

Abstract
Factor XIII catalyzes formation of γ-glutamyl-ε-lysyl crosslinks within fibrin clots. FXIII A2 can be activated proteolytically with thrombin and low mM Ca2+ or nonproteolytically with high monovalent/divalent cations along with low mM Ca2+. Physiologically, FXIII A2 is poised to respond to transient influxes of Ca2+ in a Na+ containing environment. A successful strategy to monitor FXIII conformational events is hydrogen-deuterium exchange (HDX) coupled with mass spectrometry. FXIII A2 was examined in the presence of different cations (Ca2+, Mg2+, Ba2+, Cu2+, Na+, TMAC+, and EDA2+) ranging from 1–2 mM, physiological Ca2+ concentration, to 50–500mM for nonproteolytic activation. Increases in FXIII solvent exposure could already be observed at 1 mM Ca2+ for the dimer interface, the catalytic site, and glutamine substrate regions. By contrast, solvent protection was observed at the secondary cleavage site. These events occurred even though 1mM Ca2+ is insufficient for FXIII activation. The metals 1mM Mg2+, 1mM Ba2+, and 1mM Cu2+ each led to conformational changes, many in the same FXIII regions as Ca2+. FXIII could also be activated nonproteolytically with 500mM tetramethylammonium chloride (TMAC+) and 500mM ethylenediamine (EDA2+), both with 2mM Ca2+. These different HDX studies help reveal the first FXIII segments that respond to physiological Ca2+ levels.
Keywords: Factor XIII, hydrogen-deuterium exchange, mass spectrometry, calcium, coagulation, transglutaminase
Introduction
Blood coagulation involves a cascade of enzyme activations that ultimately concludes in a soluble fibrin network. This network is covalently crosslinked by the transglutaminase factor XIII (FXIII)1 to form an insoluble clot [1–3]. Due to the critical role of FXIII in coagulation, there is a need to better understand the conditions that influence FXIII activation/activity as well as the structural dynamics of this enzyme system.
In plasma, FXIII is a protransglutaminase with a tetrameric structure A2B2, where the B subunits act as a carrier for the catalytically active A subunits [4]. By contrast, cellular FXIII exists in platelets, placenta, and monocytes as an A2 dimer. Recombinant FXIII A2 has been successfully crystallized [5], and the resultant structure exhibits well-defined and sequentially folded domains that include the activation peptide (1–37), the β sandwich (38–184), the catalytic core (185–515), β-barrel 1 (516–628), and β-barrel 2 (629–730). See Figure 1A and B.
Figure 1.

The FXIII A2 zymogen 2.1 Å crystal structure 1F13 [39]. (A) A cartoon model to illustrate the intimate contact at the dimer interface with the domains being color coded (gray) activation peptide, (white)β-Sandwich, (black) catalytic core, (white) β-barrel 1 and (black) β-barrel 2. (B) Ribbon view of one FXIII-A monomer with the domains represented by the same colors as (A). The regions of interest are labeled: activation peptide (1–37), Gln recognition peptide 4 (72–97), Gln recognition peptide 7 (190–230), catalytic triad (C314, H373 and D396), Tyrosine 560, secondary cleavage site (513–514) and the proposed lysine recognition region (646–658). These figures were created using VMD [52].
When FXIII is activated physiologically, thrombin (IIa) cleaves the N-terminal activation peptide (residues 1–37). The presence of low mM Ca2+ assists in the dissociation of the B subunits from plasma FXIII. In addition, Ca2+ promotes exposure of the catalytic C314 from both plasma and cellular FXIII resulting in an active A2 dimer (FXIIIa*) [6–8]. Like other transglutaminases (TGases), the active site of FXIII consists of a thiol-containing catalytic triad (C314, H373, and D396), but FXIII is unique in the fact that it exists as a dimer. In the presence of a suitable acyl-donor glutamine containing substrate, C314 of FXIIIa forms a thioester bond and ammonia is released. The acyl-donor substrate is then covalently linked to a primary amine (lysine) acyl-acceptor forming γ-glutamyl-ε-lysyl cross-links involving the α- and γ-chains of fibrin which stabilizes the growing clot [9].
In addition to IIa proteolytic activation, FXIII A2 can be non-proteolytically activated when the Ca2+ concentration is greater than 50 mM (FXIIIa° or FXIIIaCa) [10–11]. FXIII A2 has also been observed in an activated state in the presence of >150 mM NaCl and low mM Ca2+ (FXIIIaNa) [12]. A related form of nonproteolytic FXIII activation has been documented in platelets [12–14]. Thrombin-stimulated platelets may exhibit increased Ca2+ levels that promote formation of an active intracellular FXIII species which has not been hydrolyzed at the R37-G38 peptide bond. In plasma, these different forms of nonproteolytic activation are minimized by the presence of the B2 subunits [15]. Regardless of the mode of activation, FXIIIa function can be hindered following hydrolysis at the secondary cleavage site (K513-S514) by thrombin [16–17]. Occupation of a nearby site with Ca2+ and other select metals can help protect this secondary hydrolysis from occurring [5, 18].
Even though the catalytic C314 is only alkylated by iodoacetamide (IAA) after activation, [19–21] the crystal structures for FXIIIaIIa and FXIIIaCa lack any major structural changes when compared to that of zymogen [5, 22–23]. We have used MALDI-TOF mass spectrometry approaches coupled with hydrogen-deuterium exchange or chemical modification to probe FXIII conformation changes occurring in solution. During activation, portions of the FXIII catalytic core and the A2 dimer interface were found to be more accessible to solvent [24–25]. By contrast, the addition of an inhibitory peptide with a glutamine isostere caused selected FXIII regions to become more protected from solvent [26]. As expected from the secondary cleavage studies, Ca2+ binding hindered solvent exposure in a FXIII segment around 513–514. Inhibition at the active site could lead to further long-range decreases in HDX in this region.
A recent paper by Kristiansen and Andersen questions our use of borate buffer supplemented with CaCl2 for mass spectrometry based projects. Our HDX studies have not, however, been carried out under deactivated concentrations (0.3 mM CaCl2) as they presume, but at 1 mM CaCl2 following either proteolytic or nonproteolytic activation. According to their paper, FXIII A2 that is activated nonproteolytically at 50 mM CaCl2 and then buffer exchanged into 1mM CaCl2 maintains 90% of its enzyme activity [27]. Moreover, many of the conformational events that we reported as being important for FXIII activation have been confirmed by HDX studies by Andersen and Faber using a buffer system of 200 mM Hepes, 150 mM NaCl, and 50 mM CaCl2 [28].
The focus of our present HDX work was to further investigate the conformational changes that occur to FXIII due to monovalent and divalent cation binding. FXIII A2 was monitored in the presence of Ca2+, Mg2+, Ba2+, Cu2+, Na+, tetramethylammonium chloride (TMAC+) or ethylenediamine (EDA2+) with concentrations ranging from physiological Ca2+ concentration, 1–2 mM, up to 50–500 mM. On its own, the low mM Ca2+ condition is unable to support FXIII activity. The HDX effects observed in this environment thus provide valuable information on early conformational changes needed in preparation for activation. Often these effects further increased as the Ca2+ concentration was raised to the levels required for nonproteolytic activation (50–500 mM). Several of the conformational changes observed with physiological 1 mM Ca2+ also occurred with the other divalent metals whereas other effects were distinct for a particular metal. In the current investigation, we also characterized a novel method of nonproteolytic FXIII activation utilizing high concentrations of organic cations TMAC+ or EDA2+. Knowledge gained from these different HDX studies will help identify the elusive structural roles of different divalent and monovalent cations. A greater understanding of the conformational dynamics of FXIII will aid in the development of new therapeutic strategies to control excessive bleeding, thrombosis, and/or atherosclerosis.
Materials and Methods
Factor XIII Preparation and Synthetic Peptides
Recombinant human cellular factor XIII A2 (FXIII A2) was generously provided by Dr. Paul Bishop (ZymoGenetics, Inc., Seattle, WA). After reconstituting the lyophilized FXIII in 18 MΩ deionized water, FXIII was buffer exchanged into 6.67 mM borate at pH 8.3. The concentration of FXIII was determined on a Cary 100 UV/vis spectrophotometer. The absorbance was monitored at 280 nm and concentration calculated with the FXIII extinction coefficient of 1.49 ml/mg cm. Aliquots (36 μl) of 16.7 μM FXIII in 6.67 mM borate were dried in a SpeedVac (Savant) and stored at −70 °C until future use.
The β casein derived FXIII substrate peptide K9 (Ac-LGPGQSKVIG-OMe) was synthesized by Peptides International (Louisville, KY). K9 was reconstituted in 18 MΩ deionized water and the concentration was confirmed by quantitative amino acid analysis (AAA Service Laboratory, Inc., Boring, OR). Purity was assessed by HPLC and mass spectrometry approaches.
Transglutaminase Activity Assay
Factor XIII activity was determined using a modified version of the Dade-Behring Berichrom Assay [29–30]. Briefly, this assay utilizes a coupled reaction involving both FXIII and glutamate dehydrogenase (GDH). FXIIIa reacts with the acyl-donor K9 releasing NH3 and the transglutaminase reaction concludes when the primary amine acyl-acceptor, glycine ethyl ester, forms an isopeptide bond with the K9 peptide. This transglutaminase activity is monitored via the conversion of α-ketoglutarate and NH3 into glutamate in the presence of reducing equivalents of NADH. The oxidation of NADH results in decreased absorbance at 340 nm.
Dry FXIII aliquots were activated nonproteolytically in a total volume of 12 μl dH2O. The final working concentration was 50 μM FXIII in 20 mM Borate at pH 8.3 with (50 mM CaCl2, 50 mM MgCl2 with 2 mM CaCl2, 50mM BaCl2 with 2 mM CaCl2, 50 mM CuCl2 with 2 mM CaCl2, 500 mM ethylenediamine (EDA2+) with 2 mM CaCl2, 500 mM NaCl with 2 mM CaCl2, and 500 mM tetramethylammonium chloride (TMAC+) with 2 mM CaCl2). FXIII was activated for 10 min at 37 °C before adding 3 μl to the assay. For each assay, the volume of activator reagent (163 μl) and detector reagent (250 μl) as well as the concentration of FXIII (300 nM) and K9 (500 μM) were held constant [31]. The assay contents were placed in a Cary 100 UV/vis spectrophotometer for 2 min at 37 °C for equilibration before the K9 substrate peptide was added. After introducing the K9 peptide, the oxidation of NADH was monitored for 25 minutes at 340 nm. Due to the coupled nature of this FXIII activity assay, there is a brief delay before consumption of NADH can be detected. The FXIII transglutaminase activity was later determined by the steepest part of the slope which correlates with enzyme catalyzed velocity (Δabs/min). The final divalent and monovalent cation concentrations in the assays (0.3–3mM) did not interfere with GDH reactivity. Using the molar absorptivity value for NADH (6220 M−1cm−1 at 340nm), the transglutaminase based velocities could be converted into (μM/min) and the final velocity values compared. For the studies involving the novel cations EDA2+, Na+, and TMAC+, transglutaminase activity was compared relative to that of FXIIICa with % FXIIICa Activity defined as (FXIIIa activity/FXIIICa) × 100. Standard Deviations of the mean for three independent trials were presented.
HDX Experimentation
There were two different groups of HDX samples, one monitoring the effects of low mM concentrations of divalent metals and the other examining non-proteolytic activation. Both groups utilized the dry buffer exchanged FXIII aliquots. To probe the effects of low mM divalent metals, 0.6 μl of 20 mM metal-chloride (CaCl2, MgCl2, BaCl2 or CuCl2) was added to the dry FXIII aliquot and evaporated to dryness in the SpeedVac and stored at −70 °C. The following HDX protocol was then adapted from methodology established in the Komives laboratory [32–33]. The dry FXIII aliquot was allowed to reach room temp before 12 μl of 99.996% D2O (Cambridge Isotope Laboratories) was added yielding a final working concentration of 50 μM FXIII and 1mM Ca2+, Mg2+, Ba2+ or Cu2+ in 20 mM Borate at pH 8.3. The samples were incubated at room temperature for 10 minutes before the HDX was quenched by adding 120 μl of chilled 0.1% TFA at pH 2.5. The quenched reaction was then immediately transferred to a tube containing activated pepsin bound to 6% agarose (Thermo Scientific, Rockford, IL). Pepsin digestion occurred for 10 min on ice. Following digestion, the reaction mix was centrifuged for 30 sec to separate the FXIII digest from the pepsin beads. Three 8.2 μl aliquots were immediately frozen in liquid N2 and each reaction condition was performed three times.
When investigating nonproteolytically activated FXIII, aliquots were prepared as described previously. Divalent and monovalent cations (< 3 μl) were added to the dry FXIII aliquot for each of the activation conditions (50 mM Ca2+, 500 mM ethylenediamine (EDA) with 2 mM Ca2+, 500 mM Na+ with 2 mM Ca2+, and 500 mM tetramethylammonium chloride (TMAC+) with 2 mM Ca2+) and evaporated to dryness in a SpeedVac and stored at −70°C. After allowing the sample to reach room temperature, 12 μl 99.996% D2O was added and the HDX occurred at 37°C for 10 min. The activated FXIII was digested and quenched as stated above.
HDX Analysis
A FXIII(a) HDX aliquot was thawed at room temperature and immediately mixed with an equal volume of 10 mg/ml α-cyano-hydroxycinnamic acid matrix (α-CHCA) (Aldrich) in 1:1:1 ethanol/CH3CN/0.1%TFA at pH 2.2, and 0.5 μl was spotted on a chilled MALDI plate. The sample spot was then quickly dried by placing the MALDI plate into a SpeedVac. The plate was then immediately inserted into the MALDI-TOF-MS (Voyager DE-Pro, Applied Biosystems). By limiting time for this procedure to < 5 min, HDX back exchange is kept at a minimum. Spectra were collected in reflector mode with 256 shots/spectrum. All peptides in the peptic digest were previously identified by Brian T. Turner, Jr. [24] and/or confirmed by MS/MS analysis on an Applied Biosystems 4700. New peptides reported in this work were derived from MS/MS studies on previously unidentified MALDI peaks that had been detected on the Voyager DE-Pro. Peptides to note are 1–23, 300–314 and 533–550. After digestion by pepsin, the FXIII A2 peptides identified represent 40 % coverage (Figure 2). These peptides focus on key FXIII regions found within the β-sandwich, the catalytic core, and the β-barrel 1 region. Additional studies with acid dependent type XIII protease did not significantly improve sequence coverage. All the MALDI spectra derived from pepsin digests were analyzed using Data Explorer (Applied Biosystems) and calibrated using two singly protonated reference peptides; monoisotopic mass 850.4787 Da (residues 535–541) and quadraisotopic mass of 1375.7097 Da (residues 220–230).
Figure 2.

FXIII peptides observed by MALDI-MS after 10 minute pepsin digestion are underlined. The underlined peptides represent 40 % coverage.
Deuterium incorporation for each isotopic cluster was quantified as described by Sabo et al. [34]. To determine the change in deuteration for FXIII under different conditions the percent deuteration for each peptide was calculated using equation 1:
where DFXIII is the amount of deuterium incorporated in FXIII zymogen, D is the amount of deuterium incorporation in FXIII under different conditions and Dmax is the theoretical maximum amount of deuterium incorporation for the given peptide under these conditions. The theoretical maximum depends on the final percentage of D2O under quench conditions (4.5 %) and accounts for all exchangeable backbone amide protons as well as a fraction of N-terminal, C-terminal and side chain exchangeable protons. In accordance with previous HDX data analysis, percent differences greater than 4.5 % are considered significant [24, 34–36], 3–4.5 % is moderate and < 3 % is modest.
Results
1 mM Divalent Metals and FXIII – HDX-MS
Human plasma contains 1–2 mM calcium, therefore we compared the solvent accessibility of FXIII zymogen (without metals) to that of FXIII in the presence of 1 mM Ca2+, Mg2+, Ba2+ or Cu2+. All studies would thus be carried out in the presence of a physiological cation concentration environment. Prior knowledge about these divalent cations includes the following derived from studies carried out at concentrations from 0.1 to 10mM. Mg2+ is smaller in size than Ca2+ and does not promote the conformation-based fluorescence changes that are observed upon Ca2+ binding to FXIII [37]. Mg2+ also does not support FXIIIaIIa activity but does protect against thrombin-catalyzed proteolysis at the secondary cleavage site [18, 37]. Larger in size than Ca2+, Ba2+ exhibits no protection against secondary cleavage and modest ability to support FXIIIaIIa activity [18, 20]. Cu2+ is a transition metal that is closer in size to Ca2+ and exerts no protection of the K513-S514 site but can support some FXIIIaIIa activity [18, 20].
For the current conformational studies on FXIII A2, Table 1 displays changes in deuterium incorporation of FXIII zymogen compared to FXIII-(Ca, Mg, Ba and Cu). The total deuterium incorporated for each pepsin-derived peptide is displayed in Figure 3. Note that the top of the chart highlights the different FXIII regions where the peptides reside.
Table 1.
Changes in Percent Deuteration: FXIII with 1 mM Metal Relative to FXIII Zymogena
| Residues | Theo Dmaxb | 1 mM Ca2+ | 1 mM Mg2+ | 1 mM Ba2+ | 1 mM Cu2+ |
|---|---|---|---|---|---|
| 1–23 | 23.3 | −19.1c | −14.6 | −22.9 | −20.7 |
| 32–40 | 8.9 | −12.7 | −6.6 | −10.9 | −8.5 |
| 83–99 | 23.2 | −0.4 | −5.6 | −6.3 | −6.5 |
| 88–98 | 10.9 | 9.2 | 7.3 | 4.6 | 3.5 |
| 100–111 | 11.8 | −4.1 | −3.4 | −5.0 | −3.6 |
| 214–230 | 18.8 | 8.8 | n/ad | 6.2 | 2.5 |
| 220–230 | 12.3 | 5.6 | 0.7 | 4.3 | 3.1 |
| 240–247 | 7.5 | 5.3 | 2.2 | 6.3 | 8.7 |
| 248–264 | 15.9 | 2.9 | 3.3 | 3.6 | 4.0 |
| 248–265 | 18.8 | 2.1 | 0.3 | 1.4 | 2.8 |
| 300–314 | 15.8 | 8.7 | 0.8 | 3.0 | −7.4 |
| 364–372 | 8.4 | 3.5 | −1.0 | −2.9 | −2.3 |
| 407–424 | 18.8 | 3.6 | 4.0 | 9.7 | −2.8 |
| 513–522 | 9.7 | −9.0 | −9.7 | −11.3 | −11.6 |
| 533–550 | 20.1 | 0.5 | −6.0 | −6.4 | −6.8 |
| 632–646 | 14.7 | 3.2 | −2.8 | −1.9 | −1.1 |
The % change for a particular peptide was calculated by the following equation: % difference = ((D − DFXIII)/Dmax) × 100 %, where D is the amount of deuterium incorporated in FXIII with metal, DFXIII is the amount of deuterium incorporated in the zymogenic state (no metal), and Dmax is the theoretical maximum number of exchangeable protons within the indicated peptide.
The maximum number of exchangeable protons within the indicated peptide, assuming 100 % deuteration. This value accounts for all exchangeable backbone amide protons and a slight fraction of N-terminal, C-terminal, and side chain exchangeable protons, which are dependent on the final percentage of D2O in solution under quench conditions (~4.5 %). A fully deuterated peptide would theoretically have acquired this amount of deuterons.
The values in bold represent significant changes in deuteration greater than ~4.5 %.
n/a refers to a peptide that was either not observed in the peptic digest or was not of sufficient intensity to quantify.
Figure 3.

The number of deuterons incorporated at 10 min for FXIII under the following conditions zymogenic (no metal) FXIII (black), 1 mM Ca2+ (up diagonal), 1 mM Mg2+ (gray), 1 mM Ba2+ (down diagonal) and 1 mM Cu2+ (white). The top of the graph illustrates regions of interest in FXIII. Error bars represent the standard deviation of the mean for 3 independent trials.
Within the β sandwich domain, the two main regions of interest are the activation peptide (1–37) and the putative Gln substrate recognition site (72–97), also known as peptide 4. After 10 minutes of HDX, the activation peptide became more protected from solvent relative to its values in the zymogen state of FXIII. Such protection was observed for all the metals tested and ranged from −6.6 % for FXIII-Mg to −22.9 % for FXIII-Ba. The peptide 4 segment [38], is represented by the pepsin derived peptides 83–99 and 88–98. For peptide 83–99, changes in deuteration ranged from −0.4 % for FXIII-Ca to −6.5 % for FXIII-Cu. By contrast, the region 88–98 became more exposed for all metals ranging from 3.5 % for FXIII-Cu to 9.2 % for FXIII-Ca. The different responses for 83–99 versus 88–98 suggest that the amide protons from residues 83–87 and/or 99 may be responsible for the HDX protection observed for 83–99 in the presence of divalent metals (Figure 3). Further support for the importance of residue 99 comes from the observation that the next FXIII segment 100–111 exhibits solvent protection in the presence of all the cations tested.
The catalytic core contains several regions of interest. Peptide 7 (190–230), another putative glutamine-substrate region [38], displayed increased deuterium incorporation for metals when compared to zymogen. FXIII-Ca was the most exposed for peptides 214–230 and 220–230 followed by FXIII-Ba, FXIII-Cu, and finally FXIII-Mg. The dimer interface between the two FXIII-A monomers (residues 240–265) was another region where increases in deuterium incorporation for all the metals was observed. Exposure within the dimer interface (240–247) for FXIII-(Ca, Mg, Ba and Cu) when compared to zymogen was 5.3, 2.2, 6.3, and 8.7 % respectively. As with the peptide 7 region, FXIII-Mg exhibited the smallest effect. Interestingly, protection from solvent (−3.4 to −5.0%) was observed for the residues 100–111. The C-terminal portion of this segment is located at the dimer interface; however, the more N-terminal portion is found within the β-sandwich region.
The catalytic C314 is located in segment 300–314 and the catalytic H373 is found one residue outside 364–372. For the 364–372 segment containing the H373, 1 mM Ca2+ showed a moderate 3.5 % increase in HDX exposure whereas the other metals all showed a modest protection. By contrast, FXIII-Ca displayed considerable exposure (8.7 %) for 300–314 when compared to FXIII-(Mg and Ba) with 0.8 % and 3.0 % exposure, respectively. FXIII-Cu was the only condition found to be protected in this region (−7.4 %). FXIII-Cu also became modestly protected in the region of 407–424 which is N-terminal to the FXIII Ca2+ binding site (−2.84 %) and makes contacts across the dimer interface. FXIII-(Ca, Mg, and Ba), however, displayed increases in deuterium incorporation for this segment. The 407–424 segment may be influenced by a nearby non-prolyl cis peptide bond that is proposed to play a role in FXIII activation [39].
The β-barrels contain the secondary cleavage site (513–514) and the lysine recognition site (646–658) [40–41]. FXIII-(Ca, Mg, Ba and Cu) all displayed protection within 513–522 when compared to zymogen (−9.0 to −11.6%). The nearby FXIII 533–550 is not influenced by 1 mM Ca whereas it is significantly solvent protected in FXIII-(Mg, Ba, and Cu) at levels of −6.0 to −6.8%. Peptide 632–646 is on the N-terminal end of the proposed lysine recognition site and is moderately exposed in FXIII-Ca (3.6 %) [41]. All other conditions showed little if any protection in this region of β-barrel 2.
TGase Activity of Nonproteolytically Activated FXIII
FXIII was nonproteolytically activated under a series of conditions and TGase activity monitored using a modified version of the Dade-Behring Berichrom assay [31]. When incubated in the presence of several different divalent metals including 50 mM Ca2+, Mg2+, Ba2+ or Cu2+, only Ca2+ displayed activity (data not shown). After changing the activation conditions to include 2 mM Ca2+ with 50 mM Mg2+, Ba2+ or Cu2+, minor activity was observed in Mg2+ and Ba2+ but not Cu2+ (Figure 4). Factor XIII activity was then monitored using a series of other nonproteolytic techniques. FXIIIaCa was compared to FXIII activated in the presence of 500 mM EDA2+ (FXIIIaEDA) with 2 mM Ca2+, 500 mM Na+ with 2 mM Ca2+ (FXIIIaNa) and 500 mM TMAC+ with 2 mM Ca2+ (FXIIIaTMAC). Using the slope (μM NADH consumed/min) as an indicator of activity, EDA2+, Na+ and TMAC+ displayed an increase in FXIII activity of 291 ± 27.7 %, 156.7 ± 16.3 % and 194.8 ± 5.5 %, respectively when compared to FXIIIaCa.
Figure 4.

TGase activity of nonproteolytically activated FXIII. Samples were incubated at 37 °C for 10 minutes then added to the assay components. At 2 minutes (12 min total total) the K9 substrate was added to the assay and the oxidation of NADH is depicted as a function of time. The experimental groups are as follows: 50 mM Cu2+ with 2 mM Ca2+ (0.0 μM/min), 50 mM Ba2+ with 2 mM Ca2+ (1.98 μM/min), 50 mM Mg2+ with 2 mM Ca2+ (9.5 μM/min) and 50 mM Ca2+ (10.7 μM/min).
Nonproteolytically activated FXIIIa – HDX-MS
The conformational dynamics of nonproteolytically activated FXIII were also probed using HDX-MS. Due to ionization issues in the MALDI, it was not possible to quantify the deuterium incorporated into FXIIIaEDA and FXIIIaTMAC. Table 2 thus displays changes in deuterium incorporation of FXIII zymogen compared to FXIII in 2 mM Ca2+, FXIIIaCa and FXIIIaNa. The total deuterium incorporated in each peptide is displayed in Figure 5.
Table 2.
Changes in Percent Deuteration: Nonproteolytically Activated FXIII Relative to FXIII Zymogena
| Peptide | Theo Dmaxb | 2 mM Ca2+ | 50 mM Ca2+ | 50 mM Na+ with 2 mM Ca2+ |
|---|---|---|---|---|
| 1–23 | 23.3 | −16.3c | −8.3 | n/a |
| 32–40 | 8.9 | −12.1 | −12.7 | −12.7 |
| 88–98 | 10.9 | −0.9 | −1.9 | −1.3 |
| 100–111 | 11.8 | −4.5 | −6.2 | −5.9 |
| 220–230 | 12.3 | 9.3 | 13.2 | 17.1 |
| 240–247 | 7.5 | 7.2 | 18.2 | 19.8 |
| 248–264 | 15.9 | 1.4 | n/ad | 2.5 |
| 248–265 | 18.8 | 4.7 | 2.2 | 4.5 |
| 300–314 | 15.8 | 7.1 | 7.7 | 11.7 |
| 407–424 | 18.8 | 0.2 | −0.1 | −0.8 |
| 513–522 | 9.7 | 0.0 | 1.7 | 1.9 |
| 533–550 | 20.1 | 2.6 | 16.8 | n/a |
For superscript a, b, c and d definitions please refer to Table 1.
Figure 5.

The number of deuterons incorporated after 10 min at 37 °C for FXIII under the following conditions zymogenic (no metal) FXIII (black), 2 mM Ca2+ (up diagonal), FXIIIaCa 50 mM Ca2+ (gray), FXIIIaNa 500 mM Na+ with 2 mM Ca2+ (whites). The top of the graph illustrates regions of interest in FXIII. Error bars represent the standard deviation of the mean for 3 independent trials.
To ensure activity was achieved during HDX, the same activation conditions used to confirm nonproteolytic activity were utilized for HDX analysis, except D2O was substituted for H2O. The extent of deuterium incorporation may vary relative to previous work since this HDX project was conducted at 37°C. When activated in the presence of 50 mM Ca2+ or in 500 mM Na+ with 2 mM Ca2+, the β sandwich activation peptide of FXIIIaCa and FXIIIaNa was more protected than in FXIII zymogen. This effect was seen in the −12.7 % change found for residues 32–40 in both FXIIIaCa and FXIIIaNa. Residues 100–111 fall within 4 Å of the dimer interface and they too displayed protection of −6.2 % and −5.9 % for FXIIIaCa and FXIIIaNa, respectively when compared to zymogen.
Peptide 7 within the catalytic core became notably exposed when nonproteolytically activated leading to a 13.2 % increase in deuterium incorporation for FXIIIaCa and a 17.1 % increase for FXIIIaNa at residues 220–230. The FXIII dimer interface (240–247) within the catalytic core experienced an 18.2 % (FXIIIaCa) and 19.8 % (FXIIIaNa) increase in solvent accessibility when activated. Increased accessibility was also evident within the catalytic cysteine containing segment (300–314). This region changed 7.7 % and 11.7 % for FXIIIaCa and FXIIIaNa respectively when compared to zymogen (Table 2). These various changes in solvent exposure were all greater than what could be observed under a more physiological 1–2 mM Ca2+ concentration. Such results suggest that these two FXIII regions (peptide 7 and dimer interface) become more solvent accessible as the cation concentration is increased (Figure 5). In a Na+-containing environment, low mM calcium is required for activation.
Events occurring within the β barrels are also important to consider. Unlike the studies done in the presence of 1 mM Ca2+, no substantial changes in solvent exposure were observed for β-barrel 1 residues 513–522 under the nonproteolytic activation conditions at 37 °C. By contrast, accessibility to solvent increased steadily from 2 to 50 mM Ca2+ for the neighboring segment 533–550 (2.6 to 16.8%). Within β-barrel 2, peptide 632–646 is located N-terminal to the lysine recognition region (646–658) and exhibited an increased deuterium incorporation of 6.2 % with FXIIIaNa.
Discussion
Calcium plays a vital role in the activation and regulation of cellular (A2) and plasma (A2B2) FXIII [18–20, 42]. HDX provides an effective solution-based approach for dissecting out how Ca2+ and other divalent and monovalent species (Mg2+, Ba2+, Cu2+, Na+, EDA2+, TMAC+) participate in the conformational dynamics of this transglutaminase.
Impact on Ca2+ binding site and surrounding regions
The FXIII ion binding helix (485–501) is located within 10 Å of β-barrel 1 and also makes contact with the other FXIII-A monomer across the dimer interface [23, 43–44]. See golden helix in Figure 6A. The primary calcium binding site involves FXIII residues Asn436, Asp438, Ala457, Glu485 and Glu490 (Figure 6A pink sticks) and site-directed mutagenesis studies have demonstrated that when the glutamate residues are removed, FXIII sensitivity to Ca2+ decreases [5, 17]. According to Lewis et al., when Ca2+ concentrations are < 2.5 mM, there is one Ca2+ bound per FXIII-A, but there are up to 8 low affinity sites when the concentration is raised above that threshold [37]. The presence of strong and weak Ca2+ binding sites has also been confirmed by 43Ca NMR studies [45]. Our studies to determine the conformational dynamics of FXIII A2 in 1 mM metal were performed to mimic physiological Ca2+ concentrations and to monitor events where a single Ca2+ should target the higher affinity metal site on FXIII. The other divalent metals were examined under these same 1 mM concentration values.
Figure 6.
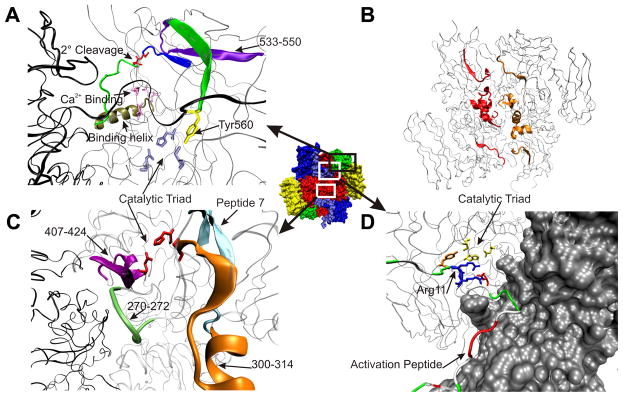
Illustration of the regions mentioned in the conformational dynamics of FXIII A2 (1F13). Central FXIII A2 has each domain labeled: β-sandwich (blue), catalytic core (red), β-barrel 1 (green) and β-barrel 2 (yellow). (A) Within the catalytic core, the Ca2+ binding site (pink) and the ion binding helix (gold) are connected to the secondary cleavage site 513–514 (red sticks) within peptide 513 to 522 (dark blue) through the (green) connector. The influence on the catalytic triad (light blue sticks) is seen through the (green β-sheet) connector between peptide 533–550 (purple) and Tyr560 (yellow). FXIII monomer A is (black) and monomer B is (silver). (B) All pepsin peptides which yield sequence coverage along the dimer interface (100–111, 240–265 and 407–424) are labeled (red) for FXIII monomer A and (gold) for monomer B. (C) Illustrates the sequence coverage around the peptide 7 (cyan) the catalytic triad (red) and their close proximity to the dimer interface and the Yb binding site 270–272 (green). Pepsin peptide 300–314 is (orange) and 407–424 is (magenta). FXIII monomer A is (black) and monomer B is (silver). (D) Depiction of the N-terminal activation peptide of FXIII monomer B (silver surface) labeled according to residue type (green = polar, white = nonpolar, red = acidic and blue = basic) and Arg11 and Arg12 are shown as (blue) sticks. The AP crosses the dimer interface and Asp373′ from FXIII monomer A is hydrogen bonded to Arg11 in close proximity to the catalytic triad (yellow) and Tyr560 (orange). All images were constructed utilizing VMD [52].
Although we do not have sequence coverage for the FXIII calcium binding site, we do cover 40 % of the enzyme including a segment containing (513–514), a secondary cleavage site that is protected from hydrolysis in the presence of calcium [18]. This site of proteolytic degradation (red sticks) is located just 12 residues down from the ion binding helix (485–510). See Figure 6A. All metals tested at 1 mM concentration (Ca2+, Mg2+, Ba2+, and Cu2+) demonstrated solvent protection around peptide 513–522. This finding was surprising considering that Mary et al. reported Ba2+ and Cu2+ provided no protection against 2° cleavage [18].
Unlike the 513–522 region, 1 mM Ca2+ did not show any deuterium protection for the nearby 533–550 segment which is part of an anti-parallel β-sheet that runs adjacent to 513–522 (Figure 6A purple). By contrast, the other divalent metals Mg2+, Ba2+ and Cu2+ all showed substantial protection within 533–550 when compared to FXIII in 1 mM Ca2+. The HDX results suggest that Ba2+ and Cu2+ exert a greater influence on 533–550 with additional effects on 513–522; however, these events are not sufficient to protect FXIII from proteolysis. 1 mM Ca2+ may exert a more direct influence on the secondary cleavage site 513–514.
The ability of 1 mM Mg2+, Ba2+ and Cu2+ to influence FXIII dynamics brings up the question of whether such divalent metals could support nonproteolytically-derived TGase activity as already observed with Ca2+. Solutions containing 50 mM Mg2+ or Ba2+, however, all required the added presence of 2 mM Ca2+ to produce FXIII activity following 10 minutes of nonproteolytic activation at 37°C. Furthermore, their levels of enzymatic activity were all less than those for 50 mM Ca2+ (Figure 4). By contrast, Factor XIII in 50 mM Cu2+, even in the presence of 2 mM Ca2+, was not able to yield TGase activity. From these studies, it is evident that a series of divalent metals can support FXIII activation and TGase activity but low mM Ca2+ is still needed to occupy what is likely the high affinity ion binding site on FXIII.
As mentioned previously, the FXIII segment 513–522 exhibits a decrease in deuteration in the presence of 1 mM Ca2+ and no change is observed for the segment 533–550. These results led to an interest in monitoring FXIII conformational dynamics after incubation in an environment of 2–50 mM Ca2+ at physiological 37°C. Curiously, the protective effect on 513–522 was lost and replaced with a steady increase in the exposure of 533–550. In the X-ray crystals, the FXIII active site is occluded by Y560 (Figure 6A yellow sticks) which resides only 10 residues from 533–550. Steady increases in Ca2+ concentration might promote movement of Y560 thus assisting C314 exposure to substrate. X-ray crystallographers have previously predicted that β-barrel 1 must move somewhat away from the catalytic core during the activation process [23]. Moreover, Y560 will need to be displaced from its H-bonding interaction with catalytic C314. Since the active site region becomes more exposed at higher Ca2+ levels, it is possible the FXIII 533–550 segment contributes a line of communication to this process. In further support of this proposal, Andersen and Faber observed their own increases in HDX based solvent exposure for FXIII peptides spanning 533–551, 556–559, and 560–573 upon formation of FXIIICa [28].
Metal Influence on Dimer Interface
The ion binding helix has direct interactions with the FXIII A2 dimer interface. According to surface calculations, the dimer interface covers 2280 Å2 and there is a 10.3 – 14.4 kcal/mol decrease in FXIII-A free energy upon dimerization [23, 46]. Key interactions between the two FXIII-A monomers are found between K113-D367′, K257-E401′ and R260-D404′. See Figure 6B for HDX coverage of the dimer interface. We find that the exposure reported previously [26] around the dimer interface for peptide 240–247 is also seen in the presence of just 1 mM Ca2+. This dimer interface exposure is further supported by increases in deuterium incorporation now recorded for 248–264 and 248–265. Solvent exposure around the dimer interface is also observed with 1 mM Ba2+ and Cu2+ but to a lesser extent by Mg2+. Although 100–111 is also positioned along the dimer interface, this peptide segment exhibits protection from deuterium exchange. Only the most C-terminal portion is actually along the dimer interface. The remaining segment is directed into the β-sandwich region which exhibits protection. These results indicate that a series of different divalent ions can disturb interactions between the two FXIII-A monomers. Such events occur even at Ca2+ levels insufficient to support FXIII activation.
Further increases in solvent accessibility along the A2 dimer interface (aa 240–264) could be observed when the FXIII was nonproteolytically activated at 37°C with 50 mM Ca2+ (FXIIICa) or 500mM Na+/2mM Ca2+ (FXIIINa). Under different HDX conditions, this same region was seen to increase in accessibility when activated by IIa as well as when inhibited by IAA and K9 DON [26]. The disruption along the dimer interface due to increasing levels of cations may also be due to the non canonical Ca2+ binding site reported for TG2 (S1 226–233) which is homologous to 264–271 found on the dimer interface of FXIII [47]. Interestingly, a Yb binding site (Figure 6C green) has been shown by X-ray crystallography to be in the vicinity of FXIII residue 271 [23]. Yee et al. proposed that an opening in the FXIII A2 dimer interface may allow for the lysine acyl-acceptor to access the catalytic core [5].
Catalytic C314 Region and Substrate Recognition
The exposure of the catalytic C314 is imperative for FXIII activity. Of the1 mM metals tested, Ca2+ promoted the largest increase in solvent accessibility for the FXIII 300–314 region. See orange peptide in Figure 6C. Ba2+ exerted a mild increase on this region and Mg2+ remained essentially the same as zymogen. Cu2+ was unique in that it demonstrated substantial protection of 300–314 when compared to zymogen. These HDX results substantiate the lack of TGase activity observed in the presence of Cu2+ and suggest that the observed solvent protection might thwart substrate interactions around the catalytic cysteine.
Additional HDX results at physiological temperature revealed that 2 mM Ca2+ had nearly the same effect on exposure of the 300–314 region as activating FXIII with 50 mM Ca2+ (FXIIIaCa). Such results suggest that low mM Ca2+ contribute to initial exposure of C314. Further conformational changes likely located within the catalytic core, the dimer interface, and/or β-barrel 1 must provide the vital link to achieve full FXIII activation and thus ability to target actions at C314.
Interestingly, segment 407–424 exhibits increased deuteration at 1mM Ca2+ but loses this solvent exposure when the temperature is raised to 37°C and then 2–50 mM Ca2+ is employed. The 407–424 segment may be influenced by a nearby non-prolyl cis peptide bond Q425-F426 that is in proximity to the dimer interface. Earlier work by our group [25] demonstrated that that FXIII C409 could be alkylated with NEM when the enzyme was nonproteolytically activated with 50 mM Ca2+ and then re-equilibrated back to 1 mM. This ability was, however, lost when the exposed FXIII active site was blocked with an inhibitory Q-containing peptide [26]. Weiss and coworkers proposed that conversion of a FXIII non-prolyl cis peptide bond to the more energetically favored trans could help drive larger scale conformational changes to fully expose the FXIII active site region [39]. The current increases in Ca2+ concentration and temperature may have aided in establishing an environment that helps promote the next alterations in FXIII conformation. Such an environment may contain a less solvent exposed FXIII 407–424 segment.
Peptide 4 (72–97) in the β-sandwich and peptide 7 (190–230) in the catalytic core are proposed glutamine substrate recognition regions [38]. Peptide 7 is represented by the cyan peptide in Figure 6C. FXIII A2 in 1 mM Ca2+ displayed enhanced solvent accessibility relative to zymogen for both 88–98 and 214–230. This FXIII exposure around the glutamine recognition region also appears to be Ca2+ specific as it induces the greatest effect compared to the other metals. By contrast, Cu2+ exhibited the smallest amount of exposure and thus appears to be least likely to allow for glutamine recognition. Within peptide 4, there are differences in solvent exposure depending on the metal tested. In the peptide segment 83–99, the percent incorporation with 1 mM Ca2+ is essentially the same as with zymogen whereas Mg2+, Ba2+, and Cu2+ show protection. Within the peptide segment 88–98, Ca2+ promoted more exposure than Mg2+ and Cu2+. Another region of interest is the Lysine recognition site 646–658 in β-barrel 2 [40]. A peptide located N-terminally to this site, 632–646, is only exposed in the presence of 1 mM Ca2+, whereas Mg2+, Ba2+ and Cu2+ all display protection in this region.
The proposed glutamine substrate binding site, peptide 7 (190–230), is located in close proximity to the Ca2+ binding site and comes within 3 Å of a loop which connects D438 to E485 and E490 within the Ca2+ binding pocket. For the nonproteolytically activated forms FXIIIaCa and FXIIIaNa, the residues 220–230 exhibited further increases in deuterium incorporation in comparison to the 1–2 mM Ca2+ conditions. The peptide 7 substrate recognition sequence is another example of a FXIII region that undergoes initial conformational changes at physiological Ca2+ levels and then exhibits greater exposure at higher ion concentrations.
Low mM Metals: Activation Peptide
The activation peptide (1–37) is cleaved by IIa during physiological activation. Residues 1–23 became protected in the presence of 1 mM Ca2+ as well as 1 mM Mg2+, Ba2+ and Cu2+. The activation peptide was also protected after nonproteolytic activation with 50 mM Ca2+ at 37°C, albeit somewhat less than 1 mM Ca2+ at room temperature. Both the FXIII 1–23 and 32–40 segments exhibited protection in the HDX studies.
It was reported recently that the FXIII AP is free in solution after IIa cleavage and available for binding to monoclonal antibodies specific to FXIII-AP [48]. Our HDX results suggest that in the presence of Ca2+ the unhydrolyzed AP remains tightly associated with FXIII A2. Figure 6D shows how the activation peptide segment straddles across both monomer units and that this segment contains a combination of polar and nonpolar residues. Moreover, there are acidic and basic side chains that can participate in stabilizing interactions with the FXIII surface. Interestingly, R11 (7B blue sticks) from the activation peptide segment is hydrogen bonded to D373′ on the opposite unit of the dimer. This R11 – D373′ interaction is also in close proximity to the FXIII catalytic triad and the vital Y560. Other hydrogen bonds also seem to anchor the FXIII AP above the catalytic core.
Earlier studies by Lewis et al reported that an additional 8 weak metal sites could be found on FXIII at higher Ca2+ concentrations. The AP region and/or a complementary segment on the β-sandwich domain may accommodate some of these divalent ions [37]. Further studies will be needed to assess whether the AP segment may aid in channeling Gln substrates towards the catalytic core. An alternative possibility is that the binding of large physiological substrates will later assist in displacing this cleaved portion of FXIII from the vicinity of the transglutaminase active site.
Further Characterizing Nonproteolytic Activation
Polgar et al. demonstrated that FXIII A2 can be activated albeit slowly by 150 mM NaCl and 2 mM Ca2+ [12]. More efficient activation occurs when NaCl is raised to the 500 – 1000mM level. Low calcium, however, is still necessary. In the current work, the organic mono and divalent cations, TMAC+ and EDA2+, were investigated for the first time. As observed with Na+, TMAC+ and EDA2+ each required some calcium to elicit FXIII transglutaminase activity. Curiously, TGase activity for the TMAC+, Na+, and EDA2+ conditions (all containing 2 mM Ca2+) were greater than that of 50 mM Ca2+, our standard nonproteolytic activation method. Such results further emphasize the important role of Ca2+. Moreover, the novel use of organic cations EDA2+and TMAC+ suggests that activation is not only driven by metal ion binding interactions. Unfortunately, ionization issues in the MALDI hindered ability to carry out HDX projects with FXIIIaEDA and FXIIIaTMAC.
Overview of Results
In the current project, the FXIII A2 was monitored in the presence of Ca2+, Mg2+, Ba2+, Cu2+, Na+, EDA2+, and TMAC+ with concentrations ranging from 1–2 mM up to 50–500 mM for nonproteolytic activation. We found several regions where physiological Ca2+concentrations initiate the changes needed for activity. As expected, 1 mM Ca2+ does lead to solvent protection for the FXIII region containing the 513–514 secondary cleavage site. Surprisingly, Ba2+ and Cu2+ could protect FXIII 513–522 and 533–550 from solvent even though these two divalent cations do not shield against secondary cleavage [18]. By contrast, increasing concentrations of Ca2+ promoted exposure of 533–550. Such Ca2+ induced effects may play a role in exposing the FXIII reactive thiol group by helping to disrupt the H-bond between C314 and Y560. When considering other cations, Mg2+ and Ba2+ could both promote nonproteolytic activation in the presence of low mM Ca2+ whereas Cu2+ could not. The presence of Cu2+ seems to hinder exposure of the active site C314 as well as the Gln substrate recognition region. Our HDX studies also revealed that 1 mM Ca2+ increased deuterium incorporation at the dimer interface, around the lysine recognition region (632–646), and at two glutamine substrate recognition regions, peptide 4 (83–99) and peptide 7 (214–230). These trends continued in nonproteolytically activated FXIII.
There has been much speculation about whether FXIII A2 can adopt an open conformation similar to that reported by Pinkas et al. for TG2 [49]. Conformational changes to activated FXIII A2 can clearly be observed by mass spectrometry approaches. The gross increases in solvent accessibility that would be expected if the β-barrels had pulled fully away from contacting the catalytic core surface, however, are not observed. The fact that FXIII functions as a dimer may be a contributing element. The dynamic nature of the FXIII in solution is also important to consider.
Calcium appears to play an intriguing regulatory role in FXIII [19–20, 50–51]. Low mM levels already begin to initiate important conformational changes needed to expose the active site and make the enzyme ready to accommodate incoming substrates. Often as divalent cation concentration increases, the effects are also enhanced. Physiologically, FXIII is poised to respond to transient influxes of Ca2+ in the presence of a Na+ containing environment [12–14]. Platelet FXIII A2 is quite sensitive to such changes in solution environment and can be more readily activated nonproteolytically [12–14]. The initial conformation events that have been observed in the current study help set the stage for the larger conformational changes that are anticipated in the presence of a physiological substrate or inhibitor.
Acknowledgments
Funding for this project is supported by NIH R01 HL68440. We are grateful for the guidance of T. Michael Sabo with the HDX procedure as well as his previous FXIII dynamics contributions. We would also like to thank Paul Bishop of Zymogenetics, Inc., Seattle, WA for supplying our recombinant FXIII A2. Dr. Michael Merchant and Daniel Wilkey in the University of Louisville Clinical Proteomics Laboratory assisted us tremendously in our MS/MS analysis. Finally, we thank P. Diophode, M. Jadhav and M. Malovichko for their critical review of the current project.
Footnotes
Abbreviations: FXIII, recombinant human cellular Factor XIII; FXIII(a), unactivated or activated Factor XIII; FXIIIaCa, calcium activated Factor XIII; FXIIIaNa, sodium activated Factor XIII; FXIIIaIIa, thrombin activated Factor XIII; IIa, thrombin; TGase, transglutaminse; TG2, transglutaminase 2; IAA, iodoacetamide; MALDI-TOF MS, matrix-assisted laser desorption-ionization time-of-flight mass spectrometry; HDX, hydrogen-deuterium exchange; GDH, glutamate dehydrogenase; GEE, glycine ethyl ester; TMAC+, tetramethylammonium chloride; EDA2+, ethylenediamine
References
- 1.Muszbek L, Bagoly Z, Bereczky Z, Katona E. Cardiovascular & Hematological Agents in Medicinal Chemistry. 2008;6:190–205. doi: 10.2174/187152508784871990. [DOI] [PubMed] [Google Scholar]
- 2.Lorand L. Annals of the New York Academy of Sciences. 2001;936:291–311. doi: 10.1111/j.1749-6632.2001.tb03516.x. [DOI] [PubMed] [Google Scholar]
- 3.Weisel JW. Advances in Protein Chemistry. 2005;70:247–299. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- 4.Yorifuji H, Anderson K, Lynch G, Van de Water L, McDonagh J. Blood. 1988;72:1645–1650. [PubMed] [Google Scholar]
- 5.Yee VC, Pedersen LC, Letrong I, Bishop PD, Stenkamp RE, Teller DC. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:7296–7300. doi: 10.1073/pnas.91.15.7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis CG, Brown KL, Credo RB, Domanik RA, Gray A, Stenberg P, Lorand L. Biochemistry. 1974;13:3774–3780. doi: 10.1021/bi00715a024. [DOI] [PubMed] [Google Scholar]
- 7.Chung SI, Lewis MS, Folk JE. Journal of Biological Chemistry. 1974;249:940–950. [PubMed] [Google Scholar]
- 8.Schwartz ML, Pizzo SV, Hill RL, Mckee PA. Journal of Biological Chemistry. 1973;248:1395–1407. [PubMed] [Google Scholar]
- 9.Pedersen LC, Yee VC, Bishop PD, Letrong I, Teller DC, Stenkamp RE. Protein Science. 1994;3:1131–1135. doi: 10.1002/pro.5560030720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorand L, Credo RB, Janus TJ. Methods in Enzymology. 1981;80:333–341. doi: 10.1016/s0076-6879(81)80029-8. [DOI] [PubMed] [Google Scholar]
- 11.Credo RB, Curtis CG, Lorand L. P Natl Acad Sci USA. 1978;75:4234–4237. doi: 10.1073/pnas.75.9.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polgar J, Hidasi V, Muszbek L. Biochemical Journal. 1990;267:557–560. doi: 10.1042/bj2670557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muszbek L, Polgar J, Boda Z. Thrombosis and Haemostasis. 1993;69:282–285. [PubMed] [Google Scholar]
- 14.Muszbek L, Haramura G, Polgar J. Thrombosis and Haemostasis. 1995;73:702–705. [PubMed] [Google Scholar]
- 15.Mary A, Achyuthan KE, Greenberg CS. Biochimica Et Biophysica Acta. 1988;966:328–335. doi: 10.1016/0304-4165(88)90082-7. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi N, Takahashi Y, Putnam FW. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:8019–8023. doi: 10.1073/pnas.83.21.8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai TS, Achyuthan KE, Santiago MA, Greenberg CS. Journal of Biological Chemistry. 1994;269:24596–24601. [PubMed] [Google Scholar]
- 18.Mary A, Achyuthan KE, Greenberg CS. Archives of Biochemistry and Biophysics. 1988;261:112–121. doi: 10.1016/0003-9861(88)90110-5. [DOI] [PubMed] [Google Scholar]
- 19.Hornyak TJ, Shafer JA. Biochemistry. 1991;30:6175–6182. doi: 10.1021/bi00239a014. [DOI] [PubMed] [Google Scholar]
- 20.Curtis CG, Brown KL, Credo RB, Domanik RA, Gray A, Stenberg P, Lorand L. Biochemistry. 1974;13:3774–3780. doi: 10.1021/bi00715a024. [DOI] [PubMed] [Google Scholar]
- 21.Hornyak TJ, Bishop PD, Shafer JA. Biochemistry. 1989;28:7326–7332. doi: 10.1021/bi00444a027. [DOI] [PubMed] [Google Scholar]
- 22.Yee VC, Pedersen LC, Bishop PD, Stenkamp RE, Teller DC. Thromb Res. 1995;78:389–397. doi: 10.1016/0049-3848(95)00072-y. [DOI] [PubMed] [Google Scholar]
- 23.Fox BA, Yee VC, Pedersen LC, Le Trong I, Bishop’ PD, Stenkamp RE, Teller DC. Journal of Biological Chemistry. 1999;274:4917–4923. doi: 10.1074/jbc.274.8.4917. [DOI] [PubMed] [Google Scholar]
- 24.Turner BT, Maurer MC. Biochemistry. 2002;41:7947–7954. doi: 10.1021/bi025630n. [DOI] [PubMed] [Google Scholar]
- 25.Turner BT, Sabo TM, Wilding D, Maurer MC. Biochemistry. 2004;43:9755–9765. doi: 10.1021/bi049260+. [DOI] [PubMed] [Google Scholar]
- 26.Sabo TM, Brasher PB, Maurer MC. Biochemistry. 2007;46:10089–10101. doi: 10.1021/bi700579z. [DOI] [PubMed] [Google Scholar]
- 27.Kristiansen GK, Andersen MD. Journal of Biological Chemistry. 2011;286:9833–9839. doi: 10.1074/jbc.M110.174128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen MD, Faber JH. International Journal of Mass Spectrometry. 2011;302:139–148. [Google Scholar]
- 29.Fickenscher K, Aab A, Stuber W. Thrombosis and Haemostasis. 1991;65:535–540. [PubMed] [Google Scholar]
- 30.Karpati L, Penke B, Katona E, Balogh I, Vamosi G, Muszbek L. Clinical Chemistry. 2000;46:1946–1955. [PubMed] [Google Scholar]
- 31.Marinescu A, Cleary DB, Littlefield TR, Maurer MC. Archives of Biochemistry and Biophysics. 2002;406:9–20. doi: 10.1016/s0003-9861(02)00407-1. [DOI] [PubMed] [Google Scholar]
- 32.Mandell JG, Falick AM, Komives EA. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandell JG, Falick AM, Komives EA. Analytical Chemistry. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 34.Sabo TM, Farrell DH, Maurer MC. Biochemistry. 2006;45:7434–7445. doi: 10.1021/bi060360k. [DOI] [PubMed] [Google Scholar]
- 35.Wang LT, Lane LC, Smith DL. Protein Science. 2001;10:1234–1243. doi: 10.1110/ps.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JW, Smith DL. Protein Science. 2001;10:1079–1083. doi: 10.1110/ps.53201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis B, Freyssinet J-M, Holbrook J. Biochem J. 1978;169:397–402. doi: 10.1042/bj1690397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Achyuthan KE, Slaughter TF, Santiago MA, Enghild JJ, Greenberg CS. Journal of Biological Chemistry. 1993;268:21284–21292. [PubMed] [Google Scholar]
- 39.Weiss MS, Metzner HJ, Hilgenfeld R. FEBS Letters. 1998;423:291–296. doi: 10.1016/s0014-5793(98)00098-2. [DOI] [PubMed] [Google Scholar]
- 40.Mitkevich OV, Sobel JH, Shainoff JR, Vlasik TN, Kalantarov GF, Trakht IN, Streltsova ZA, Samokhin GP. Blood Coagulation & Fibrinolysis. 1996;7:85–92. doi: 10.1097/00001721-199601000-00011. [DOI] [PubMed] [Google Scholar]
- 41.Mitkevich OV, Shainoff JR, DiBello PM, Yee VC, Teller DC, Smejkal GB, Bishop PD, Kolotushkina IS, Fickenscher K, Samokhin GP. Journal of Biological Chemistry. 1998;273:14387–14391. doi: 10.1074/jbc.273.23.14387. [DOI] [PubMed] [Google Scholar]
- 42.Credo RB, Curtis CG, Lorand L. Biochemistry. 1981;20:3770–3778. doi: 10.1021/bi00516a016. [DOI] [PubMed] [Google Scholar]
- 43.Ichinose A, Davie EW. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5829–5833. doi: 10.1073/pnas.85.16.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi N, Takahashi Y, Putnam FW. Proc Natl Acad Sci USA. 1986;83:8019–8023. doi: 10.1073/pnas.83.21.8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ambrus A, Banyai I, Weiss MS, Hilgenfeld R, Keresztessy Z, Muszbek L, Fesus L. Journal of Biomolecular Structure & Dynamics. 2001;19:59. doi: 10.1080/07391102.2001.10506720. [DOI] [PubMed] [Google Scholar]
- 46.Souri M, Yee VC, Kasai K, Kaneshiro T, Narasaki K, Castaman G, Ichinose A. British Journal of Haematology. 2001;113:652–654. doi: 10.1046/j.1365-2141.2001.02797.x. [DOI] [PubMed] [Google Scholar]
- 47.Király R, Csosz E, Kurtán T, Antus S, Szigeti K, Simon-Vecsei Z, Korponay-Szabó IR, Keresztessy Z, Fésüs L. The FEBS Journal. 2009;276:7083–7096. doi: 10.1111/j.1742-4658.2009.07420.x. [DOI] [PubMed] [Google Scholar]
- 48.Ortner E, Schroeder V, Walser R, Zerbe O, Kohler HP. Blood. 2010;115:5089–5096. doi: 10.1182/blood-2009-11-253062. [DOI] [PubMed] [Google Scholar]
- 49.Pinkas DM, Strop P, Brunger AT, Khosla C. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai TS, Achyuthan KE, Santiago MA, Greenberg CS. J Biol Chem. 1994;269:24596–24601. [PubMed] [Google Scholar]
- 51.Cooke RD, Pestell TC, Holbrook JJ. Biochemical Journal. 1974;141:675–682. doi: 10.1042/bj1410675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]