


Abstract
During the past years, exogenous DNA molecules have been used in gene and molecular therapy. At present, it is not known how these DNA molecules reach the cell nucleus. We used an in cell single-molecule approach to observe the motion of exogenous short DNA molecules in the cytoplasm of eukaryotic cells. Our observations suggest an active transport of the DNA along the cytoskeleton filaments. We used an in vitro motility assay, in which the motion of single-DNA molecules along cytoskeleton filaments in cell extracts is monitored; we demonstrate that microtubule-associated motors are involved in this transport. Precipitation of DNA-bound proteins and mass spectrometry analyses reveal the preferential binding of the kinesin KIFC1 on DNA. Cell extract depletion of kinesin KIFC1 significantly decreases DNA motion, confirming the active implication of this molecular motor in the intracellular DNA transport.
INTRODUCTION
In eukaryotic cells, the DNA is organized in chromatin and mostly confined in the nucleus. However, during mitosis, chromosomes are in contact with the cytoplasm: the positioning of the chromosomes along the mitotic spindle and the segregation of sister chromatids occur thanks to microtubule-associated molecular motors (1–5). Exogenous DNA molecules may also be delivered to the cytoplasm of a eukaryotic cell during a viral or bacterial infection. Gene therapy and DNA-based therapy (6,7) imply the transit of DNA fragments in the cytoplasm to reach the nucleus of eukaryotic cells, as the dissociation of the DNA molecule from its vector occurs in the cytoplasm (8).
Although naked DNA is mostly degraded in the cytoplasm by nuclease enzymes, some DNA molecules enter the nucleus and may be included in the genome. Vaughan and Dean (9) observe that the DNA plasmids bearing the nuclear targeting sequence simian vacuolating virus 40 (SV40) DTS (DNA targeting sequence) bind to cytoplasmic dynein and use the microtubule network to reach the nucleus. The authors hypothesize that such an active transport is due to the affinity of SV40 DTS sequences for a transcription factor containing the nuclear localization signals (NLSs) that would promote the trafficking along the microtubules through cytoplasmic dynein (10). Other work shows that the extreme intracellular crowding hampers the mobility of relatively long DNA molecules (>250 bp), which would need an active transport to traffic within the cytoplasm. In particular, the actin network acts as a physical obstacle to DNA diffusion (11,12). With the exception of these few pioneering experiments and despite the relevance of cytoplasmic DNA transport in the context of gene therapy, there are no longtime observations of the motion of DNA fragments towards the nucleus or towards the plasma membrane. Analysis of DNA traffic in the cell encounters two major limitations: (i) proteins can specifically bind some sequences and affect the DNA location as demonstrated with SV40 DTS sequence (9), and (ii) DNA is progressively degraded in the cytoplasm resulting in a mixture of different molecules lengths and degradation products (13). To overcome these limitations, we use a short double-stranded DNA molecule (32 bp), the Dbait, developed as an adjuvant of anti-cancer therapies (14). This molecule is relatively stable in living systems [several hours in cell extract and in blood (15)]. Moreover, it has been designed to contain no homology with any human sequence (16,17) to reduce the binding of DNA-binding proteins in cytoplasm. Therefore, Dbait is a unique tool for studying the general traffic of DNA in cytoplasm.
The scope of our work is to determine whether the DNA in cytoplasm is simply driven to the nucleus by thermal diffusion (Brownian motion) or whether an active transport by molecular motors is needed. Using an in cell single-molecule approach, we observe the intracellular motion of individual naked DNA molecules in real-time. To visualize the single DNA and acquire long trajectories with high-spatial resolution, DNA molecules are conjugated with single-fluorescent quantum dots (QDs). Fluorescent QDs are bright inorganic nanoparticles with an extreme photo-stability (no photo-bleaching) and have been already successfully used to follow for long periods (minutes) the motion of single proteins both at the cell membrane (18) and inside the cytoplasm (19–23). Eventually, the trajectory of each DNA is recorded and analyzed to discriminate between a pure diffusive behavior and an active directed motion. In addition to in cell experiments, we develop an in vitro assay to mimic the intracellular transport of the DNA on the cytoskeleton filaments. In this well-controlled environment, we probe the specific motion of DNA with isolated microtubules. These experiments suggest an active transport of DNA fragments caused by microtubule-associated molecular motors; therefore, we complete our study by a DNA/protein co-purification assay and mass spectrometry (MS) analysis. We identify an interaction between DNA and four kinesins (KIF1C, KIF4A, KIF14 and KIFC1) and the cytoplasmic dynein. In vitro experiments in the presence or in the absence of molecular motors suggest a prominent role of KIFC1 protein in the intracellular transport of bare DNA. Eventually, immunofluorescence co-localization experiments also confirm a specific binding of double-stranded bare DNA to KIFC1 molecular motors.
MATERIALS AND METHODS
Choice of the DNA molecule
Dbait is 32-bp double-stranded oligonucleotide protected from nucleophilic degradation by a carboxylic acid linker loop on one extremity and phosphorothioate nucleotides on the other end. The sequence is 5′-bGCTGTGCCCACAACCCAGCAAACAAGCCTAGA-(H)-TCTAGGCTTGTTTGCTGGGTTGTGGGCACAGC-3′, where H is a hexaethylene glycol linker, b is a biotin and underlined marks the position of phosphorothioate nucleotides. Dbait is obtained by automated solid-phase oligonucleotide synthesis and purified by denaturing reversed-phase high-performance liquid chromatography (HPLC) as previously describe (16). The 5′-end is functionalized with a biotin to allow conjugation with the QD. It has been demonstrated that neither the hexaethylenglycol nor the phosphorothioate terminations have a biological effect (17,24). The possibility to irreversibly associate the Dbait to the QD and its stability with respect to nuclease enzymes make the Dbait a good candidate to allow long observations in the cytoplasm.
DNA:QD conjugation
Single-particle imaging in living cells is a powerful tool to investigate the dynamics of individual biological molecules (or a small number of them) directly in their natural environment and without averaging effects (19,20,25–28). We use semiconductor QDs to visualize the DNA inside living cells, as the high brightness and photo-stability of QD enable long observations at single-particle level, with high-spatial and -temporal resolution and in an optically noisy environment (21–23,29–32). We use streptavidin-coated QD (Qdot nanocrystals, Molecular Probe, Emission: 655 nm) to form an irreversible bond with the biotinylated DNA. The ratio DNA:QD is adjusted to ∼10 DNA per QD. After mixing, DNA molecules and the QDs are incubated for ∼10 min in the presence of a bovine serum albumin (BSA) to avoid unspecific interactions.
Intracellular loading of DNA:QD complexes
The DNA:QD complexes are internalized in HeLa cells by osmotic lysis of pinocytic vesicles (33). Cells cultured on 30-mm glass coverslips are immersed in the hypertonic medium (94% Dulbecco’s Modified Eagle’s Medium (D-MEM), 5% fetal bovine serum, 1% HEPES and glucose at saturating concentration—GIBCO) with the DNA:QD constructions at 10 nM concentration (40 µl in 360 µl of hypertonic medium) for 12 min, then immersed in the hypotonic medium (60% D-MEM and 40% water) for 2 min and kept in the recovery medium (90% Opti-MEM and 10% fetal bovine serum) for the rest of the observation time. All experiments are performed at 37°C and 5% CO2.
A control test with HeLa cells with disrupted microtubule network is performed. Cells are processed as following. After internalization of DNA:QD complexes, cells are kept for 10 min at 37°C and 5% CO2. Then, cells are incubated with the microtubule-depolymerizing agent nocodazole (1 µM) and kept for 40 min at 37°C and 5% CO2. We observe the motion of DNA:QD complexes in these conditions.
Cytoplasmic extracts
Cytoplasmic extracts (CE) are prepared from HeLa cells, grown in complete D-MEM (89% D-MEM, 10% fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml of streptomycin—GIBCO), under controlled conditions (37°C, 5% CO2). At 100% confluence, the cells are washed with phosphate-buffered saline (PBS) and then detached from the surface with trypsin solution (0.05% Trypsin–EDTA, GIBCO). The cells are collected in a pellet by centrifugation (3 min at 600g). The cell pellet is suspended in CL buffer (50 mM Tris, 150 mM NaCl, 0.1% Triton X-100 and 10 mM EDTA, pH 7.2) supplemented with protease inhibitor cocktail and gently mixed for 10 min at 4°C. The Triton X-100 compromises the integrity of cell membrane, facilitating cell lysis. As an alternative to lysis by Triton X-100, we use a Potter-Elvehjem homogenizer or the complete lysis buffer (Roche). Soluble proteins are then separated by two repeated centrifugations (600g for 10 min and 15 000g for 20 min at 4°C). CE for in vitro motility assays are stored at −80°C (20% of glycerol added) after flash freezing. We use fresh CE for co-purification experiments because the protein characterization in MS is strongly limited by protein quantity: the freezing step could cause a protein loss.
In vitro motility assay
Cytoplasmic proteins–DNA–QD complexes (CyProt:DNA:QD)
To prepare the complexes of DNA and cytoplasmic proteins, conjugated with the QDs (CyProt:DNA:QD), the DNA molecules (final concentration 200 nM) are first incubated for 20 min at 4°C with the CE (final concentration 0.4 mg/ml). Afterwards, the streptavidin-coated QDs are added to this mix at the final concentration of 20 nM. To remove the excess of CE, the complexes are centrifuged (25 000g for 20 min at 4°C) and washed in a buffer containing 25 mM imidazole (pH 7.5), 25 mM KCl and 4 mM MgCl2.
Microtubules
Microtubules are polymerized at 37°C from bovine brain tubulin (Cytoskeleton) diluted at 2 mg/ml in the buffer BRB (32 mM PIPES, 4 mM MgCl2 and 4 mM ethylene glycol tetraacetic acid (EGTA), pH 6.8) with 10% of glycerol, in the presence of GTP at 1 mM. After 30 min of incubation, the microtubules are stabilized with paclitaxel at 50 µM (previously dissolved in DMSO) then separated from the non-polymerized tubulin by centrifugation (10 min at 25 000g, 37°C). The microtubules are re-suspended in the BRB buffer supplemented with paclitaxel (20 µM) and kept at room temperature (23°C) up to 48 h.
Actin filaments
Actin filaments are made using commercial rabbit muscle G-actin (Cytoskeleton) diluted at 2.5 mg/ml. The polymerization is induced by mixing 2 µl of G-actin and 2 µl of ATP (6 mM) in 8 µl of AB buffer (25 mM imidazole, 25 mM KCl, 4 mM MgCl2 and 1 mM EGTA) and incubating for 30 min at room temperature. Actin filaments are stabilized with Alexa Fluorescent 488 phalloidin (2 µl at 66 µM).
Flow chambers
Flow chambers for in vitro experiments are (18 mm × 6 mm × 100 µm) made of two coverslips separated by double-sided adhesive tapes. To allow the binding of microtubules to the coverslip, the flow chambers are coated with poly-l-lysine [solution 0.1% (w/v) in H2O—Sigma Aldrich] before the injection of the microtubules. The excess of microtubules (not bound to the poly-l-lysine layer) is removed by rinsing the chamber with large volumes of buffer supplemented with 10 mg/ml of casein to prevent non-specific interactions. Similarly, we prepare flow chambers coated with actin filaments. In this case, the coverslip is previously coated with myosin-II treated with N-ethylmaleimide to inhibit the ATPase site, which promotes the binding of actin filaments (34). The chamber is rinsed with AB buffer supplemented with 5 mg/ml of casein to prevent non-specific interactions.
Motility buffer
In-vitro experiments are performed in BRB buffer (with 20 μM of paclitaxel) for microtubules and AB buffer for actin filaments, supplemented with nucleotides [ATP, ADP or Adenylyl Imidodiphosphate (AMP.PNP)], casein (5 mg/ml), an oxygen-scavenging system (20 mM d-glucose, 20 mg/ml glucose–oxydase and 8 mg/ml catalase) and, when needed, an ATP regeneration system (40 mM phospho-creatine and 0.1 mg/ml creatine–phosphokinase). All observations are done at room temperature. To study the interaction between CyProt:DNA:QD complexes and filaments, we introduce the complexes without nucleotides or in presence of ADP (for actin filaments) or AMP.PNP (for microtubules) and we wait for 20 min. Then, the chamber is rinsed to remove the excess of complexes in solution. To study the motion, the CyProt:DNA:QD complexes are introduced in the presence of ATP and we observe them immediately.
Data collection and analysis
In cell experiments are performed using an epifluorescence microscope. The movies are acquired on an inverted Zeiss Axiovert 100 equipped with a 100× Zeiss objective (NA 1.40) and an EMCCD camera (Andor, Neo sCMOS 2560 × 2160, pixel size 6.5 µm). Movies are acquired at 100 frames per second.
In vitro experiments are performed on the same customized set-up using an epifluorescence microscope for the study of the interaction and a total internal reflection fluorescence microscope (TIRFM) with a 63 × objective (Olympus, NA 1.49), a laser diode as light source (532 nm, 40 mW) and a CCD camera (Watek, 756 × 512 pixel) for the observations of the CyProt:DNA:QD motion. These experiments are performed with an acquisition rate of 25 frames per second.
The analysis of the trajectories is performed using two different algorithms for the in cell and in vitro experiments. The in cell experiments are tracked using a customized version of the MTT algorithm (35) adapted to our purpose. For the in vitro experiments, the tracking is performed with a version of particle tracking algorithm developed by Crocker et al. (36) and adapted for MATLAB by Daniel Claire (Georgetown University) and Eric Dufresne (Yale University). We estimate the pointing accuracy of the tracking by following a QD stuck on the coverslip for 1000 frames and measuring the standard deviation of its position. We find that the error is ∼30 nm.
DNA-binding protein purification
We perform three independent co-purification experiments. CE are first pre-cleared (8 times) using streptavidin-magnetic beads (MyOne Streptavidin T1—Dynabeads) for 30 min at 4°C under rotation. Then, 2.5 mg of pre-cleared CE is mixed with 4 µg of DNA and incubated overnight at 4°C under rotation. Streptavidin-magnetic beads are added to the mix ‘DNA-cytoplasmic extracts’ for 30 min at 4°C under rotation to form a bond with biotinylated DNA. After three washes in cold lysis buffer, proteins bound to the beads are incubated in sample buffer with reducing agent and then boiled for 2 min. Proteins are loaded and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) using Nupage (4–12%) Bis–Tris gels (Invitrogen).
Liquid chromatography-MS/MS analysis
In gel digests are performed as described in standard protocols. Briefly, after SDS–PAGE and washing of the excised gel slices, proteins are reduced by adding 10 mM 1,4-Dithiothreitol (DTT; Sigma Aldrich) before alkylation with 55 mM iodoacetamide (Sigma Aldrich). After washing and shrinking of the gel pieces with 100% acetonitrile, trypsin (Sequencing Grade Modified, Roche Diagnostics) is added and proteins are digested overnight in 25 mM ammonium bicarbonate at 30°C. We achieve peptide concentration and separation using an actively split capillary HPLC system (Ultimate 3000 system, Dionex, Germering, Germany) connected to the LTQ Orbitrap XL MS (Thermo Scientific). The MS is set to acquire a single-MS scan followed by up to five data-dependent scans (dynamic exclusion repeat count of 1, repeat duration of 30 s, exclusion duration of 180 s and lock-mass option is enabled). The resulting spectra are then analyzed via the Mascot Software created with Proteome Discoverer (version: 1.2.0.92, Thermo Scientific) using the SwissProt Homo sapiens Protein Database (18 April 2012, 20 255 sequences). The resulting Mascot result files are loaded into the myProMS (37) server for further processing. In myProMS, we fix the estimated false discovery rate (FDR) of all peptide and protein identifications to <1%, by automatically filtering on peptide length, mass error and Mascot score of all peptide identifications.
Immunodepletion molecular motors assay
Cells are processed for immunodepletion as following. CE is pre-cleared using protein G agarose beads for 30 min at 4°C under rotation. Supernatant is collected and incubated with protein G agarose beads and 1 μg of irrelevant rabbit anti-human IgG antibody for 30 min at 4°C. Supernatant is then incubated (2 h at 4°C) in parallel with beads pre-coated with 1 μg of antibodies against each of the four kinesins identified by MS. We use rabbit polyclonal antibodies (Bethyl Laboratories): (i) anti-KIFC1 antibody (A300–952A), (ii) anti-KIF14 (A300–233A), (iii) anti-KIF1C (A301–069A) and (iv) anti-KIF4A (A301–074A). After three washes in cold lysis buffer, immunoprecipitated proteins bound to the beads are incubated in sample buffer with reducing agent and then boiled for 2 min. Immunoprecipitated proteins are loaded and separated by SDS–PAGE using Nupage (4–12%) Bis–Tris gels (Invitrogen). Material is transferred on nitrocellulose membrane (Millipore) to XCell IItm Blot Module (Invitrogen) and detected using the specific antibody for each kinesin. Depleted cell lysates are used to perform the in vitro experiments: the DNA molecules are mixed with depleted cell lysates (separately for every kinesin) and then with the streptavidin-coated QDs, as described earlier in the text. The complexes obtained in every depleted lysate are diluted in motility buffer and injected in the flow chamber covered by microtubules.
In vitro immunofluorescence co-localization
Dbait 32-bp molecules functionalized with Cy3 fluorophore are used for immunofluorescence co-localization experiment. The complexes of DNA and cytoplasmic proteins are prepared by mixing the DNA molecules (final concentration 400 nM) and CE (final concentration 0.7 mg/ml) at 4°C for 20 min. Afterwards, complexes are incubated with microtubules in a flow chamber for 20 min in BRB buffer supplemented with casein (5 mg/ml), paclitaxel (20 μM) and AMP–PNP (1 mM). Then, the chamber is rinsed to remove the excess of complexes in solution. Primary anti-KIFC1 and fluorescent secondary antibody [goat anti-rabbit antibodies conjugated to Alexa Fluor 488 (Invitrogen)] are diluted in ImmunoFluorescence (IF) buffer [BRB buffer containing casein (5 mg/ml), paclitaxel (20 μM), BSA (2 mg/ml) and an oxygen-scavenging system (20 mM d-glucose, 20 mg/ml of glucose–oxydase and 8 mg/ml of catalase)]. Complexes bound to the microtubules are incubated with anti-KIFC1 for 5 min at room temperature. The chamber is then washed twice with IF buffer, and the fluorescent secondary antibody is incubated for 6 min at room temperature. The chamber is washed twice with IF buffer and one time with BRB-containing paclitaxel (20 μM) and an oxygen-scavenging system. The co-localization observations are performed by Förster resonance energy transfer (FRET) microscopy. The donor is the secondary antibody (Alexa488), and the acceptor is the DNA (Cy3). Control experiment is performed by incubating CE with microtubules in absence of DNA. Kinesin KIFC1 is revealed by immunofluorescence as describe earlier in the text.
RESULTS
Active and cytoskeleton-dependent transport of DNA revealed by in cell single-particle tracking
The DNA:QD complexes were internalized into the HeLa cells (see ‘Materials and Methods’ section), and their motion was followed by real-time epifluorescence. Each HeLa cell was observed both by phase contrast microscopy (Figure 1A) and fluorescence. The fluorescent spots correspond to DNA:QD complexes or small aggregates. We noticed that most of the DNA:QD complexes were localized in the cytoplasm, whereas few remained trapped at cell membrane. None seemed to be distributed within the nucleus. From real-time observations, two populations of cytoplasmic DNA:QD complexes were observed: most of them exhibited a diffusive motion (∼90%), whereas a small fraction had a directional motion with constant speed (∼10%). Examples of such directional motions are given, respectively, in Figure 1B and C showing two stacks of images acquired with a time resolution of 10 ms. These directed movements are incompatible with pure diffusion and more likely because of an active transport. Interestingly, only half of the observed movements were directed towards the nucleus. This does not suggest any preferential direction of this active transport.
Figure 1.

In cell single-particle experiments with DNA:QD complexes. (A) Bright field image of a HeLa cell plated onto a glass coverslip: we have superposed the trajectories of the DNA:QD complexes observed in fluorescence (in red). The blue boxes correspond to the particles in the sequences shown in (B) and (C). Stars indicate the beginning of the tracks. The scale is 10 micron. (B) and (C) Two examples of directed trajectories. Left: frame sequence of two DNA:QD complexes extracted from a movie acquired at 100 frames per second. The scale is 2 micron. Right: MSD for both trajectories. The MSD is fitted to the power law MSD(t) = a·tb. The coefficients are b = 1.8 and b = 2 in (B) and (C), respectively, indicating a directed motion.
To quantify the speed and the duration of such directed motion, all the DNA:QD complexes were tracked using an automatic tracking algorithm (see ‘Materials and Methods’ section). For each fluorescent spot, the tracking algorithm provides the trajectory in xy plane with respect to time (red lines in Figure 1B and C). To discriminate between the directed movements and the diffusive ones, we computed the mean-square displacement: MSD(t) = < [r(t) − r(t0)]2 > (Figure 1B and C, right). The MSD of each trajectory was fitted to the power law MSD(t) = a·tb. As we are interested in directional movements, we only kept the super-diffusive trajectories where b is >1. Further, to avoid false positives, such as short portions of diffusive movements that may appear as directed runs, we manually checked each track and discarded the trajectories shorter than 0.5 μm. This arbitrary threshold obviously biases the average distance covered by the DNA:QD complexes, but prevents us from unwanted artifacts. Eventually, the xy trajectories were projected along the major axis of displacement as described previously (22).
We examined the duration of the DNA:QD complexes movements and their speed from 157 independent directed trajectories extracted from 40 min of real-time videos (25 Hz). We found that the DNA:QD complexes move with an average speed v = 0.60 ± 0.06 μm/s (the error is the standard error of the mean) and a duration d = 2.0 ± 0.1 s. Those values are compatible with the typical velocity and duration exhibited by microtubule-associated molecular motors. Then, we incubated cells with the microtubule-depolymerizing agent nocodazole (see ‘Materials and Methods’ section): the fraction of directional DNA:QD motions decreased down to 2%, suggesting that DNA:QD intracellular movements require an intact microtubule network. To control whether the QDs alone were responsible for this movement, we analyzed the intracellular movement of naked QDs (not conjugated with the DNA). Even though more rare as compared with DNA:QD complexes, directed movements were also observed in the absence of DNA. This is probably because of an unspecific interaction between the streptavidin coating and some proteins or organelles in the cytoplasm. From a sample of 45 events, we observed that the QDs move faster in the absence of DNA (v = 1.04 ± 0.1 μm/s) and for shorter periods (1.3 ± 0.15 s). This difference in speed and residence time between naked and DNA-covered QDs may be occasioned by a different type of interaction with cytoplasmic proteins. However, because of the complexity of in cell experiments and the intrinsic low statistics of single-molecule observations, the in cell experiments were not sufficient to conclude that the active transport is specific of the DNA molecules. To rule out the possibility that the active transport of the DNA:QD complexes is due to an unspecific interaction of the DNA or the QD with a molecular motor or a vesicle or an organelle, we set-up an in vitro motility assay to evaluate the affinity between the DNA:QD complexes and the cytoskeleton.
Active DNA traffic is microtubule-dependent
We designed an in vitro assay to mimic the intracellular transport of the DNA molecules in a well-controlled environment to understand whether the DNA trafficking is specifically dependent on the cytoskeleton. The DNA molecules were first incubated with the CE to allow interactions of the biotinylated-DNA with the cytoplasmic proteins, then mixed with the streptavidin-QDs to form the complexes CyProt:DNA:QD (see ‘Materials and Methods’ section). The CyProt:DNA:QD complexes were isolated by centrifugation to remove the excess of CE. Simultaneously, we prepared a flow chamber coated either with microtubules or with actin filaments (see ‘Materials and Methods’ section) and passivated with either casein or BSA to reduce the non-specific binding of CyProt:DNA:QD complexes to the coverslip. The CyProt:DNA:QD complexes were diluted in the motility buffer (see ‘Materials and Methods’ section) and injected in the flow chamber. To discard the signal coming from the QDs diffusing in solution (far from the surface) and increase the signal-to-noise ratio, motility experiments were performed by TIRFM.
The interaction between the CyProt:DNA:QD complexes and the actin filaments was investigated in two chemical states: in the absence of nucleotides and with 2 mM of ADP. In none of these cases we observed any co-distribution between the CyProt:DNA:QD and the actin filaments. The observation is illustrated in the Figure 2A, where we do not observe any correlation between the positions of QDs (spots) and actin network (filaments). No motion was observed in the presence of ATP (2 mM), whereas in the same experimental conditions, the QDs covered with the actin-based myosin V molecules exhibited a clear directional motion (38). This result strongly indicates that the CyProt:DNA:QD complexes do not have any affinity for the actin filaments and suggests that the intracellular transport of cytoplasmic DNA fragments is independent of myosin activity.
Figure 2.

In-vitro system to mimic the intracellular DNA transport. (A) and (B) Study of the interaction between the cytoskeleton filaments and the CyProt:DNA:QD complexes. (A) Fluorescence image of an experiment with the actin filaments. The filaments were prepared with the fluorescent phalloidin (Atto 488). We used a high-pass emission filter system to observe the fluorescence of the filaments and of the QDs simultaneously. In this image, we observe no co-localization between the filaments and the spots. The scale is 5 micron. (B) Fluorescence image of an experiment with the microtubules. The microtubules are not fluorescent because they can be observed in RICM. In this fluorescent image, we observe the filaments (drawn in blue) covered by CyProt:DNA:QD complexes. The scale is 5 micron. Velocity (C) and processivity (D) distributions. We impose a cut-off of 0.5 micron for the processivity. (E) and (F) Two examples of motion of the CyProt:DNA:QD complexes along the microtubules in presence of ATP (1 mM). The trajectories are indicated in orange and red. Left: the sequences are extracted from two movies acquired at 25 frames per seconds. Right: MSD allows selecting only the directed trajectories along the filaments. In (E) b = 1.92 and in (F) b = 1.59. The scale bar is 1 micron in both sequences.
On the contrary, when the CyProt:DNA:QD complexes were incubated with microtubules, we observed a clear co-localization between spots and filaments. Figure 2B shows a large number of CyProt:DNA:QD complexes bound to the microtubules in the presence of a non-hydrolysable analog of ATP, the AMP.PNP (1 mM). The light-blue lines represent a fraction of the microtubules, which are visualized separately by reflection interference contrast microscopy (RICM). The fact that many spots stand along the filaments indicates a strong association between the CyProt:DNA:QD complexes and the microtubules. We noticed that this association is dependent on the ionic strength, as a higher strength (>150 mM NaCl) disrupts the interaction between the DNA complexes and the microtubules. As a control, we verified that QDs alone, without the DNA fragments, incubated in the same cytoplasmic extracts, do not co-localize with microtubules, excluding any non-specific interaction between the QDs and the cytoplasmic proteins that might induce an affinity for the microtubules. Moreover, when the DNA:QD complexes were incubated with microtubules in the absence of cytoplasmic proteins, we observed no interaction between the DNA:QD complexes and the microtubules, excluding any direct interaction between the DNA and the microtubules. CyProt:DNA:QD complexes were introduced in the flow chamber together with 1 mM ATP. Before each motility experiment, an image of the microtubules on the surface was acquired by RICM. We observed a large fraction (60%) of immobile CyProt:DNA:QD complexes, which are either bound to the microtubules or stuck to the surface. Conversely, 40% of the CyProt:DNA:QD complexes exhibited a clear movement (over a movie of 2 min). This ATP-based activity supports the hypothesis that the DNA fragments are actively transported along the microtubules by dynein or kinesin molecular motors. Among the 40% of moving spots, we distinguished three behaviors: (i) directional motion, typical of molecular motors (50%), (ii) confined diffusion (20%) and (iii) bidirectional motion, probably because of a competition of two or more molecular motors (30%). Examples of trajectories for each behavior are showed in Supplementary Data (Supplementary Figures S1–S4). In the following, we will focus on the unidirectional movements.
The motion of the CyProt:DNA:QD complexes along the microtubules was tracked to determine their trajectory (x,y)(t) (see ‘Materials and Methods’ section). We extracted the mean-square displacement MSD(t) = atb, as done for the particle tracked inside the HeLa cells (Figure 2E and F). The exponent b extracted from the MSD was used to select the directed trajectories. Among the whole set of trajectories, we selected the tracks for which the MSD exhibits an exponent b > 1.5 to reject the immobile spots (b = 0) and the purely diffusive ones (b = 1). We also discarded the trajectories shorter than ∼0.5 μm, as the MSD is affected by a large error for short tracks. A total of 784 tracks satisfied these criteria, and they are used to determine the average speed and processivity of the trajectories. We found an average velocity v = 1.32 ± 0.2 μm/s (Figure 2C), a mean processivity p = 0.80 ± 0.01 μm (Figure 2D) and a mean duration d = 0.84 ± 0.03 s.
We noticed that the velocity is twice higher in vitro than in cell, and the duration is lower in vitro than in cell. This could be due to the choice of the buffer for in vitro experiments that does not perfectly reproduce the cytosol in terms of for example ionic strength and pH.
These in vitro experiments on a reconstituted system show the implication of the microtubule network in the cytoplasmic transport of DNA fragments. Indeed, they suggest the implication of one or several molecular motors of kinesin or dynein families.
Identification of kinesin and dynein binding to DNA
To identify molecular motors interacting with the DNA fragments, we performed the co-purification experiments of biotinylated-DNA incubated with CE. Pre-cleared cytoplasmic lysates were mixed to the DNA molecules overnight, and then streptavidin-magnetic beads were added to the mix (see ‘Materials and Methods’ section). The proteins bound to the DNA fragments were separated according to their electrophoretic mobility by using the SDS–PAGE. Biotinylated-alkaline phosphatase and pre-cleared cytoplasmic lysates without DNA were used as control. Figure 3A shows an example of a gel. From the left: the alkaline phosphatase, the proteins in the control condition (CTRL) and in the co-purification with the DNA molecules (DNA).
Figure 3.
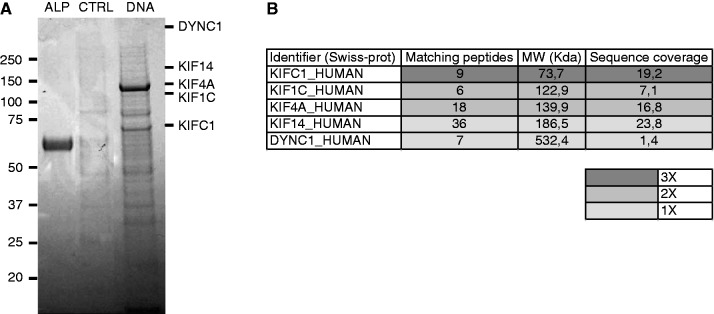
(A) and (B) Co-purification experiment. (A) Gel example. Proteins are loaded and separated by SDS–PAGE. Alkaline phosphatase (ALP), used as control of streptavidin-biotin bound, control using streptavidin-magnetic beads (CTRL) and proteins bound to the DNA. (B) Molecular motors identified by LC-MS/MS using comparative proteomics. Hits are represented according to the number of peptides identified, the Molecular Weight (MW) and the protein coverage and the number of times that they were found in all three.
Because of the large number of proteins involved, we decided to identify all the proteins both in control and DNA samples. Comparative proteomics is a powerful method to eliminate contaminant proteins obtained with affinity approaches. All proteins that were also found in the control condition were regarded as contaminants and eliminated from the hit list (Supplementary Table S1). In three independent screening experiments, five molecular motors were identified that were specifically enriched in the presence of DNA among hundreds of proteins specifically bound to DNA: three plus-directed kinesins (KIF1C, KIF4A and KIF14), the minus-directed C-terminal kinesin KIFC1 and the cytoplasmic dynein-1 (see Table in Figure 3B). Hits are represented according to the protein coverage and the number of times that they are found in the three experiments. Notice also that KIFC1 is the only molecular motor present in each co-purification. Thus, KIFC1 is the more probable candidate for the active motion of DNA fragments in the cytoplasm.
Interestingly, no myosin was found in the co-purification experiments. This result corroborates the in vitro assays and rules out any possible affinity between the double-stranded naked DNA and the actin filaments.
We note that the presence of minus-directed motors (C-terminal kinesin and dynein) is compatible with the fact that exogenous DNA will eventually reach the nucleus. Moreover, the three plus-end–directed kinesins (KIF1C, KIF4A and KIF14) could explain the small fraction of bidirectional motions observed in the in vitro experiments. As the kinesins sequences are better covered than dynein, we focused our attention on these molecular motors.
In vitro DNA transport is dependent on the kinesin KIFC1
To evaluate the implication in the intracellular DNA transport of the kinesins identified in co-purification experiments, we performed in vitro experiments on microtubules in presence and in absence of each kinesin. The DNA molecules were incubated with depleted cell lysates prepared as described in ‘Materials and Methods’ section and then mixed with the streptavidin QDs. Immunoprecipitation efficiency was controlled by western blot using specific antibodies for every kinesin (Figure 4A–D). The control test (CTRL) was performed using the cell extracts incubated with irrelevant rabbit anti-human IgG. The complexes obtained in control and depleted conditions were diluted in the motility buffer and injected in the flow chamber pre-coated with microtubules (see ‘Material and Methods’ section). The movements of the complexes were observed by TIRFM to discard the signal coming from the QDs in solution. The motions were tracked, and the trajectories were analyzed as described in the in vitro experiments (see ‘Materials and Methods’ section). We focused only on the directed motions selected by imposing the MSD exponent b > 1.5 and the trajectory length > 0.5 μm. Figure 4E shows the percentages of directed motions reported to the control test for every depleted condition. We observe that KIFC1 significantly influences the motion of DNA fragments along the microtubules, with a reduction of 74 ± 26% of the DNA motion in the absence of KIFC1. The immunodepletion of the three other kinesins (KIF1C, KIF4A and KIF14) also induces a reduction of the DNA motility in the order of 30%. Considering the large dispersion between different runs (also in the order of ±30%), this reduction cannot be considered as statistically relevant. Nevertheless, a possible role of these kinesins in the transport of cytoplasmic DNA cannot be ruled out a priori.
Figure 4.

(A–D) Immunoprecipitation of HeLa cell lysates using rabbit anti-human IgG as a control (CTRL) or antibodies against the four kinesins. Input and immunoprecipitated materials are revealed by western blot using specific antibodies for the four kinesins. (E) Directed movements observed in the control test and in depleted tests reported to the control test.
These results suggest the possible involvement of the minus-directed kinesin KIFC1 in the intracellular transport of DNA.
Kinesin KIFC1 co-localizes with DNA molecules in vitro
Using FRET microscopy, we investigated the co-localization between the kinesin KIFC1 and DNA molecules in vitro. Complexes DNA/cytoplasmic proteins were prepared by mixing fluorescently labeled DNA-Cy3 molecules with CE (see ‘Materials and Methods’ section). These complexes were incubated with microtubules in a flow chamber, in presence of 1 mM of AMP–PNP. Afterwards, the KIFC1 proteins were labeled with anti-KIFC1 primary antibody and a fluorescent secondary antibody (Alexa488). To verify that we do not observe any signal coming from the secondary antibody in the FRET channel, we performed a control experiment without DNA: KIFC1 was revealed by immunofluorescence as described in ‘Materials and Methods’ section. Figure 5 shows an example of FRET image. On the left is the fluorescein isothiocyanate (FITC) channel to visualize the secondary antibody binding KIFC1; on the right is the FRET channel. We do not observe any signal in the FRET channel in absence of DNA (Figure 5, top). On the contrary, in presence of DNA (Figure 5, bottom), we observe a FRET signal of DNA-Cy3 molecules excited from the secondary antibody, supporting the hypothesis of a direct interaction between KIFC1 and DNA molecules.
Figure 5.
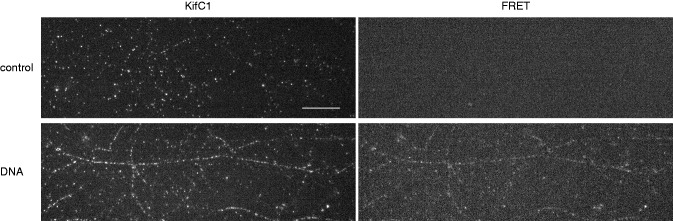
Immunofluorescence co-localization experiment. Top: control experiment is performed in absence of DNA molecules. Kinesin KIFC1 bound to the microtubules is visualized in the FITC channel (image on the left). On the right is the FRET channel to confirm that there is no fluorescence passage from KIFC1. Bottom: DNA experiment is performed by using a DNA marked with Cy3. On the left is the KIFC1 in the FITC channel. On the right we observe the DNA molecules excited by the secondary antibody bound to KIFC1. Scale bar is 10 μm.
DISCUSSION
We studied the transport of exogenous DNA fragments in the cytoplasm of human cells using a synergistic approach that includes (i) intracellular tracking of naked DNA molecule, (ii) the characterization of this motion in a reconstituted in vitro assay that mimics the cytoplasmic transport of DNA molecules and (iii) an affinity co-purification assay to find the candidate molecules interacting with the bare DNA, among the large number of cytoplasmic molecular motors (39–41). Our results are in good agreement with the observations by Vaughan and Dean (9), which show the implication of a motor directed towards the minus-end of the microtubules.
MS analysis points to the possible implication of five molecular motors: two molecules belonging to the kinesin-3 family, the KIF1C that participates in the retrograde vesicular transport from the Golgi apparatus to the endoplasmic reticulum (42) and the KIF14 that is involved in the intracellular organelles transport (43) and in the alignment and congression of the chromosomes (44). KIF4A (member of kinesin-4 family) is a chromo-kinesin involved in the chromosomes alignment in the spindle midzone. KIF4A contains an NLS and a DNA-binding cargo domain in the sequence allowing a direct interaction with the DNA (45,46). Recently, Wu et al. (47) also showed the implication of KIF4A in DNA damage response. Thus, its role in the DNA transport cannot be excluded, even though we find its presence only in one co-purification experiment. The cytoplasmic dynein-1 is also identified by the heavy chain DYNH1C1 (48). It is a minus-directed motor implicated in different cellular processes, such as chromosomes segregation (3,49), virus retrograde transport (50,51) and the transport of the DNA molecules containing a NLS (10,52) or a DTS (9). For these reasons, it also might be involved in the transport of DNA fragments in the cytoplasm. Eventually, MS analyses always reveal the presence of the C-terminal kinesin KIFC1 (member of the kinesin-14 family). This is the only molecular motor found in the three co-purification experiments. During spermatogenesis, the motor KIFC1 participates to the formation of the acrosome and is associated with nuclear transport factors, such as the importin-β (53). Similar to the cytoplasmic dynein-1, the kinesin KIFC1 moves towards the minus-end of microtubules and is mostly associated to vesicular structures. As a result, KIFC1 proteins are generally localized in the perinuclear region. From our perspective, it is also interesting to note that the protein KIFC1 is involved in chromosome alignment and congression (44) and has a specific affinity for nucleoporins NUP50 and NUP153 (54). This suggests that KIFC1 may have a post-mitotic role because of this double affinity for nuclear pores and DNA molecules, either directly or mediated by DNA-associated proteins. Moreover, Nath et al. (55) found that KIFC1 is required for the motility of early endocytic vesicles in mouse liver. The authors observed a mean velocity of (0.60 ± 0.04) μm/s, which is in agreement with the average velocity of DNA in cell. Even though studies on the human kinesin KIFC1 are still limited, KIFC1 is homologous to the Drosophila Ncd protein (53,56–59,60,61), a more widely investigated minus-ended kinesin. Single-molecule experiments on Ncd fragments show that this protein exhibits a low-duty ratio (59) with a consequent weak processivity (56). An apparent processivity is obtained when several Ncd molecules are simultaneously bound to the same microtubule and work in a coordinated manner (57). Such a variable processivity may explain the low number of directed movements observed in our in cell and in vitro experiments, where most of the DNA:QD complexes either diffuse or transiently bind to the microtubules without moving. In fact, only the DNA:QD complexes with several active KIFC1 molecules would processively move along the microtubules. A recent study on single–GFP-Ncd molecules observed by TIRFM shows that ∼50% of the full-length Ncd molecules remain attached to the microtubules, probably via a microtubule-binding site in the N-terminal tail domain (60). This study also highlights that Ncd behavior strongly depends on the ionic strength: >55 mM of monovalent salt, the Ncd proteins diffuse along the microtubules with small bias towards the minus-end. This finding is in good agreement with the salt-dependent affinity of the DNA:QD complexes for the microtubules observed in our in vitro assay; however, a much lower velocity is found in vitro for Ncd (which might be due also to different experimental conditions). Fink et al. (61) confirm the role of the tail domain in the diffusive motion of Ncd proteins and the motor activity linked to the head domain. The evidence of a passive interaction between the Ncd tail and the microtubules is compatible with our in vitro observations, where most of the DNA:QD complexes remain bound to the microtubules without moving or with a locally confined diffusive movement. All those experimental evidences point to a possible implication of the human kinesin KIFC1 in the transport of naked double-stranded DNA towards the minus-end of microtubules. It also indicates that the activity of KIFC1 may be essential to carry exogenous DNA fragments to the cell nucleus in gene therapy or other therapies based on the delivery of nucleic acids.
In conclusion, in this work, we have shown how the combined effort of in cell single-particle tracking, in vitro reconstituted system and proteomics has allowed us to identify one of the possible DNA transport mechanisms in the cytoplasm. In the future, the role of microtubule-associated proteins, cytoplasmic receptors and effectors of the motor will need to be identified. The technique presented here constitutes a new and promising approach to investigate the intriguing and broad field of intracellular transport.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–4.
FUNDING
EU FW7 NMP programme (CAMINEMS project); Agence Nationale pour la Recherche (DYNREC project); Association pour la Recherche sur le Cancer. Funding for open access charge: Agence Nationale pour la Recherche.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Maxime Deforet for his help with single-particle tracking and Wolfgang Faigle for his initial contribution to MS experiments. They also thank Aurélien Roux, Fabien Montel and Karine Guevorkian for the useful discussions and critical reading of the manuscript. They thank Maria-Isabel Yuseff for having kindly read and corrected the manuscript. The authors greatly acknowledge the Nikon Imaging Centre and the imaging platform Pict-IBiSA at Institut Curie-CNRS.
REFERENCES
- 1.Heald R. Motor function in the mitotic spindle. Cell. 2000;102:399–402. doi: 10.1016/s0092-8674(00)00044-1. [DOI] [PubMed] [Google Scholar]
- 2.Mazumdar M, Misteli T. Chromokinesins: multitalented players in mitosis. Trends Cell Biol. 2005;15:349–355. doi: 10.1016/j.tcb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Yang Z, Tulu US, Wadsworth P, Rieder CL. Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr. Biol. 2007;17:973–980. doi: 10.1016/j.cub.2007.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunet S, Vernos I. Chromosome motors on the move. From motion to spindle checkpoint activity. EMBO Rep. 2001;2:669–673. doi: 10.1093/embo-reports/kve158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tokai N, Fujimoto-Nishiyama A, Toyoshima Y, Yonemura S, Tsukita S, Inoue J, Yamamota T. Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 1996;15:457–467. [PMC free article] [PubMed] [Google Scholar]
- 6.Patil SD, Rhodes DG, Burgess DJ. DNA-based therapeutics and DNA delivery systems: a comprehensive review. AAPS J. 2005;7:E61–E77. doi: 10.1208/aapsj070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutreix M, Cosset JM, Sun JS. Molecular therapy in support to radiotherapy. Mutat. Res. 2010;704:182–189. doi: 10.1016/j.mrrev.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan EE, Dean D. Intracellular trafficking of plasmids during transfection is mediated by microtubules. Mol. Ther. 2006;13:422–428. doi: 10.1016/j.ymthe.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salman H, Abu-Arish A, Oliel S, Loyter A, Klafter J, Granek R, Elbaum M. Nuclear localization signal peptides induce molecular delivery along microtubules. Biophys. J. 2005;89:2134–2145. doi: 10.1529/biophysj.105.060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukacs GL, Haggie P, Seksek O, Lechardeur D, Freedman N, Verkman AS. Size-dependent DNA mobility in cytoplasm and nucleus. J. Biol. Chem. 2000;275:1625–1629. doi: 10.1074/jbc.275.3.1625. [DOI] [PubMed] [Google Scholar]
- 12.Dauty E, Verkman AS. Actin cytoskeleton as the principal determinant of size-dependent DNA mobility in cytoplasm: a new barrier for non-viral gene delivery. J. Biol. Chem. 2005;280:7823–7828. doi: 10.1074/jbc.M412374200. [DOI] [PubMed] [Google Scholar]
- 13.Lechardeur D, Sohn KJ, Haardt M, Joshi PB, Monck M, Graham RW, Beatty B, Squire J, O’Brodovich H, Lukacs GL. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6:482–497. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 14.Dutreix M, Sun JS. Dbait and uses thereof. Patent US7476729 B2. 2009 [Google Scholar]
- 15.Schlegel A, Buhler C, Devun F, Agrario C, Urien S, Lokiec F, Sun JS, Dutreix M. Pharmacokinetics and toxicity in rats and monkeys of coDbait: a therapeutic double-stranded DNA oligonucleotide conjugated to cholesterol. Mol. Ther. Nucleic Acids. 2012;1:1–10. doi: 10.1038/mtna.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, Alberti C, Josserand V, Coll JL, Sastre-Garau X, et al. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin. Cancer Res. 2009;15:1308–1316. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- 17.Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PloS One. 2009;4:1–11. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahan M, Lévi S, Luccardini C, Rostaing P, Riveau B, Triller A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 19.Yang W, Gelles J, Musser SM. Imaging of single-molecule translocation through nuclear pore complexes. PNAS. 2004;101:12887–92. doi: 10.1073/pnas.0403675101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courty S, Luccardini C, Bellaiche Y, Cappello G, Dahan M. Tracking individual kinesin motors in living cells using single quantum-dot imaging. Nano Lett. 2006;6:1491–1495. doi: 10.1021/nl060921t. [DOI] [PubMed] [Google Scholar]
- 22.Pierobon P, Achouri S, Courty S, Dunn AR, Spudich JA, Dahan M, Cappello G. Velocity, processivity, and individual steps of single myosin V molecules in live cells. Biophys. J. 2009;96:4268–4275. doi: 10.1016/j.bpj.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierobon P, Cappello G. Quantum dots to tail single bio-molecules inside living cells. Adv. Drug Deli. Rev. 2012;64:167–178. doi: 10.1016/j.addr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Quanz M, Herbette A, Sayarath M, De Koning L, Dubois T, Sun JS, Dutreix M. Heat shock protein 90α (Hsp90α) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 2012;287:8803–8815. doi: 10.1074/jbc.M111.320887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schütz GJ, Kada G, Pastushenko VP, Schindler H. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 2000;19:892–901. doi: 10.1093/emboj/19.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iino R, Koyama I, Kusumi A. Single molecule imaging of green fluorescent proteins in living cells: e-cadherin forms oligomers on the free cell surface. Biophys. J. 2001;80:2667–2677. doi: 10.1016/S0006-3495(01)76236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seisenberger G, Ried MU, Endress T, Büning H, Hallek M, Bräuchle C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- 28.Deich J, Judd EM, McAdams HH, Moerner WE. Visualization of the movement of single histidine kinase molecules in live Caulobacter cells. PNAS. 2004;101:15921–15926. doi: 10.1073/pnas.0404200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson SR, Ali MY, Trybus KM, Warshaw DM. Random walk of processive, quantum dot-labeled myosin Va molecules within the actin cortex of COS-7 cells. Biophys. J. 2009;97:509–518. doi: 10.1016/j.bpj.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowe AR, Siegel JJ, Kalab P, Siu M, Weis K, Liphardt JT. Selectivity mechanism of the nuclear pore complex characterized by single cargo tracking. Nature. 2010;468:600–603. doi: 10.1038/nature09285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajan SS, Liu HY, Vu TQ. Ligand-bound quantum dot probes for studying the molecular scale dynamics of receptor endocytic trafficking in live cells. ACS Nano. 2008;2:1153–1166. doi: 10.1021/nn700399e. [DOI] [PubMed] [Google Scholar]
- 32.Yoo J, Kambara T, Gonda K, Higuchi H. Intracellular imaging of targeted proteins labeled with quantum dots. Exp. Cell Res. 2008;314:3563–3569. doi: 10.1016/j.yexcr.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Okada CY, Rechsteiner M. Introduction of macromolecules into cultured mammalian cells by osmotic lysis of pinocytic vesicles. Cell. 1982;29:33–41. doi: 10.1016/0092-8674(82)90087-3. [DOI] [PubMed] [Google Scholar]
- 34.Clemen AE, Vilfan M, Jaud J, Zhang J, Bärmann M, Rief M. Force-dependent stepping kinetics of myosin-V. Biophys. J. 2005;88:4402–4410. doi: 10.1529/biophysj.104.053504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sergé A, Bertaux N, Rigneault H, Marguet D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods. 2008;5:687–694. doi: 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- 36.Crocker J, Grier DG. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 1996;179:298–310. [Google Scholar]
- 37.Poullet P, Carpentier S, Barillot E. myProMS, a web server for management and validation of mass spectrometry-based proteomic data. Proteomics. 2007;7:2553–2556. doi: 10.1002/pmic.200600784. [DOI] [PubMed] [Google Scholar]
- 38.Cappello G, Pierobon P, Symonds C, Busoni L, Gebhardt JCM, Rief M, Prost J. Myosin V stepping mechanism. PNAS. 2007;104:15328–15333. doi: 10.1073/pnas.0706653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 40.Höök P, Vallee RB. The dynein family at a glance. J. Cell Sci. 2006;119:4369–4371. doi: 10.1242/jcs.03176. [DOI] [PubMed] [Google Scholar]
- 41.Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. PNAS. 2006;103:3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorner C, Ciossek T, Müller S, Møller PH, Ullrich A, Lammers R. Characterization of KIF1C, a new kinesin-like protein involved in vesicle transport from the Golgi apparatus to the endoplasmic reticulum. J. Biol. Chem. 1998;273:20267–20275. doi: 10.1074/jbc.273.32.20267. [DOI] [PubMed] [Google Scholar]
- 43.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–476. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Zhu C, Zhao J, Bibikova M, Leverson JD, Bossy-wetzel E, Fan J, Abraham RT, Jiang W, Jolla L, Diego S, et al. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. Cell. 2005;16:3187–3199. doi: 10.1091/mbc.E05-02-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SZ, Adler R. Chromokinesin: a DNA-binding, kinesin-like nuclear protein. J. Cell Biol. 1995;128:761–768. doi: 10.1083/jcb.128.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazumdar M, Sundareshan S, Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J. Cell Biol. 2004;166:613–620. doi: 10.1083/jcb.200401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu G, Zhou L, Khidr L, Guo XE, Kim W, Lee YM, Krasieva T, Chen PL. A novel role of the chromokinesin Kif4A in DNA damage response. Cell Cycle. 2008;7:2013–2020. doi: 10.4161/cc.7.13.6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfister KK, Shah PR, Hummerich H, Russ A, Cotton J, Annuar AA, King SM, Fisher EMC. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1. doi: 10.1371/journal.pgen.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharp DJ, Rogers GC, Scholey JM. Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat. Cell Biol. 2000;2:922–930. doi: 10.1038/35046574. [DOI] [PubMed] [Google Scholar]
- 50.Suomalainen M, Nakano MY, Keller S, Boucke K, Stidwill RP, Greber UF. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leopold PL, Kreitzer G, Miyazawa N, Rempel S, Pfister KK, Rodriguez-Boulan E, Crystal RG. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum. Gene Ther. 2000;165:151–165. doi: 10.1089/10430340050016238. [DOI] [PubMed] [Google Scholar]
- 52.Mesika A, Kiss V, Brumfeld V, Ghosh G, Reich Z. Enhanced intracellular mobility and nuclear accumulation of DNA plasmids associated with a karyophilic protein. Hum. Gene Ther. 2005;16:200–208. doi: 10.1089/hum.2005.16.200. [DOI] [PubMed] [Google Scholar]
- 53.Yang WX, Sperry AO. C-terminal kinesin motor KIFC1 participates in acrosome biogenesis and vesicle transport. Biol. Reprod. 2003;69:1719–1729. doi: 10.1095/biolreprod.102.014878. [DOI] [PubMed] [Google Scholar]
- 54.Maliga Z, Junqueira M, Toyoda Y, Ettinger A, Mora-Bermúdez F, Klemm RW, Vasilj A, Guhr E, Ibarlucea-Benitez I, Poser I, et al. A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat. Cell Biol. 2013;15:325–334. doi: 10.1038/ncb2689. [DOI] [PubMed] [Google Scholar]
- 55.Nath S, Bananis E, Sarkar S, Stockert RJ, Sperry AO, Murray JW, Wolkoff AW. Kif5B and Kifc1 interact and are required for motility and fission of early endocytic vesicles in mouse liver. Mol. Biol. Cell. 2007;18:1839–1849. doi: 10.1091/mbc.E06-06-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allersma MW, Gittes F, DeCastro MJ, Stewart RJ, Schmidt CF. Two-dimensional tracking of ncd motility by back focal plane interferometry. Biophys. J. 1998;74:1074–1085. doi: 10.1016/S0006-3495(98)74031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stewart RJ, Semerjian J, Schmidt CF. Highly processive motility is not a general feature of the kinesins. Eur. Biophys. J. 1998;27:353–360. doi: 10.1007/s002490050142. [DOI] [PubMed] [Google Scholar]
- 58.deCastro MJ, Fondecave RM, Clarke LA, Schmidt CF, Stewart RJ. Working strokes by single molecules of the kinesin-related microtubule motor ncd. Nat. Cell Biol. 2000;2:724–729. doi: 10.1038/35036357. [DOI] [PubMed] [Google Scholar]
- 59.Pechatnikova E, Taylor EW. Kinetics processivity and the direction of motion of Ncd. Biophys. J. 1999;77:1003–1016. doi: 10.1016/S0006-3495(99)76951-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furuta K, Toyoshima YY. Minus-end-directed motor Ncd exhibits processive movement that is enhanced by microtubule bundling in vitro. Curr. Biol. 2008;18:152–157. doi: 10.1016/j.cub.2007.12.056. [DOI] [PubMed] [Google Scholar]
- 61.Fink G, Hajdo L, Skowronek KJ, Reuther C, Kasprzak AA, Diez S. The mitotic kinesin-14 Ncd drives directional microtubule-microtubule sliding. Nat. Cell Biol. 2009;11:717–723. doi: 10.1038/ncb1877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.