

Summary
The gtaI gene of Rhodobacter capsulatus encodes an N-acyl-homoserine lactone (acyl-HSL) synthase. Immediately 5′ of the gtaI gene is ORF rcc00328 that encodes a potential acyl-HSL receptor protein. A combination of genetic and biochemical approaches showed that rcc00328 (renamed gtaR) modulates the production of a genetic exchange element called the gene transfer agent (RcGTA), and regulates the transcription of gtaI. Although gtaI mutants exhibited decreased levels of RcGTA production, mutagenesis of gtaR did not, whereas a gtaR/gtaI double mutant produced wild-type levels of RcGTA. Because mutagenesis of gtaR suppressed the effect of the gtaI mutation, we suggest that the GtaR protein is a negative transcriptional regulator of RcGTA gene expression. We discovered that the gtaR and gtaI genes are co-transcribed, and also negatively regulated by the GtaR protein in the absence of acyl-HSL. A His-tagged GtaR protein was purified, and DNA-binding experiments revealed a binding site in the promoter region of the gtaRI operon. This GtaR protein did not bind to the RcGTA promoter region, and therefore modulation of RcGTA production appears to require at least one additional factor. We found that RcGTA production was stimulated by spent media from other species, and identified exogenous acyl-HSLs that induce RcGTA.
Introduction
Many bacteria utilize a LuxI/LuxR-type of system where a LuxI homologue synthesizes an N-acyl-homoserine lactone (acyl-HSL) signalling molecule, and a LuxR homologue functions as the cognate receptor protein that modulates the transcription of a set of target genes, when a threshold concentration of acyl-HSL is reached (Bassler, 1999; Miller and Bassler, 2001; Waters and Bassler, 2005). The resultant modulation of behaviour in response to changing population densities is called quorum sensing (Fuqua et al., 1994; Tsai and Winans, 2010).
Quorum sensing allows bacteria to sense members of their own species, in some cases other species, and even bacterial sensing of eukaryotes has been proposed (Gonzalez and Keshavan, 2006). This type of cell density-dependent gene regulation is involved in a range of behaviours, such as regulation of symbiosis (Rhizobium leguminosarum), plant pathogenesis (Agrobacterium tumefaciens), and in human disease (Burkholderia mallei, Vibrio cholerae, Pseudomonas aeruginosa) (Piper et al., 1993; Zhu et al., 2002; Duerkop et al., 2007; Sanchez-Contreras et al., 2007). Quorum sensing also plays a significant role in biofilm formation in situ and in vitro (Davies et al., 1998).
There are several reports indicating that quorum sensing is involved in horizontal gene transfer. For example, transfer of the tumour-inducing Ti conjugative plasmid from A. tumefaciens to plant cells is regulated by an octanoyl-homoserine lactone-dependent system (Piper et al., 1993), and production of the gene transfer agent (RcGTA) of the photosynthetic α-proteobacterium Rhodobacter capsulatus is controlled by quorum sensing via a C16-acyl-HSL, produced by the GtaI protein (Schaefer et al., 2002). The RcGTA horizontal gene transfer agent resembles a defective, tailed bacteriophage, which packages and transfers ~ 4.3 kb of random genomic DNA to R. capsulatus cells (Lang and Beatty, 2000; Leung, 2010). The RcGTA structural gene cluster resembles a prophage in the R. capsulatus chromosome, but RcGTA does not form plaques in phage assay protocols, and is unable to fit the entire 15 kb structural gene cluster into a single RcGTA particle. The RcGTA gene cluster codon usage is similar to the average codon usage of the rest of the R. capsulatus genome. RcGTA-like genes are widespread throughout the α-proteobacteria and appear to have a long evolutionary history in concert with the host genomes (Lang and Beatty, 2000; 2007).
Some organisms that possess LuxI/R-types of quorum-sensing systems respond to multiple N-acylhomoserine lactones, including those not synthesized by the organism itself (Wagner-Dobler et al., 2005; Patankar and Gonzalez, 2009). Some well-documented examples include the LasR protein in P. aeruginosa (Winson et al., 1995; Savka et al., 2011), and luminescence genes in Vibrio fischeri (Winson et al., 1998). Most LuxR-type of proteins require a bound acyl-HSL to bind to a target DNA sequence and either stimulate or repress transcription. However, it is becoming increasingly evident that there is a second family of LuxR homologues that appear to fold correctly and bind to a target DNA sequence in the absence of an acyl-HSL (Tsai and Winans, 2010).
The GtaI acyl-HSL synthase of R. capsulatus is 26% identical (43% similar) in a full-length alignment with CerI, a LuxI homologue in the related bacterium Rhodobacter sphaeroides (Schaefer et al., 2002). In conjunction with the discovery of the gtaI gene, sequences encoding three CerR homologues (a LuxR-type of protein in R. sphaeroides) were located in the R. capsulatus genome (Schaefer et al., 2002). One of these CerR homologues is encoded by the ORF rcc00328 (located directly 5′ of gtaI) and was predicted to be the cognate signal receptor of the acyl-HSL synthesized by GtaI. The other two CerR homologues are encoded by genes at distant genomic locations, are less than 24% identical to GtaR or other LuxR proteins in sequence alignments, and their possible functions are unknown.
We used combined genetic and biochemical approaches to test the hypothesis that the ORF rcc00328 (herein named gtaR) encodes a cognate receptor of the acyl-HSL produced by the GtaI protein. We found that gtaR and gtaI are co-transcribed, and therefore comprise the gtaRI operon. On the basis of the phenotypes of single mutants of gtaR and gtaI, as well as a gtaR/gtaI double mutant, we suggest that the GtaR protein acts as a transcriptional repressor of the gtaRI operon in the absence of acyl-HSL. Purified GtaR protein was found to bind specifically to DNA fragments containing the promoter region of the gtaRI operon, and we identified a DNA sequence protected from DNase digestion by GtaR. In addition, we show that GtaR is involved in regulating RcGTA gene expression, as part of the gtaI-dependent quorum-sensing system. However, the GtaR protein does not bind to the RcGTA promoter region, indicating that this quorum-sensing system regulates at least one other factor that directly modulates RcGTA gene transcription. To evaluate whether R. capsulatus is capable of communicating with other species, we tested crude, cell-free spent culture media and a variety of pure acyl-HSLs for effects on RcGTA gene expression. We found that RcGTA production is induced by spent growth media from other bacterial species, and identified a variety of acyl-HSLs that induce RcGTA production in a gtaI mutant, all of which appear to modulate the activity of GtaR.
Results
Co-transcription of gtaR and gtaI
The gtaI putative start codon is only 49 bp from the gtaR stop codon, and it was speculated that they are transcribed as an operon (Schaefer et al., 2002) because many LuxI/LuxR-type quorum-sensing pairs are co-regulated at the transcriptional level. To test this hypothesis, reverse-transcription PCR was employed. Total RNA from R. capsulatus B10 was used to generate cDNA, which was subsequently used as a template in PCR reactions. Primers were designed to amplify the intergenic regions between the gtaR and gtaI genes, and between gtaR, gtaI and the genes located immediately upstream (rcc00327; rplQ) and downstream (rcc00330) (Fig. 1A). A series of PCR reactions was performed using the cDNA, genomic DNA, or total RNA as template. An amplification product of the region between gtaR and gtaI was obtained using cDNA or genomic DNA as template (Fig. 1B). In contrast, neither the upstream nor the downstream primer pairs yielded a PCR product from the cDNA template, although products were seen using DNA template (Fig. 1B). None of the primer pairs yielded a product in the RNA negative control. These results show that the gtaR and gtaI genes are co-transcribed, and that this transcription unit does not include either of the flanking genes.
Fig. 1.

RT-PCR of the gtaRI bicistronic transcript.
A. Schematic representation of gtaR, gtaI and the two flanking genes rplQ and rcc00330; locations of primer sites designed to amplify across intergenic regions are indicated by the small arrows.
B. PCR results using genomic DNA, unfractionated RNA, or cDNA produced from total RNA as the template.
Autoregulation of the gtaRI operon
To determine whether the expression of the gtaRI operon is regulated by GtaR and GtaI, we initially measured promoter activity using the gtaR::lacZ fusion plasmid p601-P2R, which contains about 1 kb of sequences 5′ to the gtaR start codon. The wild-type, ΔgtaI, ΔgtaR and ΔgtaRI strains containing p601-P2R were grown in RCV medium under phototrophic conditions to early stationary phase, and the β-galactosidase specific activity determined. The β-galactosidase activity of the ΔgtaI strain containing p601-P2R was found to be decreased relative to wild type, whereas the expression in the ΔgtaR mutant was not significantly altered (Fig. 2). The β-galactosidase activity of the ΔgtaRI mutant strain containing p601-P2R was similar to that of the wild-type and ΔgtaR strains (Fig. 2), indicating that, indeed, GtaI and GtaR function in the same system to regulate gtaRI expression. Furthermore, addition of exogenous C16-HSL (2 μM) to the ΔgtaI strain containing p601-P2R restored wild-type levels of gtaR::lacZ expression, and trans-complementation of the ΔgtaRI mutant containing p601-P2R with an extrachromosomal copy of the gtaR gene [ΔgtaRI(R)] reverted gtaR::lacZ expression to ΔgtaI levels (Fig. 2).
Fig. 2.

Expression of a gtaR::lacZ fusion in R. capsulatus gtaI, gtaR and gtaRI mutants. β-galactosidase activities of strains containing the gtaR::lacZ gene fusion plasmid, p601-P2R, are indicated on the vertical axis. The wild-type strain B10 is indicated by WT, mutant strains are indicated as ΔgtaI, ΔgtaR and ΔgtaRI. The label C16 indicates the activity obtained when the gtaI mutant culture was supplemented with C-16 acyl-HSL. The label ΔgtaRI(R) indicates the activity obtained when the gtaRI mutant was trans-complemented with a copy of the gtaR gene in plasmid pIND4R. Error bars represent the standard deviation of the mean between samples (n ≥ 3).
Identification of the gtaR promoter region and GtaR-binding sequences
Two ATGs in the same reading frame were identified as possibly encoding the start codon of the gtaR gene. The 5′-most ATG (annotated as the start codon in the genome sequence) is 102 bp upstream of the other ATG, and there are no compelling Shine–Dalgarno sequences for either ATG. To determine whether either of these ATGs encodes the true start codon, two translationally in-frame gtaR::lacZ fusions were constructed. The plasmid p601-P1R contains a fusion to the 5′ (upstream) ATG and plasmid p601-P2R contains a fusion to the 3′ (downstream) ATG, both with ~ 1 kb of sequence extending to the same 5′ end, well into the region that was found not to be co-transcribed with gtaRI. The wild-type strain containing each of these plasmids was grown in RCV medium under phototrophic conditions, harvested at early stationary phase, and evaluated for β-galactosidase specific activity. The β-galactosidase specific activity from p601-P1R was about 15% of the activity from p601-P2R (Fig. 3), which leads us to assign the downstream ATG as the genuine start codon for gtaR, in contrast to the genome annotation (GenBank CP001312).
Fig. 3.
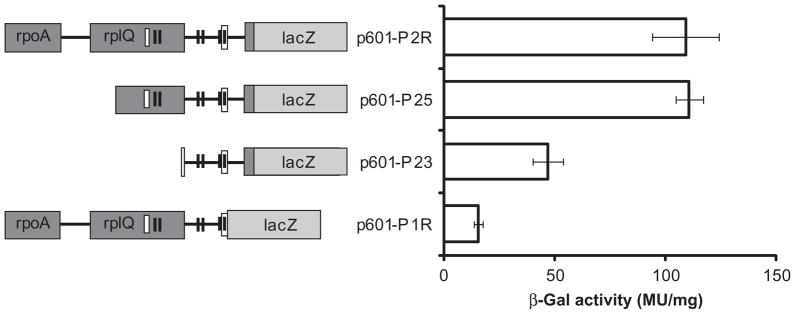
Effects of deletions in the gtaR promoter region on initiation of transcription. On the left are representations of the gtaR gene 5′ segments and the gtaR::lacZ fusions used to investigate sequences important for gtaR expression. Predicted −10 and −35 sites are represented by pairs of black boxes, whereas the empty boxes represent putative lux boxes. On the right are the β-galactosidase specific activities of wild-type cells containing each gtaR::lacZ gene fusion plasmid. Error bars represent the standard deviation of the mean between samples (n = 3).
In silico analysis of the gtaR upstream region identified three predicted promoters (Figs 3 and S1). The locations of these putative −10 and −35 sites are centred approximately 134, 230 and 494 bp upstream of the gtaR start codon identified above. The 5′-most predicted −10 and −35 sites were found within rcc00327 (the rplQ homologue), but can be eliminated as a promoter because our RT-PCR data showed that there is no readthrough from rplQ to gtaR. To identify potential GtaR-binding sites, the gtaR promoter region was scanned for lux box homologues (Experimental procedures). A homologous sequence, and hence a potential GtaR-binding site, was found to overlap the −10 site of the 3′-most predicted promoter (Fig. 3).
A series of gtaR::lacZ promoter deletion fusions was made to identify regulatory sequences of the gtaR promoter. The plasmids p601-P2R, p601-P25 and p601-P23 contain 1093, 595 and 209 bp, respectively, of sequences upstream of the gtaR start codon. The wild-type strain containing plasmid p601-P2R, p601-P25 or p601-P23 was grown in RCV medium under phototrophic conditions, harvested at early stationary phase, and assayed for β-galactosidase specific activity.
We found that the activity from p601-P25 (595 bp of upstream sequences) was the same as that from p601-P2R (1093 bp of upstream sequences) (Fig. 3), indicating that under these growth conditions the 595 bp upstream sequences are sufficient for gtaR expression. The activity of p601-P23 (209 bp of upstream sequences) was ~ 40% of p601-P2R (Fig. 3), showing that sequences important for gtaR expression are located between 209 and 595 bp upstream of the gtaR start codon. These sequences include the 5′-most predicted lux box.
To evaluate whether the regulation of the gtaRI operon by gtaR was direct or indirect, electrophoresis mobility shift assays (EMSAs) were used to determine the binding of a 6 histidine-tagged GtaR protein (GtaR-6xHis) to a DNA fragment containing the gtaRI promoter region. The DNA fragments are depicted in Fig. 4A, and were designed to include both predicted lux boxes (681 bp fragment), the 5′-most predicted lux box (800 bp fragment), or the 3′-most predicted lux box (345 bp fragment).
Fig. 4.

Specific binding of the GtaR-6xHis protein to sequences 5′ to gtaR.
A. Representation of the gtaR gene and 5′ sequences. The black boxes indicate putative-10 and −35 sites of three predicted promoters, and the empty boxes are predicted lux box homologues. Brackets indicate the DNA fragments used for EMSAs.
B. EMSA of the 681 bp fragment of the gtaR 5′ region in the presence of a 210 bp non-specific competitor DNA fragment (from plasmid pUC19).
C. EMSA of the 345 bp fragment of the gtaR 5′ region in the presence of a 210 bp non-specific competitor DNA fragment (from plasmid pUC19).
D. EMSA of the 800 bp fragment of the gtaR 5′ region.
Electrophoresis mobility shift assays using the 681 bp fragment (100 nM) containing both predicted lux boxes of the gtaR promoter region showed a band with altered mobility when GtaR-6xHis protein concentrations were 90 nM or higher (Fig. 4B). The proportion of shifted to non-shifted DNA increased with increased GtaR-6xHis protein concentration, and 100% of the DNA appeared to be shifted at 1.44 μM GtaR-6xHis. The negative control, the 210 bp competitor DNA band, showed no change in mobility even in the presence of 1.44 μM GtaR-6xHis. These results indicate that GtaR directly regulates the transcription of the gtaRI operon by binding to the promoter region in the absence of acyl-HSL. EMSAs using the 345 bp fragment containing the 3′-most predicted lux box showed results similar to the 681 bp fragment data (Fig. 4C), whereas no shift of the 800 bp fragment was seen (Fig. 4D), indicating that the 5′-most predicted lux box is not a site for GtaR-binding under these conditions.
To evaluate the effect of acyl-HSL on the interaction of GtaR-6xHis with the gtaR promoter region, we attempted EMSAs using the 345 bp amplicon in a number of ways. Acyl-HSLs were added to the EMSA binding reaction, or pre-incubated with the GtaR-6xHis protein before the binding reaction in excess (2- to 3-fold) compared with the concentrations found in cultures (Schaefer et al., 2002). Both of these methods were repeated with the 6-His tag cleaved off the GtaR-6xHis protein. Furthermore, the GtaR-6xHis gene was expressed with HSL present in the Escherichia coli culture, and at all stages of protein purification, but there was no change in either the number of retarded bands seen in the EMSA or in the fraction of total DNA retarded by a given concentration of GtaR-6xHis in any of these experiments (data not shown).
DNA sequence bound by GtaR
To identify the GtaR binding sequence, DNase footprinting experiments were performed using GtaR-6xHis and a 245 bp fragment of the 345 bp gtaR promoter region, previously verified in EMSAs as containing a GtaR binding site (Fig. 4C). 32P-5′-end-labelled primers complementary to the coding and non-coding strands were used in two different amplifications. GtaR-6xHis was bound to each of these labelled fragments and the mixture exposed to DNase I.
The footprinting results showed a protected region and hypersensitive sites (Fig. 5A and B). Such hypersensitive sites are indicative of bending of the DNA by GtaR binding (Travers, 1989). The protected region overlaps the 3′-most predicted lux box and the 3′-most predicted −10 site, both of which are about 102 bp upstream of the gtaR start codon. The position of this protected region and hypersensitive sites on the two strands are shifted by ~ 3 bp, indicating that GtaR binds DNA in the minor groove (Suck et al., 1988; Travers, 1989). The binding of GtaR within a predicted −10 sequence is consistent with our suggestion that this protein represses transcription of the gtaRI operon.
Fig. 5.

DNase I footprint analysis of the interaction between GtaR-6xHis and the gtaR promoter region.
A. Labelled DNA fragments were incubated with different concentrations of GtaR-6xHis as indicated above the autoradiograms. ATGC designate DNA-sequencing lanes, grey bars indicate the limits of protection, and arrows indicate bases that were hypersensitive to DNase I.
B. Sequence of the DNA protected by GtaR-6xHis. The black bars indicate the limits of protection, the predicted −10 region is boxed, and the hatched bar shows the position of the sequences similar to lux boxes (Table S1). The arrows indicate bases that were hypersensitive to DNase I.
The site protected by GtaR-binding was confirmed in EMSAs using a series of sequentially truncated DNA fragments. These fragments were generated by PCR amplification using primers that made successive 5′ deletions from the 345 bp fragment of the gtaR promoter. A 186 bp amplicon, named DR9, contains the 3′-most predicted lux box and the 3′-most predicted −10 and −35 sites. EMSAs of DR9 showed a retarded band when 90 nM or more GtaR-6xHis protein was used (Fig. 6). The proportion of bound to unbound DNA increased with GtaR-6xHis protein concentration, which is similar to the EMSAs of the 345 bp fragment (Fig. 4C). These results indicate that GtaR binding sequences are found within the 186 bp fragment. The 160 bp amplicon, named DR7, is missing all sequences 5′ of the GtaR binding sites. EMSAs of DR7 showed a shifted band (Fig. 6), indicating that GtaR-6xHis is still capable of binding this DNA fragment. EMSAs of the 152, 142 and 137 bp amplicons (named DR5, DR3 and DR1 respectively) contained deletions of one, two or all three of the GtaR binding sites, respectively, and showed little or no evidence of a retarded band even at the highest concentration of GtaR-6xHis protein (1.44 μM) (Fig. 6). The absence of a clear and strong band shift in the EMSAs of DR5 and DR3 indicates that GtaR requires all three sites for binding under the assay conditions used here. The EMSAs of DRR, which contains the sequence 5.′ of DR7, indicated that GtaR could not bind to this fragment. These observations confirm that there are no GtaR binding sites 5′ of the site identified in the footprinting assays.
Fig. 6.

Effects of deletions in the gtaR promoter region on the binding of GtaR-6xHis. On the left are representations of the DNA fragments used in the EMSAs. The black boxes represent putative −10 and −35 sites, the empty box surrounding the left-most −10 site is the predicted lux box, and the grey bar indicates the protected region identified by DNase I footprinting. The size and name of each DNA fragment is given, and the corresponding EMSAs are on the right, with the concentration of the GtaR-6xHis protein in each reaction given above.
Effects of mutations in gtaR and gtaI on RcGTA production
We investigated the possibility that gtaR and gtaI function as a cognate quorum-sensing pair that regulates RcGTA gene expression by studying separate knockouts of gtaR (ΔgtaR strain) and gtaI (ΔgtaI strain), and a double knockout of gtaI and gtaR (ΔgtaRI strain). RcGTA expression was evaluated by using the orfg1::lacZ fusion plasmid p601-g65, Western blots probed with anti-RcGTA capsid protein antibodies, and at the functional level by RcGTA transduction bioassays, as described in the Experimental procedures section.
We found that the orfg1::lacZ expression was lower in the ΔgtaI strain, but not significantly different in the ΔgtaR and ΔgtaRI strains, relative to the wild-type strain (Fig. 7A). Furthermore, supplying the ΔgtaI strain with exogenous C16-HSL restored wild-type levels of orfg1::lacZ expression, and trans-complementation of the gtaR mutation restored the gtaI mutant phenotype. This is similar to the gtaR gene expression profile in these strains (see above section), and indicates that RcGTA is regulated by the GtaR/GtaI quorum-sensing system.
Fig. 7.

Comparison of RcGTA gene expression in quorum-sensing mutant strains of R. capsulatus.
A. β-Galactosidase activities of strains containing the orfg1::lacZ promoter fusion plasmid p601-g65.
B. Western blots of cells probed with RcGTA capsid protein antiserum.
C. Frequency of RcGTA-mediated gene transfer. The wild-type strain B10 is indicated by WT, mutant strains are indicated as ΔgtaI, ΔgtaR and ΔgtaRI, and the GtaR complement is indicated as ΔgtaRI(R); C16 indicates the ΔgtaI culture supplemented with C16 acyl-HSL. Error bars represent the standard deviation of the mean between samples (n = 3).
The amount of RcGTA capsid protein detected in Western blots was decreased in the ΔgtaI mutant compared with the wild-type strain, whereas the ΔgtaR and ΔgtaRI mutants were similar to the wild-type strain (Fig. 7B). Addition of exogenous C16-HSL (2 μM) to the ΔgtaI strain restored wild-type levels of RcGTA production, and trans-complementation of the ΔgtaRI mutant with an extrachromosomal copy of the gtaR gene [ΔgtaRI(R)] reverted this strain to the ΔgtaI phenotype [Fig. 7B; ΔRI(R)].
The gene transduction assays showed a pattern of results that paralleled the other approaches. The ΔgtaI, ΔgtaR and ΔgtaRI strains yielded 18 ± 10%, 81 ± 9% and 116 ± 43%, respectively, as many transductants as the wild-type control, whereas the ΔgtaI strain supplemented with C16-HSL and the ΔgtaRI(R) strain yielded 103 ± 23% and 7 ± 3% as many transductants respectively (Fig. 7C).
The similarity in the results of these three independent measures of RcGTA gene expression strongly argues that GtaI and GtaR are part of the same signalling system, because the gtaR mutation reproducibly suppresses the effect of the gtaI mutation, and introduction of the gtaR gene on a plasmid into the double knockout (ΔgtaRI) strain restores the single knockout (ΔgtaI) phenotype. Therefore, in this signalling system, the gtaR mutation is dominant over the gtaI mutation. To evaluate whether the regulation of the RcGTA structural genes by GtaR might be direct or indirect, EMSAs were used. EMSAs using the 650 bp PCR amplicon of the RcGTA promoter region present in plasmid p601-g65 showed no retardation of the 650 bp DNA band (Fig. S2). These experiments were also performed in the presence of acyl-HSLs in the same manner as with the gtaRI promoter region (see above), with no observed shift in band mobility. Because the gtaRI promoter region showed a mobility shift in EMSAs using the GtaR-6xHis protein (see above), the absence of a shift using the RcGTA promoter region indicates that GtaR does not directly interact with the RcGTA promoter, at least under these conditions.
RcGTA production is stimulated by substances produced by other species
We investigated whether RcGTA production could be stimulated by diffusible signal molecules from other bacterial species by adding 2 ml of sterile, cell-free media from stationary phase cultures of different organisms (Table 1) into 17 ml phototrophic ΔgtaI cultures. Cultures were grown to the stationary phase and evaluated for RcGTA capsid protein production in Western blots probed with antiserum against the capsid protein. Partial restoration of capsid production was observed with addition of culture media from R. capsulatus B10, R. sphaeroides 2.4.1, Paracoccus denitrificans and E. coli MG1655 (Fig. 8A), whereas Rhodopseduomonas palustris CGA009 and uninoculated Luria–Bertani (LB) medium failed to elicit a response. These data were confirmed in an RcGTA transduction assay, where ΔgtaI cultures supplemented with cell-free media from R. sphaeroides 2.4.1, P. denitrificans and E. coli MG1655 cultures yielded 56 ± 13%, 59 ± 14% and 34 ± 6%, respectively, as many transductants as the wild-type control, whereas an unsupplemented ΔgtaI culture generated only 9 ± 6% as many transductants (Fig. 8B). R. sphaeroides 2.4.1 and P. denitrificans are both known to produce long-chain acyl-HSLs (Puskas et al., 1997; Schaefer et al., 2002), and so our results represent a bioassay indicating that a variety of acyl-HSLs stimulate the production of RcGTA. However, E. coli MG1655 is not known to synthesize an autoinducer of similar size and structure to long-chain acyl-HSLs, but rather produces a boron-containing substance called AI-2 (Surette et al., 1999; Chen et al., 2002). To evaluate whether AI-2 stimulates RcGTA production, we tested the ability of the ΔgtaI strain to respond to growth medium from an E. coli MG1655 ΔluxS strain (produces no AI-2) (Tavender et al., 2008), and observed a response similar to medium from wild-type E. coli MG1655 (data not shown). Regardless, these results indicate that R. capsulatus is capable of communicating with a variety of other bacteria by reception of more than one type of signal, and responding by inducing the production of RcGTA.
Table 1.
Species used as culture supernatant donors.
| Species | Class | Produces an N-acylhomoserine lactone |
|---|---|---|
| Rhodobacter capsulatus SB1003 | α-proteobacteria | Yes; N-hexadecanoyl-L-homoserine lactone and N-tetradecanoyl-L-homoserine lactone (Schaefer et al., 2002) |
| Rhodobacter sphaeroides 2.4.1 | α-proteobacteria | Yes; 7,8-cis-N-tetradecenoyl homoserine lactone (Puskas et al., 1997) |
| Paracoccus denitrificans PD1222 | α-proteobacteria | Yes; N-hexadecanoyl-L-homoserine lactone (Schaefer et al., 2002) |
| Rhodopseduomonas palustris CGA009 | α-proteobacteria | No; produces a p-Coumaroyl-homoserine lactone (Hirakawa et al., 2011) |
| Escherichia coli MG1655 | γ-proteobacteria | No; produces an AI-2 (Xue et al., 2009) |
Fig. 8.

RcGTA production in R. capsulatus acyl-HSL synthase mutant strain ΔgtaI in response to the addition of cell-free media from stationary phase cultures. On the left are data from wild-type strain B10 and ΔgtaI mutant controls with no addition, and on the right are data from addition of C16-acyl-HSL or sterile LB medium to the ΔgtaI mutant. In the middle are data when added media were from: wild-type R. capsulatus B10 (+ WT); R. sphaeroides 2.4.1 (R. sph); P. denitrificans (P. den); R. palustris CGA009 (R. pal ); E. coli MG1655 (E. col ).
A. Western blot evaluating RcGTA capsid production.
B. Frequency of RcGTA-mediated gene transfer relative to the wild-type strain B10. Error bars represent the standard deviation of the mean between samples (n = 3).
RcGTA production is stimulated by multiple, specific acyl-HSLs
To confirm and extend the results obtained by inducing RcGTA gene expression with signals from other species, we tested a wide variety of commercially available acyl-HSLs in bioassays of the ΔgtaI mutant, using the three methods described above for analysis of gtaR and gtaI mutants.
The β-galactosidase activity in cells containing the RcGTA promoter fusion plasmid p601-g65 was decreased in the ΔgtaI mutant compared with the wild-type strain (Fig. 8A), consistent with a prior report (Schaefer et al., 2002). A significant restoration of β-galactosidase activity in the ΔgtaI strain was observed with the addition of C12-, C14-, C16-, C16c- and C18-HSL (Fig. 9A). The ability of R. capsulatus to respond to C16c-HSL (N-cis-hexadec-9Z-enoyl-L-homoserine lactone), which has a double bond in the acyl tail, indicates that the length rather than the shape of the HSL acyl tail is important for detection by the R. capsulatus quorum-sensing system. These findings show that R. capsulatus is capable of responding to multiple exogenous acyl-HSLs by inducing transcription of RcGTA orfg1.
Fig. 9.

Comparison of RcGTA gene expression in R. capsulatus wild-type B10, and the ΔgtaI mutant in the absence and presence of exogenous acyl-HSLs.
A. β-Galactosidase activities of strains containing the orfg1::lacZ promoter fusion plasmid p601-g65.
B. Western blots of cells probed with RcGTA capsid protein antiserum.
C. Frequency of RcGTA-mediated gene transfer. WT, wild-type strain B10, and ΔgtaI, the gtaI knockout, both with no addition of acyl-HSL. C4-C18, chain length of acyl-HSLs added to cultures of the ΔgtaI mutant. Error bars represent the standard deviation of the mean between samples (n = 3).
The transcriptional changes in response to multiple acyl-HSLs were paralleled by restoration of RcGTA capsid protein production to approximately the wild-type level in Western blots of ΔgtaI cells from cultures supplemented with C12-, C14-, C16-, C16c- and C18-HSL (Fig. 9B). RcGTA production may be weakly stimulated by short-chain acyl-HSLs, as faint bands are visible in response to acyl-HSLs with chain lengths of C4 to C10.
A quantitative measure of the amount of mature, functional RcGTA particles released from ΔgtaI cells in response to the addition of acyl-HSLs was obtained in gene transduction assays. We found that cell-free culture medium from the ΔgtaI strain generated 13 ± 14% the number of transductants as the wild-type strain, whereas supplementation of ΔgtaI cultures with acyl-HSLs yielded 102 ± 13% (C12), 115 ± 35% (C14), 103 ± 23% (C16), 102 ± 25% (C16c) and 105 ± 31% (C18) of the number obtained with a wild-type donor (Fig. 9C).
Experiments on ΔgtaRI cultures supplemented with C12-, C16-, and C18-HSL showed little difference in RcGTA capsid protein production and transduction efficiencies, compared with unsupplemented ΔgtaRI cultures (Fig. S3). Taken together, these data show that R. capsulatus responds to a wide range of N-acylhomoserine lactones, with a maximal response to endogenous acyl-HSLs and those with a similar acyl chain length. Because the ΔgtaI mutant increased RcGTA gene expression in response to acyl-HSLs whereas the ΔgtaRI mutant did not, it appears that these multiple signals modulate the activity of the GtaR protein, and that the ORFs rcc01088 and rcc01823 do not affect the expression of RcGTA genes.
Discussion
We discovered that the R. capsulatus gtaR and gtaI genes are co-transcribed, and that the gtaRI operon is autoregulated by binding of GtaR to the promoter region to repress expression. These conclusions arise in part from the finding that the ΔgtaI mutant had decreased expression of the gtaR gene, whereas the ΔgtaR mutant had the wild-type level of expression. We initially considered three interpretations of these data: (i) GtaR and GtaI do not function in a common regulatory system; (ii) the three luxR-type genes gtaR, rcc01088 and rcc01823 are all functional and redundant, and therefore a triple knockout would be required to produce a mutant phenotype; and (iii) GtaR functions by binding DNA in the absence of the acyl-HSL signal. However, the finding that in the ΔgtaRI mutant the gtaR mutation suppresses the phenotypic effects of the gtaI mutation argues in favour of a mechanism in which the GtaR protein is capable of binding to DNA in the absence of the acyl-HSL synthesized by GtaI, as was confirmed in the EMSA and footprinting experiments. Taken together, our results are consistent with a model in which GtaR represses transcription of gtaRI, and this repression is antagonized by acyl-HSL synthesized by GtaI. The observation that the gtaRI double mutant did not respond to long-chain acyl-HSLs provides additional support for this model, and the fact that gtaI and gtaR are co-transcribed lends support to the idea that they act as a quorum-sensing pair. If this model is correct, autoregulation of the gtaRI operon by GtaR would produce an autoinducing circuit observed in many quorum-sensing systems. Generally, LuxR-type proteins positively regulate expression of both their own and their cognate luxI-type gene when bound by an acyl-HSL. Hence, when acyl-HSL is detected by the cell, an autoinducing loop is initiated where the acyl-HSL signal is continuously amplified (Lazdunski et al., 2004). In our model, both the gtaR and gtaI genes are negatively regulated by the GtaR protein, which when bound by acyl-HSL, releases DNA and allows transcription of the operon to occur. This system would allow for amplification of the acyl-HSL signal, although by derepression rather than activation of transcription, which is an unusual mechanism of action in LuxI/R-type quorum-sensing systems. To our knowledge no other example of a gene encoding a LuxR-type protein that is able to bind DNA in the absence of the acyl-HSL is co-transcribed with an acyl-HSL synthase gene. Other luxR-type genes encoding proteins that bind DNA as an apoprotein and repress target genes are convergently transcribed with the adjacent luxI-type of gene (Tsai and Winans, 2010).
Using an in silico analysis, we identified several putative lux boxes upstream of orfg1 and gtaR, but only the sequence in the intergenic region between gtaR and the upstream rplQ gene bound to GtaR-6xHis. The percentage identity between these sequences and lux box sequences identified by others was low (Table S1), and in general lux box sequences are not well-conserved (Fuqua et al., 1994; Horng et al., 2002). We attempted to locate other sequences that might be bound by the GtaR protein by scanning the R. capsulatus SB1003 genome using the GtaR footprint and degenerate sequences derived thereof, with negative results. A previous analysis of the P. aeruginosa LasR protein, which is a LuxR-type protein that binds a C12-HSL, found that there was no correlation between observed LasR DNA-binding and the presence of proposed ‘las-boxes’ (the predicted LasR DNA-binding consensus sequence) (Whiteley and Greenberg, 2001; Schuster et al., 2004). In fact, the authors observed that promoters containing a predicted ‘las-box’ were no more likely to be bound by LasR than promoters lacking such a consensus sequence. In general, it was noted that LasR-specific promoters exhibited little overall sequence conservation and a notable lack of sequences with dyad symmetry, which suggested to the authors that LasR might contact recognition sites at positions of degenerate composition (Whiteley and Greenberg, 2001; Schuster et al., 2004). Similarly, it appears that more work is needed to clarify the key bases that define a GtaR binding site.
Like GtaR, CarR, a LuxR-type protein in Erwinia carotovora, binds to its target DNA sequence in the absence of the signal molecule, N-(3-oxohexanoyl)-L-HSL (Welch et al., 2000). In the presence of sufficient N-(3-oxohexanoyl)-L-HSL concentrations, the affinity of CarR for DNA decreases until it is no longer able to bind (Welch et al., 2000). Although we suggest that GtaR functions in a similar manner, it has not yet been possible to demonstrate the release of DNA fragments containing the 345 bp gtaR promoter region in the presence of C12- or C16-HSL. There are technical difficulties associated with experiments using long-chain acyl-HSLs, in part due to poor solubility in aqueous solution, and so the lack of an effect of adding acyl-HSL to the EMSAs may be due to low concentration in vitro. In vivo, the cell membrane could accumulate long-chain acyl-HSLs. An alternative explanation is that GtaR is capable of binding DNA in both the absence and presence of acyl-HSL, with opposing effects on gene expression, although in this case an alternative binding site (not blocking RNA polymerase binding to the promoter) would appear to be required. There are no known quorum-sensing systems that function in this manner, although such behaviour has been observed in response regulators that respond to phosphorylation and dephosphorylation. A study of the EsaR protein, a LuxR-type of transcription repressor in Pantoea stewartii, found that interaction with its cognate acyl-HSL signal altered the EsaR protein’s affinity for DNA (Minogue et al., 2002). Despite considerable evidence that acyl-HSL antagonizes EsaR activity, the protein retained its ability to bind EMSA target DNA in the presence of excess acyl-HSL, and it is possible that a similar phenomenon is occurring with GtaR.
Previous analysis of the R. capsulatus SB1003 genome sequence identified gtaR and two other ORFs (rcc01088 and rcc01823) that are homologous to CerR, a LuxR-type transcriptional regulator in the related species R. sphaeroides 2.4.1 (Foster and Cairns, 1994; Puskas et al., 1997; Schaefer et al., 2002). To clarify the relationship of GtaR to quorum-sensing response regulators of known structure and function, a phylogenetic analysis of GtaR, rcc01088, rcc01823, and representative LuxR-type proteins was done (Fig. S4). Based on this analysis, GtaR does not cluster with any well-characterized LuxR-type proteins. Instead, GtaR groups in an apparent clade of LuxR-type proteins found in Rhizobium and Rhodobacter species, all of which are known to synthesize long-chain acyl-HSLs. To our knowledge the experiments on GtaR described herein are the first to address a mode of action for any member of this group (Puskas et al., 1997; Marketon and Gonzalez, 2002; Schaefer et al., 2002; Wisniewski-Dye et al., 2002). Furthermore, the C16c-HSL, which contains a double bond in the acyl tail, had an effect similar to C16-HSL on RcGTA production. This may indicate that it is the acyl tail length rather than stereochemistry that determines detection of acyl-HSLs in R. capsulatus and, conceivably, the species containing a LuxR-type of protein that clusters with GtaR.
In a previous study, Schaefer et al. (2002) observed that culture liquid of stationary phase R. capsulatus contains up to 390 nM C16-HSL. It was also observed that approximately half of the long-chain acyl-HSLs produced by R. capsulatus (C14 and C16-HSL) are sequestered within cells, and are likely membrane-associated. It was determined that C16-HSL is the preferred signal for RcGTA expression, as a half-maximal response to exogenous signal was observed at a lower concentration for C16-HSL than C14-HSL, and the half-maximal responses ranged from 2.5 to 10 nM (Schaefer et al., 2002). In this study, acyl-HSLs were added to cultures to a final concentration of 2 μM, which is approximately a fivefold excess over what is found in wild-type culture liquids and considerably in excess of what was previously observed to generate a maximal response in R. capsulatus. Although the precise concentration of exogenous acyl-HSLs required to stimulate RcGTA production is unknown, we speculate that the more similar an autoinducer structure is to the native R. capsulatus acyl-HSL, the lower the concentration required to generate a response will be, as such a phenomenon was observed with the LasR protein in P. aeruginosa (Savka et al., 2011).
The investigation of R. capsulatus responses to crude, spent media from other bacterial species showed significant responses to signals produced by R. sphaeroides 2.4.1 and P. denitrificans, which produce the long-chain acyl-HSLs 7,8-cis-N-tetradecanoyl homoserine lactone and N-hexadecanoyl-L-homoserine lactone respectively (see Table 1). Surprisingly, R. capsulatus also appeared to respond to a signal produced by E. coli MG1655, albeit it to a lesser degree. E. coli MG1655 produces AI-2, which is used in interspecies quorum sensing (Surette et al., 1999). Some species that do not synthesize AI-2 have response regulators that detect AI-2 produced by other species as a means to ‘spy’ on communication by other bacteria (Bassler, 2002). We speculated that R. capsulatus also uses this strategy because, although a search of the R. capsulatus SB1003 genome revealed no luxS (involved in AI-2 synthesis) homologue, homologues of proteins that function in the import of and response to AI-2 were found. There are homologues of Lsr proteins R, K, A, C, D and B (encoded by rcc02883, rcc02884, rcc02880, rcc02879, rcc02878 and rcc02877 respectively) in the genome sequence, with a minimum of 39% identity to the prototypical Lsr proteins found in E. coli. However, because the effect of culture liquid from wild-type E. coli MG1655 and an isogenic ΔluxS strain did not differ significantly, it appears that induction of RcGTA production in response to sterile E. coli MG1655 growth medium is not due to the presence of AI-2. Nevertheless, the production of RcGTA is induced by non-endogenous acyl-HSLs and cell-free media from other species, and so R. capsulatus is capable of sensing the surrounding population of other as well as its own species.
In summary, we propose a model in which GtaR is autoregulatory and binds to the gtaRI operon promoter to repress transcription in the absence of long-chain acyl-HSL. We also suggest the existence of a clade of LuxR-type transcriptional regulators that respond to long-chain acyl-HSLs. Although GtaR appears to regulate RcGTA transcription, the effect is indirect, therefore implicating another transcription regulator of RcGTA that remains unknown. Finally, we speculate that R. capsulatus is able to ‘listen in’ on the surrounding population, and that in times of stress resulting from competition with large populations of other species, R. capsulatus senses molecules produced by other species and activates RcGTA production as a mechanism to enhance genetic diversity, by creating new combinations of alleles that could confer a selective advantage over its competitors.
Experimental procedures
Bacterial strains and growth of cultures
The E. coli strain DH10B (Invitrogen) was used for cloning, BL21(DE3) (Invitrogen) for protein expression, S17-1 (Simon et al., 1983) and HB101(pRK2013) (Ditta et al., 1985) for conjugation of plasmids into R. capsulatus, and MG1655 (Blattner et al., 1997) was used as a source for diffusible signals. E. coli strains were grown at 37°C or 30°C (for protein expression) in LB medium (Sambrook et al., 1989) supplemented with the appropriate antibiotics at the following concentrations (μg ml−1): ampicillin, 150; tetracycline-HCl, 10; kanamycin sulphate 50; and gentamicin sulphate, 10.
The R. capsulatus strains B10 (wild type) (Marrs, 1974), BLKI (ΔgtaI), BLKR (ΔgtaR) and BLKO (ΔgtaRI) were grown in either rich YPS (Wall et al., 1975) or minimal RCV (Beatty and Gest, 1981) media under phototrophic (anaerobic, illuminated) conditions at ~ 30°C (Beatty and Gest, 1981). For use in the several assays, unless otherwise stated cells were harvested in the early stationary phase, 4 h after culture density increased less than 10 Klett units in 4 h. When appropriate, media were supplemented with tetracycline-HCl at 0.5 μg ml−1, gentamicin sulphate at 3 μg ml−1 and rifampicin at 80 μg ml−1. In the experiments testing for R. capsulatus response to various non-endogenous N-acylhomoserine lactones, exogenous N-acylhomoserine lactones (Cayman Chemical) were dissolved in dimethyl sulphoxide (DMSO) and added to ΔgtaI mutant cultures to a final concentration of 2 μM.
To test for exogenous acyl-HSL produced during bacterial growth, R. sphaeroides 2.4.1 was grown in LB medium under phototrophic conditions at ~ 30°C and harvested in stationary phase. P. denitrificans and E. coli MG1655 were grown aerobically at 37°C in LB medium and harvested in stationary phase, Rhodopseduomonas palustris CGA009 was grown in YPS medium under phototrophic conditions at ~ 30°C and harvested in stationary phase. Cells were removed by centrifugation and the resultant supernatant passed through a 0.2 μM filter. Approximately 2 ml of each filtrate was added to 17 ml phototrophic ΔgtaI cultures, which were then grown to stationary phase and evaluated for RcGTA production.
Culture turbidity was used as a measure of the number of cells per millilitre, and monitored by measuring light-scattering with a Klett-Summerson photometer (filter #66; red); 100 Klett units represent approximately 4 × 108 R. capsulatus colony-forming units per millilitre.
Recombinant DNA techniques and plasmids
Standard methods of DNA purification, restriction enzyme digestion, and other modification techniques were used (Sam-brook et al., 1989). The plasmid pXCA601 contains a promoterless lacZ allele with a BamHI site in the 8th codon (Hui et al., 1984; Adams et al., 1989) and was used for the construction of orfg1::lacZ, gtaR::lacZ fusion plasmids and the gtaR::lacZ promoter deletion plasmids.
The orfg1::lacZ promoter fusion plasmid p601-g65, which contains a translationally in-frame fusion between the R. capsulatus orfg1 of the RcGTA gene cluster and the E. coli lacZ coding sequence, with ~ 600 bp of sequence 5′ of the orfg1 start codon present, was used to evaluate RcGTA transcription. The gtaR::lacZ promoter fusion plasmid p601-PR2, which contains an in-frame fusion between the 9th codon of the R. capsulatus gtaR gene and the 8th codon of the E. coli lacZ gene, extending ~ 1 kb 5′ of the gtaR start codon was used to evaluate transcription of the gtaRI operon, and to create the promoter deletion plasmids p601-P25, p601-P23 and p601-P1R for gtaR promoter mapping.
The full-length inserts and deletion inserts were made by PCR amplification using wild-type R. capsulatus genomic DNA as the template and the primer sets GTA5 and GTA2.6, KOR1F and LHR2.2, LHR5 and LHR2.2, LHR3 and LHR2.2, and KOR1F and LHR2 (Table S2). The resulting amplicons were digested with PstI and BamHI (Invitrogen) and ligated into pXCA601. This ligation resulted in translationally in-frame fusions between either orfg1or gtaR and the lacZ genes in the plasmids p601-g65, p601-P2R, p601-P25, p601-P23 and p601-P1R (in corresponding order to the primer sets above).
An N-terminal 6xHis-tag was added to the GtaR protein for purification using a chelated nickel resin in column chromatography. This was done by cloning the gtaR gene into the expression vector pET-28a(+) (Novagen). The gtaR gene was amplified by PCR using R. capsulatus B10 chromosomal DNA as template, with the 5′ (upstream) primer PR28n and the 3′ (downstream) primer PR28c (Table S2), which introduced an NdeI and BamHI site respectively. This amplicon was ligated into pET-28a(+) as an NdeI to BamHI (Invitrogen) fragment, resulting in the plasmid pET28R. The plasmid pET28R was then transformed into the E. coli BL21(DE3) strain.
Construction of chromosomal deletion mutant strains
All gene disruptions made in this study were markerless in-frame deletions of the majority of the gene. All mutant strains were constructed from the R. capsulatus B10 strain. The first five and the last two codons of the gtaR gene and the first eight and last seven codons of the gtaI gene are present in the chromosome of the BLKR (ΔgtaR) and BLKI (ΔgtaI) mutants respectively. The BLKO (ΔgtaRI) mutant has both the first five and the last two codons of the gtaR gene, the gtaR–gtaR intergenic sequences, and the first eight and last seven codons of the gtaI gene present on the chromosome. The construction of these mutants is described by Leung (Leung, 2010), and was confirmed by PCR and sequencing.
Complementation of gtaR
The primers gtaR_comp_up and gtaR_comp_down were used to amplify the region ~ 600 bp upstream of the gtaR start codon, the entire gtaR gene itself, as well as the first several codons of the gtaI gene. The resultant amplicon was ligated as a KpnI to BamHI fragment into the vector pIND4 (Ind et al., 2009) to produce the plasmid pIND4R, which was then conjugated in to R. capsulatus BLKO (ΔgtaRI), resulting in the strain ΔgtaRI(R).
GtaR phylogenetic and in silico promoter analysis
Phylogenetic analysis of GtaR, rcc01088, rcc01823, and representative LuxR-type proteins was generated using a MUSCLE multiple sequence alignment (Edgar, 2004) and a Bayesian MCMC tree building algorithm (Huelsenbeck and Ronquist, 2001; Dereeper et al., 2008; 2010).
The BPROM program from Softberry (http://linux1.softberry.com/berry.phtml) was used to identify potential −10 and −35 sites in the gtaR and orfg1 promoter regions. The sequence alignment software Multalin version 5.4.1 (Corpet, 1988) and the lux box sequence (ACCTGTAGGA TCGTA CAGGT) (Fuqua et al., 1994) were used to find lux box-homologous sequences in an attempt to identify potential GtaR-binding sites.
RNA isolation and RT-PCR
Chromosomal DNA was isolated from R. capsulatus B10 using a phenol-chloroform DNA extraction as previously described (Wilson, 2001). Total RNA was extracted from early stationary phase cultures of R. capsulatus B10 using the RNeasy Mini Kit (Qiagen), and residual DNA contamination removed by treatment with DNaseI (Ambion). A cDNA library was generating using Superscript III (Invitrogen) using random hexamers as primers, and total R. capsulatus B10 RNA as a template. Three sets of primers were designed to amplify intergenic regions between the genes rplQ and gtaR, gtaR and gtaI, and gtaI and rcc00330. The primers rplQ-gtaR_up and rplQ-gtaR_down were used to amplify the region between rplQ and gtaR; gtaR-gtaI_up and gtaR-gtaI_down were used to amplify the region spanning gtaR and gtaI; and gtaI-rcc00330_up and gtaI-rcc00330_down were used to amplify the intergenic region between gtaI and rcc00330 (see Table S2 for primer sequences). PCRs were performed utilizing each primer set using chromosomal DNA, total RNA or cDNA as a template. Amplification products were separated on a 1% agarose gel at a constant voltage (100 V) for approximately 30 min and stained with ethidium bromide to visualize DNA bands.
β-Galactosidase assays
The β-galactosidase specific activity of cells containing the orfg1::lacZ, gtaR::lacZ and gtaR::lacZ promoter deletion fusions were assayed as described (Leung, 2010) using sonication to break cells and total protein measured using the Lowry method (Peterson, 1983) with bovine serum albumin as the standard. The units of β-galactosidase specific activities are given in Miller units per milligram of total protein (MU mg−1) (Miller, 1992; Leung, 2010). The statistical significance of results was evaluated by a two-tailed t-test assuming unequal variance. A P-value of 0.05 (95% confidence interval) was set as a cut-off for significance.
Western blot
Rhodobacter capsulatus cell cultures were grown under phototrophic conditions in YPS medium and harvested at stationary phase, as determined by monitoring culture turbidity. The cells were harvested by centrifugation and lysed by addition of SDS to 2% and boiling for 10 min. Aliquots of lysate from equivalent number of cells, according to optical density at 660 nm, were separated using 12% SDS-PAGE gels and blotted onto a nitrocellulose membrane (Perkin Elmer). The blotting was done using a Mini Trans-Blot apparatus (Bio-Rad) according to manufacturer’s specifications in Electroblot Buffer [27.5 mM Tris-Base, 192 mM glycine, 20% methanol] at 100 V (constant voltage) for ~ 1.5 h.
The primary antibody (rabbit) was raised against the R. capsulatus RcGTA capsid protein (Taylor, 2004). Primary antibody binding was detected using a peroxidase-linked anti-rabbit Ig secondary antibody (from donkey; Amersham) as part of the enhanced chemiluminescence (ECL) kit according to the manufacturer’s instructions (Amersham).
RcGTA transduction assay
Bioassays for RcGTA activity were performed as described (Taylor, 2004) with minor modifications. Wild-type R. capsulatus B10 (sensitive to rifampicin) was used as an RcGTA recipient strain. Donor strains of B10RifR, ΔgtaIRifR, ΔgtaRRifR and ΔgtaRIRifR were grown in YPS medium under phototrophic conditions and collected at stationary phase. Donor strain cultures were normalized to 450 Klett units by addition of YPS medium, and 200 μl of 0.2 μm filtered culture liquid was mixed with 100 μl of the indicator strain cells and 400 μl of G-buffer [10 mM Tris-Cl (pH 7.8), 1 mM MgCl2, 1 mM CaCl2, 1 mM NaCl, 500 μg ml−1 BSA] (Solioz et al., 1975). The entire mixture was incubated for 90 min at 30–35°C with gentle agitation and then 900 μl of RCV medium was added and incubation under the same conditions was continued for 3–4 h. The cells were spread on RCV rifampicin plates and incubated aerobically at 30°C for 2–3 days. Resulting rifampicin resistant colonies were counted (typically over 30 per plate) and the average was corrected by subtracting the number of spontaneous rif resistant colonies (no addition of RcGTA) (generally between 0 and 3). Because of variability in total numbers of transductants between individual experiments, RcGTA transductions efficiencies are expressed as % wild type (B10), with each experimental strain being compared with the wild-type control within the same experiment. Statistical significance of results was evaluated by a two-tailed t-test assuming unequal variance. A P-value of 0.05 (95% confidence interval) was set as a cut-off for significance.
GtaR-6xHis protein purification
Protein was purified following the QIA expressionist kit (Qiagen) suggested protocol to yield native protein with little modification. Cultures of BL21(DE3)(pET28R) were grown in LB medium supplemented with 50 μg ml−1 kanamycin sulphate at 37°C with shaking at 250 r.p.m. to an OD660 = 0.55, induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), transferred to 30°C with shaking at 250 r.p.m. until the OD660 = 1.0, and harvested by centrifugation. Cell pellets were resuspended in 4 ml of lysis buffer [20 mM HEPES (pH 8.0), 150 mM NaCl, 5 mM imidazole, 10% glycerol, 1× Roche Complete Mini Protease Inhibitor (according to manufacturer’s protocol)], and treated with 5 mg of lysozyme and 1 mg DNaase I for 1 h, on ice. The cells were then lysed by French press and cellular debris separated by centrifugation at 12 100 g for 25 min, followed by centrifugation of the resultant supernatant fluid at 541 000 g for 15 min at 4°C. The protein in the supernatant fluid was purified by affinity chromatography over a column packed with Ni2+-nitrilotriacetic acid agarose resin (Qiagen) and eluted with elution buffer 1 [20 mM HEPES (pH 8.0), 50 mM NaCl, 200 mM imidazole, 10% glycerol, 1× Roche Complete Mini Protease Inhibitor]. The protein was then stored in elution buffer 1 at −80°C.
EMSA
The DNA fragments used for EMSA experiments were generated by PCR using R. capsulatus B10 chromosomal DNA as the template, and the primer sets LF9 and LHR2.2, LF7 and LHR2.2, LF5 and LHR2.2, LF3 and LHR2.2, LF1 and LHR2.2, and LHR3 and LHRrf, LHR3 and LHR2.2, and KOR1F and 3RPL (Table S2). They were named DR9, DR7, DR5, DR3, DR1, DRR, P23 and R23 respectively. The competitor DNA fragment was generated by PCR using pUC19 as template and the primers −21M13F and M200R (Table S2).
The GtaR-6xHis protein was diluted in dilution buffer [20 mM HEPES (pH 8.0), 50 mM NaCl, 10% glycerol] before the binding reaction to equalize volumes added to each reaction. Binding of GtaR-6xHis protein (concentrations from 0 to 1.44 μM) to 100 nM DNA fragments was done in 5 μl reaction volumes containing 10 mM HEPES (pH 8.0) at 37°C for 30 min. Glycerol was added to each reaction to yield a final concentration of 10%. The reactions were separated on 4–5% native polyacrylamide gels with the Mini-PROTEAN II systems (Bio-Rad) according to the manufacturer’s recommendation in 1× TBE [446 mM Tris-Base, 445 mM boric acid, 10 mM EDTA (pH 8.0)] for 2–4 h at 60 V (constant voltage) at room temperature. The gels were then stained with ethidium bromide and photographed with UV illumination.
For EMSAs testing for altered DNA binding by GtaR-6xHis in the presence of acyl-HSLs, 2 μM C12- or C16-HSL was present in the E. coli culture during recombinant gene expression, all stages of GtaR-6xHis protein purification, and EMSA.
DNase I footprinting assay
The primers LHR2.2 and PRcheck were 5′ end-labelled using [γ-32P]-ATP (7000 Ci mmol−1; Perkin Elmer) and T4 polynucleotide kinase (Invitrogen). The excess-[γ-32P]-ATP was removed using the Nucleotide Removal Kit (Qiagen), and the primers concentrated by ethanol precipitation. The DNA fragments (labelled on the coding or non-coding strand) used for the footprinting assays were generated by performing PCR, using previously generated amplicon as template with either labelled LHR2.2 or PRcheck, and unlabeled PRcheck or LHR2.2 (respectively) as primers. These 245 bp labelled amplicons were isolated on a 5% polyacrylamide gel, extracted by electroelution, and concentrated by ethanol precipitation. Binding of 0 to 1.44 μM GtaR-6xHis protein to 200 000 CPM of the DNA fragments was done in a 25 μl reaction volume containing 10 mM HEPES (pH 8.0) at 37°C for 30 min. DNase I (6 × 10−4 U; Invitrogen) was added to each reaction followed by incubation at 37°C for 15 s. The DNase I reaction was stopped by adding 75 μl of a 27 mM solution of EDTA, protein was removed from the DNA by phenol extraction, and the DNA concentrated by ethanol precipitation. The DNA was dissolved in formamide buffer [90% formamide, 0.5× TBE, 0.05% (w/v) bromophenol blue, 0.05% (w/v) xylene cyanol], and separated on a 6% polyacrylamide sequencing gel containing 7 M urea (Sambrook et al., 1989) at 25 W (constant power) for 2.5 h. The gels were dried and exposed to X-ray film.
Supplementary Material
Acknowledgments
This research was funded by a grant to J.T.B. from the Canadian Institutes of Health Research. We thank J. P. Armitage for providing plasmid pIND4.
Footnotes
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams CW, Forrest ME, Cohen SN, Beatty JT. Structural and functional analysis of transcriptional control of the Rhodobacter capsulatus puf operon. J Bacteriol. 1989;171:473–482. doi: 10.1128/jb.171.1.473-482.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL. How bacteria talk to each other: regulation of gene expression by quorum sensing. Curr Opin Microbiol. 1999;2:582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- Bassler BL. Small talk. Cell-to-cell communication in bacteria. Cell. 2002;109:421–424. doi: 10.1016/s0092-8674(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Beatty JT, Gest H. Biosynthetic and bio-energetic functions of citric acid cycle reactions in Rhodopseudomonas capsulata. J Bacteriol. 1981;148:584–593. doi: 10.1128/jb.148.2.584-593.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Dorsselaer AV, Pelczer I, Bassler BL, Hughson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditta G, Schmidhauser T, Yakobson E, Lu P, Liang XW, Finlay DR, et al. Plasmids related to the broad host range vector, pRK290, useful for gene cloning and for monitoring gene expression. Plasmid. 1985;13:149–153. doi: 10.1016/0147-619x(85)90068-x. [DOI] [PubMed] [Google Scholar]
- Duerkop BA, Ulrich RL, Greenberg EP. Octanoyl-homoserine lactone is the cognate signal for Burkholderia mallei BmaR1-BmaI1 quorum sensing. J Bacteriol. 2007;189:5034–5040. doi: 10.1128/JB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Cairns J. The occurrence of heritable Mu excisions in starving cells of Escherichia coli. EMBO J. 1994;13:5240–5244. doi: 10.1002/j.1460-2075.1994.tb06855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JE, Keshavan ND. Messing with bacterial quorum sensing. Microbiol Mol Biol Rev. 2006;70:859–875. doi: 10.1128/MMBR.00002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa H, Oda Y, Phattarasukol S, Armour CD, Castle JC, Raymond CK, et al. Activity of the Rhodopseudomonas palustris p-coumaroyl-homoserine lactone-responsive transcription factor RpaR. J Bacteriol. 2011;193:2598–2607. doi: 10.1128/JB.01479-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng YT, Deng SC, Daykin M, Soo PC, Wei JR, Luh KT, et al. The LuxR family protein SpnR functions as a negative regulator of N-acylhomoserine lactone-dependent quorum sensing in Serratia marcescens. Mol Microbiol. 2002;45:1655–1671. doi: 10.1046/j.1365-2958.2002.03117.x. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Hui A, Hayflick J, Dinkelspiel K, de Boer HA. Mutagenesis of the three bases preceding the start codon of the beta-galactosidase mRNA and its effect on translation in Escherichia coli. EMBO J. 1984;3:623–629. doi: 10.1002/j.1460-2075.1984.tb01858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ind AC, Porter SL, Brown MT, Byles ED, de Beyer JA, Godfrey SA, Armitage JP. Inducible-expression plasmid for Rhodobacter sphaeroides and Paracoccus denitrificans. Appl Environ Microbiol. 2009;75:6613–6615. doi: 10.1128/AEM.01587-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AS, Beatty JT. Genetic analysis of a bacterial genetic exchange element: the gene transfer agent of Rhodobacter capsulatus. Proc Natl Acad Sci USA. 2000;97:859–864. doi: 10.1073/pnas.97.2.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AS, Beatty JT. Importance of widespread gene transfer agent genes in alpha-proteobacteria. Trends Microbiol. 2007;15:54–62. doi: 10.1016/j.tim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Lazdunski AM, Ventre I, Sturgis JN. Regulatory circuits and communication in Gram-negative bacteria. Nat Rev Microbiol. 2004;2:581–592. doi: 10.1038/nrmicro924. [DOI] [PubMed] [Google Scholar]
- Leung MM-L. PhD Thesis. Vancouver: Department of Microbiology & Immunology, University of British Columbia; 2010. CtrA and GtaR: two systems that regulate the Gene Transfer Agent in Rhodobacter capsulatus; p. 168. [Google Scholar]
- Marketon MM, Gonzalez JE. Identification of two quorum-sensing systems in Sinorhizobium meliloti. J Bacteriol. 2002;184:3466–3475. doi: 10.1128/JB.184.13.3466-3475.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs B. Genetic recombination in Rhodopseudomonas capsulata. Proc Natl Acad Sci USA. 1974;71:971–973. doi: 10.1073/pnas.71.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetics : A Laboratory Manual and Handbook for Escherichia Coli and Related Bacteria. Plainview, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Minogue TD, Wehlandvon Trebra M, Bernhard F, von Bodman SB. The autoregulatory role of EsaR, a quorum-sensing regulator in Pantoea stewartii ssp. stewartii: evidence for a repressor function. Mol Microbiol. 2002;44:1625–1635. doi: 10.1046/j.1365-2958.2002.02987.x. [DOI] [PubMed] [Google Scholar]
- Patankar AV, Gonzalez JE. Orphan LuxR regulators of quorum sensing. FEMS Microbiol Rev. 2009;33:739–756. doi: 10.1111/j.1574-6976.2009.00163.x. [DOI] [PubMed] [Google Scholar]
- Peterson G. Determination of total protein. Methods Enzymol. 1983;91:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- Piper KR, Beck von Bodman S, Farrand SK. Conjugation factor of Agrobacterium tumefaciens regulates Ti plasmid transfer by autoinduction. Nature. 1993;362:448–450. doi: 10.1038/362448a0. [DOI] [PubMed] [Google Scholar]
- Puskas A, Greenberg EP, Kaplan S, Schaefer AL. A quorum-sensing system in the free-living photo-synthetic bacterium Rhodobacter sphaeroides. J Bacteriol. 1997;179:7530–7537. doi: 10.1128/jb.179.23.7530-7537.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning : A Laboratory Manual New York. Plainview: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanchez-Contreras M, Bauer WD, Gao M, Robinson JB, Downie JA. Quorum-sensing regulation in rhizobia and its role in symbiotic interactions with legumes. Philos Trans R Soc Lond B Biol Sci. 2007;362:1149–1163. doi: 10.1098/rstb.2007.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savka MA, Le PT, Burr TJ. LasR receptor for detection of long-chain quorum-sensing signals: identification of N-acyl-homoserine lactones encoded by the avsI locus of Agrobacterium vitis. Curr Microbiol. 2011;62:101–110. doi: 10.1007/s00284-010-9679-1. [DOI] [PubMed] [Google Scholar]
- Schaefer AL, Taylor TA, Beatty JT, Greenberg EP. Long-chain acyl-homoserine lactone quorum-sensing regulation of Rhodobacter capsulatus gene transfer agent production. J Bacteriol. 2002;184:6515–6521. doi: 10.1128/JB.184.23.6515-6521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Urbanowski ML, Greenberg EP. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc Natl Acad Sci USA. 2004;101:15833–15839. doi: 10.1073/pnas.0407229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol. 1983;1:784–791. [Google Scholar]
- Solioz M, Yen HC, Marris B. Release and uptake of gene transfer agent by Rhodopseudomonas capsulata. J Bacteriol. 1975;123:651–657. doi: 10.1128/jb.123.2.651-657.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suck D, Lahm A, Oefner C. Structure refined to 2A of a nicked DNA octanucleotide complex with DNase I. Nature. 1988;332:464–468. doi: 10.1038/332464a0. [DOI] [PubMed] [Google Scholar]
- Surette MG, Miller MB, Bassler BL. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for auto-inducer production. Proc Natl Acad Sci USA. 1999;96:1639–1644. doi: 10.1073/pnas.96.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender T, Halliday N, Hardie K, Winzer K. LuxS-independent formation of AI-2 from ribulose-5-phosphate. BMC Microbiol. 2008;8:98. doi: 10.1186/1471-2180-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TA. MSc Thesis. Vancouver: Department of Microbiology & Immunology, University of British Columbia; 2004. Evolution and regulation of the Gene Transfer Agent (GTA) of Rhodobacter capsulatus; p. 66. [Google Scholar]
- Travers AA. DNA conformation and protein binding. Annu Rev Biochem. 1989;58:427–452. doi: 10.1146/annurev.bi.58.070189.002235. [DOI] [PubMed] [Google Scholar]
- Tsai CS, Winans SC. LuxR-type quorum-sensing regulators that are detached from common scents. Mol Microbiol. 2010;77:1072–1082. doi: 10.1111/j.1365-2958.2010.07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner-Dobler I, Thiel V, Eberl L, Allgaier M, Bodor A, Meyer S, et al. Discovery of complex mixtures of novel long-chain quorum sensing signals in free-living and host-associated marine alphaproteobacteria. Chembiochem. 2005;6:2195–2206. doi: 10.1002/cbic.200500189. [DOI] [PubMed] [Google Scholar]
- Wall JD, Weaver PF, Gest H. Gene transfer agents, bacteriophages, and bacteriocins of Rhodopseudomonas capsulata. Arch Microbiol. 1975;105:217–224. doi: 10.1007/BF00447140. [DOI] [PubMed] [Google Scholar]
- Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- Welch M, Todd DE, Whitehead NA, McGowan SJ, Bycroft BW, Salmond GP. N-acyl homoserine lactone binding to the CarR receptor determines quorum-sensing specificity in Erwinia. EMBO J. 2000;19:631–641. doi: 10.1093/emboj/19.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley M, Greenberg EP. Promoter specificity elements in Pseudomonas aeruginosa quorum-sensing-controlled genes. J Bacteriol. 2001;183:5529–5534. doi: 10.1128/JB.183.19.5529-5534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2001;27Chapter 2(Unit 2):2.4.1–2.4.5. 4. doi: 10.1002/0471142727.mb0204s56. [DOI] [PubMed] [Google Scholar]
- Winson MK, Camara M, Latifi A, Foglino M, Chhabra SR, Daykin M, et al. Multiple N-acyl-L-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 1995;92:9427–9431. doi: 10.1073/pnas.92.20.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winson MK, Swift S, Fish L, Throup JP, Jorgensen F, Chhabra SR, et al. Construction and analysis of luxCDABE-based plasmid sensors for investigating N-acyl homoserine lactone-mediated quorum sensing. FEMS Microbiol Lett. 1998;163:185–192. doi: 10.1111/j.1574-6968.1998.tb13044.x. [DOI] [PubMed] [Google Scholar]
- Wisniewski-Dye F, Jones J, Chhabra SR, Downie JA. raiIR genes are part of a quorum-sensing network controlled by cinI and cinR in Rhizobium leguminosarum. J Bacteriol. 2002;184:1597–1606. doi: 10.1128/JB.184.6.1597-1606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T, Zhao L, Sun H, Zhou X, Sun B. LsrR-binding site recognition and regulatory characteristics in Escherichia coli AI-2 quorum sensing. Cell Res. 2009;19:1258–1268. doi: 10.1038/cr.2009.91. [DOI] [PubMed] [Google Scholar]
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci USA. 2002;99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.