induced peritonitis
Since uncontrolled inflammatory response is considered to play a major role in the pathogenesis of sepsis (3, 33), and SRC-3−/− mice are markedly susceptible to LPS-induced endotoxic shock due to increased production of proinflammatory cytokines such as TNF-a, IL-1β, and IL-6 (22), we hypothesized that the increased mortality and multiple organ damages observed in SRC-3−/− mice during E. coli-induced peritonitis are caused by an overwhelming inflammatory response. We therefore measured the levels of proinflammatory cytokines after SRC-3−/− and wild-type mice were infected by E. coli. The concentrations of TNF-α and IL-6 in plasma of both types of mice were low at 6 h post-infection and they remained low in wild-type mice at 20 h post-infection, but the concentrations of TNF-α and IL-6 in plasma of SRC-3−/− mice at 20 h after infection were increased 12- and 30-fold, respectively (Fig. 2, A and B). The concentrations of IL-1β in plasma of both types of mice were increased to a similar degree at 6 h. However, sustained levels of IL-1β were observed in SRC-3−/− mice in contrast to the substantial decrease of IL-1β in wild-type mice at 20 h (Fig. 2C). The concentrations of TNF-α, IL-1β, and IL-6 in peritoneal fluid of both types of mice were relatively low at 6 h post-infection, but their concentrations in SRC-3−/− mice were markedly increased at 20 h compared to wild-type mice (Fig. 2, D–F). The higher levels of proinflammatory cytokines observed in SRC-3−/− mice at 20 h indicates that SRC-3 deficiency is responsible for the much more robust local and systemic inflammatory responses.
FIGURE 2.

SRC-3−/− mice produce more proinflammatory cytokines than wild-type mice during E. coli-induced peritonitis. ELISA of TNF-α, IL-1β and IL-6 in plasma (A–C) and peritoneal fluid (D–F) from SRC-3−/− and wild-type mice at 6 h and 20 h after infection. Data are the means + SD of five mice per time point. ** p<0.01.
SRC-3−/− mice exhibit impaired bacterial clearance during E. coli-induced peritonitis
It is suggested that the increased local inflammation may be beneficial to bacterial clearance during E. coli-induced peritonitis (3). To determine whether SRC-3 deficiency, which augments inflammation, can enhance bacterial clearance, we assessed the number of E. coli in peritoneal fluid, blood, and tissue homogenates of SRC-3−/− and wild-type mice at 6 h and 20 h after infection. Bacterial loads had no differences in both groups of mice at 6 h after infection, unexpected, SRC-3−/− mice had significantly more bacteria in peritoneal fluid compared with wild-type mice at 20 h post-infection (Fig. 3A). Similarly, blood, liver, spleen, and lung of SRC-3−/− mice had more bacterial loads than wild-type mice at 20 h post-infection (Fig. 3, B–E). These data demonstrate that SRC-3 deficiency impairs bacterial clearance during E. coli-induced peritonitis.
FIGURE 3.

SRC-3−/− mice exhibit more severe bacterial outgrowth and dissemination during E. coli-induced peritonitis. Bacterial loads in peritoneal fluid (A), blood (B), liver (C), spleen (D) and lung (E) at 6 and 20 h after infection with E. coli. Data are the means + SD of five mice per time point. **p<0.01. F, SRC-3 deficiency had no effect on antibacterial activity of serum. G and H, Expression of antimicrobial protein lipocalin-2 (Lcn2) in the liver (G) or spleen (H) from both types of mice with or without infection of E. coli was analyzed by real-time PCR. Data are the means + SD of four mice per group.
Host possesses many kinds of defensive systems against bacterial infection. Infection-induced acute phase serum represents an iron-restricted environment for bacteria, and contains many substances related to antibacterial activity such as antimicrobial peptides, complements and cytokines (27). Therefore, it plays an important role in host defense against bacterial infection. Bacteriostatic effects of serum were determined by measuring the growth of E. coli in sera from SRC-3−/− and wild-type mice. Both types of acute phase sera effectively inhibited bacterial growth as compared to normal sera, but no differences were observed, indicating that the differences in bacteria outgrowth and disseminations between infected SRC-3−/− and wild-type mice are not due to any differences in bacteriostatic effects of acute phase serum (Fig. 3F). Consistent with this, there were no significant differences in the expression of lipocalin-2, an iron-sequestering bacteriostatic peptide expressed in the liver and spleen but secreted into the blood circulation (27), between SRC-3−/− and wild-type mice in the absence and presence of E. coli infection (Fig. 3, G and H).
Phagocytosis of E. coli by SRC-3−/− peritoneal macrophages and neutrophils is decreased
Because no significant differences were detected in the peritoneal leukocyte counts between SRC-3−/− and wild-type mice during E. coli-induced peritonitis (table I), we speculated other defects than leukocyte recruitments may be responsible for the impaired bacterial clearance observed in SRC-3−/− mice. Phagocytosis of microorganisms by macrophages is a key component of the host defense against bacterial infections (34, 35). To determine whether there are any differences in phagocytosis between SRC-3−/− and wild-type macrophages, we examined the phagocytosis of E. coli and fluospheres by peritoneal macrophages from SRC-3−/− and wild-type mice. Phagocytosis of E. coli by SRC-3−/− macrophages was significantly decreased compared with that by wild-type macrophages (Fig. 4, A and B), while phagocytosis of fluospheres by both types of macrophages was comparable (Supplementary Fig. 2). Furthermore, we found that phagocytosis of E. coli by SRC-3−/− peritoneal neutrophils was also significantly decreased (Fig. 4, C and D). To determine whether phagocytosis of other types of bacteria by SRC-3−/− macrophages is impaired, we investigated the phagocytosis of gram-positive bacteria S. aureus and found that phagocytosis of S. aureus by SRC-3−/− macrophages was significantly decreased (Supplementary Fig. 3). These results suggest that SRC-3−/− leukocytes have defects in phagocytosis of bacteria.
Table I.
Cell counts in peritoneal fluid during E. coli-induced peritonitis
| Cells (×105/ml) | 6h | 20h | ||
|---|---|---|---|---|
|
| ||||
| +/+ | −/− | +/+ | −/− | |
| Total cells | 9.2±0.3 | 10.3±1.5 | 42.0±21.8 | 43.9±24.6 |
| Macrophages | 1.9±0.7 | 1.5±0.1 | 12.7±10.7 | 10.7±2.5 |
| Neutrophils | 7.3±0.4 | 8.7±1.6 | 29.6±13.6 | 33.2±26.3 |
Data are the means ± SD of five mice per time point at 6 and 20 h after infection
FIGURE 4.

Phagocytosis of E. coli by peritoneal macrophages and neutrophils is decreased in SRC-3−/− mice. A and B, Phagocytosis of E. coli by peritoneal macrophages. Macrophages were incubated with E. coli-FITC at an m.o.i. of 100. FACS histogram is a representative of three independent experiments. C and D, Phagocytosis of E. coli by peritoneal neutrophils. FACS histogram is a representative of three independent experiments. Relative mean fluorescence intensity (MFI) was expressed in fold change relative to wild-type (+/+) values assigned a unit of 1. Data are the means + SD of four mice per group. E and F, The expression of SR-A (E) and CR3 (F) in macrophages was determined by real-time PCR. Data are the means + SD of four mice per group. G, The expression of SR-A protein on the surface of macrophages was analyzed by flow cytometry. Data shown are representative of three independent experiments. H, Bactericidal activity of macrophages from SRC-3−/− and wild-type mice. Data are the means + SD of four mice per group. *p<0.05, **p<0.01.
Macrophages express several surface receptors such as scavenger receptor A (SR-A) and complement receptor C3 (CR3) to aid in the recognition of microorganisms (36). The expression of SR-A and CR3 mRNAs in macrophages was analyzed by real-time PCR. SR-A expression was significantly lower in SRC-3−/− macrophages than in wild-type macrophages, but CR3 expression had no difference (Fig. 4, E and F). In agreement with mRNA expression data, the expression of SR-A protein on the surface of SRC-3−/− macrophages was lower than that of wild-type macrophages (Fig. 4G). These results suggest that SRC-3 deficiency-caused decrease in SR-A expression may be partially responsible for the impaired phagocytosis of bacteria observed in SRC-3 null macrophages. Once engulfing bacteria, SRC-3−/− and wild-type macrophages displayed a similar bactericidal activity (Fig. 4H), suggesting that SRC-3−/− macrophages have no defect in bacterial killing.
SRC-3 deficiency increases apoptosis of peritoneal macrophages
Bacterial pathogens are ingested and destroyed by macrophages, which is a mechanism used by host to combat bacterial infections. Although most bacteria are killed by macrophages, some intracellular bacteria, such as Shigella and Listeria have been shown to directly trigger apoptosis of macrophages in order to evade the host immune response (37, 38). Recent studies have demonstrated that apoptosis in macrophages is also induced after phagocytosis of E. coli (39–41). Therefore, the extents of apoptosis in peritoneal macrophages from SRC-3−/− and wild-type mice were examined by flow cytometric measurement of macrophage subG1 populations which represents the percentage of apoptotic cells, as well as annexin-V staining. As shown in Fig. 5A, the subG1 population of SRC-3−/− macrophages was significantly larger than that of wild-type macrophages in both absence and presence of E. coli. The results of annexin-V staining confirmed that SRC-3−/− macrophages had higher apoptotic rates (Fig. 5B and Supplementary Fig. 4). Furthermore, we investigated the percentage of apoptotic cells in peritoneal fluid from mice after infected by E. coli. The results in Fig. 5C showed that peritoneal cells from E. coli-infected SRC-3−/− mice had higher apoptotic rates compared with that from E. coli-infected wild-type mice.
FIGURE 5.

Deficiency of SRC-3 increases apoptosis of peritoneal macrophages due to reduced expression of catalase. A and B, Apoptosis of macrophages from SRC-3−/− mice was significantly higher than that of wild-type mice in the absence and presence of E. coli. The percentage of fragmented DNA (subG1 populations) or Annexin V+/PI− population are indicated in the graphics. Data are the means + SD of four mice per group. ** p<0.01. C, The percentage of apoptotic cells in peritoneal cells from SRC-3−/− mice at 20 h after infection by E. coli is significantly higher than that of wild-type mice. Data are the means + SD of three mice per group. ** p<0.01. D, Deficiency of SRC-3 enhanced the levels of ROS in macrophages. Data shown are representative of three independent experiments. E, Apoptosis of macrophages from both types of mice in the presence of E. coli was decreased after treatment with NAC. Data are the means + SD of three mice per group. ** p<0.01. F, The expression of catalase was determined by real-time PCR. Data are the means + SD of four mice per group. ** p<0.01. G, The catalase activity of wild-type macrophage was inhibited by 3-AT (50 mM). Data are the means + SD of three mice per group. * p<0.05. H, Wild-type macrophages accumulated more ROS after treated with a catalase specific inhibitor 3-AT. Data shown are representative of three independent experiments. I, Apoptosis of wild-type macrophages was increased after treatment with 3-AT. Data are the means + SD of three mice per group, ** p<0.01. J, Pre-treating wild-type mice with 3-AT led to more bacterial loads in peritoneal fluid at 20 h after infection by E. coli. Data are the means + SD of three mice per group, * p<0.05. K, The expression of anti-apoptotic gene Bcl-2 was decreased in wild-type macrophage after treatment with 3-AT. Data are the means + SD of three mice per group, * p<0.05. L, The expression of Bcl-2 was decreased in SRC-3−/− macrophages. Data are the means + SD of four mice per group. * p<0.05.
ROS are known to regulate cell death in a variety of cell types (43). We therefore examined the levels of ROS in macrophages from both types of mice. The levels of ROS were dramatically increased in SRC-3−/− macrophages compared with wild-type cells in the presence of E. coli (Fig. 5D). To further investigate whether the intracellular production of ROS was involved in apoptosis, we treated cells with an antioxidant scavenger N-Acetylcysteine (NAC) to abrogate the intracellular ROS. The rates of apoptosis in SRC-3−/− and wild-type macrophages were significantly decreased to similar levels after NAC treatment (Fig. 5E), indicating that ROS is indeed responsible for the increased macrophage apoptosis.
Several antioxidant enzymes, including catalase, superoxide dismutase (SOD), and glutathione peroxidase (GPX) are involved in protecting cells against ROS. The levels of catalase, SOD1, and GPX mRNAs in both genotypic macrophages were measured, but only catalase expression in SRC-3−/− macrophages was significantly lower than that of wild-type cells (Fig. 5F and Supplementary Fig. 5). Consistent with reduced catalase expression, the enzymatic activity of catalase was also significantly reduced in SRC-3−/− macrophages compared with wild-type macrophages (Supplementary Fig. 6). To examine whether reduced catalase activity can result in an accumulation of ROS and a concomitant increase of cell apoptosis, a catalase-specific inhibitor 3-amino-1,2,4-triazole (3-AT) was used to inhibit catalase activity (Fig. 5G). The levels of ROS and apoptosis were notably increased in cells treated with 3-AT as compared to untreated group (Fig. 5, H and I), suggesting that catalase plays an important role in defending against ROS and apoptosis. Because 3-AT can also inhibit catalase activity in vivo (44), we injected 3-AT into wild-type mice to determine whether reduction of catalase activity in vivo could result in the impaired bacterial clearance. As shown in Fig. 5J, 3-AT treatment resulted in more bacterial loads in peritoneal fluid compared with the untreated group.
Several lines of evidence demonstrate that ROS can suppress the expression of anti-apoptotic gene Bcl-2 (45, 46). Consistently, we found that the catalase-specific inhibitor 3-AT, which was shown to be able to increase ROS accumulation (Fig. 5H), effectively suppressed the expression of Bcl-2 in macrophages (Fig. 5K). Since ROS accumulation was increased in SRC-3−/− macrophages, we examined whether the expression of Bcl-2 is decreased in these cells. As anticipated, deficiency of SRC-3 led to a significant decrease of Bcl-2 expression (Fig. 5L).
SRC-3 regulates the expression of catalase in macrophages
Catalase is an antioxidant enzyme whose expression is transcriptionally regulated by some factors such as SP1, NF-Y and WT1/Egr-related factor (31, 47). Since the expression of catalase is reduced in SRC-3−/− macrophage, we hypothesize that catalase expression is regulated by SRC-3. To determine whether SRC-3 regulates de novo transcription of the catalase gene, we examined the effects of SRC-3 up-regulation and down-regulation on the transcriptional activity of the catalase promoter in a transient transfection and luciferase reporter assay. Catalase promoter activity was increased in RAW264.7 cells co-transfected with SRC-3 expression vector and decreased in RAW264.7 cells co-transfected with SRC-3 specific siRNA (Fig. 6, A and B), suggesting that SRC-3 is required for transcriptional activation of catalase. Furthermore, the results of ChIP assay demonstrated that SRC-3 was recruited to the catalase promotor (Fig. 6C).
FIGURE 6.
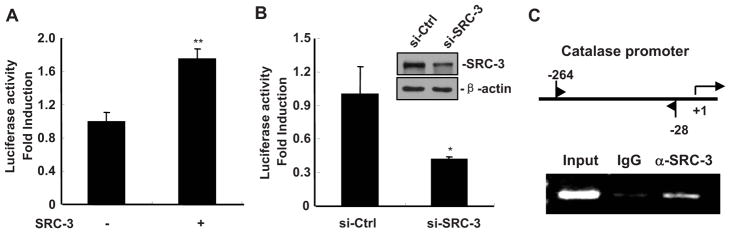
SRC-3 regulates the expression of catalase in macrophages. A, Catalase promoter activity was increased in RAW264.7 cells co-transfected with SRC-3 expression. Data are presented as the means + SD (n=3). ** p<0.01. (B) Catalase promoter activity was decreased in RAW264.7 cells co-transfected with SRC-3 specific siRNA. Inserted: western blot analysis revealed a down-regulated of SRC-3 protein by SRC-3 siRNA but not control siRNA. Data are presented as the means + SD (n=3). * p<0.05. C, SRC-3 was associated with the promoter of catalase. Upper panel: positions of the subfragments detected in ChIP assay; Lower panel: ChIP assay showed that SRC-3 was associated with the promoter of catalase. IgG: goat IgG, α-SRC-3: anti-SRC-3 goat IgG.
Discussion
We have previously shown that SRC-3−/− mice are highly susceptible to LPS-induced lethality and SRC-3 is a negative regulator of inflammatory response (22). In the present study, we demonstrate that SRC-3−/− mice are markedly susceptible to the lethality caused by E. coli infection-induced peritonitis. Therefore, the enhanced inflammation as indicated by increased production of proinflammatory cytokines should be at least in part responsible for these phenotypes observed in SRC-3−/− mice. An uncontrolled systemic inflammation is harmful to the host, but inflammation is beneficial to the effective clearance of bacteria (3). For example, lack of IL-10, an anti-inflammatory cytokine, increases systemic inflammatory response and causes multiple organ failures and death, while it enhances bacterial clearance in mice during E. coli peritonitis (48). We therefore initially hypothesize that enhanced inflammatory responses in SRC-3−/− mice are better for clearing bacteria during E. coli peritonitis. Surprisingly, SRC-3−/− mice display more bacterial loads and systemic dissemination despite the increased inflammatory responses. It indicates that SRC-3−/− mice have defects in controlling both bacterial outgrowth and inflammatory responses. Intensively increased bacterial loads, which provide more potent inflammatory stimulus, may be able to further enhance inflammation and increase mortality rates of SRC-3−/− mice. On the other hand, the exaggerated bacterial loads alone are able to cause the increased mortality. Therefore, the impaired capacity of SRC-3−/− mice to clear a bacterial infection and the overproduction of host proinflammatory cytokines are both responsible for the increased mortality observed.
Macrophages are required for the innate immune response to bacterial infection. Our results demonstrate that SRC-3 deficiency impaired phagocytosis of E. coli by macrophage through reducing the expression of SR-A, an important receptor for Fc receptor-independent phagocytosis (49). The apoptotic response of macrophages plays a vital role in the pathogenesis of sepsis (40, 50). For example, liver X receptors-null macrophages undergo accelerated apoptosis upon infection and exhibit defective bacterial clearance in vivo (38). Therefore, macrophage survival intimately concerns the host defense against infectious pathogens. Our data demonstrate that macrophages from SRC-3−/− mice are more sensitive to E. coli induced apoptosis compared with the wild-type macrophages. In addition, increased apoptosis of SRC-3−/− macrophages may explain the slightly decreased peritoneal macrophage count in SRC-3−/− mice (Table I), despite the levels of MCP-1, MIP-2 and KC, three major mediators of leukocyte recruitment (42), are significantly increased in the peritoneal cavity at 20 h after infection (Supplementary Fig. 7).
ROS and the resulting oxidative stress play a pivotal role in apoptosis (43, 51), and a state of severe oxidative stress is encountered in sepsis, with host endogenous antioxidant defenses overcome (52). Alterations in the redox status of host by various anti-oxidants, such as NAC, block apoptosis in cells (43) and blunt the inflammatory response to sepsis (53). Our data are consistent with this notion that SRC-3−/− macrophages exhibit higher levels of ROS, and the apoptosis induced by E. coli can be repressed by NAC. Catalase, an antioxidant enzyme, has a direct antioxidant activity through catalyzing the conversion of hydrogen peroxide to water. We find that SRC-3 is involved in the regulation of catalase transcription. Therefore, the decreased expression of catalase underlies higher level of ROS and more apoptotic rate observed in SRC-3−/− macrophages. Overexpression of catalase increases the Bcl-2 expression (45), which prevents cells from apoptosis by an anti-oxidative mechanism (43). In this study, a decrease in catalase expression and a concomitant decrease in Bcl-2 expression as well as an increase in apoptosis are observed in SRC-3−/− macrophages. Thus, down-regulation of the Bcl-2 expression further sensitizes SRC-3−/− macrophages to E. coli-induced apoptosis.
We have previously shown that in response to LPS stimulation, SRC-3 interacts with translational repressor TIA-1 and TIAR to suppress the protein synthesis of proinflammatory cytokines in the cytoplasm, which limits excessive inflammatory response, reduces systemic organ damage, and improves survival (22). In this study, we further demonstrate that during E. coli-induced peritonitis, macrophage SRC-3 not only suppresses proinflammatory cytokine synthesis to limit an inflammatory response, but also enhances the bacterial clearance by macrophages through acting as a transcriptional coactivator in the nucleus to up-regulate the expression of SR-A and catalase, leading to an enhanced phagocytosis of bacteria and a reduced macrophage apoptosis. Therefore, the highly coordinated cytoplasmic and nuclear functions of SRC-3 effectively prevent animal from sepsis-induced lethality (Fig. 7). Taken together, these findings clearly demonstrate that SRC-3 is a master regulator of sepsis and plays an essential role in protecting the host against bacterial infection.
FIGURE 7.

Schematic model for the mechanism by which SRC-3 protects mice from E. coli peritonitis. Upon E. coli infection, macrophage SRC-3 suppresses proinflammatory cytokine synthesis to limit inflammatory responses and multiple organ damage. On the other hand, SRC-3 enhances bacterial clearance by macrophages through up-regulating the expression of catalase, Bcl-2, and SR-A to reduce macrophage apoptosis and to enhance phagocytosis of E. coli. Both actions help to prevent animal from E. coli peritonitis-induced lethality.
Supplementary Material
Acknowledgments
We thank Yang Li and Xiaoliang Wang for their technical assistance.
Footnotes
This work was supported by grants from National Natural Science Foundation of China (30600566 and 30770455), National Basic Research Program of China (973 Program, 2008CB517311 and 2009CB522200), the Science Planning Program of Fujian Province (2009J1010), and National Institutes of Health of USA (NIH, DK058242).
Abbreviations used in this paper: SRC-3, steroid receptor coactivator 3; AST, aspartate aminotransferase; ALT, alanine aminotransferase; BUN, blood urea nitrogen; m.o.i., multiplicity of infection; ROS, Reactive oxygen species; DCFH-DA, 2′, 7′-dichlorofluorescein diacetate; SR-A, macrophage scavenger receptor class A; CR3, complement receptor C3; SOD, superoxide dismutase; GPX, glutathione peroxidase; NAC, N-Acetylcysteine; 3-amino-1,2,4-triazole, 3-AT.
References
- 1.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 2.van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 3.van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13:413–426. ix. doi: 10.1016/s0891-5520(05)70083-0. [DOI] [PubMed] [Google Scholar]
- 4.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov. 2003;2:736–746. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340:207–214. doi: 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- 6.Dalrymple SA, Slattery R, Aud DM, Krishna M, Lucian LA, Murray R. Interleukin-6 is required for a protective immune response to systemic Escherichia coli infection. Infect Immun. 1996;64:3231–3235. doi: 10.1128/iai.64.8.3231-3235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinheiro da Silva F, Aloulou M, Skurnik D, Benhamou M, Andremont A, Velasco IT, Chiamolera M, Verbeek JS, Launay P, Monteiro RC. CD16 promotes Escherichia coli sepsis through an FcR gamma inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med. 2007;13:1368–1374. doi: 10.1038/nm1665. [DOI] [PubMed] [Google Scholar]
- 8.Shuster DE, Kehrli ME, Jr, Rainard P, Paape M. Complement fragment C5a and inflammatory cytokines in neutrophil recruitment during intramammary infection with Escherichia coli. Infect Immun. 1997;65:3286–3292. doi: 10.1128/iai.65.8.3286-3292.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 11.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci U S A. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW. TRAM-1, A novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem. 1997;272:27629–27634. doi: 10.1074/jbc.272.44.27629. [DOI] [PubMed] [Google Scholar]
- 14.Zhou G, Hashimoto Y, Kwak I, Tsai SY, Tsai MJ. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol Cell Biol. 2003;23:7742–7755. doi: 10.1128/MCB.23.21.7742-7755.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuang SQ, Liao L, Wang S, Medina D, O’Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005;65:7993–8002. doi: 10.1158/0008-5472.CAN-05-1179. [DOI] [PubMed] [Google Scholar]
- 16.Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, Ittmann M, Tsai SY, Tsai MJ. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–7983. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
- 17.Coste A, Antal MC, Chan S, Kastner P, Mark M, O’Malley BW, Auwerx J. Absence of the steroid receptor coactivator-3 induces B-cell lymphoma. EMBO J. 2006;25:2453–2464. doi: 10.1038/sj.emboj.7601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ying H, Willingham MC, Cheng SY. The steroid receptor coactivator-3 is a tumor promoter in a mouse model of thyroid cancer. Oncogene. 2008;27:823–830. doi: 10.1038/sj.onc.1210680. [DOI] [PubMed] [Google Scholar]
- 19.Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O’Malley BW, Xu J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937–5950. doi: 10.1128/MCB.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li HJ, Haque Z, Lu Q, Li L, Karas R, Mendelsohn M. Steroid receptor coactivator 3 is a coactivator for myocardin, the regulator of smooth muscle transcription and differentiation. Proc Natl Acad Sci U S A. 2007;104:4065–4070. doi: 10.1073/pnas.0611639104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha} Proc Natl Acad Sci U S A. 2008;105:17187–17192. doi: 10.1073/pnas.0808207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu C, York B, Wang S, Feng Q, Xu J, O’Malley BW. An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell. 2007;25:765–778. doi: 10.1016/j.molcel.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClean KL, Sheehan GJ, Harding GK. Intraabdominal infection: a review. Clin Infect Dis. 1994;19:100–116. doi: 10.1093/clinids/19.1.100. [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O’Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379–6384. doi: 10.1073/pnas.120166297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saresella M, Roda K, Speciale L, Taramelli D, Mendozzi E, Guerini F, Ferrante P. A rapid evaluation of phagocytosis and killing of Candida albicans by CD13+ leukocytes. J Immunol Methods. 1997;210:227–234. doi: 10.1016/s0022-1759(97)00196-8. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–361. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- 27.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 28.Lai XH, Xu JG, Melgar S, Uhlin BE. An apoptotic response by J774 macrophage cells is common upon infection with diarrheagenic Escherichia coli. FEMS Microbiol Lett. 1999;172:29–34. doi: 10.1111/j.1574-6968.1999.tb13445.x. [DOI] [PubMed] [Google Scholar]
- 29.Woo CH, Eom YW, Yoo MH, You HJ, Han HJ, Song WK, Yoo YJ, Chun JS, Kim JH. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J Biol Chem. 2000;275:32357–32362. doi: 10.1074/jbc.M005638200. [DOI] [PubMed] [Google Scholar]
- 30.Sinha AK. Colorimetric assay of catalase. Anal Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7. [DOI] [PubMed] [Google Scholar]
- 31.Luo D, Rando TA. The regulation of catalase gene expression in mouse muscle cells is dependent on the CCAAT-binding factor NF-Y. Biochem Biophys Res Commun. 2003;303:609–618. doi: 10.1016/s0006-291x(03)00397-8. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Meng X, Kuhlman JR, Nelin LD, Nicol KK, English BK, Liu Y. Knockout of Mkp-1 enhances the host inflammatory responses to gram-positive bacteria. J Immunol. 2007;178:5312–5320. doi: 10.4049/jimmunol.178.8.5312. [DOI] [PubMed] [Google Scholar]
- 33.van Westerloo DJ, I, Giebelen A, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der Poll T. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis. 2005;191:2138–2148. doi: 10.1086/430323. [DOI] [PubMed] [Google Scholar]
- 34.Knapp S, Matt U, Leitinger N, van der Poll T. Oxidized phospholipids inhibit phagocytosis and impair outcome in gram-negative sepsis in vivo. J Immunol. 2007;178:993–1001. doi: 10.4049/jimmunol.178.2.993. [DOI] [PubMed] [Google Scholar]
- 35.Thomas CA, Li Y, Kodama T, Suzuki H, Silverstein SC, El Khoury J. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J Exp Med. 2000;191:147–156. doi: 10.1084/jem.191.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwiatkowska K, Sobota A. Signaling pathways in phagocytosis. Bioessays. 1999;21:422–431. doi: 10.1002/(SICI)1521-1878(199905)21:5<422::AID-BIES9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Hilbi H, Chen Y, Thirumalai K, Zychlinsky A. The interleukin 1beta-converting enzyme, caspase 1, is activated during Shigella flexneri-induced apoptosis in human monocyte-derived macrophages. Infect Immun. 1997;65:5165–5170. doi: 10.1128/iai.65.12.5165-5170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell M, Cheng RG, Saez E, Miller JF, Tontonoz P. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 39.Hacker H, Furmann C, Wagner H, Hacker G. Caspase-9/-3 activation and apoptosis are induced in mouse macrophages upon ingestion and digestion of Escherichia coli bacteria. J Immunol. 2002;169:3172–3179. doi: 10.4049/jimmunol.169.6.3172. [DOI] [PubMed] [Google Scholar]
- 40.Albee L, Shi B, Perlman H. Aspartic protease and caspase 3/7 activation are central for macrophage apoptosis following infection with Escherichia coli. J Leukoc Biol. 2007;81:229–237. doi: 10.1189/jlb.0506358. [DOI] [PubMed] [Google Scholar]
- 41.Wang JH, Zhou YJ, He P, Chen BY. Roles of mitogen-activated protein kinase pathways during Escherichia coli-induced apoptosis in U937 cells. Apoptosis. 2007;12:375–385. doi: 10.1007/s10495-006-0623-6. [DOI] [PubMed] [Google Scholar]
- 42.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 43.Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 44.Heim WG, Appleman D, Pyfrom HT. Production of catalase changes in animals with 3-amino-1, 2, 4-triazole. Science. 1955;122:693–694. doi: 10.1126/science.122.3172.693. [DOI] [PubMed] [Google Scholar]
- 45.Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci U S A. 2003;100:15035–15040. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li D, Ueta E, Kimura T, Yamamoto T, Osaki T. Reactive oxygen species (ROS) control the expression of Bcl-2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci. 2004;95:644–650. doi: 10.1111/j.1349-7006.2004.tb03323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nenoi M, Ichimura S, Mita K, Yukawa O, Cartwright IL. Regulation of the catalase gene promoter by Sp1, CCAAT-recognizing factors, and a WT1/Egr-related factor in hydrogen peroxide-resistant HP100 cells. Cancer Res. 2001;61:5885–5894. [PubMed] [Google Scholar]
- 48.Sewnath ME, Olszyna DP, Birjmohun R, ten Kate FJ, Gouma DJ, van Der Poll T. IL-10-deficient mice demonstrate multiple organ failure and increased mortality during Escherichia coli peritonitis despite an accelerated bacterial clearance. J Immunol. 2001;166:6323–6331. doi: 10.4049/jimmunol.166.10.6323. [DOI] [PubMed] [Google Scholar]
- 49.Peiser L, Gough PJ, Kodama T, Gordon S. Macrophage class A scavenger receptor-mediated phagocytosis of Escherichia coli: role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect Immun. 2000;68:1953–1963. doi: 10.1128/iai.68.4.1953-1963.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tapfer H, Liigant A, Simovart HE, Poldoja E, Kokk K, Naaber P, Talvik R. Dissemination of bacteria in multiple organs associated with apoptosis and macrophage activity in different stages of experimental sepsis. Scand J Surg. 2003;92:163–170. doi: 10.1177/145749690309200210. [DOI] [PubMed] [Google Scholar]
- 51.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 52.Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. Br J Anaesth. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- 53.Paterson RL, Galley HF, Webster NR. The effect of N-acetylcysteine on nuclear factor-kappa B activation, interleukin-6, interleukin-8, and intercellular adhesion molecule-1 expression in patients with sepsis. Crit Care Med. 2003;31:2574–2578. doi: 10.1097/01.CCM.0000089945.69588.18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.