

Abstract
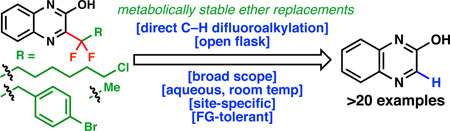
This work delineates a method for the modular synthesis of reagents that are capable of direct incorporation of difluoroalkyl groups onto heterocycles. The scope and generality of this method is exemplified with the difluoroethyl group (along with the introduction of a new reagent for difluoroethylation, DFES-Na) and a proof of principle is shown for a general synthesis of fluorinated heteroarylether bioisosteres.
Keywords: heterocycle, fluoroalkylation, difluoroethyl, C–H functionalization, bioisostere
Medicinal chemists have a love-hate relationship with aryl ethers, which are generally enlisted to explore SAR and to modulate the properties of bioactive scaffolds. Yet, aryl ethers (such as the methoxy group) represent a metabolic liability and thus much effort is expended to replace such moieties with metabolically stable derivatives or bioisosteres.[1] The goal of this work was to develop a rapid and direct means for medicinal chemists to introduce stable, fluorinated mimics of alkoxy ethers into their scaffolds. A tried-and true method to achieve this is a point mutation of the oxygen atom to a difluorinated carbon atom. As shown in Figure 1A for the case of a methoxy ether, the difluoroethyl group mimics the steric and electronic features of a methoxy group albeit with a conformational preference of the former that places the methyl group out of the plane of the aromatic ring.[2] Numerous applications of this difluoroethyl-methoxy replacement can be found in the literature. For example, two recent reports (1[3] and 2[4]) have demonstrated the remarkable advantage that such a modification can have on both potency and metabolic stability. Unfortunately, the only known route to such compounds uses fluorinating agents such as DAST (diethylaminosulfur trifluoride) or its derivatives to convert a ketone into the corresponding difluoroalkyl group thus requiring a significant investment of resources since direct methods for latestage appendage are not known. Even programmed approaches involving cross coupling are unprecedented, thus rendering routes to these types of compounds a serious unmet need that could have an immediate impact in the synthesis of medicines. In this Communication, the invention of reagents and methods for the direct installation of fluorinated heteroarylether bioisosteric replacements is reported.
Figure 1.

A. The difluoroethyl group mimics a methoxy group and improves the bioactivity and metabolic stability of medicinally important molecules, B. Invention of DFES-Na and a modular approach to difluoroalkylsulfinate preparation. %metab = % of parent compound metabolized after 30 min of incubation with rat liver microsomes.3
Sulfinate salts have recently emerged as a promising method for the direct incorporation of alkyl groups onto heteroaromatic systems through a formal C–H functionalization via a radical addition process.[5, 6] Thus, a difluoroethylsulfinate salt was pursued as shown in Figure 1B. Analogous to the synthesis of the difluoromethylsulfinate salt (which is prepared from the corresponding sulfonyl chloride),[6b] 5 was targeted. After extensive experimentation and evaluation of numerous routes, a multi-step path to 5 via 3 and 4 was devised only to find that this material was a low boiling liquid that, in our hands, was problematic to manipulate. Furthermore, this approach is not modular and would require a lengthy synthesis for each difluoroalkyl group that one wished to transfer. Inspiration was thus drawn from two independent reports, one from the Prakash-Olah group[7] and one from the Hu group.[8] The former group elegantly demonstrated that Hu’s reagent (7) could be efficiently alkylated and cleaved to liberate sodium sulfinate salts (which were immediately oxidized to sulfonates). This route was adapted for the scalable preparation[9] of Nadifluoroethylsulfinate (9, DFES-Na, a new chemical entity), a stable white solid that has been commercialized by Sigma-Aldrich (catalog # L511404).
With a reliable synthesis of the requisite sulfinate salt (9) in hand, optimization of the C–H alkylation was conducted as shown in Table 1A. Using caffeine (10), in a 2.5:1 mixture of CH2Cl2:H2O with TBHP as oxidant, and in the absence of any additives, only trace amounts of the desired product 11 were observed (entry 1). Although Brønsted acids such as TFA improved the conversion (entry 2), zinc-based Lewis acids (entries 3–13) dramatically enhanced the reaction. Ultimately, ZnCl2 was identified as the optimal additive along with TsOH•H2O (entry 13).
Table 1.
A. Optimization of the reaction. B. Scope of C–H difluoroethylation of heteroarene substrates. C. Comparison to current state-of-the-art.
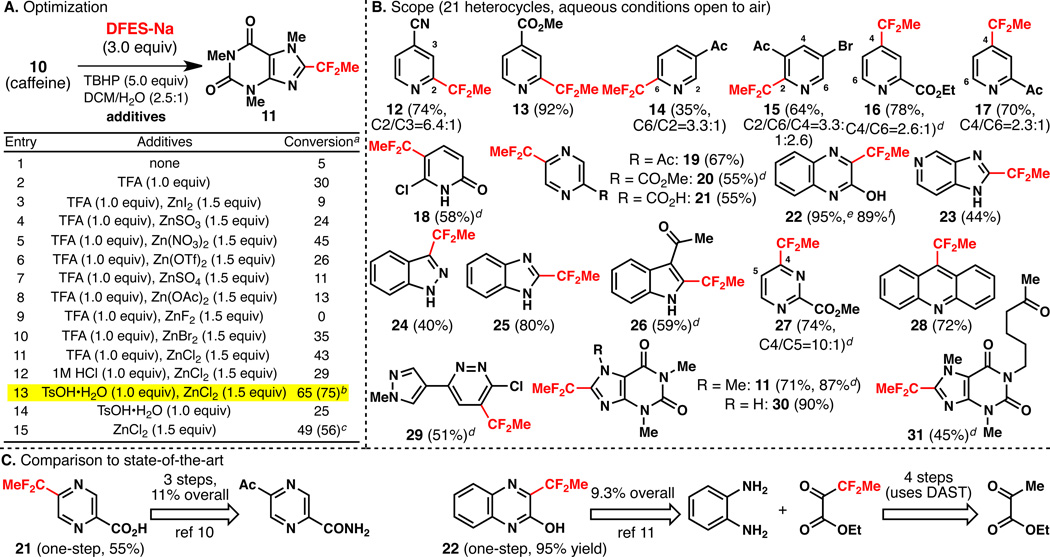 |
Using these optimized conditions, a wide range of heterocycles were examined as depicted in Table 1B. The scope, site-selectivity, and functional group tolerance are notable aspects of the method. Thus, pyridines (12–17), pyridones (18), pyrazines (19–21), quinoxalines (22), pyridylbenzimidazoles (23), indazoles (24), benzimidazoles (25), indoles (26), pyrimidines (27), benzoquinolines (28), pyridazines (29), and xanthines (11, 30, 31) are all amenable to fluoroalkylation. Out of the 21 examples shown, 18 could potentially result in mixtures of regioisomers. Remarkably, 12 of these react with DFES-Na to deliver a single regioisomer, two react with good regioselectivity (6.4:1 or higher), and four achieve modest but synthetically useful selectivity (2.3:1 or higher). Particularly notable examples are 18, 23, and 29, all of which have 3 or more C–H bonds that could conceivably be substituted. The functional group tolerance of this reaction, which is conducted with water as a co-solvent and open to the air if desired, is also notable. Ketones, esters, nitriles, chlorides, bromides, free N–H heterocycles, and even free carboxylic acids are tolerated as well as free amines and alcohols (vide infra). These reactions are also scalable, with heterocycle 22 being difluoroethylated on a gram-scale in 89% isolated yield.
Cursory analysis of the patent literature indicates that the current method can dramatically simplify the way such fluoroalkylated compounds are prepared. As shown in Table 1C, the known route to pyrazine 21[10] involves a three-step procedure proceeding in 11% overall yield, all of which involve laborious functional group manipulations (and DAST). The known route to 22[11] requires a 5-step sequence proceeding in 9.3% overall yield (using DAST and KCN). In contrast, the one-step routes to 21 and 22 proceed in 55 and 95% yields respectively and represent ideal[12] syntheses.
Difluoroethyl radicals generated from DFES-Na are also capable of adding to Michael acceptors and thiols as shown in Figure 2A. In an extreme example (Figure 2B) of site specificity and functional group tolerance, an advanced intermediate from a program at Bristol-Myers Squibb (36) can be directly difluoroethylated to deliver 37 without protecting the reactive and oxidizable benzylic alcohol and amine.
Figure 2.

A. Direct difluoroethylation of heteroaromatic Michael acceptors and thiols. B. Direct difluoroethylation of an advanced intermediate from the Bristol-Myers Squibb compound library. C. A general synthesis of fluorinated heteroarylether bioisosteric reagents and a proof of concept on 2-quinoxalinol. Reagents and conditions: (a) reactions performed on 0.2 mmol scale, 40 (2.0 equiv), TBHP (5.0 equiv), TsOH•H2O (1.0 equiv), ZnCl2 (1.0 equiv), 0 to 23 °C, reaction completed in 24 h; (b) reactions performed on 0.1 mmol scale, 41 (1.5 equiv), TBHP (5.0 equiv), TsOH•H2O (1.0 equiv), ZnCl2 (1.5 equiv), reaction completed in 4 h at 0 °C after second addition of 41 (1.5 equiv), TBHP (5.0 equiv).
Perhaps the most significant finding of this work is that the current strategy for introducing a difluoroethyl group (methoxy bioisostere) can also be enlisted for the synthesis of virtually any difluoroalkyl group (alkoxy bioisostere) of interest. As shown in Figure 2C, the same approach used to create 9 (Figure 1B) could be used for the synthesis of sulfinate salts 40 and 41. Thus, subjecting Hu’s sulfone (7) to the Olah-Prakash alkylation conditions delivered 38 and 39. Subsequent cleavage furnished 40 and 41, which were used to accomplish the direct difluoroalkylation of 2-quinoxalinol to deliver the alkyl ether and benzyl ether bioisosteres 42 and 43 in 83 and 56% yields, respectively. There are currently no other known methods for making compounds of this sort.
This work establishes a simple, reliable, and scalable platform for the direct synthesis of fluorinated bioisosteres of alkoxy ethers. Such moieties are highly sought after in the context of drug discovery and agroscience but the chemical synthesis of such compounds severely hampers their incorporation. Recognition that Hu’s reagent 7 can be employed in concert with the Olah-Prakash protocol led to a programmable synthesis of sulfinate reagents such as 9 (DFES-Na), 40, and 41. Aside from direct radical functionalization, sulfinate reagents have already begun to find use in other types of useful transformations.[13] The addition of zinc chloride was critical for the success of the radical alkylation method that, in the case of 9, demonstrates admirable conversion, site selectivity, functional group tolerance, and extreme operational simplicity. While caveats to this work involve the use of excess reagent, the use of inexpensive but stoichiometric additives, as well as a reliance on innate selectivity, the features of this work bode well for the widespread adoption of this chemistry in a medicinal chemistry setting.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the NIH/NIGMS (GM-073949), Eisai, the Binational Science Foundation, and the Frost Foundation (to D.S). We are grateful to P. Siva (Bristol-Myers Squibb) for the calculations shown in Figure 1A, to J. Kadow (Brisol-Myers Squibb) for a sample of 36, and to T. Ryba and C. Thomas (Sigma-Aldrich) for a donation of chemicals. The Uehara Memorial Foundation and SIOC provided funding for postdoctoral fellowships to Y. F. and Q. Z., respectively.
References
- 1.Meanwell NA. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.Calculations performed using Suite 2012: Maestro, version 9.3, Schrödinger, LLC, New York, NY, 2012 and Q-Chem (used for torsional scan). See supporting information for full details of calculations
- 3.Anderson MO, Zhang J, Liu Y, Yao C, Phuan P-W, Verkman AS. J. Med. Chem. 2012;55:5942–5950. doi: 10.1021/jm300491y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jiménez-Díaz MB, Gamo F-J, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. J. Med. Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langlois BR, Laurent E, Roidot N. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]
- 6.a) Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc. Natl Acad. Sci. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fujiwara Y, Dixon JA, Rodriguez RA, Baxter RD, Dixon DD, Collins MR, Blackmond DG, Baran PS. J. Am. Chem. Soc. 2012;134:1494–1497. doi: 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fujiwara Y, Dixon JA, O'Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herle B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95–99. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prakash GKS, Ni C, Wang F, Hu J, Olah GA. Angew. Chem. Int. Ed. 2011;50:2559–2563. doi: 10.1002/anie.201007594. . For an elegant synthesis of 1,1-difluoroethyl groups by nucleophilic addition to aldehydes, see Mogi R, Morisaki K, Hu J, Prakash GKS, Olah GA. J. Fluorine Chem. 2007;128:1098–1103.
- 8.Zhao Y, Huang W, Zhu L, Hu J. Org. Lett. 2010;12:1444–1447. doi: 10.1021/ol100090r. [DOI] [PubMed] [Google Scholar]
- 9.The synthesis starts from two commercially available starting materials, 2-mercaptopyridine (Sigma-Aldrich catalog number M5852, US$2.67/g) and diethyl (bromodifluoromethyl)phosphonate (Matrix Scientific, catalog number 007430, US$3.29/g).
- 10.Ellard JM, Farthing CN, Hall A. WO 2011/09898
- 11.Chen H, Mcdaniel KF, Green BE, Shanley JP, Kruger AW, Gandarilla J, Welch DS, Cink RD, Gai Y, Wang G, Yat S. WO 2011/156337
- 12.Gaich T, Baran PS. J. Org. Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- 13.a) Ye Y, Künzi SA, Sanford MS. Org. Lett. 2012;14:4979–4981. doi: 10.1021/ol3022726. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Beller M, Neumann H, Li Y, Wu L. Chem. Commun. 2013 doi: 10.1039/c2cc36554e. [DOI] [PubMed] [Google Scholar]; c) Li Z, Cui Z, Liu Z-Q. Org. Lett. 2013;15:406–409. doi: 10.1021/ol3034059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.