

Abstract
Fanconi anemia (FA) is a rare genomic instability disorder characterized by progressive bone marrow failure and predisposition to cancer. FA-associated gene products are involved in the repair of DNA interstrand crosslinks (ICLs). Fifteen FA-associated genes have been identified, but the genetic basis in some individuals still remains unresolved. Here, we used whole-exome and Sanger sequencing on DNA of unclassified FA individuals and discovered biallelic germline mutations in ERCC4 (XPF), a structure-specific nuclease-encoding gene previously connected to xeroderma pigmentosum and segmental XFE progeroid syndrome. Genetic reversion and wild-type ERCC4 cDNA complemented the phenotype of the FA cell lines, providing genetic evidence that mutations in ERCC4 cause this FA subtype. Further biochemical and functional analysis demonstrated that the identified FA-causing ERCC4 mutations strongly disrupt the function of XPF in DNA ICL repair without severely compromising nucleotide excision repair. Our data show that depending on the type of ERCC4 mutation and the resulting balance between both DNA repair activities, individuals present with one of the three clinically distinct disorders, highlighting the multifunctional nature of the XPF endonuclease in genome stability and human disease.
Main Text
Fanconi anemia (FA) is characterized by bone marrow failure (BMF), congenital malformations, hypersensitivity to DNA interstrand crosslink (ICL)-inducing agents, chromosome fragility, and a high susceptibility to cancer. Since the discovery of the first FA-associated gene 20 years ago, all together, 15 genes associated with FA have been identified; these include FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, FANCE, FANCF, FANCG (XRCC9), FANCI, FANCJ (BRIP1), FANCL (PHF9), FANCM, FANCN (PALB2), FANCO (RAD51C), and FANCP (SLX4)1,2 (MIM 227650, 300514, 227645, 605724, 227646, 600901, 603467, 614082, 609053, 609054, 614083, 614087, 610832, 613390, and 613951, respectively). Studies to unravel the genetic basis of this rare disorder uncovered a genome-maintenance pathway that protects dividing cells against replication-blocking DNA lesions. To identify additional FA-associated genes, we used the SOLiD 4 platform for whole-exome sequencing on peripheral-blood DNA from a Spanish FA individual (FA104) who was previously excluded from all known FA complementation groups (this study was approved by the Institutional Committee on Ethical Research in Human Samples, and proper informed consent was obtained). FA104 was born to unrelated parents and was diagnosed neonatally with a malformative syndrome suggestive of FA, the symptoms of which included bilateral absent thumbs, microsomy, esophageal atresia, a ventrally translocated anus, and dysplastic and low-set ears. She did not show any dermatological abnormality such as skin hyperpigmentation, photosensitivity, sunlight-induced scarring, or atrophy. FA104 developed BMF at the age of 2 years and died as a result of a hemorrhagic shock after bone marrow transplantation at the age of 4 years. A positive chromosome-breakage test unambiguously confirmed the FA diagnosis: 92% of the cells showed on average 4.4 diepoxybutane (DEB)-induced breaks. Lymphoblasts from this individual were hypersensitive to mitomycin-C (MMC) and melphalan but were insensitive to the topoisomerase I inhibitor camptothecin and the PARP inhibitor KU58948 (data not shown) and showed normal FANCD2 monoubiquitination and RAD51 focus formation.3 This suggests a defect downstream within the FA pathway, which does not involve homologous recombination. On the basis of a recessive mode of inheritance, exome sequencing identified 17 candidate disease genes for FA104 (Table S1, available online); of these, ERCC4 (MIM 133520; also known as XPF) immediately caught our attention given the involvement of the XPF-ERCC1-structure-specific nuclease in ICL repair.4 Both ERCC4 mutations were predicted to be pathogenic: a 5 bp deletion in exon 8 (c.1484_1488delCTCAA) was predicted to lead to a frameshift and a premature stop codon (p.Thr495Asnfs*6), and a missense mutation in exon 11 (c.2065C>A [p.Arg689Ser]; RefSeq accession numbers NG_011442.1, NM_005236.2, and NP_005227.1) was predicted to change a highly conserved arginine within the nuclease active site of XPF. Sanger sequencing on blood DNA confirmed these mutations (Figure 1A) and their correct segregation (data not shown). In MMC-resistant FA104 lymphoblasts (FA104R) obtained after long-term exposure to a low dose of MMC, we detected a mutation that restored the ERCC4 reading frame (Figure S1A), supporting the notion that MMC sensitivity is due to ERCC4 mutations. Consistently, XPF levels were reduced in FA104 lymphoblasts but were normalized in the reverted FA104R lymphoblasts (Figure S1B). Immunoblotting did not detect a truncated XPF, indicating that only the p.Arg689Ser altered XPF was present in the FA104 cell line.
Figure 1.
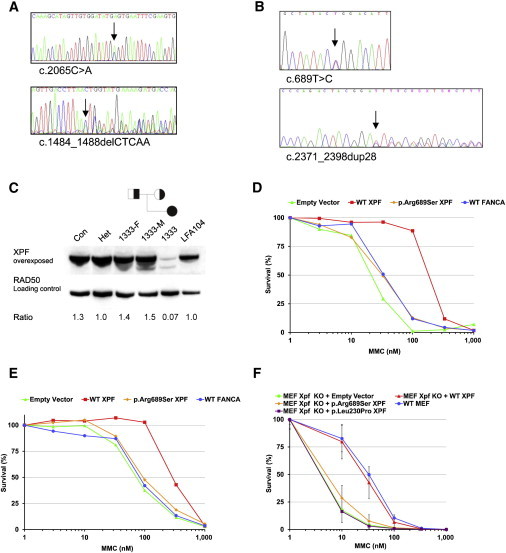
ERCC4 Mutations and XPF Deficiency in FA Individuals
(A) Sequence analysis of blood DNA from FA104 revealed a missense mutation in exon 11 (c.2065C>A [p. Arg689Ser]) (upper panel) and a 5 bp deletion in exon 8 leading to a frameshift and premature termination of translation (c.1484_1488delCTCAA [p. Thr495Asnfs*6]) (lower panel).
(B) Sequence analysis of blood DNA from 1333 revealed a missense mutation in exon 4 (c.689T>C [p.Leu230Pro]) (upper panel) and a 28 bp duplication in exon 11 (lower panel) leading to a frameshift and a premature stop codon (c.2371_2398dup28 [p.Ile800Thrfs*24]).
(C) Immunoblot analysis showing XPF expression in lymphoblasts from 1333 and FA104. Lymphoblasts from a healthy individual (Con), the parents of 1333 (1333-F and 1333-M), and an unrelated ERCC4 mutation carrier (Het) were used as controls. XPF levels are expressed as a ratio relative to the loading control (RAD50).
(D) Genetic complementation of MMC sensitivity in FA104 lymphoblasts by wild-type XPF, but not by p.Arg689Ser altered XPF. Site-directed mutagenesis was used for introducing point mutations into the pWPXL-XPF hemagglutinin (HA)-tagged plasmid with the QuickChange method (Stratagene) as described.5 Lentiviral supernatant production and transduction were done as previously described,6 and cells were grown for 10 days in the presence of MMC. Data represent a typical result of at least three independent experiments.
(E) Genetic complementation of MMC sensitivity of 1333 lymphoblasts by wild-type XPF (experiments were performed as in D).
(F) MMC-induced growth inhibition of Ercc4-knockout MEFs transduced with lentiviral particles coding for GFP (negative control vector), wild-type XPF, and p.Arg689Ser and p.Leu230Pro altered XPF. Data represent means and SD of at least three independent experiments.
Sanger sequencing on 18 unclassified FA individuals from Germany revealed biallelic ERCC4 mutations in another individual (1333). Individual 1333 was born in 2002 and was unambiguously diagnosed with FA at the age of 5 years as a result of multiple FA-related features, such as perinatal growth retardation, short stature, pronounced microcephaly, café-au-lait spots, an ostium-primum defect, biliary atresia with fibrosis of the liver, BMF, and a positive chromosome-fragility test (0.2, 6.7, and 9.4 breaks per cell at 0, 50, and 100 ng/ml MMC, respectively). Individual 1333 is redheaded and has pale skin color, but no spontaneous or UV-light-induced skin lesions were reported at the age of 10 years. Similar to those of FA104, lymphoblasts from individual 1333 were normal with regard to FANCD2 monoubiquitination and RAD51 focus formation and were sensitive to MMC and melphalan but insensitive to the topoisomerase I inhibitor camptothecin and to the PARP inhibitor KU58948 (data not shown). Individual 1333 carries a 28 bp duplication in exon 11 of the maternal allele (c.2371_2398dup28 [p.Ile800Thrfs*24]; Figure 1B), and this duplication is predicted to result in a truncated XPF that lacks the double helix-hairpin-helix (HhH2) domain involved in heterodimerization with ERCC1 and DNA binding.7 The paternal allele contains a missense mutation that changes a highly conserved amino acid residue within the helicase-like domain (c.689T>C [p. Leu230Pro]; Figure 1B). Immunoblot analysis showed that a missense altered XPF and a truncated 90–95 kDa XPF are present at very low levels (Figure 1C). As expected, the truncated XPF was undetectable with an antibody against the C-terminal HhH2 domain of XPF (amino acids 866–916, data not shown). Interestingly, the truncated XPF was absent in a MMC-resistant lymphoblastoid cell line (1333R) generated by long-term exposure to MMC, and near-normal XPF levels were detected in this reverted cell line (Figure S1C). PCR amplification and sequence analysis revealed that the 28 bp duplication had disappeared in 1333R (Figure S1D) and had thus restored the wild-type sequence. Both the inherited duplication and the somatic reversion might have been triggered by an inverted 5 bp repeat flanking the region.
Genetic complementation of MMC sensitivity in lymphoblasts from both FA individuals was achieved by lentiviral transduction of wild-type ERCC4 cDNA (Figures 1D and 1E). In addition, we expressed wild-type and mutant human ERCC4 cDNAs in embryonic fibroblasts (MEFs) from Ercc4 (Xpf)-null mice. We found that ectopic expression of ERCC4 mutants encoding p.Leu230Pro and p.Arg689Ser did not complement MMC sensitivity of these MEFs (Figure 1F), providing additional evidence that the ERCC4 missense mutations found in both FA individuals inactivate XPF. The genetic and functional data show that mutations in ERCC4 cause FA in two unrelated nonconsanguineous individuals. Because mutations in ERCC4 cause an additional FA subtype (FA-Q), we propose FANCQ as an alias for ERCC4.
ERCC4 mutations have been linked to the skin-photosensitive and nucleotide excision repair (NER)-deficient disorders xeroderma pigmentosum (XP [MIM 278700, 610651, 278720, 278730, 278740, 278760, 278780, and 278750])8 and XFE progeroid syndrome (MIM 610965),9 and we therefore tried to understand why the identified ERCC4 variants specifically lead to FA. We hypothesized that these mutants cause an FA phenotype because of a strong deficiency in ICL repair but have sufficient NER activity to prevent clinically relevant skin photosensitivity and other NER-related features. Compared to an XP complementation group C (XP-C) lymphoblast line, FA104 lymphoblasts were indeed not sensitive to UVC light (Figure 2A). Given that UV-light survival experiments are challenging in lymphoblastoid cell lines, we studied skin fibroblasts from individual 1333 (FA104 fibroblasts were not available) and found that the UV-light sensitivity in FA individual 1333 was milder than that in XP complementation group F (XP-F) individual XP2YO (Figure 2B). In addition, the FA-specific XPF alterations p.Leu230Pro and p.Arg689Ser rescued 100% of the UVC sensitivity of XP2YO fibroblasts (Figure S2A) and approximately 80% of the UVC-light sensitivity of Ercc4-null MEFs (Figure S2B) but were both unable to complement MMC sensitivity (Figure 1F). Furthermore, XFE and 1333 fibroblasts responded typically like FA cells upon MMC-induced survival (Figure 2C), DEB-induced chromosome breakage (Figure 2D), and MMC-induced G2-phase arrest (Figure 2E), whereas XP-F cells showed milder MMC sensitivity and lacked DEB-induced chromosome fragility and MMC-induced cell-cycle arrest (Figure 2C–2E). Previous experiments in Chinese hamster ovary cells also demonstrated that the XFE-specific p.Arg153Pro altered XPF does not rescue MMC or UV-light sensitivity.11 Therefore, we conclude that XP, XFE, and FA cells with ERCC4 mutations clearly have a distinct response to UV light and MMC (Table S2).
Figure 2.
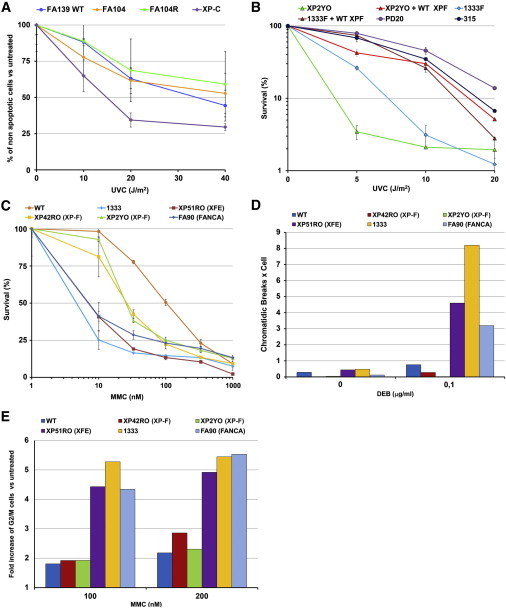
UV-Light and ICL Sensitivities of ERCC4 Mutants Leading to FA
(A) UVC-light-induced apoptosis in FA104 lymphoblasts. Cells were analyzed for UVC-light-induced apoptosis 24 hr after irradiation with the use of the Annexin-V-FLUOS Staining Kit (Roche). Data represent means and SD of at least three independent experiments.
(B) UVC-light-induced growth inhibition of human ERCC4-deficient immortal fibroblast cell lines (from XP individual XP2YO and FA individual 1333) transduced with lentiviral particles carrying cDNA coding for wild-type XPF. The results are expressed as a percentage of viable UVC-light-treated cells relative to untreated controls. Data represent means and SD of two independent experiments.
(C) MMC sensitivity of human ERCC4-deficient primary fibroblasts from XP, FA, and XFE individuals (XP42RO, 1333, and XP51RO, respectively). Data represent means and SD of two independent experiments.
(D) DEB-induced chromosome-fragility test in human ERCC4-deficient primary fibroblasts from XP, FA, and XFE individuals (XP42RO, 1333, and XP51RO, respectively).
(E) MMC-induced G2/M cell-cycle arrest in the same cells as in (D). Experiments presented in (D) and (E) were performed as reported earlier.10
To further investigate the extent of NER deficiency in the FA-affected individuals, we measured UV-light-induced unscheduled DNA synthesis (UDS) in primary skin fibroblasts from individual 1333 and from an XP-F individual (XP42RO) with mild clinical UV-light sensitivity and found 24 ± 4% and 21 ± 3% residual UDS activity, respectively (Figure 3A). We also determined UDS in Ercc4-null MEFs expressing the FA-specific XPF alterations p.Leu230Pro or p.Arg689Ser. The levels of UDS activity were 39.7% and 48.4% of the normal mean for p.Leu230Pro and p.Arg689Ser altered XPF, respectively (Figure 3B), enough to complement 80% of UVC-light sensitivity of these MEFs (Figure S2B). In XPF-deficient human XP2YO fibroblasts, p.Leu230Pro and p.Arg689Ser altered XPF rather efficiently corrected the defective removal of 6-4 photoproducts (PPs) at sites of local UV damage (Figure 3C). In contrast, XP2YO cells expressing the ERCC4 mutant with the 28 bp duplication were completely deficient in NER activity, as predicted from the disruption of the ERCC1- and DNA-binding domain of this truncated protein. The studies presented in Figures 2 and 3 demonstrate that FA cells with ERCC4 mutations are fully deficient in ICL repair but retain significant levels of NER activity.
Figure 3.
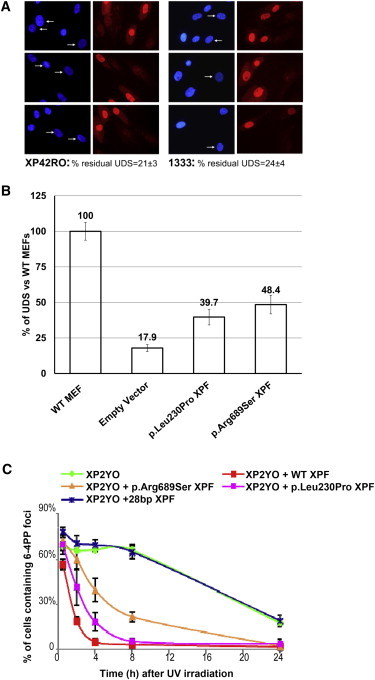
NER Analysis of ERCC4 Mutants In Vivo
(A and B) In primary fibroblasts, unscheduled DNA synthesis (UDS) representing global NER activity was measured with 5-ethynyl-deoxyuridine grossly as previously described.12
(A) XP-F (XP42RO) and FA (1333) cells (arrows) were compared to mixed-in normal fibroblasts preloaded with polystyrene microbeads (no arrows), used as an internal control. UDS signal was quantified from 20–40 random XP-F or FA G1/G2 nuclei and expressed as a percentage of control wild-type cells.
(B) UDS signals in Ercc4−/− MEFs measured as in (A) are expressed as a percentage of control wild-type MEFs. Ercc4−/− cells were stably expressing an empty vector or one of various ERCC4 cDNAs (wild-type or encoding p.Leu230Pro or p.Arg689Ser).
(C) Repair kinetics of UV-light-induced DNA damage by FA-specific ERCC4 mutants in ERCC4- and NER-deficient human cells (XP2YO). Cells expressing wild-type XPF, p.Arg689Ser or p.Leu230Pro altered XPF, or XPF resulting from the 28 bp duplication were locally irradiated with UV light, cultured for the indicated times, fixed and stained for 6-4 PPs, and tagged with HA with the use of specific antibodies. Data represent the percentage of cells with 6-4 PP spots at various time points; means and SD of at least two independent experiments are shown. For each experiment, 100 cells were counted.
Cell lines from XP-F individuals show a characteristic failure of the altered XPF to properly translocate to the nucleus through aggregation of the protein in the cytoplasm.11 This feature is evident for XP-causing mutations and accentuated in cells from the individual with XFE syndrome. However, FA-causing XPF missense altered proteins can actually translocate to the nucleus, where they are recruited to sites of active NER (Figures S3A and S3B) and can interact with SLX4 and ERCC1 (Figures S3C and S3D). These results might be functionally important, given that a recent article reports that SLX4 interaction with XPF is crucial for ICL repair and that SLX4-knockout mice phenocopy FA.13 Using Xenopus extracts, J.C. Walter’s group reported that the FA upstream pathway genes are required to regulate a nuclease that makes DNA incisions near the ICL.14 Given that FA-specific altered XPF proteins can reach the site of damage, we then investigated their ability to cleave DNA. For this aim, the p.Arg689Ser altered XPF was purified as a heterodimer with ERCC1 as previously described.5 Subsequently, NER reactions were performed with the purified altered protein, extracts from XPF-deficient XP2YO cells, and a plasmid containing an NER substrate (1,3-cisplatin intrastrand crosslink).15 Consistent with the functional data above, the purified heterodimer composed of ERCC1 and p.Arg689Ser XPF is proficient in the excision step of NER similarly to wild-type XPF, given that it restored the ability to cleave and remove the site-specific intrastrand crosslink from the plasmid in XP2YO cell extracts (Figure 4A). Nevertheless, the excision reaction is not perfect given that the excised fragments are, on average, 1 nucleotide longer than expected from a normal reaction with wild-type-XPF-ERCC1 dimer (Figure 4A, lane 4). We also performed in vitro nuclease activity assays with purified ERCC1-p.Arg689Ser-XPF and ERCC1-p.Arg689Ala-XPF on a stem-loop model DNA substrate. Unlike wild-type XPF and altered XPF proteins causing XP (p.Arg799Trp) or XFE progeroid syndrome (p.Arg153Pro),11 p.Arg689Ser XPF is unable to cleave such a substrate (Figure 4B), indicating that the nuclease-type activity of p.Arg689Ser XPF is grossly abnormal. Unfortunately, we could not perform these biochemical experiments with the p.Leu230Pro altered XPF because we were unable to express and purify ERCC1-p.Leu230Pro-XPF as a result of its low stability and tendency to aggregate. We finally checked whether the FA-specific altered XPF proteins ectopically expressed in Ercc4-null MEFs can perform the incision step of ICL repair. Both p.Leu230Pro and p.Arg689Ser altered XPF completely restored the incision defect of Ercc4-null MEFs, as measured by the COMET assay (data not shown), but the cells remained hypersensitive to ICLs (Figure 1F). Although additional biochemical experiments are required, our results suggest that the ICL sensitivity of individuals FA104 and 1333 is not directly linked to the absence of XPF nuclease activity. It seems unlikely that the defect is a downstream step of homologous recombination because FA104 and 1333 cells are not sensitive to PARP inhibitors and are normal in Rad51 focus formation. Given that the nuclease activity of the FA-specific p.Arg689Ser altered XPF is grossly abnormal, it is tempting to speculate that the ICL-unhooking step in these FA cells leaves an intermediate aberrant substrate that is irreparable by subsequent ICL-repair factors.
Figure 4.
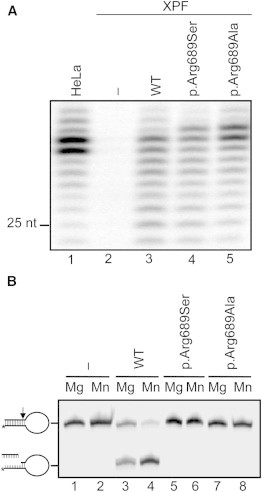
Nuclease Activity of Altered XPF
(A) NER activity of wild-type and altered ERCC1-XPF dimer. A plasmid containing a site-specific 1,3-intrastrand cis-Pt DNA crosslink was incubated with whole-cell extracts from HeLa cells or XPF-deficient cells (XP2YO) complemented with recombinant ERCC1-XPF purified from Sf9 insect cells as reported.13 The excised DNA fragments of 24–32 nucleotides are shown. The position of a 25-mer is indicated.
(B) Incision of a stem-loop substrate with wild-type and altered XPF. The 3′ Cy5-labeled substrate was incubated with recombinant ERCC1-XPF in the presence of 2 mM MgCl2 or 0.4 mM MnCl2, and the products were analyzed by denaturing PAGE. The incision reaction was performed essentially as described earlier.5,11
Our genetic, biochemical, and functional studies, along with the characterization of previous ERCC4 mutations causing XP-F and XFE, provide a model for the mechanistic understanding of how mutations in ERCC4 lead to three distinct diseases (Table S2). Most of the presently known XP-F individuals suffer from a relatively mild form of XP.16 Cells from these individuals have a reduced level of XPF in the nucleus because the altered XPF has a tendency to aggregate in the cytoplasm.11 This reduced level of nuclear XPF is insufficient to mediate complete NER, but it still has enough ICL-repair-specific functions to prevent chromosome fragility, cell-cycle arrest, and subsequent FA clinical manifestations. A second set of ERCC4 mutations, characterized in this study, allow localization of the protein to the nucleus, where they exert a certain level of NER activity but are fully deficient in ICL repair. p.Arg689Ser XPF is a stable and NER-proficient protein with an active site structure that prevents it from properly processing ICL-repair intermediates. p.Leu230Pro XPF is more similar to the products of previously described ERCC4 mutations in that it is less stable and might have a tendency to aggregate in the cytoplasm. However, sufficient amounts of the protein are properly folded and reach chromatin, where it appears to have some activity in the removal of 6-4 PPs. Residual NER activity in the skin tissue of individual 1333 in vivo might explain why this individual has no clinically relevant skin photosensitivity, although we cannot exclude that dermatological problems will arise later in life. A final category of ERCC4 mutations is associated with XFE progeroid syndrome, which is characterized by very low levels of nuclear XPF, apparently insufficient to support either NER or ICL repair. Importantly, the only XFE-affected individual described suffered from both skin photosensitivity and anemia9,16 and shared some cellular features with XP (NER defect and UV-light sensitivity) and FA (extreme ICL sensitivity, DEB-induced chromosome fragility, and MMC-induced cell-cycle arrest), suggesting that XFE syndrome is characterized by a combination of XP and FA manifestations (Table S2). Exhaustion of hematopoietic stem cells is also an attribute of ERCC1-XPF hypomorphic mice that mimic XFE (Laura Niedernhofer, personal communication). Microsomy, microcephaly, and liver fibrosis were likewise observed in FA individual 1333, in Ercc1- and Ercc4-deficient mice, and in the unique ERCC1-deficient individual, who all lack ICL-repair functions.17–21
In a broader sense, this study demonstrates that depending on the type of ERCC4 mutation and the balance between NER and ICL-repair activities, affected individuals present with one of three clinically distinct disorders. This resembles the case of XPD, which is involved in XP complementation group D, trichothiodystrophy (MIM 601675), or Cockayne syndrome (MIM 216400) depending on the type of mutation,22 and highlights the value of characterizing rare genetic disorders for gaining insight into the mechanisms of genome maintenance and human disease. XPF has a central role in preventing genome instability, cancer, BMF, developmental abnormalities, and premature aging. Like those of other breast and ovarian cancer susceptibility genes mutated in FA,23,24 the product of ERCC4 also acts downstream of FANCD2 monoubiquitination. Therefore, it is important to study FANCQ as a candidate gene in hereditary breast and ovarian cancer.
Acknowledgments
The use of FANCQ as an alias for ERCC4 was approved by the HUGO Gene Nomenclature Committee. We would like to thank the families affected by Fanconi anemia and their clinicians for providing samples and clinical data, as well as María A. Blasco (Centro Nacional de Investigaciones Oncológicas, Madrid) for providing Ercc4-deficient mouse embryonic fibroblasts. The J.A.B. laboratory is funded by grants from European Program “7FWP, Health” (PERSIST; agreement 222878), the Spanish Ministry of Science and Innovation (Refs110-90.1 and SAF 2009-07164), Programa RETICS-RD06/0010/0015 ISCIII, and Fundación Botín. O.D.S. acknowledges funding from the National Institutes of Health (GM080454 and CA092584). C.S. is funded by CCA/V-ICI Amsterdam. D.S. and B.S. received grants from the Deutsche Fanconi-Anaemie-Hilfe, Aktionskreis Fanconi-Anaemie, and the Schroeder-Kurth Fund. J.S.’s laboratory is funded by the Generalitat de Catalunya (SGR0489-2009), the ICREA-Academia award, the Spanish Ministry of Science and Innovation (Centre for Biomedical Network Research on Rare Diseases [CIBERER] CB06/07/0023, SAF2009-11936, and SAF2012-31881), and the European Regional Development FEDER Funds. CIBERER is an initiative of the Instituto de Salud Carlos III, Spain.
Contributor Information
Detlev Schindler, Email: schindler@biozentrum.uni-wuerzburg.de.
Jordi Surrallés, Email: jordi.surralles@uab.es.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M.A., Steltenpool J., Oostra A.B., Eirich K., Korthof E.T., Nieuwint A.W. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 2.Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonio Casado J., Callén E., Jacome A., Río P., Castella M., Lobitz S., Ferro T., Muñoz A., Sevilla J., Cantalejo A. A comprehensive strategy for the subtyping of patients with Fanconi anaemia: conclusions from the Spanish Fanconi Anemia Research Network. J. Med. Genet. 2007;44:241–249. doi: 10.1136/jmg.2006.044719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deans A.J., West S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell. 2009;36:943–953. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Enzlin J.H., Schärer O.D. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002;21:2045–2053. doi: 10.1093/emboj/21.8.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almarza E., Río P., Meza N.W., Aldea M., Agirre X., Guenechea G., Segovia J.C., Bueren J.A. Characteristics of lentiviral vectors harboring the proximal promoter of the vav proto-oncogene: a weak and efficient promoter for gene therapy. Mol. Ther. 2007;15:1487–1494. doi: 10.1038/sj.mt.6300213. [DOI] [PubMed] [Google Scholar]
- 7.de Laat W.L., Sijbers A.M., Odijk H., Jaspers N.G., Hoeijmakers J.H. Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res. 1998;26:4146–4152. doi: 10.1093/nar/26.18.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sijbers A.M., de Laat W.L., Ariza R.R., Biggerstaff M., Wei Y.F., Moggs J.G., Carter K.C., Shell B.K., Evans E., de Jong M.C. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 9.Niedernhofer L.J., Garinis G.A., Raams A., Lalai A.S., Robinson A.R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 10.Trujillo J.P., Mina L.B., Pujol R., Bogliolo M., Andrieux J., Holder M., Schuster B., Schindler D., Surrallés J. On the role of FAN1 in Fanconi anemia. Blood. 2012;120:86–89. doi: 10.1182/blood-2012-04-420604. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad A., Enzlin J.H., Bhagwat N.R., Wijgers N., Raams A., Appledoorn E., Theil A.F., J Hoeijmakers J.H., Vermeulen W., J Jaspers N.G. Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet. 2010;6:e1000871. doi: 10.1371/journal.pgen.1000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Limsirichaikul S., Niimi A., Fawcett H., Lehmann A., Yamashita S., Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU) Nucleic Acids Res. 2009;37:e31. doi: 10.1093/nar/gkp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crossan G.P., van der Weyden L., Rosado I.V., Langevin F., Gaillard P.H., McIntyre R.E., Gallagher F., Kettunen M.I., Lewis D.Y., Brindle K., Sanger Mouse Genetics Project Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knipscheer P., Räschle M., Smogorzewska A., Enoiu M., Ho T.V., Schärer O.D., Elledge S.J., Walter J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moggs J.G., Yarema K.J., Essigmann J.M., Wood R.D. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J. Biol. Chem. 1996;271:7177–7186. doi: 10.1074/jbc.271.12.7177. [DOI] [PubMed] [Google Scholar]
- 16.Gregg S.Q., Robinson A.R., Niedernhofer L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst.) 2011;10:781–791. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weeda G., Donker I., de Wit J., Morreau H., Janssens R., Vissers C.J., Nigg A., van Steeg H., Bootsma D., Hoeijmakers J.H. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 18.McWhir J., Selfridge J., Harrison D.J., Squires S., Melton D.W. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat. Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 19.Tian M., Shinkura R., Shinkura N., Alt F.W. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell. Biol. 2004;24:1200–1205. doi: 10.1128/MCB.24.3.1200-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaspers N.G., Raams A., Silengo M.C., Wijgers N., Niedernhofer L.J., Robinson A.R., Giglia-Mari G., Hoogstraten D., Kleijer W.J., Hoeijmakers J.H., Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 2007;80:457–466. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregg S.Q., Gutiérrez V., Robinson A.R., Woodell T., Nakao A., Ross M.A., Michalopoulos G.K., Rigatti L., Rothermel C.E., Kamileri I. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55:609–621. doi: 10.1002/hep.24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleaver J.E., Lam E.T., Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009;10:756–768. doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- 23.Meindl A., Hellebrand H., Wiek C., Erven V., Wappenschmidt B., Niederacher D., Freund M., Lichtner P., Hartmann L., Schaal H. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 24.Levy-Lahad E. Fanconi anemia and breast cancer susceptibility meet again. Nat. Genet. 2010;42:368–369. doi: 10.1038/ng0510-368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.