

Abstract
The 26S proteasome is a 2.5-MDa, ATP-dependent multisubunit proteolytic complex that processively destroys proteins carrying a degradation signal. The proteasomal ATPase heterohexamer is a key module of the 19S regulatory particle; it unfolds substrates and translocates them into the 20S core particle where degradation takes place. We used cryoelectron microscopy single-particle analysis to obtain insights into the structural changes of 26S proteasome upon the binding and hydrolysis of ATP. The ATPase ring adopts at least two distinct helical staircase conformations dependent on the nucleotide state. The transition from the conformation observed in the presence of ATP to the predominant conformation in the presence of ATP-γS induces a sliding motion of the ATPase ring over the 20S core particle ring leading to an alignment of the translocation channels of the ATPase and the core particle gate, a conformational state likely to facilitate substrate translocation. Two types of intersubunit modules formed by the large ATPase domain of one ATPase subunit and the small ATPase domain of its neighbor exist. They resemble the contacts observed in the crystal structures of ClpX and proteasome-activating nucleotidase, respectively. The ClpX-like contacts are positioned consecutively and give rise to helical shape in the hexamer, whereas the proteasome-activating nucleotidase-like contact is required to close the ring. Conformational switching between these forms allows adopting different helical conformations in different nucleotide states. We postulate that ATP hydrolysis by the regulatory particle ATPase (Rpt) 5 subunit initiates a cascade of conformational changes, leading to pulling of the substrate, which is primarily executed by Rpt1, Rpt2, and Rpt6.
Keywords: AAA ATPase, ubiquitin–proteasome pathway, hybrid methods in structural biology
The 26S proteasome is the executive arm of the ubiquitin-proteasome system (UPS), the major pathway for intracellular protein degradation in eukaryotic cells (1, 2). It degrades proteins that are marked for destruction by the covalent attachment of a polyubiquitin chain. The 2.5-MDa complex comprises a core particle (CP), the 20S proteasome, harboring the proteolytically active sites and one or two regulatory particles (RPs), which associate with the barrel-shaped CP. A key component of the RP is the AAA-ATPase module (ATPase associated with various cellular activities), which associates with the α-rings of the CP and prepares substrates for degradation. Whereas the 26S proteasome is the only soluble ATP-dependent protease in the eukaryotic cytosol, bacteria possess several different ATPdependent proteases like the ClpXP, HslUV, and the eubacterial proteasome-ARC/Mpa (AAA ATPase forming ring-shaped complexes/mycobacterial proteasomal ATPase) system (3).
The CP consists of four seven-membered rings (4). The two adjacent heteroheptameric β-rings form a cavity, which harbors the active sites (5, 6). The β-rings are sandwiched between two heteroheptameric α-rings, whose C-termini form a gate controlling access to the central cavity. Both ends of the CP associate with 19S RPs, which are responsible for the recognition of substrates, their unfolding, and translocation into the CP. Although the CP crystal structure became available almost two decades ago (5, 6), the molecular architecture of the entire 26S proteasome has been determined only recently by cryoelectron microscopy (cryo-EM) approaches (7–9). We refer to it as an ATP-hydrolyzing (ATPh) structure in the following because it has been determined with samples prepared in the presence of saturating ATP concentrations. Surrounded by the RP non-ATPases (Rpn) 1–3, 5–13, and 15, the RP AAA-ATPases (Rpt1–6) form heterohexameric rings that attach to both α-rings. Collectively, the six subunits enclose a translocation channel, which substrates have to pass before they can enter the CP via the gate formed by the CP α-subunits. By forcing their passage through the narrow channel of the AAA ring, they become unfolded as in other protease-associated AAA-ATPases (e.g., ClpXP, HslUV, and ARC). The AAA-ATPase hexamer consists of two stacked rings: The AAA folds of the Rpt subunits assemble into the AAA ring, which binds to the CP. The AAA folds are preceded by oligonucleotide-binding (OB) domains, which form the OB ring (10). The N-terminal coiled coils of the Rpt subunits form three distinct heterodimers, which radiate outwards from the OB ring. The AAA-ATPase has reverse-chaperone or unfoldase activity (10). The unfolding of substrates occurs as they pass the narrow channel of the AAA ring. Substrates are assumed to be pulled by conserved aromatic residues lining the channel, which belong to the “pore loops” (11). Translocation of substrates that do not require unfolding has been reported to be facilitated by ATP binding as indicated by accelerated degradation of peptides in the presence of ATP-γS (12). ATP-γS is a slowly hydrolysable ATP analog, which effectively traps the 26S proteasome in an ATP-bound form without hydrolysis.
Here, we used cryo-EM single-particle analysis to obtain insights into the nucleotide-dependent structural changes of the 26S proteasome. The subnanometer resolution reconstruction of the 26S proteasome in the presence of ATP-γS reveals that the RP undergoes a major conformational change. We interpreted the ATP-γS–bound conformation beyond the nominal resolution by using an atomic model that was built by fitting essentially rigid domains of the ATPh conformation. The structural changes we observe offer a possible explanation for the increased activity of the 26S proteasome upon ATP binding. The arrangement of RP subunits with respect to the CP suggests that the ATP-γS structure represents a translocation-competent conformation and provides insight into the mechanism of force generation and protein unfolding by the proteasomal ATPases.
Results
Pseudoatomic ATP-γS–Bound 26S Proteasome Structure.
We used automated data acquisition to collect a dataset of ∼280,000 Saccharomyces cerevisiae 26S proteasome particles in the presence of 1 mM ATP-γS. Three-dimensional reconstructions were computed without imposing any symmetry (Fig. S1). When C2 symmetry was imposed, the density became more blurred and the RP is resolved significantly worse (with Fourier shell correlation of 0.5 at 13.2 Å and 9.9 Å at 0.3, compared for 9.8 Å and 8.1 Å for C1 symmetry; Fig. S2). The asymmetry of the holocomplex is also apparent from the distinctly different structures of the two RPs (Fig. S3). Analysis of the variance map corresponding to the reconstruction suggests that the particles exhibit a substantial degree of structural heterogeneity, which is higher than that observed for the ATPh map (Fig. S4 A and B). For both maps, the variance is highest in the vicinity of Rpn1, which is known to interact with various proteasome interacting proteins.
For further analysis, we used the proteasome half with less variability near Rpn1, as judged by the local resolution of the reconstruction (Fig. S4C). Rod-like densities indicated that the resolution is sufficient to localize protein helices in the map. A pseudoatomic model of the 26S proteasome molecule in the ATP-γS–bound conformational state (Fig. 1) was obtained by using the ATPh structure as a starting model (PDB ID code: 4B4T) (9). Because of the relatively low resolution of the map, the subunits were strongly restrained by an elastic network, which kept subunit domains essentially rigid during the fitting process. The colocalization of α-helices in the final model and the rod-shaped features in the EM map suggests that the subunit models are positioned accurately and that the conformational change does not involve major structural changes within the subunits or domains (Fig. S5). In the following, the conformational changes of the ATP-γS structure are compared with the ATPh structure.
Fig. 1.
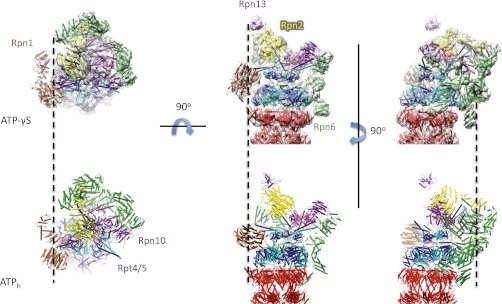
Comparison of the ATP-γS–bound structure and ATPh structure. (Upper) Pseudoatomic ATP-γS model fitted into the cryo-EM reconstruction of the 26S proteasome (gray) in three different views. (Lower) Corresponding views of ATPh model (PDB ID code: 4B4T). The dashed lines mark the positions of Rpn13 (Left and Center) and Rpn6 (Right) to facilitate the comparison between Upper and Lower.
Large-Scale Conformational Change of RP.
Substantial conformational changes were observed in the 26S proteasome upon exchanging ATP to ATP-γS in the buffer (Fig. 1 and Movies S1 and S2). Although the CPs are essentially identical for the two nucleotide states, the RPs differ notably. Not only does the ATPase ring shift in the plane orthogonal to the CP axis, it is also remodeled. These conformational changes propagate to the Rpn subunits. All Rpn subunits undergo a substantial rotation between the two different nucleotide states. This rotation is particularly apparent at the distal ends of the RP, where the N- and C-terminal segments of Rpn2 form an elongated domain. This domain undergoes a rotation of ∼15 degrees between the two different nucleotide states around the long axis of the CP (Fig. 1).
The rotation of the Rpn subunits brings the ubiquitin receptor Rpn10 into closer proximity of the coiled-coil of the AAA-ATPase dimer Rpt4/Rpt5. A concomitant conformational change of this coiled coil induces contact of it with Rpn10. Interestingly, cross-links between this coiled coil and Rpn10 had been observed (8). These cross-links were considered as false positives based on the ATPh structure, but they comply with the ATP-γS–bound structure.
Interdomain Arrangement of AAA-ATPase Subunits.
We analyzed the intersubunit domain arrangement within the AAA ring for both the ATP-γS and ATPh conformations (Fig. 2 A and B). It has been postulated for ClpX that the contacts formed by the large subunit of one ATPase molecule and the small subunit of its neighbor remain rigid during the ATP-hydrolysis cycle (13) (Fig. 2 A and B). Movements of such modules (from now on referred to as intersubunit modules; ISMs) are responsible for the mechanical outcome of the ATPase cycle. The six ISMs constituting the ClpX ring are structurally similar to each other across the hexamer.
Fig. 2.

Intersubunit modules in the AAA ring. (A and B) Structures of the AAA-ATPase for the ATP-γS structure and the ATPh structure, respectively. The subunits Rpt1, Rpt6, and Rpt4 are shown in blue and Rpt2, Rpt3, and Rpt5 in cyan. Large and small AAA domains of each subunit are shown in darker and lighter shades, respectively. ISMs are annotated by black (closed conformation) and red (open) borders. (C and D) Structure of ISM1–5 in its closed and open conformations, respectively. Rotation of the small domain by ∼21° is required for the conformational transition between the two states. (E) Nucleotide-binding sites of Rpt5 and Rpt4 in the ATP-γS structure. For the closed ISM (ISM4–5) the Arg finger (magenta triangle) is placed in the proximity of nucleotide-binding site of Rpt4 (shown in red), allowing for interaction. For the open conformation (ISM5–1), the Arg finger rotates away from the nucleotide-binding site of Rpt5 (white); this rearrangement prevents an engagement of the Arg finger in binding. (F and G) The two lockwasher-like topologies of the AAA-ATPase hexamer. Continuity of interactions between the neighboring subunits is illustrated by the involvement of the Arg finger in nucleotide binding at the adjacent large domain. The introduction of the open ISM (enclosed in red) allows closure of the ring.
Different from ClpX, the ISMs of the proteasomal ATPases adopt two distinct conformations. In the ATP-γS–bound structure, the ISM formed by the large AAA-domain of Rpt1 and the small AAA domain of Rpt5 (ISM1–5) deviate significantly from the other five ISMs (Fig. 2 A and D), which resemble the structure observed in ClpX (PDB ID code: 3HWS) (14). In contrast, ISM1–5 is structurally similar to the ISM formed by crystal packing in the crystal structure of the proteasome-activating nucleotidase (PAN) bound to ADP (15) (PDB ID code: 3H4M). PAN is the homohexameric homolog of the Rpt subunits in archaea; however, it does not form the hexamer in the crystal because of the OB domain deletion. In the ATPh structure, ISM3–6 is similar to PAN-ADP, whereas the other five subunits again resemble the ClpX structures. During its transition from the PAN-ADP–like conformation in the ATP-γS structure to the ClpX-like conformation in the ATPh structure, the large domain of ISM1–5 undergoes a rotation of 21 degrees (Fig. 2 C and D). From now on, we will refer to the PAN-ADP–like conformations (ISM3–6 for the ATPh structure and ISM1–5 for the ATP-γS–bound structure) as “open” ISMs, and to the ClpX-like conformation as “closed” ISM.
Nucleotide-Binding Sites.
In the AAA-ATPases, the nucleotide is bound at the interface of the large and the small domain of any given ATPase subunit. However, the major recognition motifs—Walker A and Walker B—are both located in the large domain. The arginine finger, an important nucleotide recognition feature composed of two arginine residues, reaches from the neighboring large subunit toward the phosphates of the ATP, contributing to the ATP binding (16). The Walker A motif is involved in nucleotide binding, whereas the Walker B motif is essential for ATP hydrolysis. The role of the Arg finger is less well understood, but mutations of this region have been shown to interfere with effective ATP hydrolysis and are often lethal (17). Furthermore, the Arg finger has been hypothesized to act as a nucleotide sensor between the neighboring subunits (18, 19).
We examined the structures of these nucleotide-binding clefts for both conformations. At the resolution of both the ATP-γS–bound and ATPh maps, it is not possible to directly visualize the bound nucleotide. However, some insight into the nucleotide state of each subunit can be obtained from the three canonical features of the ATP binding site: Walker A and B motifs and arginine finger. The subunits whose small domains are involved with the closed ISMs have all three features in proximity to form a complete nucleotide-binding site. In contrast, the subunits whose small domains form the open ISMs (Rpt6 in ATPh structure and Rpt5 in ATP-γS structure) lack the proximal Arg finger (Fig. 2E). This effect is due to the rotation of the large subunit involved in an open ISM, which removes the Arg finger from the neighboring nucleotide-binding site. Thus, because of the presence of two ISMs, two different types of the nucleotide-binding sites exist in the ring.
Topology of AAA-ATPase Ring.
The AAA-ATPase rings in both conformations deviate from a sixfold pseudosymmetry and the subunits arrange in a helical staircase/lockwasher-like structure (Fig. 2 F and G), similar to the ones previously seen for helicases (7, 20). In such a hexamer, the pore loops are positioned on different altitudes with respect to the plane of the CP α-subunits following a lockwasher-like course. The contacts between the neighboring subunits are mediated by the Arg fingers reaching to the neighboring nucleotide-binding sites (contact between the two adjacent large subunits), and the direct contact between the neighboring large and small subunits forming the ISMs.
The helical arrangement is broken when one of the ISMs adopts an open conformation and the distance between the two neighboring large domains increases. Such conformational feature coincides with the retraction of the Arg finger leaving the nucleotide-binding site incomplete. This lack of continuity of contacts around the ring causes a deviation from the planar structure and leads to a lockwasher-like arrangement.
Interaction of AAA-ATPases with Neighboring Subunits.
Because of the symmetry mismatch between the CP (pseudo-D7) and the AAA-ATPase ring (pseudo-C6), the individual Rpt-α subunit interactions differ (Fig. 3). Strikingly, the pseudosymmetry axes of CP and each AAA-ATPase hexamer do not align in the ATPh conformation; the AAA-ATPase rings are laterally shifted with respect to the CP (21). In the ATPh structure, Rpt1, Rpt4, and Rpt5 have the largest interaction surface with the CP. In the ATP-γS structure, the small domains of two ATPases, Rpt1 and Rpt5, remain in contact with the alpha ring (α5 and α6 subunits, respectively). In contrast, the contact between Rpt4 and the α7-α1 interface is lost upon the conformational transition.
Fig. 3.

Displacement of small AAA domains of Rpt subunits with respect to the two adjacent α-subunits upon the change of the nucleotide state. Arrangements in the ATPh conformation are shown in color and the ATP-γS arrangements in gray.
There are also a number of interactions between AAA ring and the lid subunits. For example, the small subunit of Rpt6 maintains its contact to Rpn6 in the ATP-γS structure. Rpn6 retains the interaction with the small domain of Rpt6, detaching from the CP upon the conformational transition. In this transition the N-terminal tetratricopeptide repeat domain of Rpn6 rotates by ∼21 degrees with respect to its PCI (proteasome, COP9, and initiation factor 3) domain (Fig. S6) Concomitantly, the surrounding PCI subunits move with respect to the CP. In particular, a contact of Rpn5 with Rpt4 and the CP is established in the ATP-γS–bound structure. Interestingly, numerous cross-links between Rpt4 and Rpn5 were observed (8), which are in better agreement with the model of the ATP-γS structure than with the ATPh structure (Fig. S7).
Channels and Pore Loops in ATP-γS Structure.
To obtain insights into the consequences of the conformational changes for substrate translocation into the CP, we analyzed the relative positions of the central openings for the α, AAA, and OB rings in both conformations. In the presence of ATP-γS, the AAA ring moves by ∼15 Å and the OB ring follows this motion (Fig. 4). As a consequence, the channel axes of the ATPase rings (both AAA and OB) align much better with the gate of the CP in the ATP-γS–bound conformation compared with the ATPh conformation. The association of Rpn11 to the OB ring appears to be preserved during the conformational change, remaining close to the substrate entry path.
Fig. 4.

Alignment of the AAA-ATPase translocation channels and the α-ring gate. (A and B) The AAA ring and the α ring in ATP-γS–bound (A) and ATPh conformation (B). (C and D) OB ring and α ring. (E and F) Schematic of channels of α, AAA, and OB ring for ATP-γS (E) and ATP (F) conformations.
We furthermore analyzed the movements of the pore loops with respect to the plane of the alpha ring. Because the resolution of our reconstructions is too low to resolve the loops experimentally, we decided to infer their location from the tip of helix 3, which we can resolve in our maps. Upon transition from the ATP-γS conformation to the ATPh conformation, the pore loops of different subunits move differently. The vertical motion of Rpt3, Rpt4, and Rpt5 is rather small (3–4 Å), whereas Rpt1, Rpt2, and Rpt6 move much more (∼8–18 Å) toward the gate of the alpha ring (Fig. 5).
Fig. 5.

Effects of transition from γS- (Left) to ATPh conformation on substrate translocation. (Left) Differences of vertical position of pore loops that are thought to pull the substrate. (Center and Right) Cut-through views through ATP-γS and ATPh structures show how alignment of the rings affects the substrate path. The pore loops of Rpt1 and Rpt3 are marked with stars.
Discussion
Nucleotide-Dependent Conformational Transition Between the Two Lockwasher-Like Conformations.
In both structures, ATPh and ATP-γS, the subunit at the bottom of the lockwasher lacking the proximal Arg finger is topologically distinct from the other five AAA-ATPase subunits in the ring. Thus, it is conceivable that these subunits (Rpt6 for the ATPh structure and Rpt5 for the ATP-γS structure) have different nucleotide-binding properties and, possibly, a nucleotide state different from that of the other subunits. It is not possible to determine the nucleotide state of Rpt1–4, but because the binding sites appear accessible and complete, they all are likely able to bind ATP.
There is no strict rule known for the correlation of ISM conformation and its nucleotide state. However, some insight into the protein–protein interactions can be obtained from the analysis of crystal packing interactions. The ISMs seen in the PAN homotrimer bound to ADP are similar to the open ISMs in our structures (Fig. S8). The Arg fingers do not engage the nucleotide in the PAN crystal structure. We therefore postulate that the Arg finger selectively engages the γ-phosphate of ATP and that the ATPase adopting such an open ISM is either empty or ADP bound (product-bound state). Therefore, we suggest that Rpt5 and Rpt6 are ADP bound or nucleotide free in the ATP-γS–bound and ATPh structure, respectively.
Recent biochemical studies suggested that the roles of mammalian Rpt5 and Rpt6 in the nucleotide cycle are distinct from those of the other four Rpt subunits (22). Although mutations in the Walker A motifs of Rpt1–4, which prevent nucleotide binding, completely precluded efficient proteasome assembly, it was not the case for such mutants of Rpt5 and Rpt6. The resulting 26S proteasome mutants were fully capable of hydrolyzing small peptides, but they were unable to degrade substrates that required ATP-dependent unfolding. If proteasomes can assemble with either Rpt5 or Rpt6 being unable to bind the nucleotide, there should be stable conformations of proteasomal ATPases without nucleotides at these subunits. This observation is consistent with our hypothesis of Rpt5 and Rpt6 being not ATP bound in the two stable conformations of the 26S proteasome, the ATP-γS and ATPh structures.
The presence of Arg fingers engaging the nucleotides (closed ISM) induces the helical lockwasher like topology of the AAA ring. Closing this ring requires incorporation of an open ISM at one site of the hexamer. The use of ATP-γS allowed us to trap the proteasomal ATPases in their high-energy prehydrolysis conformation. We propose that transition between the two lockwasher conformations depends on ATP-hydrolysis by Rpt5 or Rpt6. We suggest that the Rpt5 subunit is empty, and Rpt6 is ATP bound in the ATP-γS structure. Upon hydrolysis of an ATP molecule bound at Rpt6 to ADP, the Arg finger in the ISM3–6 would be no longer required to engage the nucleotide and could rearrange into an open conformation. That would release the topological strain in the ring and allow the ISM1–5 to rearrange into the closed conformation, which allows ATP binding. Thus, the ATPh structure would effectively represent a lower-energy conformation of the hexamer. The reverse transition would require a reversal of all these steps. It may well be possible that the transition between the ATP-γS–bound and ATPh conformations involves intermediate steps, e.g., open ISM conformations at subunits other than Rpt5 and Rpt6 possibly giving rise to a rotary mechanism suggested for V-ATPases (23) and DnaB-like helicases (24) that both belong to the same superfamily as proteasomal ATPases (25). The observed structural heterogeneity of the ATP-γS data supports such a notion, and further efforts in classification will be required to identify additional intermediate states.
Distinct Roles of Different ATPases in Substrate Handling.
In PAN, the archaeal homolog of the proteasomal AAA-ATPase hexamer, the pore loop is thought to be responsible for substrate pulling (11). It has also been postulated to have such a role in other ATPases (26). Although the change in the ATPase ring conformation appears to be symmetrical in the sense that the general lockwasher topology is preserved, the motions of the pore loops with respect to the alpha ring central pore are strikingly different. The difference of the motions indicate distinct roles of the subunits in substrate translocation. Rpt1, Rpt2, and Rpt6 generate most of the pulling motion that unfolds the substrate and translocates it into the CP. The other subunits, Rpt3, Rpt4, and Rpt5, mostly move horizontally, following rearrangement of the whole ATPase ring and not obstructing the path of the substrate (Fig. 5). We suggest that these three subunits move in to hold the substrate in place and prevent its slipping backward, without exerting any direct pulling force. Upon transition to the low energy ATPh conformation, the loops of Rpt1, Rpt2, and Rpt6 are in their lowest positions (after the pulling of the substrate) and the subunits Rpt3, Rpt4, and Rpt5 are lifted out of the path of the substrate.
Translocation Competent Conformation of 26S Proteasome.
The conformational changes seen in the ATP-γS structure suggest that they facilitate translocation of the substrate, which would explain the accelerated degradation of peptides by the 26S proteasome in the presence of ATP-γS compared with ATP (12). Therefore, we propose that this high-energy conformation is translocation competent. When the pulling ATPases are primed for pulling the substrate, the channels of the three concentric rings (OB, AAA, and α) become aligned, clearing its path to the core particle. Rpn11, the proteasome-incorporated deubiquitylase, follows the motions of the OB ring, always remaining close to the substrate entry path. Upon substrate processing, polyubiquitin covalently attached to the protein substrate chain will eventually be moved close to the ATPase channel, where it will be cleaved by Rpn11. In this postpulling conformation, the three rings become misaligned once again, holding the substrate in place and preventing its slipping backward.
Conclusions
The use of ATP-γS has enabled us to stall the 26S proteasome in a hitherto unknown conformation. We postulate that this structure is a translocation-competent conformation representing the state before pulling of the substrate into the CP. The conformational changes involve ATPase remodeling, alignment of the channels in the ATPase and CP gate, and the rearrangement of RP non-ATPase subunits. The transition between the two lockwasher-like conformations of the AAA-ATPase is key to exerting force on the substrate. Although so far only two different conformational states of the proteasome have been elucidated, they already give a general idea of ATPase-dependent motion. The high variance observed in our cryo-EM data (Fig. S4) in the presence of ATP-γS suggests that the 26S proteasomes adopts additional low-abundance conformational states beyond the one described here. Further biochemical and structural studies will be required to address the issues of kinetics of ATP hydrolysis and of substrate binding and processing in more detail.
Materials and Methods
Sample Preparation and Cryo-EM.
The 26S proteasomes were purified from 3xFLAGRpn11 S. cerevisiae cells as described (27). In brief, cells are lysed, proteasomes are pulled down by using anti-FLAG-beads (Sigma) and are further purified by using a sucrose gradient. In contrast to the original protocol, nucleotide was exchanged for 1 mM ATP-γS during the affinity purification step. Afterward, every buffer contained 1 mM ATP-γS. The sample was applied to holey carbon grids (Quantifoil Micro Tools), vitrified, and imaged by using a Titan Krios electron microscope (FEI). Data were automatically acquired by using TOM2 (28). Images were corrected for the contrast transfer function in TOM (29), particles were localized automatically based on ref. 30), and reconstruction was performed by using XMIPP (31). Variance maps of the ATPh and ATP-γS maps were generated as described in ref. 32.
Molecular Modeling.
For the fitting, we used the Molecular Dynamics Flexible Fitting (MDFF) approach (33) by using the ATPh structure as the initial model. To avoid overfitting due to limited resolution of the reconstruction, each proteasomal subunit was restrained by an elastic network to preserve its initial tertiary structure. Proteasomal ATPases were additionally split into the small α-helical C-terminal domain, large ATPase domain, and N-terminal OB ring domain, which were allowed to move independently of each other.
Supplementary Material
Acknowledgments
We thank Antje Aufderheide for support in generating the variance maps, Elizabeth Villa for support with MDFF, and Andreas Bracher for carefully reading the manuscript. Our research is supported by funding from the European Union Seventh Framework Programme Proteomics Specification in Space and Time Grant HEALTH-F4-2008-201648 (to W.B.), Deutsche Forschungsgemeinschaft Excellence Cluster CIPSM and SFB-1035 (to W.B.), Human Frontiers Science Program Career Development Award (to F.F.), and European Molecular Biology Organization and Marie Curie Actions Long-Term Fellowship (to P.Ś.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The single particle reconstruction and the atomic coordinates have been deposited in the Electron Microscopy Data Bank, www.ebi.ac.uk/pdbe/emdb (accession no. 2348); and the Protein Data Bank, www.pdb.org (PDB ID code 4bgr), respectively.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305782110/-/DCSupplemental.
References
- 1.Voges D, Zwickl P, Baumeister W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 2.Kish-Trier E, Hill CP. Structural biology of the proteasome. February 13, 2013. Annu Rev Biophys, 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 4.Peters JM, Cejka Z, Harris JR, Kleinschmidt JA, Baumeister W. Structural features of the 26 S proteasome complex. J Mol Biol. 1993;234(4):932–937. doi: 10.1006/jmbi.1993.1646. [DOI] [PubMed] [Google Scholar]
- 5.Löwe J, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268(5210):533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 6.Groll M, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 7.Lander GC, et al. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482(7384):186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasker K, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109(5):1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck F, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci USA. 2012;109(37):14870–14875. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djuranovic S, et al. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell. 2009;34(5):580–590. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 11.Zhang F, et al. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34(4):485–496. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell. 2011;144(4):526–538. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 2012;19(6):616–622. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139(4):744–756. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34(4):473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 2004;146(1-2):106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6(7):519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, et al. Multifunctional roles of the conserved Arg residues in the second region of homology of p97/valosin-containing protein. J Biol Chem. 2005;280(49):40515–40523. doi: 10.1074/jbc.M509636200. [DOI] [PubMed] [Google Scholar]
- 19.Karata K, Inagawa T, Wilkinson AJ, Tatsuta T, Ogura T. Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases. Site-directed mutagenesis of the ATP-dependent protease FtsH. J Biol Chem. 1999;274(37):26225–26232. doi: 10.1074/jbc.274.37.26225. [DOI] [PubMed] [Google Scholar]
- 20.Thomsen ND, Berger JM. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell. 2009;139(3):523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nickell S, et al. Insights into the molecular architecture of the 26S proteasome. Proc Natl Acad Sci USA. 2009;106(29):11943–11947. doi: 10.1073/pnas.0905081106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YC, Li X, Thompson D, Demartino GN. ATP binding by proteasomal ATPases regulates cellular assembly and substrate-induced functions of the 26 S proteasome. J Biol Chem. 2013;288(5):3334–3345. doi: 10.1074/jbc.M112.424788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arai S, et al. Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature. 2013;493(7434):703–707. doi: 10.1038/nature11778. [DOI] [PubMed] [Google Scholar]
- 24.Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell. 2012;151(2):267–277. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lupas AN, Martin J. AAA proteins. Curr Opin Struct Biol. 2002;12(6):746–753. doi: 10.1016/s0959-440x(02)00388-3. [DOI] [PubMed] [Google Scholar]
- 26.Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat Struct Mol Biol. 2008;15(2):139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- 27.Sakata E, et al. The catalytic activity of Ubp6 enhances maturation of the proteasomal regulatory particle. Mol Cell. 2011;42(5):637–649. doi: 10.1016/j.molcel.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 28.Korinek A, Beck F, Baumeister W, Nickell S, Plitzko JM. Computer controlled cryo-electron microscopy—TOM² a software package for high-throughput applications. J Struct Biol. 2011;175(3):394–405. doi: 10.1016/j.jsb.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Nickell S, et al. TOM software toolbox: Acquisition and analysis for electron tomography. J Struct Biol. 2005;149(3):227–234. doi: 10.1016/j.jsb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Hrabe T, Beck F, Nickell S. Automated particle picking based on correlation peak shape analysis and iterative classification. Int. J. Med. Biol. Sci. 2012;6:1–7. [Google Scholar]
- 31.Scheres SH, Núñez-Ramírez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat Protoc. 2008;3(6):977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penczek PA, Kimmel M, Spahn CM. Identifying conformational states of macromolecules by eigen-analysis of resampled cryo-EM images. Structure. 2011;19(11):1582–1590. doi: 10.1016/j.str.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16(5):673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.