

Abstract
Reverse cholesterol transport (RCT) can proceed through the classic hepatobiliary route or through the nonbiliary transintestinal cholesterol efflux (TICE) pathway. Scavenger receptor class B type I (SR-BI) plays a critical role in the classic hepatobiliary route of RCT. However, the role of SR-BI in TICE has not been studied. To examine the role of intestinal SR-BI in TICE, sterol balance was measured in control mice and mice transgenically overexpressing SR-BI in the proximal small intestine (SR-BIhApoCIII-ApoAIV-Tg). SR-BIhApoCIII-ApoAIV-Tg mice had significantly lower plasma cholesterol levels compared with wild-type controls, yet SR-BIhApoCIII-ApoAIV-Tg mice had normal fractional cholesterol absorption and fecal neutral sterol excretion. Both in the absence or presence of ezetimibe, intestinal SR-BI overexpression had no impact on the amount of cholesterol excreted in the feces. To specifically study effects of intestinal SR-BI on TICE we crossed SR-BIhApoCIII-ApoAIV-Tg mice into a mouse model that preferentially utilized the TICE pathway for RCT (Niemann-Pick C1-like 1 liver transgenic), and likewise found no alterations in cholesterol absorption or fecal sterol excretion. Finally, mice lacking SR-BI in all tissues also exhibited normal cholesterol absorption and fecal cholesterol disposal. Collectively, these results suggest that SR-BI is not rate limiting for intestinal cholesterol absorption or for fecal neutral sterol loss through the TICE pathway.
Keywords: reverse cholesterol transport, fecal neutral sterol excretion, scavenger receptor class B type I
Cardiovascular disease (CVD) accounts for roughly one-third of all deaths in the United States (1, 2). One of the most accurate predictors of CVD incidence is plasma concentrations of low density lipoprotein cholesterol (LDLc). Hence, lowering LDLc has been the primary therapeutic goal for decades with the introduction of statin drugs. However, even with the substantial LDLc lowering achieved with statin therapy, CVD-associated mortality and morbidity has been reduced by only ∼30% (1, 2). Given this unmet therapeutic need, major interest has thus shifted toward developing high density lipoprotein cholesterol (HDLc)-elevating agents, because HDLc was shown to be an even stronger predictor than LDLc for CVD in a number of large population studies including the Framingham Heart Study (3). There have been many mechanisms proposed to explain the cardioprotective properties of HDLc (4–6), but the most widely accepted is that HDL directly facilitates the process of reverse cholesterol transport (RCT).
In the classic model of RCT, extrahepatic (peripheral) cholesterol is delivered to the liver via HDL for secretion into bile and subsequent loss through the feces (7, 8). A critical checkpoint in this process occurs in the hepatocyte, where cholesterol may be converted to bile acids or directly secreted into bile as free cholesterol (FC) (7, 8). A fraction of the bile acid and cholesterol secreted into bile is lost in the feces, and this is the major route of disposal for cholesterol and its metabolites from the body (7, 8). Within this classic framework of RCT, HDL-mediated delivery of peripheral cholesterol to the liver directly results in biliary secretion (7, 8). Given this model, plasma HDLc levels should accurately predict both biliary sterol secretion and fecal sterol loss. However, plasma HDLc levels are not an accurate predictor of both biliary sterol secretion and fecal sterol loss. This is exemplified by the fact biliary and fecal sterol loss is quite normal in mice with extremely low HDLc levels (9–12). In addition, there is a clear disconnection between biliary and fecal sterol loss in several mouse models of genetically altered hepatic cholesterol metabolism (13–18). These findings have led to the discovery of a pathway for RCT that persists in the surgical or genetic absence of biliary secretion (12, 14–16, 19–24) called transintestinal cholesterol efflux (TICE). Therefore, a new model for conceptualizing RCT has emerged (23, 24) that involves two distinct excretory routes: 1) the classic hepatobiliary route, and 2) the nonbiliary TICE pathway (23, 24). From a therapeutic standpoint, exploiting the nonbiliary TICE pathway is a more attractive option, because increasing biliary cholesterol secretion can promote cholesterol gallstone formation (25, 26). Importantly, the major mechanisms regulating the classic hepatobiliary RCT pathway have been well defined (13, 18, 27, 28), but almost no information exists regarding mediators of the nonbiliary TICE pathway. Given the clear role of the scavenger receptor class B type I (SR-BI) in lipoprotein clearance and RCT, the purpose of this work was to define the role of SR-BI as an intestinal lipoprotein receptor facilitating the TICE pathway.
SR-BI is a membrane-associated glycoprotein, which was first identified by its close homology to the lipoprotein scavenger receptor CD36 (29, 30). SR-BI is expressed in a wide variety of tissues, with the highest levels of expression in those regulating cholesterol metabolism and steroid hormone production such as the liver and adrenal gland (29–31). It has been a matter of debate, but SR-BI seems to localize to both apical and basolateral membranes in polarized cells (32–37), and likely undergoes sterol-dependent basolateral to apical transcytosis (32–37). Importantly, SR-BI has been classified as a major HDL receptor, facilitating a unique sterol transport process called selective uptake (38–40). Direct evidence that SR-BI plays a role in hepatobiliary RCT has come from studies in mice. SR-BI overexpression results in diminished HDLc levels (41–47), increased biliary cholesterol secretion (41–47), and marked protection against atherosclerosis (45–47). Furthermore, mice lacking SR-BI accumulate large apoE-rich HDL particles in plasma (49–53), have decreased biliary sterol excretion (49–53), and develop severe atherosclerosis (52, 53). In fact, mice lacking SR-BI in the apoE-deficient background develop occlusive coronary artery atherosclerosis, myocardial infarction, and die at a very early age (52, 53). The hyperlipidemic and proatherogenic effects seen with SR-BI deficiency are thought to be primarily due to SR-BI's critical role in selective uptake of HDL cholesteryl esters (CEs) into the liver, thereby promoting biliary and fecal sterol loss. In line with this, Zhang et al. (54) reported that hepatic overexpression of SR-BI specifically promoted macrophage to feces RCT, and SR-BI−/− mice had reduced macrophage RCT. However, the role of intestinal SR-BI has not been clearly addressed.
In contrast to the liver, the role of SR-BI in the intestine has been a matter of intense debate. Several investigators have speculated that SR-BI facilitates cholesterol absorption instead of promoting basolateral to apical movement through the TICE pathway as we postulated here. The first evidence for this possibility was demonstrated by Hauser et al. (55), who showed that SR-BI was present in brush border membrane vesicles (BBMVs), and a SR-BI antibody reduced cholesterol binding to BBMVs. SR-BI can be localized to the apical membrane of the enterocyte (36), and is a high-affinity cholesterol receptor in intestinal BBMV (56). However these in vitro findings could not be substantiated in mice with targeted disruption of SR-BI. In fact, mice lacking SR-BI have normal or enhanced intestinal cholesterol absorption (57). Further, the cholesterol absorption inhibitor ezetimibe (EZE) potently inhibits cholesterol absorption in SR-BI-deficient mice, indicating that SR-BI is not likely involved in EZE-sensitive cholesterol transport (58). In contrast to SR-BI deficiency, one study has demonstrated that intestinal overexpression of SR-BI does result in a modest increase in movement of a gavaged cholesterol tracer into the plasma, but fractional cholesterol absorption and mass fecal sterol loss were not measured (59). In this current work we have attempted to clarify the role of intestinal SR-BI in RCT using both gain-of-function and loss-of-function mouse models.
MATERIALS AND METHODS
Animals
Mice overexpressing SR-BI specifically in the small intestine were generated as previously described (59). Briefly, the murine cDNA for SR-BI was cloned downstream of the apolipoprotein C-III enhancer (−500/−890 bp)-apolipoprotein A-IV promoter (−700 bp), and the resulting plasmid was linearized via SalI and AvrI restriction digestion, and used to generate transgenic mice by pronuclear microinjection into the B6D2 background. Resulting SR-BIhApoCIII-ApoAIV-Tg mice were subsequently backcrossed to the C57BL/6 background for over 12 generations, and were kindly provided by Dr. Xavier Collet (INSERM, France). These mice were intercrossed with mice transgenically overexpressing Niemann-Pick C1-like 1 (NPC1L1) specifically in the liver (NPC1L1LiverTg). Creation of NPC1L1LiverTg mice has been described previously (13). All experiments were done in the high overexpressing line NPC1L1-Tg112 (13), which had been backcrossed to the C57BL/6 background for over 9 generations. Therefore, all mice had been backcrossed to C57BL/6 background for greater than 9 generations before study, and male sibling controls were used to minimize effects of genetic heterogeneity and sex. Genotyping was confirmed by PCR analysis on genomic DNA isolated from tail snip as previously described (13, 59). For SR-BI and NPC1L1 transgenic mouse studies, at 6–8 weeks of age, mice were switched from a diet of rodent chow to a diet containing 10% of energy as palm oil-enriched fat and either low (0.015%, w/w) or high (0.2%, w/w) cholesterol for a period of 2–8 weeks. For ezetimibe treatment experiments, male wild-type (WT) and SR-BIhApoCIII-ApoAIV-Tg littermates were fed a diet containing 0.015% cholesterol (w/w) for 4 weeks. After 4 weeks on diet, mice were orally gavaged daily for three consecutive days with either vehicle or 0.3 mg EZE as previously described (13). For all SR-BI knockout studies, C57BL/6J mice were obtained from Charles River (Sulzfeld, Germany). SR-BI knockout mice were obtained from the Jackson Laboratories (Bar Harbor, ME) and subsequently backcrossed to the C57BL/6J genetic background for a total of eight generations. Genotyping was confirmed by PCR as previously described (50). All animals were housed in temperature controlled rooms (21°C) with alternating 12 h periods of light and dark and ad libitum access to water and rodent chow (Abdiets, Woerden, The Netherlands). SR-BI knockout experiments were performed in accordance with national laws in the Netherlands, and the responsible ethics committee of the University of Groningen approved all protocols. For all other studies, mice were maintained in an American Association for Accreditation of Laboratory Animal Care-approved animal facility, and all experimental protocols were approved by the institutional animal care and use committee at the Wake Forest University School of Medicine.
Plasma lipid and lipoprotein analyses
Total plasma cholesterol and plasma triglyceride (TG) concentrations were measured using a colorimetric assay as previously described (13–15, 59, 60) in mice that had been fasted for 4 h during the light cycle (9:00 AM–1:00 PM). Cholesterol concentrations in very low density lipoproteins (VLDLs), low-density lipoproteins (LDLs), and high density lipoproteins (HDLs) were determined after separation of plasma samples by high-performance liquid chromatography (HPLC) with a GE Healthcare Life Sciences Superose-6 10/300 GL column run at a flow rate of 0.5 ml/min as previously described (13–15, 60, 61).
Immunoblotting
Total liver and jejunal homogenates were made in a modified radioimmunoprecipitation assay buffer containing PBS (pH 7.5), 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 2 mM Na3VO4, 20 mM glycerophosphate, 10 mM NaF, and a protease inhibitor mixture (Calbiochem) including 500 μM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride, 1 μg/ml aprotinin, 1 μM E-64, 500 μM EDTA, and 1 μM leupeptin. Homogenates (25 μg per lane) were separated by 4–12% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and incubated with the following antibodies: 1) rabbit anti-SR-BI IgG (Novus Biologicals, #NB400-104); and 2) mouse anti-β actin antibody (Sigma, #A1978).
Immunolocalization of intestinal SR-BI
Jejunum of WT and SR-BIhApoCIII-ApoAIV-Tg mice were mounted in OCT compound (Ted Pella, Inc.), frozen in liquid nitrogen, and stored at −80°C. Sections (6 μm) were obtained via a cryostat (Lecia, #CM1510 S), immediately fixed with 10% formalin for 10 min, and subsequently air dried for 30 min. Resulting slides were washed in PBS (pH 7.4) three times, 5 min each. PBS was carefully removed by blotting and then a circle was drawn around each section with a hydrophobic pen (Vector Labs, ImmEdge cat#H-4000). Next the sections were blocked for 30 min with 4% blocking buffer (2% goat serum and 2% BSA in PBS at room temperature in a humid chamber. Thereafter, SR-BI primary antibody (rabbit anti-SR-BI IgG, Abcam®, #ab396) was diluted 1:1000 in 2% blocking buffer (1 ml PBS + 20 μl 50% BSA) and incubated for 1 h at room temperature. Sections were again washed in PBS three times for 5 min each. Goat anti-rabbit secondary antibody (FITC anti-rabbit IgG H+L, Vector catalog number BA-4000) was diluted 1:5000 in 2% blocking buffer and added dropwise on the tissue sections for 30 min. After extensive washing, the slides were stained with 4',6-diamidino-2-phenylindole (DAPI) to identify nuclei. Photomicrographs were obtained via fluorescence microscopy using the following parameters: UV filter (340–380 nm) for DAPI and blue filter (465–495 nm) for FITC.
Cholesterol absorption and fecal sterol excretion measurements
For all SR-BIhApoCIII-ApoAIV-Tg and NPC1L1LiverTg transgenic studies (Figs. 1–4) fractional cholesterol absorption was measured using the fecal dual-isotope method, and fecal neutral sterol excretion was measured by gas-liquid chromatography as previously described (13–15, 61). For SR-BI knockout studies, fractional cholesterol absorption and fecal neutral and acidic sterol loss were measured using slightly different methodology as previously described (63). Briefly, to quantify intestinal cholesterol absorption: following oral administration by gavage of a mixture of 1 μCi 14C-cholesterol and 2 μCi 3H-sitostanol in 100 μl medium chain triglyceride oil feces were collected for 48 h, dried, weighed, and ground. Sterols were extracted as detailed above and radioactivity determined by liquid scintillation counting. Cholesterol absorption was calculated according to the following formula: % absorption = [ratio 14C /3H (dosing solution) − ratio 14C/3H (sample)]/[ratio 14C/3H (dosing solution)]. For fecal neutral and acidic sterol analysis in SR-BI−/− mice, feces of individually housed mice were collected over a period of 24 h. Fecal samples were dried, weighed, and thoroughly ground. Aliquots were used for determination of bile acids and neutral sterols by gas-liquid chromatography as described (62).
Fig. 1.

Intestinal overexpression of SR-BI decreases plasma cholesterol, but does not alter intestinal cholesterol absorption. Male WT and SR-BIhApoCIII-ApoAIV-Tg (SR-BI) mice littermates were fed diets containing either 0.015% or 0.2% cholesterol (w/w) for a period of 8 weeks. A: Western blot analysis of liver and jejunal homogenates (n = 3 per group) from mice fed 0.015% cholesterol for 2 weeks. B: Immunolocalization of SR-BI in the mouse jejunum. Sections (6 μm) of mouse jejunum were fixed with 3.7% formaldehyde/PBS, permeabilized with 0.05% Tween 20, and stained with a polyclonal antibody raised against SR-BI to determine tissue and subcellular localization; blue indicates nucleus, red indicates SR-BI. C: Total plasma cholesterol and HDLc in 6 week old chow-fed mice (Chow) or mice fed 0.015% or 0.2% cholesterol (Chol) for 4 weeks. D: Fractional cholesterol absorption measured by fecal dual-isotope method, and mass fecal neutral sterol loss (FNSL) determined by gas-liquid chromatography measured after 6 weeks on diet. Data in panels (C) and (D) represent the mean ± SEM from 6 to 10 mice per group. *Significantly different from WT mice within each diet group (P < 0.05).
Fig. 4.
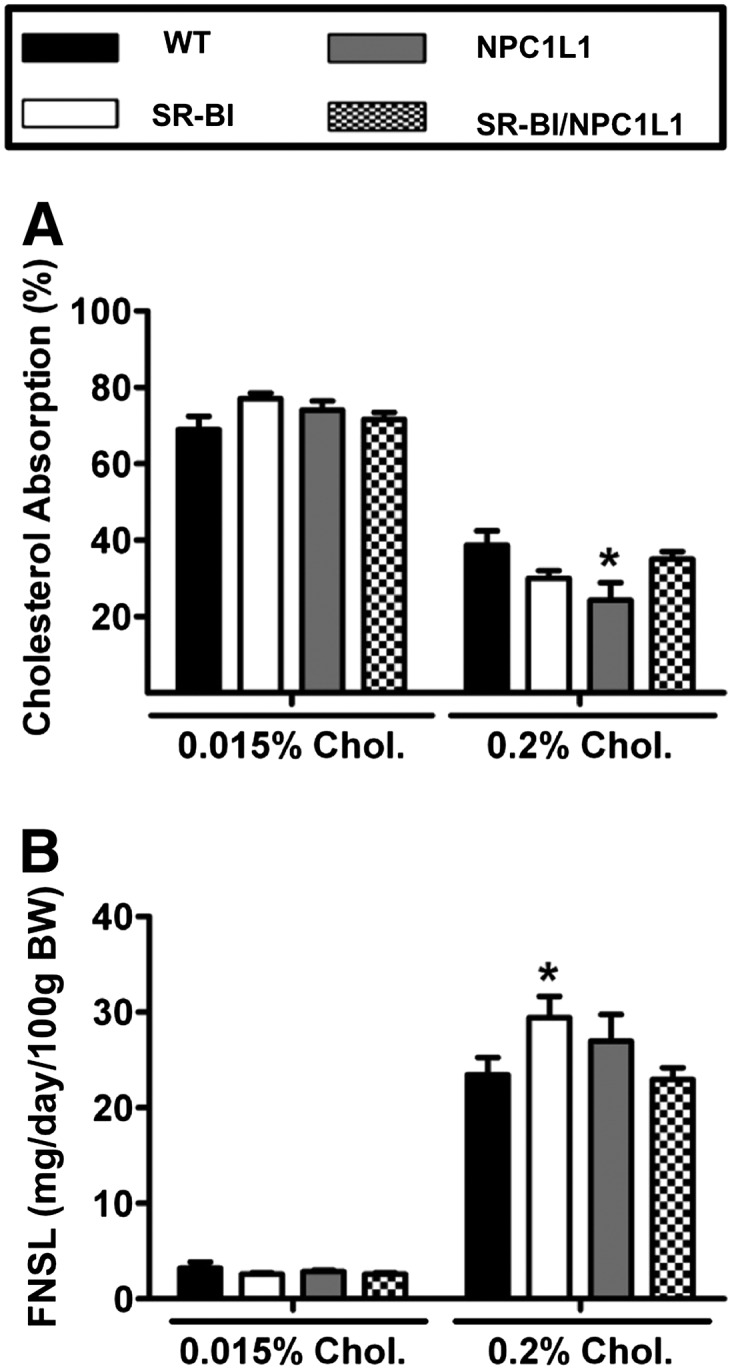
Intestinal overexpression of SR-BI does not augment TICE. WT, SR-BIhApoCIII-ApoAIV-Tg (SR-BI), liver NPC1L1 transgenics (NPC1L1), and SRBI/NPC1L1 double transgenic male mice were fed either 0.015% or 0.2% cholesterol (Chol.) (w/w) for 2 weeks. A: Fractional cholesterol absorption measured by the fecal dual-isotope method. B: Mass fecal neutral sterol loss (FNSL) determined by gas-liquid chromatography measured after 2 weeks on diet. Data shown represent the mean ± SEM from four to eight mice per group. *Significantly different from WT mice within each diet group (P < 0.05).
Hepatic lipid quantification
Liver lipid extracts were made and total cholesterol (TC), CE, FC, TG, or phospholipid mass was measured using detergent-solubilized enzymatic assays as previously described (13–15, 63).
Biliary lipid collection and analysis
Determination of lipid concentrations in gall bladder bile lipid was conducted as previously described (13–15, 61) in sedated mice that had been fasted for 4 h during the light cycle (9:00 AM–1:00 PM). Briefly, bile was collected by directly puncturing the gall bladder with a 30G1/2 needle. A measured volume (∼10 μl) of bile was placed into a glass tube and the neutral lipids were extracted and analyzed using enzymatic methods as described for liver lipids. Aliquots of the aqueous phase of the extraction were analyzed for bile acid content with an enzymatic assay using hydroxysteroid dehydrogenase (64).
Statistical analyses
All data were analyzed using either a one-way or a two-way ANOVA followed by Student's t-tests for post hoc analysis. Differences were considered significant at P < 0.05. All analyses were performed using JMP version 5.0.12 (SAS Institute, Cary, NC) software.
RESULTS
Intestinal overexpression of SR-BI decreases plasma cholesterol, but does not alter intestinal cholesterol absorption
To determine the relative protein abundance and subcellular localization of SR-BI we utilized immunoblotting and immunofluorescent techniques to detect endogenous and ectopically expressed SR-BI in the liver and small intestine of WT and SR-BI transgenic mice. As previously described SR-BIhApoCIII-ApoAIV-Tg mice have 2- to 3-fold more SR-BI in the liver, when compared with WT mice (Fig. 1A), likely due to the fact that the apo-CIII/apoA-IV enhancer region can drive modest hepatic expression. Interestingly, in the jejunum, we could barely detect endogenous SR-BI in WT mice (Fig. 1A), whereas SR-BIhApoCIII-ApoAIV-Tg mice had SR-BI protein levels similar to that found in the liver of WT mice (Fig. 1A). In agreement with Western blotting, immunofluorescence detection of endogenous SR-BI in the jejunum showed barely detectable signal, yet the SR-BIhApoCIII-ApoAIV-Tg mice had readily detectable levels of protein that localized to both the apical and basolateral membranes of enterocytes (Fig. 1B). In previous studies mice overexpressing SR-BI in the small intestine were shown to have a dramatic reduction in plasma cholesterol levels when maintained on rodent chow (59). We examined total plasma cholesterol and HDLc levels in mice fed either a standard rodent chow, or a semi-synthetic diet containing either 0.015% or 0.2% cholesterol (wt/wt). Under all dietary conditions the SR-BIhApoCIII-ApoAIV-Tg mice exhibited significantly reduced total plasma cholesterol and HDLc levels, when compared with WT littermates (Fig. 1C). Bietrix et al. (59) reported modest increases in cholesterol absorption by quantifying only the appearance of gavaged 14C-cholesterol into the plasma over a 4 h time course in SR-BIhApoCIII-ApoAIV-Tg mice. However, measuring the appearance of 14C-cholesterol in the plasma compartment using this experimental design reflects not only intestinal cholesterol absorption, but also plasma lipoprotein clearance rates and hepatic lipoprotein secretion. To more definitely measure cholesterol absorption without these other confounding factors, we utilized both the fecal dual-isotope method as well as quantifying mass fecal cholesterol loss. Importantly, on both the low and high cholesterol diets there was similar intestinal cholesterol absorption and fecal neutral sterol loss in WT and SR-BIhApoCIII-ApoAIV-Tg littermates (Fig. 1D).
Treatment with the NPC1L1 inhibitor EZE does not unmask a role for intestinal SR-BI in cholesterol absorption or fecal sterol loss
Given that fractional cholesterol absorption was high in our initial studies (Fig. 1D), we were concerned that relative contributions of the TICE pathway to fecal cholesterol loss may have been masked because most of the lumenal cholesterol was likely being recaptured by the intestinal cholesterol transporter NPC1L1. To overcome this concern we treated mice with EZE to block intestinal uptake of lumenal cholesterol by NPC1L1. EZE treatment for 2 days significantly reduced intestinal cholesterol absorption by ∼40% in both WT and SR-BIhApoCIII-ApoAIV-Tg mice (Fig. 2A). Note that in these experimental groups of mice fecal neutral sterol loss was not different between WT and SR-BIhApoCIII-ApoAIV-Tg mice before EZE treatment was initiated (Fig. 2B). Twenty-four hours after EZE treatment both WT and SR-BIhApoCIII-ApoAIV-Tg mice increased fecal neutral sterol loss by ∼5-fold (Fig. 2C). This EZE-induced increase in fecal neutral sterol loss was still apparent after 48 h of EZE treatment (Fig. 2D). Collectively, these studies suggest that regardless of whether intestinal cholesterol uptake was intact or blocked with EZE, intestinal SR-BI overexpression had no impact on the amount of cholesterol excreted in the feces.
Fig. 2.

Treatment with the NPC1L1 inhibitor EZE does not unmask a role for intestinal SR-BI in cholesterol absorption. Male WT and SR-BIhApoCIII-ApoAIV-Tg (SR-BI) littermates were fed a diet containing 0.015% cholesterol (w/w) for 4 weeks. After 4 weeks on diet, mice were orally gavaged daily for 3 consecutive days with either vehicle (VEH) or 0.3 mg EZE. A: Fractional cholesterol absorption measured by fecal dual-isotope method for feces collected during the first 48 h of the experiment. B, C: Mass fecal neutral sterol loss (FNSL) determined by gas-liquid chromatography measured after 4 weeks on diet prior to EZE treatment at baseline (B), 1 day (C), or 2 days (D) post-EZE treatment. Data represent the mean ± SEM from 6 to 10 mice per group, and values not sharing the same superscript differ significantly (P < 0.05).
Intestinal overexpression of SR-BI does not augment nonbiliary reverse cholesterol transport
The main goal of these studies was to determine the role of intestinal SR-BI in nonbiliary RCT or TICE. The rationale for studying SR-BI stems from the well-known ability of SR-BI to traffic cholesterol across hepatocytes in a unidirectional basolateral to apical manner (32–35). We hypothesized that intestinal SR-BI may likewise traffic plasma cholesterol across the enterocyte in a basolateral to apical manner thereby contributing to TICE. To study the role of intestinal SR-BI in TICE we crossed SR-BIhApoCIII-ApoAIV-Tg mice with the TICE-predominant NPC1L1LiverTg mouse model, which has been previously described as having dramatically reduced biliary cholesterol secretion (13, 14). This cross resulted in four experimental genotypes for phenotypic comparisons: WT, SR-BIhApoCIII-ApoAIV-Tg, NPC1L1LiverTg, and SR-BIhApoCIII-ApoAIV-Tg + NPC1L1LiverTg double transgenic mice (SRBI/NPC1L1). SR-BI mRNA expression in the proximal small intestine was confirmed to be increased 10- to 14-fold in SR-BIhApoCIII-ApoAIV-Tg single transgenics and double transgenics (SR-BIhApoCIII-ApoAIV-Tg + NPC1L1LiverTg), when compared with WT littermates fed 0.2% dietary cholesterol (data not shown). As expected, regardless of dietary cholesterol exposure, NPC1L1LiverTg and SRBI/NPC1L1 double transgenic mice had severely reduced biliary cholesterol levels, when compared with either WT or SR-BIhApoCIII-ApoAIV-Tg mice (Fig. 3E). Likely due to the 2- to 3-fold overexpression of SR-BI in the liver, SR-BIhApoCIII-ApoAIV-Tg mice exhibited a small increase in biliary cholesterol levels when fed a 0.2% cholesterol diet (Fig. 3E). As previously reported, NPC1L1LiverTg mice had a specific reduction in biliary cholesterol (Fig. 3E), but normal levels of biliary bile acid (Fig. 3F) and biliary phospholipid secretion (Fig. 3G). Despite alterations in biliary cholesterol levels, NPC1L1LiverTg mice did not accumulate cholesterol or triglycerides in the liver (Fig. 3A–C). Interestingly, SR-BIhApoCIII-ApoAIV-Tg mice had modest reductions in hepatic FC and CE, compared with WT mice (Fig. 3B, D), but this was only apparent in mice fed 0.2% cholesterol. Collectively, hepatic and biliary lipid levels in this experiment mimic what was anticipated for dietary cholesterol feeding in these mouse models (13, 14, 59). More importantly, when we measured fractional cholesterol absorption in the four genotypes of mice, only the NPC1L1LiverTg mice fed 0.2% dietary cholesterol were significantly different than the WT controls on the same diet (Fig. 4A). As seen in the previous two experiments (Figs. 1D, 2A), fractional cholesterol absorption was unaltered in SR-BIhApoCIII-ApoAIV-Tg mice regardless of NPC1L1 genotype (Fig. 4A). In agreement with this, fecal neutral sterol loss was also unaltered across the four genotypes (Fig. 4B). Only in the 0.2% dietary cholesterol group did we see a modest increase in fecal neutral sterol loss in the SR-BIhApoCIII-ApoAIV-Tg mice, compared with all other groups (Fig. 4B). Collectively, these studies demonstrate under several experimental conditions that intestinal overexpression of SR-BI does not impact either cholesterol absorption or TICE in mice.
Fig. 3.

Hepatic and biliary cholesterol levels in intestinal SR-BI and hepatic NPC1L1 double transgenic mice. WT, SR-BIhApoCIII-ApoAIV-Tg (SR-BI), liver NPC1L1 transgenics (NPC1L1), and SRBI/NPC1L1 double transgenic male mice were fed either 0.015% or 0.2% cholesterol (Chol.) (w/w) for 2 weeks. Liver samples were extracted and enzymatically analyzed to quantify the mass of (A) TC, (B) CEs, (C) TGs, or (D) FC. All hepatic lipid values were normalized to total tissue protein content. Panels (E–G) represent raw concentrations and molar ratios of gallbladder cholesterol (E), bile acids (F), and phospholipids (PL) (G). Data shown represent the mean ± SEM from four to eight mice per group. *Significantly different from WT mice within each diet group (P < 0.05).
Global genetic deficiency of SR-BI does not alter fecal sterol loss or cholesterol absorption
To further characterize the role of SR-BI in intestinal cholesterol flux, we examined mass fecal neutral and acidic sterol loss and fractional cholesterol absorption in chow-fed SR-BI knockout mice. As has been previously suggested (57), chow-fed SR-BI knockout mice have normal levels of both neutral and acidic sterols in feces (Fig. 5A, B). In agreement, SR-BI knockout mice have unaltered fractional cholesterol absorption (Fig. 5C). Collectively, these results do not support the notion that intestinal SR-BI is required for intestinal cholesterol absorption or movement of sterols into the feces.
Fig. 5.

Genetic deficiency of SR-BI does not alter fecal sterol loss or intestinal cholesterol absorption. Male WT and SR-BI knockout (SR-BI−/−) mice littermates were maintained on a standard chow diet. A: Fecal neutral sterol loss (FNSL). B: Fecal acidic sterol loss (FASL). C: Fractional cholesterol absorption measured by the fecal dual-isotope method. Data represent the mean ± SEM from six mice per group. No significant differences were detected.
DISCUSSION
Although several groups have hypothesized a role for SR-BI in intestinal cholesterol absorption (36, 55, 56, 59, 65), studies where cholesterol absorption was actually quantified in mouse models of SR-BI deficiency (57) and now transgenic overexpression (Figs. 1, 2, 4) have not supported such a role. Instead there is much stronger evidence that SR-BI does not play a role in intestinal cholesterol absorption. The major findings of our present studies are the following: 1) endogenous SR-BI protein expression in the small intestine is barely detectable when compared with very abundant levels in the liver (Fig. 1A, B); 2) endogenous and ectopically expressed SR-BI localizes to both apical and basolateral membranes in enterocytes (Fig. 1B); 3) SR-BIhApoCIII-ApoAIV-Tg mice have reduced total plasma cholesterol and HDLc levels compared with WT littermates (59) (Fig. 1C); 4) intestinal overexpression of SR-BI does not alter intestinal cholesterol absorption either in the absence or presence of the cholesterol absorption inhibitor EZE (Fig. 2); and 5) intestinal overexpression of SR-BI does not increase TICE in mice with substantially reduced contribution of the biliary pathway for RCT (NPC1L1LiverTg) (Fig. 4). Collectively, these findings suggest that intestinal SR-BI is not rate limiting for the trafficking of cholesterol across the enterocyte either in an apical to basolateral fashion, or through the basolateral to apical TICE pathway in mice.
Several laboratories have previously proposed a role for SR-BI as a high-affinity receptor for cholesterol at the brush border membrane (55, 56), as well as a mechanism by which cholesterol is trafficked from the intestinal lumen across the enterocyte in an apical to basolateral pattern for packaging into chylomicrons (36, 55, 56, 59, 65). Further, intestinal SR-BI has been linked to overproduction of chylomicrons in insulin-resistant states (65). However, the majority of cell-based work examining the subcellular trafficking pattern of SR-BI has agreed that in polarized cells SR-BI sorts HDL-derived cholesterol from basolateral membranes to apical membranes (32–35), and the receptor itself has shown a similar trafficking pattern (32–35). In agreement with this directional trafficking pattern in polarized cells, hepatic overexpression or knockout of SR-BI either promotes or diminishes the movement of HDL-derived cholesterol into bile, respectively (37, 42–44, 49). In fact all experimental evidence in the liver supports a role for SR-BI in moving HDL-derived cholesterol into bile (32–49). Based on both the in vitro and in vivo evidence supporting a role for SR-BI in unidirectional transcytosis of cholesterol (32–35), and the lack of evidence that SR-BI promotes intestinal cholesterol absorption (57, 58) (Figs. 1–5), we hypothesized that intestinal SR-BI may play a rate-limiting role in TICE to move plasma cholesterol across the enterocyte for efflux into feces.
Although it has been well documented that SR-BI knockout mice do not have defects in intestinal cholesterol absorption (57), a previous study in SR-BIhApoCIII-ApoAIV-Tg mice indirectly demonstrated a potential role for intestinal SR-BI in cholesterol absorption (59). In this study Bietrix et al. (59) measured intestinal cholesterol absorption by gavaging WT and SR-BIhApoCIII-ApoAIV-Tg mice with 14C-cholesterol and followed the appearance of this tracer into the plasma compartment over a 4 h period. Unfortunately, examining plasma appearance of a gavaged tracer reports on not only intestinal cholesterol absorption, but also plasma lipoprotein clearance and hepatic repackaging of intestinally derived cholesterol during the experimental period. Using the standardized fecal dual-isotope assay to measure cholesterol absorption in these same mice, we were able to show in three separate experiments that SR-BIhApoCIII-ApoAIV-Tg mice do not have increased intestinal cholesterol absorption (Figs. 1, 2, 4). In agreement with this, mass fecal loss of cholesterol is normal in SR-BIhApoCIII-ApoAIV-Tg mice (Figs. 1, 2, 4). Importantly, both cholesterol absorption and fecal neutral sterol loss were normal in SR-BIhApoCIII-ApoAIV-Tg mice fed different amounts of dietary cholesterol as well as in mice treated with the intestinal cholesterol absorption inhibitor EZE (Figs. 1, 2).
Alternatively, we hypothesized that intestinal SR-BI may play a key role in removing plasma cholesterol for delivery to the TICE pathway. To more definitely address a potential role for intestinal SR-BI in promoting TICE, we crossed SR-BIhApoCIII-ApoAIV-Tg mice to mice with genetically reduced biliary contributions to RCT (NPC1L1LiverTg). However, we found no role for intestinal SR-BI in promoting TICE. This is in agreement with previous reports using an intestinal perfusion system that demonstrated that small intestine segments from SR-BI deficient mice exhibited functional TICE (20). Furthermore, it was recently demonstrated that TICE does not depend on HDL-dependent delivery of plasma cholesterol to the intestine (12). In agreement with this, several studies have demonstrated that selective uptake of HDL-derived cholesterol is rather low in mouse small intestine, when compared with liver and adrenal, and intestinal selective uptake does not mirror alterations in fecal neutral sterol loss under different experimental conditions (66, 67). Collectively, these data indicate that other basolateral cholesterol transporters must be involved in clearance of plasma cholesterol for the TICE pathway. The intestinal low density lipoprotein receptor (LDLr) is an obvious candidate, given that the small intestine has the second most LDLr-mediated lipoprotein clearance behind the liver (68). However, we have previously shown functional TICE in LDLr-deficient mice (15). Furthermore, it has recently been shown that intestinal LDLr is proteolytically degraded in mice treated with a liver X receptor agonist (69), which is a drug class that substantially augments TICE (14, 16). Collectively, these studies do not support a rate-limiting role for the LDLr in the small intestine for mediating TICE. Given that the canonical HDL and LDL receptors (SR-BI and LDLr) are not essential for TICE, it will be critical to determine the role of other LDLr-related proteins as well as novel candidate receptors in the TICE pathway.
Although it seems quite clear that intestinal SR-BI is not involved in cholesterol absorption (57, 58) or TICE (Figs. 3, 4), it now becomes important to gain a better understanding of the role SR-BI does play in the small intestine. There is strong support for SR-BI binding cholesterol in isolated BBMVs (55, 56). However, binding of cholesterol to BBMV is not predictive of intestinal cholesterol absorption rates in mice (70). These studies strongly suggest that ex vivo binding of cholesterol to BBMV does not accurately predict the true rate of intestinal cholesterol absorption, and results using the BBMV system need to be validated using standard in vivo assays for cholesterol absorption such as the fecal dual-isotope assay or lymph duct cannulations. An alternative role for SR-BI in the small intestine was recently postulated by Béaslas et al. (71). In this study it was demonstrated that SR-BI could activate intracellular signaling pathways in response to extracellular lipid stimuli (71). These results are not inconsistent with a similar signaling role for SR-BI in endothelial nitric oxide synthase activation in endothelial cells (72). Most recently, SR-BI was shown to be a plasma membrane cholesterol sensor that governs intracellular signaling processes in both enterocytes and endothelial cells (73). Further studies are needed to definitively address such a primary signaling role for SR-BI in the enterocyte in vivo. In summary, intestinal SR-BI is not involved in cholesterol absorption or fecal neutral sterol loss through the TICE pathway in mice. Therefore, additional studies are required to identify intestinal lipoprotein receptors involved in the delivery of plasma cholesterol to the intestine for the TICE pathway, as well as to define the true physiological function of SR-BI in the enterocyte.
Acknowledgments
The authors thank Paul Dawson (Wake Forest School of Medicine) for critical insights into this work.
Footnotes
Abbreviations:
- BBMV
- brush border membrane vesicle
- CE
- cholesteryl ester
- CVD
- cardiovascular disease
- DAPI
- 4',6-diamidino-2-phenylindole
- EZE
- ezetimibe
- FC
- free cholesterol
- HDLc
- high density lipoprotein cholesterol
- LDLc
- low density lipoprotein cholesterol
- LDLr
- low density lipoprotein receptor
- NPC1L1
- Niemann-Pick C1-like 1
- NPC1L1LiverTg
- Niemann-Pick C1-like 1iver transgenic
- RCT
- reverse cholesterol transport
- SR-BI
- scavenger receptor class B type I
- TC
- total cholesterol
- TICE
- transintestinal cholesterol efflux
- TG
- triglyceride
- WT
- wild-type
This work was supported by the National Heart, Lung, and Blood Institute through a Pathway to Independence Grant (K99/R00-HL-096166 to J.M.B.) and a Program Project Grant (5P01HL-049373 to L.L.R.), and a grant from the Netherlands Organization for Scientific Research (VIDI Grant 917-56-358 to U.J.F.T.).
REFERENCES
- 1.Roger V. L., Go A. S., Lloyd-Jones D. M., Benjamin E. J., Berry J. D., Borden W. B., Bravata D. M., Dai S., Ford E. S., Fox C. S., et al. 2012. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 125: e1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). 2002. Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 106: 3143–3421. [PubMed] [Google Scholar]
- 3.Gordon T., Castelli W. P., Hjortland M. C., Kannel W. B., Dawber T. R. 1977. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62: 707–714 [DOI] [PubMed] [Google Scholar]
- 4.Rosenson R. S., Brewer H. B., Jr, Davidson W. S., Fayad Z. A., Fuster V., Goldstein J., Hellerstein M., Jiang X. C., Phillips M. C., Rader D. J., et al. 2012. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 125: 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barter P. J., Nicholls S., Rye K. A., Anantharamaiah G. M., Navab M., Fogelman A. M. 2004. Antiinflammatory properties of HDL. Circ. Res. 95: 764–772 [DOI] [PubMed] [Google Scholar]
- 6.Yvan-Charvet L., Wang N., Tall A. R. 2010. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 30: 139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietschy J. M., Turley S. D. 2002. Control of cholesterol turnover in the mouse. J. Biol. Chem. 277: 3801–3804 [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Rader D. J. 2007. Molecular regulation of macrophage reverse cholesterol transport. Curr. Opin. Cardiol. 22: 368–372 [DOI] [PubMed] [Google Scholar]
- 9.Jolley C. D., Woollett L. A., Turley S. D., Dietschy J. M. 1998. Centripetal cholesterol flux to the liver is dictated by events in the peripheral organs and not by the plasma high density lipoprotein or apolipoprotein A-I concentration. J. Lipid Res. 39: 2143–2149 [PubMed] [Google Scholar]
- 10.Groen A. K., Bloks V. W., Bandsma R. H., Ottenhoff R., Chimini G., Kuipers F. 2001. Hepatobiliary cholesterol transport is not impaired in Abca1-null mice lacking HDL. J. Clin. Invest. 108: 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie C., Turley S. D., Dietschy J. M. 2009. ABCA1 plays no role in the centripetal movement of cholesterol from peripheral tissues to the liver and intestine in the mouse. J. Lipid Res. 50: 1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vrins C. L., Ottenhoff R., van den Oever K., de Waart D. R., Kruyt J. K., Zhao Y., Van Berkel T. J., Havekes L. M., Aerts J. M., Van Eck M., et al. 2012. Trans-intestinal cholesterol efflux is not mediated through high density lipoprotein. J. Lipid Res. 53: 2017–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Temel R. E., Tang W., Ma Y., Rudel L. L., Willingham M. C., Ioannou Y. A., Davies J. P., Nilsson L. M., Yu L. 2007. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentrations and is a target of ezetimibe. J. Clin. Invest. 117: 1968–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temel R. E., Sawyer J. K., Yu L., Lord C., Degirolamo C., McDaniel A., Marshall S., Wang N., Shah R., Rudel L. L., et al. 2010. Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab. 12: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown J. M., Bell T. A., III, Alger H. M., Sawyer J. K., Smith T. L., Kelley K., Shah R., Wilson M. D., Davis M. A., Lee R. G., et al. 2008. Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss. J. Biol. Chem. 283: 10522–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruit J. K., Plosch T., Havinga R., Boverhof R., Groot P. H., Groen A. K., Kuipers F. 2005. Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology. 128: 147–156 [DOI] [PubMed] [Google Scholar]
- 17.Mauad T. H., van Nieuwkerk C. M., Dingemans K. P., Smit J. J., Schinkel A. H., Notenboom R. G., van den Bergh Weerman M. A., Verkruisen R. P., Groen A. K., Oude Elferink R. P. 1994. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 145: 1237–1245 [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L., Hammer R. E., Li-Hawkins J., Von Bergmann K., Lutjohann D., Cohen J. C., Hobbs H. H. 2002. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA. 99: 16237–16242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Velde A. E., Vrins C. L., van den Oever K., Kunne C., Oude Elferink R. P., Kuipers F., Groen A. K. 2007. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology. 133: 967–975 [DOI] [PubMed] [Google Scholar]
- 20.van der Velde A. E., Vrins C. L., van den Oever K., Seeman I., Oude Elferink R. P., van Eck M., Kuipers F., Groen A. K. 2008. Regulation of direct transintestinal cholesterol excretion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 295: G203–G208 [DOI] [PubMed] [Google Scholar]
- 21.van der Veen J. N., van Dijk T. H., Vrins C. L., van Meer H., Havinga R., Bijsterveld K., Tietge U. J., Groen A. K., Kuipers K. 2009. Activation of the liver X receptor stimulates trans-intestinal excretion of plasma cholesterol. J. Biol. Chem. 284: 19211–19219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vrins C. L., van der Velde A. E., van den Oever K., Levels J. H., Huet S., Oude Elferink R. P., Kuipers F., Groen A. K. 2009. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 50: 2046–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Temel R. E., Brown J. M. 2012. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr. Opin. Lipidol. 23: 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brufau G., Groen A. K., Kuipers F. 2011. Reverse cholesterol transport revisited: contribution of biliary versus intestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 31: 1726–1733 [DOI] [PubMed] [Google Scholar]
- 25.Cooper A. D. 1991. Metabolic basis of cholesterol gallstone disease. Gastroenterol. Clin. North Am. 20: 21–46 [PubMed] [Google Scholar]
- 26.Hayes K. C., Livingston A., Trautwein E. A. 1992. Dietary impact on biliary lipids and gallstones. Annu. Rev. Nutr. 12: 299–326 [DOI] [PubMed] [Google Scholar]
- 27.Groen A., Kunne C., Jongsma G., van den Oever K., Mok K. S., Petruzzelli M., Vrins C., Bull L., Paulusma C. C., Oude Elferink R. P. 2008. Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology. 134: 2091–2100 [DOI] [PubMed] [Google Scholar]
- 28.Annema W., Tietge U. J. 2012. Regulation of reverse cholesterol transport – a comprehensive appraisal of available animal studies. Nutr. Metab. (Lond). 9: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calvo D., Vega M. A. 1993. Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem. 268: 18929–18935 [PubMed] [Google Scholar]
- 30.Acton S. L., Scherer P. E., Lodish H. F., Krieger M. 1994. Expression cloning of SR-BI, a CD36 related class B scavenger receptor. J. Biol. Chem. 269: 21003–21009 [PubMed] [Google Scholar]
- 31.Landschulz K. T., Pathak R. K., Rigotti A., Krieger M., Hobbs H. H. 1996. Regulation of scavenger receptor, class B type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J. Clin. Invest. 98: 984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silver D. L., Wang N., Xiao X., Tall A. R. 2001. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type I results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J. Biol. Chem. 276: 25287–25293 [DOI] [PubMed] [Google Scholar]
- 33.Burgos P. V., Klattenhoff C., de la Fuente E., Rigotti A., Gonzalez A. 2004. Cholesterol depletion induces PKA-mediated basolateral-to-apical transcytosis of the scavenger receptor class B type I in MDCK cells. Proc. Natl. Acad. Sci. USA. 101: 3845–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harder C. J., Meng A., Rippstein P., McBride H. M., McPherson R. 2007. SR-BI undergoes cholesterol-stimulated transcytosis to the bile canaliculus in polarized WIF-B cells. J. Biol. Chem. 282: 1445–1455 [DOI] [PubMed] [Google Scholar]
- 35.Wüstner D., Mondal M., Huang A., Maxfield F. R. 2004. Different transport routes for high density lipoprotein and its associated free sterol in polarized hepatic cells. J. Lipid Res. 45: 427–437 [DOI] [PubMed] [Google Scholar]
- 36.Cai S. F., Kirby R. J., Howles P. N., Hui D. Y. 2001. Differentiation-dependent expression and localization of the class B type I scavenger receptor in the intestine. J. Lipid Res. 42: 902–909 [PubMed] [Google Scholar]
- 37.Sehayek E., Wang R., Ono J. G., Zinchuk V. S., Duncan E. M., Shefer S., Vance D. E., Ananthanarayanan M., Chait B. T., Breslow J. L. 2003. Localization of the PE methylation pathway and SR-BI to the canalicular membrane: evidence for apical PC biosynthesis that may promote biliary excretion of phospholipids and cholesterol. J. Lipid Res. 44: 1605–1613 [DOI] [PubMed] [Google Scholar]
- 38.Rigotti A., Trigatti B., Babitt J. 1997. Scavenger receptor BI: a cell surface receptor for high density lipoprotein. Curr. Opin. Lipidol. 8: 181–188 [DOI] [PubMed] [Google Scholar]
- 39.Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., Krieger M. 1996. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 271: 518–520 [DOI] [PubMed] [Google Scholar]
- 40.Liu B., Krieger M. 2002. Highly purified scavenger receptor class B, type I reconstituted into phosphatidylcholine/cholesterol liposomes mediates high affinity high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 277: 34125–34135 [DOI] [PubMed] [Google Scholar]
- 41.Kozarsky K. F., Donahee M. H., Rigotti A., Iqbal S. N., Edelman E. R., Krieger M. 1997. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 387: 414–417 [DOI] [PubMed] [Google Scholar]
- 42.Ji Y., Wang N., Ramakrishnan R., Sehayek E., Huszar D., Breslow J. L., Tall A. R. 1999. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J. Biol. Chem. 274: 33398–33402 [DOI] [PubMed] [Google Scholar]
- 43.Wang N., Arai T., Ji Y., Rinninger F., Tall A. R. 1998. Liver-specific overexpression of scavenger receptor BI decreases levels of very low density lipoprotein ApoB, low density lipoprotein ApoB, and high density lipoprotein in transgenic mice. J. Biol. Chem. 273: 32920–32926 [DOI] [PubMed] [Google Scholar]
- 44.Ueda Y., Royer L., Gong E., Zhang J., Cooper P. N., Francone O., Rubin E. M. 1999. Lower plasma levels and accelerated clearance of high density lipoprotein (HDL) and non-HDL cholesterol in scavenger receptor class B type I transgenic mice. J. Biol. Chem. 274: 7165–7171 [DOI] [PubMed] [Google Scholar]
- 45.Wiersma H., Gatti A., Nijstad N., Oude Elferink R. P., Kuipers F., Tietge U. J. 2009. Scavenger receptor class B type I mediates biliary cholesterol secretion independent of ATP-binding cassette transporter g5/g8 in mice. Hepatology. 50: 1263–1272 [DOI] [PubMed] [Google Scholar]
- 46.Ueda Y., Gong E., Royer L., Cooper P. N., Francone O., Rubin E. M. 2000. Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J. Biol. Chem. 275: 20368–20373 [DOI] [PubMed] [Google Scholar]
- 47.Arai T., Wang N., Bezouevski M., Welch C., Tall A. R. 1999. Decreased atherosclerosis in heterozygous low-density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J. Biol. Chem. 274: 2366–2371 [DOI] [PubMed] [Google Scholar]
- 48.Kozarsky K. F., Donahee M. H., Glick J. M., Krieger M., Rader D. J. 2000. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 20: 721–727 [DOI] [PubMed] [Google Scholar]
- 49.Rigotti A., Trigatti B. L., Penman M., Rayburn H., Herz J., Krieger M. 1997. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. USA. 94: 12610–12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varban M. L., Rinninger F., Wang N., Fairchild-Huntress V., Dunmore J. H., Fang Q., Gosselin M. L., Dixon K. L., Deeds J. D., Acton S. L., et al. 1998. Targeted mutation reveals a central role for SR-BI in hepatic selective uptake of high density lipoprotein cholesterol. Proc. Natl. Acad. Sci. USA. 95: 4619–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trigatti B., Rayburn H., Vinals M., Braun A., Miettinen H., Penman M., Hertz M., Schrenzel M., Amigo L., Rigotti A., et al. 1999. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc. Natl. Acad. Sci. USA. 96: 9322–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braun A., Trigatti B. L., Post M. J., Sato K., Simons M., Edelberg J. M., Rosenberg R. D., Schrenzel M., Krieger M. 2002. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 90: 270–276 [DOI] [PubMed] [Google Scholar]
- 53.Zhang S., Picard M. H., Vasile E., Zhu Y., Raffai R. L., Weisgraber K. H., Krieger M. 2005. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation. 111: 3457–3464 [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y., Da Silva J. R., Reilly M., Billheimer J. T., Rothblat G. H., Rader D. J. 2005. Hepatic overexpression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 115: 2870–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hauser H., Dyer J. H., Nandy A., Vega A. A., Werder M., Bieliauskaite E., Weber F. E., Compassi S., Gemperli A., Boffelli D., et al. 1998. Indentification of a receptor mediating absorption of dietary cholesterol in the intestine. Biochemistry. 37: 17843–17850 [DOI] [PubMed] [Google Scholar]
- 56.Labonté E. D., Howles P. N., Granholm N. A., Rojas J. C., Davies J. P., Ioannou Y. A., Hui D. Y. 2007. Class B type I scavenger receptor is responsible for the high affinity cholesterol binding activity of intestinal brush border membrane vesicles. Biochim. Biophys. Acta. 1771: 1132–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mardones P., Quinones V., Amigo L., Moreno M., Miquel J. F., Schwarz M., Miettinen H. E., Trigatti B., Krieger M., VanPatten S., et al. 2001. Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I-deficient mice. J. Lipid Res. 42: 170–180 [PubMed] [Google Scholar]
- 58.Altmann S. W., Davis H. R., Jr, Yao X., Laverty M., Compton D. S., Zhu L. J., Crona J. H., Caplen M. A., Hoos L. M., Tetzloff G., et al. 2002. The identification of intestinal scavenger receptor class B, type I (SR-BI) by expression cloning and its role in cholesterol absorption. Biochim. Biophys. Acta. 1580: 77–93 [DOI] [PubMed] [Google Scholar]
- 59.Bietrix F., Yan D., Nauze M., Rolland C., Bertrand-Michel J., Comera C., Schaak S., Barbaras R., Groen A. K., Perret B., et al. 2006. Accelerated lipid absorption in mice overexpressing intestinal SR-BI. J. Biol. Chem. 281: 7214–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown J. M., Chung S., Sawyer J. K., Degirolamo C., Alger H. M., Nguyen T., Zhu X., Duong M., Wibley A. L., Shah R., et al. 2008. Inhibition of stearoyl-coenzyme A desaturase 1 dissociates insulin resistance and obesity from atherosclerosis. Circulation. 118: 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Temel R. E., Lee R. G., Kelley K. L., Davis M. A., Shah R., Sawyer J. K., Wilson M. D., Rudel L. L. 2005. Intestinal cholesterol absorption is substantially reduced in mice deficient in both ABCA1 and ACAT2. J. Lipid Res. 46: 2423–2431 [DOI] [PubMed] [Google Scholar]
- 62.Wiersma H., Gatti A., Nijstad N., Kuipers F., Tietge U. J. 2009. Hepatic SR-BI, not endothelial lipase, expression determines biliary cholesterol secretion in mice. J. Lipid Res. 50: 1571–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carr T. P., Andresen C. J., Rudel L. L. 1993. Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extract. Clin. Biochem. 26: 39–42 [DOI] [PubMed] [Google Scholar]
- 64.Turley S. D., Dietschy J. M. 1978. Re-evaluation of the 3 alpha-hydroxysteroid dehydrogenase assay for total bile acids in bile. J. Lipid Res. 19: 924–928 [PubMed] [Google Scholar]
- 65.Hayashi A. A., Webb J., Choi J., Baker C., Lino M., Trigatti B., Trajcevski K. E., Hawke T. J., Adeli K. 2011. Intestinal SR-BI is upregulated in insulin-resistant states and is associated with overproduction of intestinal apoB48-containing lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 301: G326–G337 [DOI] [PubMed] [Google Scholar]
- 66.Chajek-Shaul T., Hayek T., Walsh A., Breslow J. L. 1991. Expression of the human apolipoprotein A-I gene in transgenic mice alters high density lipoprotein (HDL) particle size distribution and diminishes selective uptake of HDL cholesteryl esters. Proc. Natl. Acad. Sci. USA. 88: 6731–6735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nijstad N., Gautier T., Briand F., Rader D. J., Tietge U. J. 2011. Biliary sterol secretion is required for functional in vivo reverse cholesterol transport in mice. Gastroenterology. 140: 1043–1051 [DOI] [PubMed] [Google Scholar]
- 68.Osono Y., Woollett L. A., Herz J., Dietschy J. M. 1995. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J. Clin. Invest. 95: 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zelcer N., Hong C., Boyadjian R., Tontonoz P. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 325: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen D. V., Drover V. A., Knopfel M., Dhanasekaran P., Hauser H., Phillips M. C. 2009. Influence of class B scavenger receptors on cholesterol flux across brush border membrane and intestinal absorption. J. Lipid Res. 50: 2235–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Béaslas O., Cueille C., Delers F., Chateau D., Chambaz J., Rousset M., Carrière V. 2009. Sensing of dietary lipids by enterocytes: a new role for SR-BI/CLA-1. PLoS ONE. 4: e4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuhanna I. S., Zhu Y., Cox B. E., Hahner L. D., Osborne-Lawrence S., Lu P., Marcel Y. L., Anderson R. G., Medelsohn M. E., Hobbs H. H., et al. 2001. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 7: 853–857 [DOI] [PubMed] [Google Scholar]
- 73.Saddar S., Carriere V., Lee W. R., Tanigaki K., Yuhanna I. S., Parathath S., Morel E., Warrier M., Sawyer J. K., Gerard R.D., et al. 2013. Scavenger receptor class B type I is a plasma membrane cholesterol sensor. Circ. Res. 112: 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]