


Abstract
We report the genomic occupancy profiles of the key hematopoietic transcription factor GATA-1 in pro-erythroblasts and mature erythroid cells fractionated from day E12.5 mouse fetal liver cells. Integration of GATA-1 occupancy profiles with available genome-wide transcription factor and epigenetic profiles assayed in fetal liver cells enabled as to evaluate GATA-1 involvement in modulating local chromatin structure of target genes during erythroid differentiation. Our results suggest that GATA-1 associates preferentially with changes of specific epigenetic modifications, such as H4K16, H3K27 acetylation and H3K4 di-methylation. Furthermore, we used random forest (RF) non-linear regression to predict changes in the expression levels of GATA-1 target genes based on the genomic features available for pro-erythroblasts and mature fetal liver-derived erythroid cells. Remarkably, our prediction model explained a high proportion of 62% of variation in gene expression. Hierarchical clustering of the proximity values calculated by the RF model produced a clear separation of upregulated versus downregulated genes and a further separation of downregulated genes in two distinct groups. Thus, our study of GATA-1 genome-wide occupancy profiles in mouse primary erythroid cells and their integration with global epigenetic marks reveals three clusters of GATA-1 gene targets that are associated with specific epigenetic signatures and functional characteristics.
INTRODUCTION
The critical functions of transcription factors in establishing lineage-specific transcription programs have been firmly established through genetic studies [reviewed in (1,2)]. Furthermore, the recent development and application of high-throughput methodologies for the genome-wide mapping of transcription factor binding patterns by chromatin immunoprecipitation (ChIP) coupled to massive parallel sequencing (ChIP-seq) [reviewed in (3)] has led to an unprecedented view of the gene target networks that transcription factors regulate during differentiation.
Erythropoiesis is a dynamic multistep process involving the terminal differentiation of erythroid progenitors to enucleated red blood cells [reviewed in (4)]. Erythroid cell differentiation is a well-characterized process; thus, it makes for an ideal model system to study the molecular events driving terminal cell differentiation. The various differentiation stages of committed erythroid cells are distinguishable by the differential expression of specific cell surface markers (5) and unique morphologies (6). For example, Ter119 is one of the key cell surface erythroid-specific antigens expressed primarily by terminally differentiating erythroblasts and is widely used to separate mature erythroid cells from proerythroblasts (5,7). Lineage commitment from multipotent progenitors to committed erythroid precursors and terminally differentiated erythrocytes involves the activation of the erythroid transcription program and the repression of alternative hematopoietic lineage programs [reviewed in (8)].
GATA-1 is a critical transcription factor that is involved in both of these regulatory functions in erythroid differentiation (9,10) and is thus essential for the terminal differentiation of erythroid cells and of other hematopoietic lineages [reviewed in (11–13)]. Most, if not all, known erythroid genes include GATA-binding sites near their promoters, including those for Gata1 itself, Gata2, Klf1 and Scl/Tal1 [reviewed in (4)]. Several lines of evidence have suggested that GATA-1 binding promotes changes in the epigenetic landscape of target genes (14–16), and GATA-1 has been reported to interact with several hematopoietic transcription factors, as well as chromatin remodeling and modification factors, such as the NuRD complex, histone acetyl transferases and polycomb-group members (4,11,16). Significantly, enforced ectopic GATA-1 expression in highly purified murine progenitor cells (myeloid or lymphoid) reprograms their differentiation towards the erythroid and megakaryocytic lineages that GATA-1 normally regulates (17–19). Thus, GATA-1 is capable of imposing an erythroid transcription program in myeloid-derived hematopoietic lineages, establishing it as a ‘master’ erythroid transcription factor. Moreover, epigenetic changes and changes in gene expression profiles occur as the net result of altered GATA-1 genome wide occupancy, to allow for the completion of the erythroid maturation program [reviewed in (4,20–22)].
Several reports have previously described the GATA-1 genome-wide occupancy by ChIP-seq in mouse (16,23,24) and human (25) erythroid cell lines, or in in vitro differentiated mouse embryonic stem (ES) cells (26), in a megakaryocytic cell line (27,28) and in primary human megakaryocytes (29). These studies agree in that (i) GATA-1 binds mostly to sequences that are distal to promoters; (ii) GATA-1 targets include genes that are both activated and repressed with differentiation; (iii) GATA-1 gene targets are enriched for histone H3K4 methylation marks; and (iv) there is a strong positive correlation between activated GATA-1 target genes and binding of the SCL/TAL-1 complex (30). In addition, the integration of GATA-1 ChIP-seq data with those for SCL/TAL-1 and KLF1 led to the identification of a few hundred gene targets that are common to all three factors (26,30–32), proposed to represent a core erythroid network enriched for genes involved in erythroid differentiation (26).
The genome-wide GATA-1–binding patterns in mouse primary fetal liver-derived erythroid cells have only been reported in the context of providing limited validation for GATA-1 ChIP-seq data in erythroid cell lines (23,33), and their complete analysis, to our knowledge, has never been presented. In this report, we provide for the first time the ChIP-seq analysis of in vivo GATA-1 occupancy profiles in fetal liver-derived Ter119− proerythroblasts and Ter119+ mature erythroid cells. Furthermore, we integrate these data with publicly available genome-wide occupancy profiles in fetal liver erythroid cells for other critical erythroid transcription factors and for histone tail post-translational modifications, leading to the description of three classes of GATA-1 gene targets with distinguishable epigenetic profiles and functional associations.
MATERIALS AND METHODS
Cell culture
Fetal liver cells were dissected from day E12.5 C57/BL6 mouse embryos and expanded for 3 days in serum-free medium, as before (34). Fractionation of Ter119− erythroid progenitors and Ter119+ differentiated erythrocytes was carried out as previously described (35).
GATA-1 ChIP sequencing and data analysis
Formaldehyde–cross-linked chromatin from 1 × 107 Ter119+ or Ter119− cells was prepared as previously described (35). Pilot experiments showed that rabbit polyclonal antibody Ab11852 (Abcam) gave the highest enrichment for GATA-1 binding to the −3.5-kb HS1 of the GATA-1 gene locus (data not shown). Anti-GATA1 ChIPed DNAs from Ter119− and Ter119+ chromatin and from a ‘no antibody’ control (input DNA) were processed for deep sequencing using the Illumina Genome Analyzer II platform according to Illumina protocols (www.illumina.com). Deep sequencing was carried out in duplicate for each ChIP sample and once for input DNA controls. All 51-nt sequence reads thus produced were mapped to the NCBI37/mm9 Mouse Genome Assembly using the Eland software (Illumina). Sequence reads with multiple genome alignments and/or more than two nucleotide mismatches were excluded. Peak calling was performed using the QuEST algorithm (36). Each sample was analyzed versus the control data set using a strict fold change (sample/control >50), a false discovery rate threshold of 0.001 and a peak score threshold of 70. Sequencing data have been deposited in EBI’s European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena/), accession number: E-MTAB-1504.
GATA-1 target gene identification
Gene location data used in the mapping analyses were extracted from BioMart (37) using the Ensembl transcript database (build 59). Gene mapping analyses were carried out by custom in-house Perl and R scripts. RNA-sequencing gene expression data for Ter119− and Ter119+ erythroid cells were downloaded from the GEO database (38) using accession number GSE32110. Total gene score (TGS) was calculated for each expressed gene separately and corresponds to the sum of the enrichment values of GATA-1 peaks that overlap the gene’s transcription start site (TSS) within a given distance window. Target genes with TGS scores <100 were discarded from subsequent analyses.
Random forest regression analysis
Random forest (RF) non-linear regression analysis applied to model changes in gene expression and histone modification levels is described in greater detail in Supplementary Methods.
Mouse fetal liver genomic occupancy database
The genome-wide transcription factor (TF) occupancy and histone modification profiles presently available for Ter119− and Ter119+ fetal liver cells were downloaded from the GEO database using accession numbers GSE27893 (H3K4me2, H3K4me3, H3K9Ac, H3K27me3, H3K36me3, H3K79me2, H4K16Ac and RNApolII), GSE27918 (H3K4me1 and H3K27Ac), GSE18720 (SCL/TAL1 Ter119−), GSE30142 (SCL/TAL1 Ter119+) and GSE21950 (PU.1). For the GSE27893 data sets, genomic occupancy profiles (wig files) were provided in 25-nt genomic bins, whereas MACS (39) with default parameters was used to create density profiles for the GSE27918, GSE18720, GSE30142 and GSE21950 data sets. TGS score for each genomic feature was calculated as the sum of the background corrected number of reads present in the density profiles that mapped within a 10-kb window upstream or downstream a gene’s TSS. Subsequently, TGS scores were mean normalized in all data sets (additional file 4).
RESULTS AND DISCUSSION
In vivo GATA-1 genomic occupancy profiling in fetal liver-derived erythroid cells
To identify genome-wide differential GATA-1–binding patterns during erythroid differentiation in vivo, we performed GATA-1 ChIP on Ter119− proerythroblasts and Ter119+ mature erythroid cells fractionated from day E12.5 mouse fetal liver cells, followed by high throughput massive parallel sequencing. ChIPed DNA from Ter119− and Ter119+ cells was sequenced in duplicate to generate 18.2 and 15.3 million uniquely mapped sequence reads, respectively (Figure 1A). Using the QuEST peak-calling algorithm (36), we assembled the unique non-redundant sequence reads for each replicate into peaks that identify potential GATA-1 bound regions across the genome. For both samples, we took the union of the peaks of the two replicates, resulting in 9795 and 14 239 peaks for the Ter119− and Ter119+ samples, respectively (Figure 1A). Visualization in both the Ter119− and Ter119+ data sets of peaks in known GATA-1 target gene loci, such as β-globin, Gata1, Gata2, Klf1 or Scl/Tal1 gene loci (10,30,40,41), or in the Zbtb7 locus that was recently identified as a GATA-1 gene target (16), provided early validation for our sequencing data (Figure 1B).
Figure 1.

Determination of GATA-1 chromatin occupancy in Ter119− and Ter119+ cells by ChIP sequencing. (A) Description of the pipeline followed for the analysis of raw deep sequencing results of GATA-1 ChIP-seq, leading to the identification of occupancy sites (peaks) and potential target genes. (B) Bona fide GATA-1 binding sites as determined by GATA-1 ChIP-seq in Ter119− and Ter119+ cells. Scale refers to read counts normalized by the peak calling algorithm (QuEST).
Plotting the distances of all identified peaks from annotated gene TSSs for both Ter119− and Ter119+ data sets showed that an appreciable fraction of GATA-1 peaks cluster proximally (within 5 kb) to gene TSSs (Supplementary Figure S1A). For both Ter119− and Ter119+ samples, ∼64 and 36% of peaks fall within intergenic and intragenic regions, respectively, with 59% of the intragenic peaks falling within introns and 35% within exons (Supplementary Figure S1B). By and large, our data on GATA-1 peak distribution do not differ significantly between Ter119− and Ter119+ cells.
Peak assignment to potential GATA-1 gene targets
We next sought to assign specific genes to the GATA-1 peaks identified in the Ter119− and Ter119+ data sets. This is usually done by nearest gene assignment or by assigning peaks that fall within a given window around a gene’s TSS and/or transcription end site (TES) (42). As this has led to differences in target gene assignments in different GATA-1 ChIP-seq studies (43), we used a systematic approach to identify the gene assignment parameters that would provide the most significant association between GATA-1 occupancy and changes in the expression profile of the potential target gene. In quantifying the association between GATA-1 occupancy and the expression profiles of the target genes identified by each assignment method, we constructed a series of RF-based prediction models (44), using GATA-1 occupancy features as predictors of the target gene’s expression profile (Supplementary Methods).
We combined the gene assignment parameters with the RNA-sequencing expression data obtained in Ter119− and Ter119+ erythroid cells by Wong et al. (45). We scored for GATA-1 peaks found within windows of increasing size (i.e. ±1, ±2, ±5, ±10 and ±20 kb) around a gene’s TSS, or within a region extending from −20 kb from a gene’s TSS to +10 kb from a gene’s TES, or by assigning peaks to the nearest TSS (Figure 2A). The number of potential GATA-1 target genes thus identified varied from 919 to 4551 expressed genes in Ter119− cells and from 1008 to 5080 in Ter119+ cells, depending on the assignment parameters (Figure 2A and Supplementary Table S1).
Figure 2.

Evaluation of different GATA-1 target gene assignment parameters. (A) Schematic representation of the pipeline used to individually evaluate the association of different gene assignment methods with differential gene expression. The number of genes reported refers to the union of GATA-1 target genes identified in Ter119− and Ter119+ cells. (B) Plot of the R2 values (% of variance explained) of RF regression models trained on the data sets produced by the different gene assignment methods. The number of trees grown is reported in the x-axis.
Each potential GATA-1 target gene was next ascribed a number of features based on the GATA-1 occupancy profiles in Ter119− and Ter119+ cells. These features included the TGS, defined as the sum of the GATA-1 peak scores assigned to it, the difference in TGS score between Ter119− and Ter119+ cells, the highest GATA-1 peak score assigned to the gene, the minimum and maximum distances of assigned GATA-1 peaks from the gene’s TSS and the total number of assigned GATA-1 peaks, thus resulting 11 features per gene in Ter119− and Ter119+ cells (Figure 2A). Based on the R2 values calculated for each model (Supplementary Methods), the most accurate ensemble of GATA-1 target genes in erythroid cells was obtained by assigning genes harboring a GATA-1 peak within a ±10-kb window of their TSS (R2 = 0.14, 3651 genes; Figure 2B and Supplementary Data set S1).
Analysis of GATA-1 target genes
Based on the ±10-kb mapping, 2590 and 2826 potential GATA-1 target genes were identified in the Ter119− and Ter119+ data sets, respectively. The union of the two data sets yielded 3651 potential GATA-1 target genes, of which 1765 genes were common to both Ter119− and Ter119+ data sets, thus giving an intersection of 48.3% (Figure 3A and B). By contrast, 825 (22.6%) and 1061 (29.1%) genes were unique to the Ter119− or Ter119+ cells, respectively (Figure 3C). These data reveal a considerable conservation of GATA-1 target genes throughout erythroid differentiation.
Figure 3.

Location analysis and score distribution of GATA-1 occupancy sites in Ter119− and Ter119+ cells. (A) Venn diagram showing the overlapping and unique GATA-1 target genes identified in Ter119− and Ter119+ erythroid cells. (B) Distribution of GATA-1 TGS and minimum distance from the target gene TSS. Scatterplot of GATA-1 potential target genes identified in both cell populations (each gene is plotted once selecting for the condition with the highest TGS). Horizontal lines define score thresholds for the three classes. Most highly enriched (Class I) target genes are found in the intersection of the two data sets and comprise most of the bibliographically described GATA-1 target genes. (C) GATA-1 potential target genes identified uniquely in Ter119− or Ter119+ cells are shown in the top and bottom scatterplots, respectively. Despite the fact that target genes unique in either Ter119− or Ter119+ cells are poor in Class I genes, they still show a prominent clustering of GATA-1 occupancy sites near the identified target gene TSS. (D) Distribution of GATA-1 potential target genes in classes. Pie chart illustrating the distribution of common genes in three TGS classes and also the presence of a large quota of genes showing dynamic changes in GATA-1 binding throughout erythroid differentiation (ascending and descending).
To further facilitate the differential analysis of potential GATA-1 gene targets in Ter119− and Ter119+ cells, we arbitrarily divided all genes into three categories on the basis of their TGS (summarized in Supplementary Table S2). Inspection of the three classes of genes led to a number of observations. First, it is clear that Class I (TGS > 500) includes most of the well-established GATA-1 erythroid-specific target genes [i.e. the Gata1 locus itself, Gata2, the β-globin locus (especially the locus control region), EpoR, Nfe2, Slc4a1, Gypa, Tal1, Lrf, Klf1, Nrf2, Runx1 and Alas2] (Figure 3B). Class I target genes are also markedly enriched in erythroid-related ontologies (Supplementary Figure S2). Thus, Class I genes, corresponding to ∼15% of all identified GATA-1 target genes (Figure 3B and Supplementary Table S2), most likely represent the erythroid transcription program. A second observation arising from this analysis is that Class II (TGS of 250–500) and III (TGS of 100–250) genes include most of the GATA-1 targets that are unique to the Ter119− or Ter119+ cells (806/825 and 1055/1061 genes, respectively; Figure 3 and Supplementary Table S2). Thirdly, mobility of an appreciable fraction of GATA-1 targets within the three classes is seen as erythroid differentiation proceeds from Ter119− to Ter119+ cells (Supplementary Table S2). More specifically, of the 1765 genes that are bound by GATA-1 in both Ter119− and Ter119+ cells, 480 genes (27%) show reduced GATA-1 binding in mature Ter119+ cells compared with Ter119− cells, whereas, 353 genes (20%) transitioned to a higher class as a result of higher enrichment for GATA-1 binding with erythroid differentiation (Figure 3D and Supplementary Table S2). Gene Ontology (GO) analysis using DAVID (46) of genes transitioning to lower categories with erythroid differentiation, revealed a relative enrichment for genes involved in immune and early hematopoietic pathways, myeloid differentiation and immune response activation (Supplementary Figure S3A), for example, Kit, Hhex and Zfp36 genes (Supplementary Figure S4). Genes transitioning to a higher category showed a relative enrichment in oxygen response pathways, chromatin organization and modification and cell cycle regulation (Supplementary Figure S3B), which are all processes associated with mature erythroid physiology. Examples include the Slc4a1, Cat and Urod genes (Supplementary Figure S5). Overall, we find that genes representing the erythroid transcription program are highly enriched for GATA-1 binding throughout differentiation.
Epigenetic landscape of GATA-1 target genes
To obtain a more global insight into the regulatory events taking place during erythroid differentiation, we integrated our GATA-1 occupancy profiles with a series of publicly available genome-wide transcription factor (TF) occupancy and histone modification profiles available for Ter119− and Ter119+ fetal liver cells (Table 1). Thus, 28 ChIP-seq data sets comprising four TFs, nine histone tail modifications, RNA polymerase II, DNA methylation ratios and gene expression profiling by RNA-seq (30–33,45,47,48) were incorporated into a single database. Importantly, with the exception of two data sets (H3K27Ac and H3K4me1) obtained from Ter119+ cells only, all other data were obtained from both Ter119− and Ter119+ fetal liver erythroid cells (Table 1). For all subsequent analyses, the TGS scores were based on the read density profiles produced for each experiment within a 10-kb window around each gene’s TSS (see ‘Materials and Methods’ section).
Table 1.
Genomic features currently available for Ter119− and/or Ter119+ fetal liver erythroid cells
| ChIP seq target | Ter119− | Ter119+ | Function | |
|---|---|---|---|---|
| GATA-1 | + | + | Transcription factors | |
| SCL/TAL-1 | + | + | ||
| PU.1 | + | + | ||
| KLF1 | + | + | ||
| H3K27Ac | + | Chromatin marks | Enhancer | |
| H3K4me1 | + | Enhancer | ||
| H3K4me2 | + | + | Activation | |
| H3K4me3 | + | + | Activation | |
| H3K9Ac | + | + | Activation | |
| H4K16Ac | + | + | Activation | |
| H3K36me3 | + | + | Elongation | |
| H3K79me2 | + | + | Elongation | |
| RNA Pol II | + | + | Elongation | |
| H3K27me3 | + | + | Silencing | |
| DNA methylation | + | + | Silencing | |
| RNA-seq | + | + | Expression | |
| Total | 16 (14 both conditions) | |||
To characterize the epigenetic landscape of GATA-1 occupied regions, we calculated the linear correlation between TGS scores of GATA-1 target genes and the TGS score calculated for each of the other TF occupancy profiles and epigenetic marks (Figure 4A). Based on this analysis, we observe that GATA-1 occupancy strongly correlates with SCL/TAL-1 binding (RTer119− = 0.53, RTer119+ = 0.49), as has been previously reported (30), whereas a much weaker correlation is observed with KLF1 occupancy profiles (RTer119− = 0.15, RTer119+ =0.07) and PU.1 (RTer119− = 0.1, RTer119+ = 0.08), as also seen by Pilon et al. (32). Furthermore, most of the histone modifications show a considerable correlation with GATA-1 binding (Figure 4A). Interestingly, GATA-1 occupancy correlates highly with the levels of H4K16Ac mark in both early and late stages of erythroid differentiation (RTer119− = 0.49, RTer119+ = 0.58) and with the levels of the enhancer related H3K27Ac and H3K4me1 marks (the latter data were only available for Ter119+ cells) (RTer119− = 0.54, RTer119+ = 0.61 and RTer119− =0.46, RTer119+ = 0.5, respectively). These data are consistent with the observations by Kowalczyk et al. (48), showing that sequences enriched in H3K27Ac are predominantly bound by GATA-1 (and other transcription factors) in erythroid cells. By contrast, we do not find a linear relationship between genome-wide H3K27me3 marks and GATA-1 occupied regions (RTer119− =−0.004, RTer119+ = −0.02). Hence, the association of GATA-1 binding with the H3K27me3 mark seen by Yu et al. (16) in a subset of repressed GATA-1 target genes in mouse erythroleukemic (MEL) cells does not seem to be reflected at the genome-wide level in fetal liver-derived erythroblasts.
Figure 4.

Association of GATA-1 occupancy with specific epigenetic events. (A) Heatmap showing the pairwise Pearson correlations between GATA-1 TGS and epigenetic mark TGSs in Ter119− and Ter119+ erythroid cells. (B) Scatterplots of observed and RF regression predicted values of selected histone mark variation between Ter119− and Ter119+ cells. Black dots represent the predicted values of the GATA-1 trained model, whereas gray dots represent the values predicted by the GATA-1/TAL1/KLF1 trained model. (C) Percentage of variation explained (R2) by the GATA-1 (first column) and GATA-1/TAL1/KLF1 (second column) trained RF regression models. Third column refers the percentage of the increase in R2 values between the two models.
GATA-1 occupancy profiles can be predictive of the variation in specific histone tail modifications
Previous studies have associated GATA-1 with the acquisition of the H3K79 methylation mark (15) and with the formation of erythroid-specific histone H3 and H4 acetylation patterns (14). We thus tested for possible GATA-1 associations with changes in specific histone modifications in erythroid differentiation. We used RF (44) to build a series of regression models that can predict the changes in the levels of histone tail modifications between Ter119− and Ter119+ cells, on the basis of GATA-1 occupancies (Supplementary Methods). A highly predictive model would provide an indirect indication of GATA-1 modulating specific aspects of the epigenetic landscape in differentiating erythroid cells. The results summarized in Figure 4C show that GATA-1 occupancy can be related, to varying extents, to the variation of all tested histone tail modifications. However, the most predictive models were obtained for the H3K79me2, H3K4me2, H3K4me3 and H4K16Ac histone marks (Supplementary Results), consistent with previous observations connecting GATA-1 to specific histone-modifying enzymes, such as CBP/p300, Dot1l and HDACs (15,49,50). Our results also show that GATA-1 preferentially associates with the H4K16 acetylation mark, rather than the H3K9 acetylation (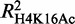 = 0.28 and
= 0.28 and 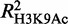 = 0.20), thus refining previous observations on GATA-1–mediated H3 and H4 acetylation patterns in erythroid-specific gene loci (14).
= 0.20), thus refining previous observations on GATA-1–mediated H3 and H4 acetylation patterns in erythroid-specific gene loci (14).
As genome-wide data for H3K27 acetylation are not presently available in Ter119− cells, we were unable to model the variation in H3K27Ac with erythroid differentiation as we did above for H4K16 and H3K9 acetylation. Thus, to include H3K27Ac in our analysis, we modeled the absolute levels of all three available histone acetylation modifications in Ter119+ cells, i.e. H3K27, H4K16 and H3K9. We found GATA-1 occupancy to be a good predictor for all three acetylation marks, with H3K27Ac showing the highest degree of correlation and H3K9Ac the lowest (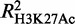 = 0.45,
= 0.45, 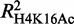 = 0.41 and
= 0.41 and 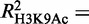 0.30). Interestingly, these observations are in agreement with GATA-1 interacting directly with the CBP/p300 acetyltransferase (49), the latter having a specificity for acetylating both H4K16 and H3K27, but not H3K9 (51,52).
0.30). Interestingly, these observations are in agreement with GATA-1 interacting directly with the CBP/p300 acetyltransferase (49), the latter having a specificity for acetylating both H4K16 and H3K27, but not H3K9 (51,52).
We also found a high correlation between GATA-1 occupancy and the variation in histone H3 methylation levels. Notably, GATA-1 seems to be associated more with changes in the di-methyl mark as opposed to those in the tri-methylation mark of lysine 4 (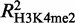 = 0.35 and
= 0.35 and 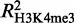 = 0.29). This observation may be related to recent findings, suggesting a tissue-specific regulatory role for H3K4me2, independently of H3K4me3 (53,54). By contrast, GATA-1 occupancy is a poor predictor of changes in the levels of the H3K27me3 mark (R2 = 0.07), suggesting that GATA-1 binding by itself is not a primary determinant for the genome-wide deposition of H3K27me3 marks during terminal erythroid differentiation.
= 0.29). This observation may be related to recent findings, suggesting a tissue-specific regulatory role for H3K4me2, independently of H3K4me3 (53,54). By contrast, GATA-1 occupancy is a poor predictor of changes in the levels of the H3K27me3 mark (R2 = 0.07), suggesting that GATA-1 binding by itself is not a primary determinant for the genome-wide deposition of H3K27me3 marks during terminal erythroid differentiation.
A second series of regression models was built by including the occupancy profiles of the hematopoietic SCL/TAL-1 and KLF1 transcription factors (30–33) with those of GATA-1 in the RF training data sets (Supplementary Methods). We noticed a higher performance for all the regression models tested compared with GATA-1 alone (Figure 4C), suggesting that SCL/TAL-1 and KLF1 may be involved together with GATA-1 in modulating epigenetic modifications. Importantly, the additional information derived from the inclusion of SCL/TAL-1 and KLF1 occupancies is more pronounced for specific histone modifications. The highest overall increase was observed for the H3K27me3 (182%), whereas the acetylation of H3K9 showed an increase of 101% (Figure 4C). These observations support the notion that distinct erythroid TF complexes are implicated in the deposition of specific epigenetic marks. Overall, our results show that GATA-1 is involved in the regulation of a large subset of target genes through the modulation of specific epigenetic events and further suggest that GATA-1 binding preferentially associates with specific histone tail modifications, such as H4K16 and H3K27 acetylation and H3K4 methylation.
Modeling gene expression of GATA-1 gene targets
Of the 3651 genes identified as GATA-1 target genes, 321 genes are upregulated by >2-fold with differentiation, 1941 genes are downregulated by >2-fold and 1390 genes show <2-fold variation between Ter119− and Ter119+ erythroid cells (45). As both GATA-1 occupancy and the epigenetic landscape are involved in the regulation of GATA-1 differentially expressed target genes (2258 genes), we integrated all of the available information (Table 1) to model the changes in their expression levels during erythroid differentiation (Supplementary Methods). This approach resulted in a remarkably highly predictive model (R2 = 0.62, r = 0.8, Figure 5A) of differential gene expression profiles by the binding signals of the four TFs, nine histone modifications, RNA pol II and DNA methylation levels measured in Ter119− and Ter119+ cells. The most predictive feature of changes in gene expression during erythroid differentiation is the change in the levels of the H3K79me2 elongation mark (Figure 5B and Supplementary Table S4), in accordance with the findings of Wong et al. (45). Changes in H3K4 methylation levels closely followed, whereas changes in GATA-1 occupancy were found to be in a group of almost equal ranking comprising H3K9Ac, RNApolII and H4K16Ac. It is interesting to note that the most predictive features (H3K79me2 and H3K4 methylation) can be, at least in part, associated with GATA-1 itself, as shown earlier in the text. This observation further consolidates the notion that part of the GATA-1 regulatory function is exerted through the modulation of the epigenetic landscape of its target genes.
Figure 5.

Evaluation of the gene expression RF regression model. (A) Scatterplot of observed and predicted values of gene expression change between Ter119− and Ter119+ cells of differentially regulated GATA-1 target genes. Red and green dots refer to values correctly predicted as decreasing or increasing mRNA levels, respectively. Blue and cyan dots refer to GATA-1 target genes with inverted predicted values. (B) Variable importance measures (%IncMSE) in predicting gene expression changes between Ter119− and Ter119+ erythroid cells. Only the 15 top ranked features are plotted. (C) Dendrogram showing clusters of GATA-1 differentially regulated target genes according to the RF calculated proximity values. (D) Barplot of corresponding gene expression fold change values of the proximity clustered GATA-1 target genes. (E) Heatmap illustrating the mean TGS values of the different occupancy profiles within the genes composing each cluster of GATA-1 differentially regulated target genes. (F) Tables showing the most highly enriched gene ontologies identified for each of the three GATA-1 target gene clusters.
To identify clusters of GATA-1 target genes bearing similar epigenetic profiles, we performed hierarchical clustering of the gene proximity values calculated by the RF regression model (Figure 5C and Supplementary Methods). This approach produced a clear separation of upregulated from downregulated genes (Figure 5D). Additionally, the branch corresponding to the downregulated genes is further dissected in two distinct gene clusters (Figure 5D). To assay for any functional distinction between these three clusters, we performed GO analysis using the DAVID online tool (46). Not surprisingly, the upregulated gene cluster (Cluster 3, 309 genes) showed high enrichment for all the heme biosynthetic processes and erythrocyte differentiation (Figure 5F) and contained genes like α- and β-globin, Scl/Tal1, Slc4a1 and Alas2. Interestingly, the analysis of the two clusters associated with downregulated genes revealed clearly distinct functional properties. Cluster 1 (1186 genes) was highly enriched for genes involved in RNA processing, the translation machinery and ribosome biogenesis, whereas Cluster 2 (763 genes) was enriched in genes involved in hematopoiesis, immune system development, myeloid and lymphoid cell differentiation and cell proliferation and included genes like PU.1, c-Kit, Lyn, Cebp, Hif1a, Runx1 and members of the Stat and Smad families.
Significantly, the three clusters show distinct epigenetic signatures (Figure 5E). Cluster 3 (upregulated genes) shows the highest levels of GATA-1, SCL/TAL-1 and KLF1 occupancy and also the highest levels of the activating and elongating histone marks. By contrast, Cluster 2, enriched in downregulated genes involved in alternative hematopoietic lineages, was associated with the highest levels of H3K27me3 histone modification and the lowest enrichment levels for all three TFs and activating and elongating histone marks. It is of interest that the majority of the GATA-1 target genes found by Yu et al. (16) to also display H3K27me3 marks, e.g. Gata2, c-kit and so forth, partition within this cluster. Cluster 1, enriched in downregulated genes involved in house-keeping processes, is characterized by the lowest levels of H3K27me3, low levels of TF occupancy and intermediate to low levels of activating and elongating marks. Even though Cluster 1 is composed of genes that exhibit decreasing mRNA levels, the lack of the H3K27me3 mark and the persistence of activating and elongating marks could be an indicator of gene expression that is maintained at low levels, or is in the process of being extinguished. By contrast, genes composing Cluster 2 show a more severe downregulation with high levels of the polycomb-group H3K27me3 repressive mark, suggesting that a repressive epigenetic memory mechanism is in place. In fact, if we compare the absolute mRNA levels through stages R2–R5 of erythroid differentiation (45), we find significantly lower mRNA levels for Cluster 2 genes (alternative lineages) compared with Cluster 1 (protein production) (P < 2.2e-16, Wilcoxon rank-sum test) (Supplementary Figure S6). Our findings significantly extend previous observations made by Cheng et al. (24) using a limited number of GATA-1 target genes in G1E cells, in which they divided repressed genes in two classes: one enriched in H3K27me3 marks and depleted for SCL/TAL-1 binding and the second class depleted for H3K27me3 marks and enriched for SCL/TAL-1 binding.
Collectively, our data reinforce previous observations for GATA-1 regulating the erythroid differentiation process at multiple levels [reviewed in (8)]. First, GATA-1 positively regulates the expression of erythroid-specific genes and genes involved in the production of mature hemoglobin molecules. Second, it negatively regulates the expression of genes involved in early hematopoietic differentiation and alternative myeloid and lymphoid lineages, by completely shutting them down to allow terminal erythroid differentiation to proceed. Third, it is directly involved in the reduced expression of the mRNA maturation and translation machinery, adjusting it to the reduced needs of the enucleated mature erythrocyte. Importantly, our work shows that specific epigenetic signatures are associated with functionally different subsets of GATA-1 target genes, thus suggesting a degree of plasticity in the regulatory functions of GATA-1.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–6 and Supplementary Data sets 1–2.
FUNDING
‘InteGeR’ FP7 Marie Curie Initial Training Network [PITN-GA-2008-214902 awarded to J.S. and J.R.]; National Institute of Diabetes and Digestive and Kidney Diseases [R01DK083389 to J.S. and J.B.]. G.L.P. is a Fellow of the ‘InteGeR’ FP7 Marie Curie Initial Training Network [PITN-GA-2008-214902]. E.K. has been a Fleming Graduate Fellow and was awarded a short term EMBO fellowship [ASTF 389-08] for visiting C.P.’s lab. Funding for open access charge: FP7 Marie Curie funding.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr George Garinis and Prof. Achilleas Gravanis (University of Crete, Greece) for helpful discussions and serving as academic advisors to G.L.P. and E.K.
REFERENCES
- 1.Magnusdottir E, Gillich A, Grabole N, Surani MA. Combinatorial control of cell fate and reprogramming in the mammalian germline. Curr. Opin. Genet. Dev. 2012;22:466–474. doi: 10.1016/j.gde.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Dillon N. Factor mediated gene priming in pluripotent stem cells sets the stage for lineage specification. Bioessays. 2012;34:194–204. doi: 10.1002/bies.201100137. [DOI] [PubMed] [Google Scholar]
- 3.Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life. 2009;61:800–830. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 5.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 6.Gutierrez L, Lindeboom F, Ferreira R, Drissen R, Grosveld F, Whyatt D, Philipsen S. A hanging drop culture method to study terminal erythroid differentiation. Exp. Hematol. 2005;33:1083–1091. doi: 10.1016/j.exphem.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Kina T, Ikuta K, Takayama E, Wada K, Majumdar AS, Weissman IL, Katsura Y. The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br. J. Haematol. 2000;109:280–287. doi: 10.1046/j.1365-2141.2000.02037.x. [DOI] [PubMed] [Google Scholar]
- 8.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–6268. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood. 2004;104:3136–3147. doi: 10.1182/blood-2004-04-1603. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez P, Bonte E, Krijgsveld J, Kolodziej KE, Guyot B, Heck AJ, Vyas P, de Boer E, Grosveld F, Strouboulis J. GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J. 2005;24:2354–2366. doi: 10.1038/sj.emboj.7600702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol. Cell. Biol. 2005;25:1215–1227. doi: 10.1128/MCB.25.4.1215-1227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crispino JD. GATA1 in normal and malignant hematopoiesis. Semin. Cell Dev. Biol. 2005;16:137–147. doi: 10.1016/j.semcdb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21:3368–3376. doi: 10.1038/sj.onc.1205326. [DOI] [PubMed] [Google Scholar]
- 14.Letting DL, Rakowski C, Weiss MJ, Blobel GA. Formation of a tissue-specific histone acetylation pattern by the hematopoietic transcription factor GATA-1. Mol. Cell. Biol. 2003;23:1334–1340. doi: 10.1128/MCB.23.4.1334-1340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell. 2009;36:682–695. doi: 10.1016/j.molcel.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heyworth C, Pearson S, May G, Enver T. Transcription factor-mediated lineage switching reveals plasticity in primary committed progenitor cells. EMBO J. 2002;21:3770–3781. doi: 10.1093/emboj/cdf368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graf T. Differentiation plasticity of hematopoietic cells. Blood. 2002;99:3089–3101. doi: 10.1182/blood.v99.9.3089. [DOI] [PubMed] [Google Scholar]
- 19.Iwasaki H, Mizuno S, Wells RA, Cantor AB, Watanabe S, Akashi K. GATA-1 converts lymphoid and myelomonocytic progenitors into the megakaryocyte/erythrocyte lineages. Immunity. 2003;19:451–462. doi: 10.1016/s1074-7613(03)00242-5. [DOI] [PubMed] [Google Scholar]
- 20.Wickrema A, Crispino JD. Erythroid and megakaryocytic transformation. Oncogene. 2007;26:6803–6815. doi: 10.1038/sj.onc.1210763. [DOI] [PubMed] [Google Scholar]
- 21.Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J. Biol. Chem. 2010;285:31087–31093. doi: 10.1074/jbc.R110.159079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajima H. Role of transcription factors in differentiation and reprogramming of hematopoietic cells. Keio J. Med. 2011;60:47–55. doi: 10.2302/kjm.60.47. [DOI] [PubMed] [Google Scholar]
- 23.Soler E, Andrieu-Soler C, de Boer E, Bryne JC, Thongjuea S, Stadhouders R, Palstra RJ, Stevens M, Kockx C, van Ijcken W, et al. The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev. 2010;24:277–289. doi: 10.1101/gad.551810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen KB, Zhang Y, Drautz D, et al. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 2009;19:2172–2184. doi: 10.1101/gr.098921.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujiwara T, O'Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, Bresnick EH. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell. 2009;36:667–681. doi: 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wontakal SN, Guo X, Smith C, MacCarthy T, Bresnick EH, Bergman A, Snyder MP, Weissman SM, Zheng D, Skoultchi AI. A core erythroid transcriptional network is repressed by a master regulator of myelo-lymphoid differentiation. Proc. Natl Acad. Sci. USA. 2012;109:3832–3837. doi: 10.1073/pnas.1121019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dore LC, Chlon TM, Brown CD, White KP, Crispino JD. Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood. 2012;119:3724–3733. doi: 10.1182/blood-2011-09-380634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chlon TM, Dore LC, Crispino JD. Cofactor-mediated restriction of GATA-1 chromatin occupancy coordinates lineage-specific gene expression. Mol. Cell. 2012;47:608–621. doi: 10.1016/j.molcel.2012.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tijssen MR, Cvejic A, Joshi A, Hannah RL, Ferreira R, Forrai A, Bellissimo DC, Oram SH, Smethurst PA, Wilson NK, et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev. Cell. 20:597–609. doi: 10.1016/j.devcel.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kassouf MT, Hughes JR, Taylor S, McGowan SJ, Soneji S, Green AL, Vyas P, Porcher C. Genome-wide identification of TAL1's functional targets: insights into its mechanisms of action in primary erythroid cells. Genome Res. 2010;20:1064–1083. doi: 10.1101/gr.104935.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tallack MR, Whitington T, Yuen WS, Wainwright EN, Keys JR, Gardiner BB, Nourbakhsh E, Cloonan N, Grimmond SM, Bailey TL, et al. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 2010;20:1052–1063. doi: 10.1101/gr.106575.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pilon AM, Ajay SS, Kumar SA, Steiner LA, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, et al. Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood. 2011;118:e139–e148. doi: 10.1182/blood-2011-05-355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu W, Cheng Y, Keller CA, Ernst J, Kumar SA, Mishra T, Morrissey C, Dorman CM, Chen KB, Drautz D, et al. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011;21:1659–1671. doi: 10.1101/gr.125088.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Lindern M, Deiner EM, Dolznig H, Parren-Van Amelsvoort M, Hayman MJ, Mullner EW, Beug H. Leukemic transformation of normal murine erythroid progenitors: v- and c-ErbB act through signaling pathways activated by the EpoR and c-Kit in stress erythropoiesis. Oncogene. 2001;20:3651–3664. doi: 10.1038/sj.onc.1204494. [DOI] [PubMed] [Google Scholar]
- 35.Schuh AH, Tipping AJ, Clark AJ, Hamlett I, Guyot B, Iborra FJ, Rodriguez P, Strouboulis J, Enver T, Vyas P, et al. ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol. Cell. Biol. 2005;25:10235–10250. doi: 10.1128/MCB.25.23.10235-10250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guberman JM, Ai J, Arnaiz O, Baran J, Blake A, Baldock R, Chelala C, Croft D, Cros A, Cutts RJ, et al. BioMart Central Portal: an open database network for the biological community. Database. 2011;2011:bar041. doi: 10.1093/database/bar041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martowicz ML, Grass JA, Boyer ME, Guend H, Bresnick EH. Dynamic GATA factor interplay at a multicomponent regulatory region of the GATA-2 locus. J. Biol. Chem. 2005;280:1724–1732. doi: 10.1074/jbc.M406038200. [DOI] [PubMed] [Google Scholar]
- 41.Valverde-Garduno V, Guyot B, Anguita E, Hamlett I, Porcher C, Vyas P. Differences in the chromatin structure and cis-element organization of the human and mouse GATA1 loci: implications for cis-element identification. Blood. 2004;104:3106–3116. doi: 10.1182/blood-2004-04-1333. [DOI] [PubMed] [Google Scholar]
- 42.MacQuarrie KL, Fong AP, Morse RH, Tapscott SJ. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet. 2011;27:141–148. doi: 10.1016/j.tig.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerenyi MA, Orkin SH. Networking erythropoiesis. J. Exp. Med. 2010;207:2537–2541. doi: 10.1084/jem.20102260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 45.Wong P, Hattangadi SM, Cheng AW, Frampton GM, Young RA, Lodish HF. Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood. 2011;118:e128–e138. doi: 10.1182/blood-2011-03-341404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 47.Shearstone JR, Pop R, Bock C, Boyle P, Meissner A, Socolovsky M. Global DNA demethylation during mouse erythropoiesis in vivo. Science. 2011;334:799–802. doi: 10.1126/science.1207306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, et al. Intragenic enhancers act as alternative promoters. Mol. Cell. 2012;45:447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 49.Blobel GA, Nakajima T, Eckner R, Montminy M, Orkin SH. CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc. Natl Acad. Sci. USA. 1998;95:2061–2066. doi: 10.1073/pnas.95.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watamoto K, Towatari M, Ozawa Y, Miyata Y, Okamoto M, Abe A, Naoe T, Saito H. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene. 2003;22:9176–9184. doi: 10.1038/sj.onc.1206902. [DOI] [PubMed] [Google Scholar]
- 51.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galvez AF, Huang L, Magbanua MM, Dawson K, Rodriguez RL. Differential expression of thrombospondin (THBS1) in tumorigenic and nontumorigenic prostate epithelial cells in response to a chromatin-binding soy peptide. Nutr. Cancer. 2011;63:623–636. doi: 10.1080/01635581.2011.539312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pekowska A, Benoukraf T, Ferrier P, Spicuglia S. A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 2010;20:1493–1502. doi: 10.1101/gr.109389.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orford K, Kharchenko P, Lai W, Dao MC, Worhunsky DJ, Ferro A, Janzen V, Park PJ, Scadden DT. Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev. Cell. 2008;14:798–809. doi: 10.1016/j.devcel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.