

Abstract
This unit describes a streamlined two-step protocol for the isolation of adult murine cardiomyocytes with subsequent Chromatin ImmunoPrecipitation (ChIP). Isolation and culturing of cardiomyocytes is a delicate process and the protocol presented here optimizes the combination of cardiomyocyte isolation with ChIP. ChIP is an invaluable method for analyzing molecular interactions occurring between a specific protein (or its post-translationally modified form) and a region of genomic DNA. ChIP has become a widely used technique in the last decade since several groundbreaking studies have focused attention on epigenetics and have identified many epigenetic regulatory mechanisms. However, epigenetics within cardiovascular biology is a new area of focus for many investigators, and we have optimized a method for performing ChIP in adult murine cardiomyocytes as we feel this will be an important aid to both the cardiovascular field and for the development of cell- and tissue-specific ChIP.
Keywords: Chromatin ImmunoPrecipitation, Adult murine cardiomyocytes, Cardiomyocytes isolation
INTRODUCTION
Only recently have the epigenetic regulators of key cardiac developmental genes begun to be discovered, and a growing body of evidence has now highlighted the effects of chromatin organization on the activity of the cardiac transcriptional network (He et al., 2012; Lee et al., 2012; Sdek et al., 2011; Wamstad et al., 2012).
Chromatin ImmunoPrecipitation (ChIP) is an invaluable method for analyzing molecular interactions occurring between a specific protein (or its post-translationally modified form) and a region of genomic DNA. When performing ChIP, cells are first fixed with formaldehyde to covalently cross-link proteins to DNA. Then chromatin is isolated from the cells, sheared and subjected to an immunoselection process, which requires the use of specific antibodies. Any DNA sequence cross-linked to the protein of interest will coprecipitate as part of the chromatin complex. After the immunoselection of chromatin fragments and purification of associated DNA, the detection of specific DNA sequences is performed by quantitative PCR (qPCR) or by standard PCR followed by gel electrophoretic analysis. Any DNA sequence specifically associated with the protein being examined will be increased (or enriched) by the immunoprecipitation process, and this will be detected by qPCR.
CHIP OF ADULT MOUSE CARDIOMYOCYTES
ChIP can be difficult when studying cardiomyocytes as it requires large cell numbers (106–107). Adult mouse cardiomyocyte isolation is challenging and often does not provide the required number of cells. In this report we present a detailed protocol for cardiomyocyte isolation followed by ChIP obtained by modification of standard protocols. The present protocol allows optimization of the cell number, buffers and timing (Figure 1). Moreover, we provide recipes to make homemade buffers thus reducing costs.
Figure 1.

Schematic of the protocol
Materials
Cardiomyocyte isolation
Adult mouse
Isofluorane
Ethanol 70%
Perfusion buffer (see recipe)
Collagenase D (Roche)
Digestion buffer (see recipe)
Stop buffer 1 (see recipe)
Stop buffer 2 (see recipe)
2,3 butanedione monoxime (BDM)
Calcium Chloride (CaCl2)
Cardiomyocyte medium (see recipe)
ChIP
16% formaldehyde solution (Electron Microscopy Sciences) or 18.5% formaldehyde solution (see recipe)
Glycine (see recipe)
Phosphate-buffered saline (PBS)
Protease inhibitor cocktail (Roche Cat #118361770001)
Cell lysis buffer (see recipe)
Nuclei lysis buffer (see recipe)
RNase (Optional, see step 45)
Proteinase k
Agarose gel (Optional, see step 45)
Gel loading buffer (Optional, see step 45)
Gel loading dye (Optional, see step 45)
Antibodies to protein of interest (preferentially ChIP-grade)
Antibody (Normal rabbit IgG)
Protein A magnetic beads (alternatively Protein G magnetic beads)
Low salt wash buffer (see recipe)
High salt wash buffer (see recipe)
LiCl wash buffer (see recipe)
TE buffer (see recipe)
Dilution buffer (see recipe)
Elution buffer (see recipe)
Molecular grade water
qPCR reagents
Surgical instrument (All surgical tools are from Fine Science Tools, we recommend Dumont #5 fine-tip forceps for the aorta cannulation)
Petri dishes
Dissecting microscope
Gel loading tips
Braided Silk Suture (6–0, metric 0.7)
Langendorff apparatus
Laminar flow culture hood
Filter (100 Micron)
Tubes (conical 15 ml, 50 ml)
Centrifuge (15 and 50 ml tube)
Hematocytometer
Incubator, humidified (37°C, 5% CO2)
Liquid nitrogen
Liquid nitrogen container
Rotating platform, at 4°C and at room temperature
Vortex
Tubes (microcentrifuge, 1.5 ml)
Centrifuge (benchtop)
Ice bucket
Ice
Sonicator
Gel electrophoresis apparatus (Optional, see step 45)
Magnetic rack
Shacking incubator, preset at 62°C
Heating block preset to 95°C
DNA purification kit
Thermal cycler
CAUTION: Formaldehyde is a biohazard. Consult MSDS for proper handling instruction.
NOTE: Any protocol using vertebrate animals must be approved by an Institutional Animal Care and Use Committee (IACUC) and must follow approved procedures for the care and use of laboratory animals.
Cardiomyocyte isolation (1 hour for preparation +1 hour for each heart isolation)
The cardiomyocyte isolation protocol is adapted from “Isolation and Culture of Adult Mouse Cardiac Myocytes“ by O'Connel TD et al (O'Connell et al., 2007).
Make 1× fresh perfusion buffer, digestion buffer without collagenase, stop buffers, and cardiomyocyte medium as described in Reagents section. Warm perfusion buffer, myocyte digestion buffer and cardiomyocyte medium to 37°C.
Prepare the perfusion and the surgical apparatus. Prepare the cannula by gently scoring a gel loading tip with a blade to create grooves (Alternatively, a 22G feeding needle, straight, with a ball tip can be used). These will prevent slipping of the heart once it is cannulated.
Tape the cannula on the lid of a Petri dish, pass two 6–0 silk suture with a half-knot around the cannula and place it under the dissecting microscope (Figure 2). Fill up a syringe with the perfusion buffer. Run perfusion buffer through the perfusion system, avoiding bubbles.
Anesthetize the mouse with isoflurane and sacrifice by cervical dislocation.
Transfer the mouse to the surgical table. Clean the chest with ethanol 70%. Open the peritoneal cavity and chest with small scissors, peel back the rib cage and cut to expose the heart. Lift the heart and cut all the vessels (Figure 3).
Transfer the heart to a Petri dish containing warm perfusion buffer and squeeze it gently to remove all the blood.
Place the heart under the dissecting microscope (Figure 3). Insert the cannula in the aorta using fine-tip forceps to slide the aorta onto the cannula. Make sure that the tip of the aorta is just above the aortic valve (not into the ventricle). Tie the treads around the aorta (Figure 3).
Connect the syringe to the cannula and inject the heart with the buffer. If the cannula is placed correctly the heart should become swollen.
Transfer the heart to the Langendorff apparatus.
Once cannulated, perfuse the heart with the perfusion buffer for 3 minutes. The heart should start turning pale (Figure 3).
- Meanwhile add 40 mg of collagenase D to the tube containing 10 ml of digestion buffer.Amount of collagenase needed may vary from different batches and for older enzyme.
- Start perfusion of the heart with the digestion buffer and digest for 8–13 min (time of digestion may vary from different batches and for older enzyme) at 4 ml/min. After 2 minutes the digestion buffer can be collected and reused for the digestion.If the heart is well perfused, it will become swollen and turn pale.
- Once enzymatic digestion of the heart is complete (heart appears swollen, pale and flaccid), cut the heart from the cannula just below the atria. Place the ventricles in a Petri dish (Figure 3) and fill them with the digestion buffer from the perfusion apparatus (roughly 10 ml).Move under a laminar flow culture hood.
Gently tease the ventricles into several small pieces with fine forceps (Figure 3). Cut a p1000 pipettetip and pipette very gently (avoid bubbles) several times (Figure 3). This process takes a few minutes.
Transfer the cells to a 15-ml conical tube with a 100 Micron filter on the top.
Add 10 ml of stop buffer A to the Petri dish and rinse it well to collect all the cells.
Transfer the solution to the 15-ml conical tube with a 100 Micron filter on the top to combine with the cell suspension.
Spin the tube at 60g for 2 minute at room temperature (RT).
Remove the supernatant and add 10 ml of stop buffer B. Pipet very slowly and transfer to a fresh Petri dish.
Add 0.3 ml of BDM.
Start a gradual calcium reintroduction: add 50 μl of 10 mM CaCl2. Mix by moving the dish forward and backward and side-to-side and incubate for 4 min at RT.
Add an additional 50 μl of 10 mM CaCl2. Mix and incubate for 4 min at RT.
Add 100 μl of 10 mM CaCl2. Mix and incubate for 4 min at RT.
Add 30 μl of 100 mM CaCl2. Mix and incubate for 4 min at RT.
Add 50 μl of 100 mM CaCl2. Mix well and incubate for 4 min at RT.
Transfer (pipetting slowly and washing the plate) the cells to a new 15 ml tube and centrifuge for 2 min at 60g.
- Remove supernatant and resuspend the cell pellet in 1 ml of media. Count the cells with a hemocytometer.A good isolation should achieve a million rod-shaped cardiomyocytes.
- Centrifuge for 2 min at 60g and remove supernatant. The cell pellet can now be processed immediately or snap frozen and stored at − 80°CA plating step on a laminin-coated plate could be performed at this point to get rid of dead cells (dead cells won't attach and can be washed away).To prepare laminin-coated plates: The stock solution is 1 mg/ml, and should be stored in 50 μl aliquots at −80°C. The day before perfusion thaw the laminin in the refrigerator and cover each 10 cm plate with 10 ml PBS + 50 ul laminin. Store the plate overnight in the incubator. Non-used plates can be stored at 4°C for several days.
Figure 2.

Cannulation setup
Figure 3.

Diagrammatic illustration of the different steps to be performed during heart perfusion
Cardiomyocytes Cross-linking (1 hour)
Preparation: Obtain ice for incubation. Place 1× PBS on ice. Remove Protease Inhibitor Cocktail and thaw it at room temperature.
-
29Resuspend the cells in 10 ml of medium (if using frozen cells add 10 ml of PBS to frozen cell pellet).If working with adherent cardiomyocytes, cells can be left attached to the laminin-coated plate for the following steps.
-
30Add 550 ul of fresh 18.5 % formaldehyde or 625 ul of 16% formaldehyde to each sample.Formaldehyde is toxic, and the preparation process involving heating results in considerable vaporization, which increases the hazard. Therefore we suggest using commercially available 16% formaldehyde solution to reduce dangerous handling.
-
31
Incubate at RT with gentle shacking for 10 minutes.
-
32
Add 1 ml of 10× Glycine to quench unreacted formaldehyde.
-
33
Swirl to mix and incubate at RT with gentle shacking for 5 minutes.
-
34Place samples on ice.If working with adherent cardiomyocytes, scrape the cells and transfer to a 15 ml tube
-
35
Spin at 800g at 4°C for 5 minutes to pellet cells.
-
36
Remove PBS/medium, being careful not to disturb the cells.
-
37
Add 10 ml of cold 1× PBS (containing 1× Protease Inhibitor Cocktail) to wash cells.
-
38
Repeat steps 35-36-37.
-
39
Spin at 800g at 4°C for 5 minutes to pellet cells
-
40
Remove PBS, being careful not to disturb the cells.
-
41
Proceed to the next step or snap freeze and store the cell pellet at -80°C
Lysis and Sonication (1 hour)
At this point more samples of the same origin can be combined (up to 3×106 cells)
-
42Resuspend cells (if frozen thaw cell pellet on ice) in cell lysis buffer (100–500 ul, enough to cover the cell pellet and have a clear solution) containing Protease Inhibitor Cocktail (PIC):
- 100 ul cell lysis buffer + 0.5 ul PIC
- 150 ul cell lysis buffer + 0.75 ul PIC
- 200 ul cell lysis buffer + 1 ul PIC
- 250 ul cell lysis buffer + 1.25 ul PIC
- 500 ul cell lysis buffer + 2.5 ul PIC
-
43
Incubate on ice for 15 minutes, vortex every 5 minutes.
-
44
Spin at 800g at 4°C for 5 minutes.
-
45Remove supernatant and resuspend cells in nuclear lysis buffer (100–500 ul, enough to cover the cell pellet and have a clear solution) containing PIC. Incubate on ice 5–30 minutes.
- 100 ul nuclei lysis buffer + 0.5 ul PIC
- 150 ul nuclei lysis buffer + 0.75 ul PIC
- 200 ul nuclei lysis buffer + 1 ul PIC
- 250 ul nuclei lysis buffer + 1.25 ul PIC
- 500 ul nuclei lysis buffer + 2.5 ul PIC
-
46Sonicate cell lysate on ice: 3 cycles of 7'30” each, 30” on/30” off at high power, using Bioruptor from Diagenode. If using a different device sonication may require optimization. (Different samples of the same origin may be combined together up to 500 ul total volume).Keep cell lysate cold. Sonication produces heat, which can denature the chromatin.
-
47
Spin at 14,000g at 4°C for 10 minutes.
-
48
Transfer supernatant to fresh microcentrifuge tube. Make 50–200 ul aliquots (chromatin from 6×105 cells is sufficient for one assay). Proceed to the next step or freeze and store the cells at −80°C
Agarose gel analysis of sonication (optional) (3 hours)
See “Critical Parameters and Troubleshooting for information on when to perform these optional steps.
-
49
Remove 10–25 ul aliquot for agarose gel analysis.
-
50
Incubate at 95°C for 10 minutes
-
51
Add 1 ul of RNase A (10 mg/ml) and incubate for 30 minutes at 37°C
-
52
Add 1 ul Proteinase K and incubate at 62°C for 30 minutes to 2 hours.
-
53Load 10 ul and 20 ul on a 1% agarose gel with a 100 bp DNA marker.The majority of sheared chromatin should run between 200 and 1000 bp (Figure 4).
Figure 4.

Agarose gel analysis of sonicated DNA
Immunoprecipitation (30 minutes+ overnight incubation)
-
54Remove PIC and thaw at RT. Prepare Dilution Buffer for the number of samples and store on ice(Remember to add a sample for negative control (Normal rabbit IgG), including a positive control is also advisable).
-
55Add dilution buffer to each IP as shown below:
- If the volume of the sample is 50 ul: 450 ul of Dilution Buffer and 2.25 ul of PIC.
- If the volume of the sample is 100 ul: 900 ul of Dilution Buffer and 4.5 ul of PIC
- If the volume of the sample is 200 ul: 800 ul of Dilution Buffer and 4 ul of PIC
-
56
Add the Dilution Buffer into each tube.
-
57
Remove 5–10 ul (1%) of the supernatant from each sample as Input and save at 4°C until step 63.
-
58Add the proper antibody (5 ug in 500 ul total volume, 10 ug in 1000 ul; amount may vary for different antibodies).Polyclonal antibodies are preferable to monoclonal. Select ChIP-grade antibody when available.
-
59
Incubate overnight at 4°C with rotation.
Following day (3–4 hours)
-
60Add Protein A magnetic beads (20 ul in 500 ul total volume, 30 ul in 1000 ul. Make sure that the beads are well mixed) to each sample and incubate for 2–3 hour at 4°C with rotation.Alternatively protein G magnetic beads can be used: please consider the antibody class to select the protein with higher affinity.
-
61
Working at 4°C, pellet magnetic beads with the magnetic separator and remove the supernatant.
-
62Working at 4°C, wash the Protein A-antibody-chromatin complex using 1 ml each of the cold buffers listed below. For each wash: add buffer, invert the tube to resuspend beads, incubate 4 minutes on a rotating platform, spin briefly and place the tube in the magnetic rack. Wait 1 minute and remove supernatant.
-
aLow Salt Wash Buffer, two washes (can reduce to one wash if cell number is low)
-
bHigh Salt Wash Buffer, two washes (can reduce to one wash if cell number is low)
-
cLiCl Wash Buffer, one wash
-
dTE Buffer, one wash
-
a
DNA isolation (2 hours and 30 minutes)
-
63
Set shaking incubator to 62°C and heating block to 95°C. Make Elution Buffer for all sample tubes and Input tubes: For each tube 100 ul Elution buffer + 1ul Proteinase K (300 ug/ml final).
-
64
Add 100 ul of elution buffer to each sample (include the input)
-
65
Incubate at 62°C for 30 minutes to 2 hours with shaking.
-
66
Incubate at 95°C for 10 minutes.
-
67
Pellet magnetic beads with the magnetic rack and transfer the supernatant to a fresh microcentrifuge tube.
DNA Purification (30 minutes)
-
68
Perform these steps according to the DNA purification kit manufacturer's guidelines.
-
69At the end dilute the DNA with 30 ul of pre-warmed water, spin, and repeat (total volume = 58 ul).Sample can be used immediately for analysis or can be frozen at − 20°C
Real-time PCR (30 minutes preparation + 2 hours run)
-
70Design primers specific for analysis of the regions of interest.Good primer design is a critical step: in general, primers should have a size of 20–24 bp, a Tm of 55–60°C and a GC content of 50–60%. Amplification products should be between 150–250 bp. Primers should be tested on genomic DNA. Design of good primers could be easily achieved by free website like Primer3 (http://frodo.wi.mit.edu/).
-
71Prepare the following mix for each sample (each sample should be run in triplicate):
- 2× Sybr green (Roche #04673484001) 7.5 ul
- Primers (F+R) 10 um 0.75 (500 nM final)
- DNA 3–4 ul
- Water up to 15 ul
-
72Start the following PCR protocol:
- 95°C for 5'
- 94°C 15”
- 60°C for 30”
- 72°C for 30” (40 Cycles)
- Melting curve from 60°C to 95°C, read every 1°C, hold 00:00:03
Data analysis
ChIP-qPCR data need to be normalized for the different sources of variability. The two most common methods are the percent input method (in which ChIP signals are divided by input signal) and the fold enrichment methods (in which ChIP signal is presented as the signal increase relative to background signal).
-
-Percent input method method (Figure 5a):
- Calculate the percent input of each sample: IP sample = 2^(Average Ct input- average Ct IP sample)
Figure 5.
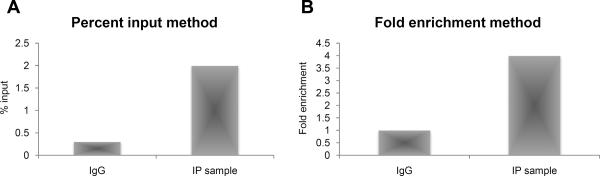
Data analysis with A) Percent input method and B) Fold enrichment method
-
-Fold Enrichment method (Figure 5b):
- Calculate the percent input of each sample: IP sample = 2^(Average Ct IP sample -average Ct IgG)
REAGENTS AND SOLUTIONS
Perfusion Buffer
Perfusion buffer 1× should be prepared fresh every day. To prepare 500 ml of buffer (enough for 5–7 isolations) combine 3.515 g of NaCl, 0.55 g of KCl, 0.041 g of KH2PO4, 0.0425 g of Na2HPO4, 0.15 g of MgSO4-7H2O, 1.3 g of Na-HEPES, 0.195 g of NaHCO3, 1.875 g of Taurine, 0.5 g of BDM and 0.5 g of D-Glucose. Adjust the pH to 7.4 and then filter the solution. If multiple isolations will be performed in a week a 10× perfusion buffer can be prepared and stored at 4°C for up to one week. In this case, prepare the 10× buffer without Taurine, BDM and D-Glucose that will be added fresh to the 1× buffer on the day of the isolation.
Digestion Buffer
For each heart, will require 10 ml (prepare a separate 50 ml tube for each heart). Combine 10 ml of Perfusion Buffer, 40 mg of Collagenase D, 55.6 ul of 10× Trypsin and 1.25 ul of 100mM CaCl2.
Stop Buffer A
For each heart, will require 10 ml. Combine 9 ml of Perfusion Buffer, 1 ml of FBS and 1.25 ul of 100mM CaCl2.
Stop Buffer B
For each heart, will require 10 ml. Combine 9.5 ml of Perfusion Buffer, 0.5 ml of FBS and 1.25 ul of 100mM CaCl2.
Cardiomyocyte Medium
For 500 ml of medium combine 500 ml of DMEM or M199 with 500 mg of BSA, 500 mg of BDM, 5 ml of Penicillin (10,000Units/ml)/Streptomycin (10,000 ug/ml) and 5 ml of Insulin-Transferrin-Selenium-A. Filter sterilize and store at 4°C.
18.5% Formaldehyde
Combine 4.8 ml of H2O with 0.925 g of paraformaldehyde and 35 ul of 1N KOH (see recipe).
1N KOH
To make 10 ml dissolve 0.56 g in 10 ml of H2O.
Glycine 10X
To prepare 200 ml of glycine 10X dissolve 18.74 g of glycine in 200 ml of PBS.
Cell Lysis Buffer
10mM TrisHCl pH 8.0, 10mM EDTA, 0.5 mM EGTA, 0.25% Triton X-100
To prepare 200 ml of Cell Lysis Buffer combine 2 ml of 1M TrisHCl pH 8.0 (see recipe), 4 ml of 0.5M EDTA (see recipe), 200 ul of 0.5M of EGTA (see recipe), 5 ml of 10% of Triton X-100 in 190.8 ml of H2O.
0.5M EGTA
To prepare 10 ml of solution combine 1.9 g of EGTA with 9 ml of H2O. Adjust the pH to 7.5/8.0 with NaOH (pellets) and adjust volume to 10 ml. EGTA will not go into solution without NaOH.
1M TrisHCl pH 8.0
For a 1 M solution, dissolve 121.1 g of Tris base in 800 ml of H2O. Adjust the pH, then adjust the volume of the solution to 1 liter with H2O. Filter or autoclave to sterilize.
0.5M EDTA
To prepare EDTA 0.5 M (pH 8.0) add 186.1 g of disodium EDTA•2H2O to 800 ml of H2O. Adjust the pH to 8.0 with NaOH (pellets). EDTA will not go into solution until the pH of the solution is correct. Filter or autoclave to sterilize.
Nuclei Lysis Buffer
50mM TrisHCl pH 8.0, 10 mM EDTA, 1% SDS
To prepare 200 ml of Nuclei Lysis Buffer combine 10 ml of 1M TrisHCl pH 8.0 (see recipe), 4 ml of 0.5M EDTA (see recipe), 20 ml of 10% of SDS in 166 ml of H2O (see recipe).
10% SDS
Dissolve 10 g of SDS in 100 ml of H2O.
Dilution Buffer
16.7 mM TrisHCl pH 8.0, 1.2 mM EDTA, 167 mM NaCl, 0.01% SDS, 1.1% Triton X-100
To prepare 200 ml of Dilution Buffer combine 4 ml of 1M TrisHCl pH 8.0 (see recipe), 440 ul of 0.5M EDTA (see recipe), 50 ul of 10% of SDS, 22ml of 10% of TritonX-100 and 8.36 ml of 4M NaCl (see recipe) in 165 ml of H2O.
Low Salt Wash Buffer
20 mM TrisHCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100
To prepare 200 ml of Low Salt Wash Buffer combine 4 ml of 1M TrisHCl pH 8.0 (see recipe), 800 ul of 0.5M EDTA (see recipe), 2 ml of 10% of SDS, 20 ml of 10% of TritonX-100 and 7.5 ml of 4M NaCl (see recipe) in 165.7 ml of H2O.
4M NaCl
To prepare a 4 M solution: Dissolve 23.37 g of NaCl in 10 ml of H2O. Filter or autoclave to sterilize.
High Salt Wash Buffer
20 mM TrisHCl pH 8.0, 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100
To prepare 200 ml of High Salt Wash Buffer combine 4 ml of 1M TrisHCl pH 8.0 (see recipe), 800 ul of 0.5M EDTA (see recipe), 2 ml of 10% of SDS, 20 ml of 10% of TritonX-100 and 25 ml of 4M NaCl (see recipe) in 148.2 ml of H2O.
LiCl Wash Buffer
10 mM Tris pH 8.0, 1 mM EDTA, 0.25M LiCl, 1% IGEPAL CA630, 1% deoxycholate
To prepare 200 ml of LiCl Wash Buffer combine 50 ml of 1M LiCl (see recipe), 2 ml of 10% of IGEPAL CA630, 2 g of 10% of deoxycholate, 2ml of 1M TrisHCl pH 8.0 (see recipe) and 400 ul of 0.5M EDTA (see recipe) in 145.6 ml of H2O.
1M LiCl
To prepare 1 M lithium chloride: Dissolve 4.24 g of LiCl in a final volume of 100 ml of H2O. Filter or autoclave to sterilize. Store the solution at 4°C.
Elution Buffer
200 mM NaCl, 1% SDS
To prepare 50 ml of Elution Buffer combine 5 ml of 10% SDS (see recipe) and 2.5 ml of 4M of NaCl (see recipe) in 42.5 ml of H2O.
COMMENTARY
Background information
ChIP is a type of immunoprecipitation used to investigate the interaction between proteins and DNA in vivo. There are two main types of ChIP: the cross-linked ChIP, which reversibly cross-links (with formaldehyde or UV light) the sheared chromatin (usually by sonication), and the native ChIP, which uses native chromatin sheared by micrococcal nuclease digestion. The cross-linked ChIP is the most common method for the analysis of DNA target of transcription factors or other chromatin-associated proteins.
The first reports of cross-linking ChIP for chromatin in vivo studies date back to the 1980's when Gilmour and Lis (Gilmour and Lis, 1984), and Varshavsky and Solomon (Solomon and Varshavsky, 1985), published their pioneering work in the field of chromatin immunoprecipitation. The two studies differed in that the cross-linking method of Gilmour and Lis utilized UV irradiation to cross-link proteins with neighboring DNA whereas Varshavsky and Solomon used formaldehyde cross-linking.
It is worth mentioning that since its discovery ChIP has constantly evolved and many “flavors” of ChIP have been elaborated (Collas, 2009). For instance, ChIP-reChIP takes advantage of two sequential immunoprecipitations to determine whether two or more proteins are close enough in a chromatin context or whether they are interacting, probably forming a complex, in a localized genomic region (Truax and Greer, 2012). More importantly, chromatin immunoprecipitation followed by microarray hybridization (ChIP-chip) or by next-generation sequencing analysis (ChIP-Seq) is currently an invaluable method with which to investigate the genome-wide distribution of chromatin-binding proteins and histone modifications in any genome with a known sequence. A detailed description of these techniques is beyond the scope of this report, and we refer the reader to exhaustive and updated reviews (Kidder et al., 2011; Park, 2009).
Epigenetics may be defined as the study of any potentially stable, and ideally, heritable change in gene expression or cellular phenotype that occurs without changes in Watson-Crick base pairing of DNA (Goldberg et al., 2007). Much of today's epigenetic research is converging on the study of covalent and non-covalent modifications of DNA and histone proteins, and the mechanisms by which such modifications influence overall chromatin structure (Bernstein and Hake, 2006; Goldberg et al., 2007). Since histones and histone PTMs are currently taking the center stage of epigenetic control, ChIP has become the election technique in the epigenetics world.
A number of recent studies highlight the role of epigenetic regulation as a key process in mechanisms of cardiovascular development and disease. The importance of the epigenetic regulatory mechanisms for transcriptional control in the cardiovascular system is just beginning to be appreciated and growing evidence demonstrates that abnormal epigenetic regulation contributes to heart disease (Wamstad et al., 2012). Therefore, in the last year we have noted increasing interest amongst cardiovascular scientists in epigenetic studies along with their associated complex methodological challenges. For example, it was recently reported that the Polycomb Repressive Complex 2 (PRC2) regulates normal mouse heart formation through its H3K27-methyltrasferase component Ezh2 (Delgado-Olguin et al., 2012; He et al., 2011). Jarid2, another component of PRC2, is also known to be fundamental during heart development (Lee et al., 2000). Brg1/Brm-associated factor (BAF) complexes also have important roles in regulating gene expression during heart development (Takeuchi et al., 2011). Moreover, Wamstad et al. found that, during cardiac differentiation, distinct chromatin patterns are coordinated with stage-specific expression of functionally related genes. They also identify stage-specific enhancers and DNA binding motifs within these regions that predict sets of transcription factors that orchestrate cardiac differentiation (Wamstad et al., 2012).
Adult cardiomyocytes are primary cells which are fundamental for many studies. Their importance is further underscored by the fact that no cell lines are available with the same properties. Cell lines divide in culture whereas a defining characteristic of cardiomyocytes is that they proliferate throughout fetal development and into the early neonatal period when division ceases and DNA replication declines quickly (Brooks et al., 1998; Parker and Schneider, 1991). Isolation and culturing of cardiomyocytes is a delicate process and the protocol presented here is a modification of a procedure presented by O'Connell et al (O'Connell et al., 2007) optimizing the combination of cardiomyocyte isolation with ChIP. The enzymes utilized for the isolation of cardiomyocytes (collagenase in this protocol) are likely not altering the histone proteins within the nuclei if the cell isolation is yielding intact cells. One cannot be absolutely certain without analyzing histone modifications in isolated cardiomyocytes and comparing them to histone modifications in intact cardiac tissue. This is difficult to do as cardiac tissue contains many cell types in addition to cardiomyocytes and this analysis has not been reported to our knowledge.
This unit describes the isolation and the ChIP protocol for adult cardiac myocytes. However, this method could be applied to other tissue-derived cells. In fact, this method provides modifications that are applicable to working with primary cells, in which cell number is a very significant limitation. Some cell- and tissue-specific modifications may be required, especially for the isolation procedure and in order to optimize the conditions for shearing crosslinked DNA.
Critical parameters and troubleshooting
Cardiomyocyte isolation
To obtain large numbers of healthy cardiomyocytes, all of the steps described above are critical and it is usually necessary to perform several experiments for practice. In general, when working with heart tissue and cardiomyocytes, always work fast but very gently and avoid vigorous agitation. In particular, cannulation should be done as quickly as possible (1 minute is desirable) (O'Connell et al., 2007); collagenase should be tested frequently to assess lot-to-lot consistency; cardiomyocytes should attach to the laminin-coated plate very quickly (less than 30 minutes); if this does not happen, prepare new laminin-coated plates and transfer the cells.
Cardiomyocyte cross-linking
In this step two factors are critical: formaldehyde has to be high quality and freshly prepared and extent of the cross-linking has to be accurate. Histones are tightly associated with DNA and excessive cross-linking can modify the lysine residues in their tails. Moreover, cells that are over-cross-linked will not produce small chromatin fragments, even by prolonged sonication, therefore resulting in lower yield (Orlando et al., 1997).
Lysis and sonication
It is very important to have efficient lysis. For this, make sure there is enough lysis buffer. Flash-freezing the cells after the incubation in the nuclear lysis buffer can help open the nuclei more efficiently. Sonication is also a critical step: perform it in a cold room and refill the water bath with ice every 5–7 minutes (or every time that the water becomes warm). Shearing the DNA to a small size (between 100 to 1000 bp) is an imperative step to achieve good resolution. Because of the importance of this step, analysis of the sheared chromatin by gel electrophoresis is strongly recommended (steps 49–53), especially when setting up the protocol and for each different cell type.
Immunoprecipitation
The quality of antibodies used for ChIP is one of the most important factors that contributes to the quality of the data generated by these studies. High sensitivity and specificity antibodies are required because they allow the detection of enrichment without substantial background noise. Many commercial antibodies that have been tested for their use in ChIP studies are available. However, results from various groups have shown that not all commercial antibodies that are designated as `ChIP grade' or `ChIP qualified' can be successfully used. Furthermore, commercially available ChIP-grade antibodies suffer from lot-to-lot variation, leading to nonspecific binding and questionable datasets (Egelhofer et al., 2011; Kidder et al., 2011; Peach et al., 2012). When possible, use polyclonal and ChIP-grade antibodies. When choosing antibodies, check the literature to select antibodies that have been successfully used for ChIP experiments. Also make sure that the subclass and the isotype of the antibody are correct for the protein (A/G) magnetic bead used (in general rabbit antibodies work for both A and G proteins while certain classes of mouse and rat immunoglobulins do not bind with protein A).
Real-time PCR
For successful PCR, the choice of primers is essential (see step 59). In some cases, it may be necessary to increase the amount of DNA added to the reaction. In general, standard PCR parameters and troubleshooting should be followed.
Anticipated results
Chip experiments usually require at least 106 cells for each immunoprecipitation. This protocol aims to increase the yield of the cardiomyocyte isolation while reducing the number of cells required to perform the ChIP experiments. Therefore we anticipate this method will optimize buffer, decrease number of steps and timing, keep the procedure simple and straightforward, and enable immunoprecipitation with a smaller number of cells than usually required for standard ChIP protocols.
Time considerations
The entire protocol can be completed in two days. On the first day, cardiomyocytes can be isolated (approximately 2–3 hours for one mouse) and then chromatin can be cross-linked (less than 2 hours), sonicated (1 hours + 3 hours for gel analysis) and incubated overnight with the antibody of interest. The following day DNA can be washed, isolated and purified (roughly 6 hours). Following this, quantitative PCR can be performed (3 hours).
For convenience, one whole day can be dedicated to cardiomyoyte isolation (5 mice or more), and the cells can be snap frozen and stored at −80C. ChIP protocol (2 days) can then performed at a convenient time.
At several points during this procedure (indicated in the above protocol) one may choose to store the samples and stop the experiment.
ACKNOWLEDGMENTS
This work was supported by NIH grant (R01-HL 088255).
LITERATURE CITED
- Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84:505–517. doi: 10.1139/o06-085. [DOI] [PubMed] [Google Scholar]
- Brooks G, Poolman RA, Li JM. Arresting developments in the cardiac myocyte cell cycle: Role of cyclin-dependent kinase inhibitors. Cardiovascular Research. 1998;39:301–311. doi: 10.1016/s0008-6363(98)00125-4. [DOI] [PubMed] [Google Scholar]
- Collas P. The state-of-the-art of chromatin immunoprecipitation. Methods Mol Biol. 2009;567:1–25. doi: 10.1007/978-1-60327-414-2_1. [DOI] [PubMed] [Google Scholar]
- Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nature Genetics. 2012;44:343–U158. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, Gu T, Kharchenko PV, Kuan S, Latorre I, Linder-Basso D, Luu Y, Ngo Q, Perry M, Rechtsteiner A, Riddle NC, Schwartz YB, Shanower GA, Vielle A, Ahringer J, Elgin SC, Kuroda MI, Pirrotta V, Ren B, Strome S, Park PJ, Karpen GH, Hawkins RD, Lieb JD. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS, Lis JT. Detecting Protein-DNA Interactions Invivo - Distribution of Rna-Polymerase on Specific Bacterial Genes. P Natl Acad Sci-Biol. 1984;81:4275–4279. doi: 10.1073/pnas.81.14.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: A landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- He A, Ma Q, Cao J, von Gise A, Zhou P, Xie H, Zhang B, Hsing M, Christodoulou D, Cahan P, Daley GQ, Kong SW, Orkin SH, Seidman CE, Seidman JG, Pu WT. Polycomb Repressive Complex 2 Regulates Normal Development of the Mouse Heart. Circ Res. 2011 doi: 10.1161/CIRCRESAHA.111.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He AB, Shen XH, Ma Q, Cao JJ, von Gise A, Zhou PZ, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Gene Dev. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder BL, Hu G, Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol. 2011;12:918–922. doi: 10.1038/ni.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee JW, Lee SK. UTX, a Histone H3-Lysine 27 Demethylase, Acts as a Critical Switch to Activate the Cardiac Developmental Program. Developmental Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Song AJ, Baker R, Micales B, Conway SJ, Lyons GE. Jumonji, a nuclear protein that is necessary for normal heart development. Circ Res. 2000;86:932–938. doi: 10.1161/01.res.86.9.932. [DOI] [PubMed] [Google Scholar]
- O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker TG, Schneider MD. Growth-Factors, Protooncogenes, and Plasticity of the Cardiac Phenotype. Annual Review of Physiology. 1991;53:179–200. doi: 10.1146/annurev.ph.53.030191.001143. [DOI] [PubMed] [Google Scholar]
- Peach SE, Rudomin EL, Udeshi ND, Carr SA, Jaffe JD. Quantitative assessment of ChIP-grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M111.015941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdek P, Zhao P, Wang YP, Huang CJ, Ko CY, Butler PC, Weiss JN, MacLellan WR. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 2011;194:407–423. doi: 10.1083/jcb.201012049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Varshavsky A. Formaldehyde-Mediated DNA Protein Crosslinking - a Probe for Invivo Chromatin Structures. P Natl Acad Sci USA. 1985;82:6470–6474. doi: 10.1073/pnas.82.19.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-Olguin P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, Zhou YQ, Yeh RF, Henkelman RM, Harvey RP, Metzger D, Chambon P, Stainier DY, Pollard KS, Scott IC, Bruneau BG. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truax AD, Greer SF. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol Biol. 2012;809:175–188. doi: 10.1007/978-1-61779-376-9_12. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and Coordinated Epigenetic Regulation of Developmental Transitions in the Cardiac Lineage. Cell. 2012 doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]